Introduction
Breast cancer (BC) is the most common malignancy
among women, with increasing incidence and cancer-associated
mortality rates between 2003 and 2013 in China (1). Clinical treatment options for BC, such
as surgery, radiotherapy and chemotherapy, are commonly used in
clinical practice (2). As a member
of the anthracycline antibiotics, Adriamycin (ADM), or doxorubicin,
exerts strong antitumor effects by targeting the DNA of cancer
cells, and has been widely used to treat numerous types of cancer,
including BC (3–5). However, the majority of women diagnosed
with advanced BC develop resistance to ADM, which limits its
further application and leads to long-term chemotherapeutic
resistance (6). Therefore, there is
an urgent need to find novel strategies to overcome drug
resistance, which will lead to better treatment outcomes for
patients.
Drug resistance can be intrinsic or acquired during
or after chemotherapy. Resistance occurs when the tumors are able
to cope with drug-induced damage; this may be through enhancing
anti-apoptotic signaling, or due to mutations in DNA damage
response pathways (7). It has been
reported that the increased apoptotic resistance of BC cells to ADM
is associated with enhanced pro-survival Bcl-2 family proteins
Bcl-2, Bcl-xL and Mcl-1 (8).
Furthermore, wild-type p53-induced phosphatase 1 (Wip1), encoded by
the PPM1D gene, has been identified as an oncoprotein overexpressed
in multiple types of human cancer (9). Recent evidence suggests that Wip1 is a
critical inhibitor in the
ataxia-telangiectasia-mutated/ataxia-telangiectasia-Rad3-related
p53 DNA damage signaling pathway (9). Because of these roles in controlling
DNA damage response and repair, the above proteins may be potential
targets for BC therapy.
MicroRNAs (miRNAs/miRs) are defined as small
non-coding regulatory RNA molecules of 18–25 nucleotides in length;
they have a profound impact on diverse biological processes,
including development, differentiation, growth and metabolism
(10,11). miRNAs serve a role in negatively
regulating gene expression at the post-transcriptional level via
binding to complementary sequences in the coding 3′untranslated
region (3′UTR) of their target mRNAs (12–14). A
single miRNA may suppress several different transcripts, pathways
and responses by regulating protein expression, and several miRNAs
may also control a single pathway (15). Calin et al (16) reported the first experimental
evidence of miR-16 involvement in mammalian oncogenesis in B-cell
chronic lymphocytic leukemia (CLL). Further studies have
demonstrated that miR-16 expression is eradicated or attenuated in
solid tumors, including BC, suggesting that miR-16 is a significant
factor in tumorigenesis (17). In
addition, DNA repair factors and oncogenes may be regulated by
miRNAs. It has been demonstrated that miR-16, as a pivotal tumor
suppressor, participates in the induction of apoptosis by targeting
Bcl-2 (18–20). Furthermore, the expression of Wip1 is
suppressed by miR-16 in the DNA damage signaling pathway (9).
Therefore, the present study aimed to investigate
whether miR-16 expression was associated with ADM resistance in BC
by comparing drug-resistant BC tumor tissues and a resistant cell
line (MCF-7/A) with ADM-sensitive tumor tissues and drug-sensitive
cell line (MCF-7/S). The expression of miR-16 was modulated to
examine its influence on Bcl-2 and Wip1 expression, and
ADM-mediated apoptosis. The potential of miR-16 targeting to be
utilized as a therapeutic approach for sensitizing drug-resistant
BC to chemotherapy was explored.
Materials and methods
Tumor sample collection
Tumor samples were collected from female patients
with BC (age range, 20–65) undergoing breast biopsy or breast mass
resection at Lianyungang Clinical College of Nanjing Medical
University (Lianyungang, China) between March 2016 and March 2018.
The study protocol was approved by the Ethics Committee of Nanjing
Medical University, and all patients involved provided written
informed consent. Resistance to ADM was defined as no response
(tumor size unchanged or increased) to treatment, or relapse within
six months of discontinuing treatment as adjuvant therapy (21). In total, 40 cases of BC (20 with ADM
resistance and 20 without) were included in the study. Patient
clinical data, including pathological grades and stages are
presented in Table I. Tumor samples
were collected before chemotherapy and divided into resistant and
sensitive tumor groups according to the response to chemotherapy.
The samples were stored in liquid nitrogen. Cancer staging was
determined as stage 1 to 4 according to the Union for International
Cancer Control TNM classification system (22). Pathological grading (G1-G3) was
performed according to the modified Scarff-Bloom-Richardson grading
system (23). HER2 positive/negative
status was determined according to the American Society of Clinical
Oncology/College of American Pathologists guidelines for HER2
testing in breast cancer (24).
 | Table I.Clinicopathological characteristics
of patients with breast cancer. |
Table I.
Clinicopathological characteristics
of patients with breast cancer.
|
| Cases, n |
|
|---|
|
|
|
|
|---|
| Variable | Adriamycin
resistance (n=20) | Adriamycin
sensitivity (n=20) | Overall (n=40) |
|---|
| Age, years |
|
<50 | 8 | 7 | 15 |
|
≥50 | 12 | 13 | 25 |
| Tumor size, cm |
|
<2 | 9 | 14 | 23 |
| ≥2 | 11 | 6 | 17 |
| Lymph node
infiltrated |
| No | 11 | 10 | 21 |
|
Yes | 9 | 10 | 19 |
| TNM
stagea |
| Stage
I/II | 3 | 15 | 18 |
| Stage
III/IV | 17 | 5 | 22 |
| Pathological
gradeb |
| G1 | 5 | 7 | 12 |
| G2 | 8 | 9 | 17 |
| G3 | 7 | 4 | 11 |
| ER
statusc |
|
Negative | 13 | 9 | 22 |
|
Positive | 7 | 11 | 18 |
| PR
statusc |
|
Negative | 11 | 8 | 19 |
|
Positive | 9 | 12 | 21 |
| HER2
statusd |
|
Negative | 8 | 9 | 17 |
|
Positive | 12 | 11 | 23 |
Cell culture
MCF-7/S and MCF-7/A cells were purchased from iCell
Bioscience Inc. Cells were cultured in Dulbecco's modified Eagle's
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% fetal bovine serum (Biological Industries), 100 U/ml penicillin
and 100 mg/ml streptomycin sulfate (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C with 5% CO2 in a 95%
humidified atmosphere.
MTT assay
The half maximal inhibitory concentration
(IC50) of ADM in MCF-7/S and MCF-7/A cells was
determined by performing MTT assays. Cells (5×103) were
seeded in 96-well plates and cultured overnight at 37°C. Next,
cells were treated with increasing concentrations of ADM (Zhejiang
Hisun Chemical Co., Ltd.) for 48 h. The concentrations of ADM for
MCF-7/S treatment were 0.01, 0.05, 0.2, 1, 5 and 10 µM, and the
concentrations of ADM for MCF-7/A treatment were 0.01, 0.05, 0.2,
1, 5, 10, 20, 50 and 100 µM. MTT reagent (neoFroxx GmbH) was added
to the culture for 4 h according to the manufacturer's protocol.
Subsequently, formazan was dissolved with DMSO. Cell viability was
determined using a microplate reader (Thermo Fisher Scientific,
Inc.) at 570 nm.
Colony-formation assay
MCF-7/S and MCF-7/A cells were seeded into 6-well
plates at 200 cells/well and maintained for 24 h before ADM
treatment (0, 1, 2, 5 and 10 µM). The treated cells were cultured
for 21 days to allow colony formation. The medium with ADM was
replaced every 7 days. Colonies were stained with 1% crystal violet
at room temperature for 10 min and subsequently counted.
Overexpression and knockdown of miR-16
by transfection with mimic and inhibitor
MCF-7/S and MCF-7/A cells (2×105) were
seeded into 6-well plates and incubated overnight. When 70–80%
confluence was reached the next day, MCF-7/S cells were transfected
with 100 nM miR-16 inhibitor or control, and MCF-7/A cells were
transfected with 50 nM miR-16 mimic or control using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). miR-16 mimic (5′-UAGCAGCACGUAAAUAUUGGCG-3′) and
corresponding negative control (mi-mNC; 5′-UUCUCCGAACGUGUCACGU-3′),
as well as the miR-16 inhibitor (5′-CGCCAAUAUUUACGUGCUGCUA-3′) and
corresponding negative control (mi-iNC;
5′-CAGUACUUUUGUGUAGUACAA-3′) were obtained from Shanghai GenePharma
Co., Ltd. The cells were collected for 24 h after transfection for
reverse transcription-quantitative PCR (RT-qPCR), and 48 h after
transfection for western blot analysis.
miRNA target prediction
The analysis of predicted miRNA targets was
performed using miRWalk 3.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/index.html)
and TargetScan 7.1 (http://www.targetscan.org/vert_71/) software.
Dual-luciferase reporter assay. To confirm that
miR-16 has binding sites in the 3′UTRs of the genes encoding Wip1
(PPM1D) and Bcl-2, MCF-7/S cells were seeded into 12-well plates
and co-transfected, using Lipofectamine® 2000, with
miR-16 or mi-mNC and pMIR-PPM1D-3′UTR (wild-type and mutant) or
pMIR-BCL-2-3′UTR (wild-type and mutant) reporter plasmids (Nanjing
Genebay Biotech Co., Ltd.). Renilla luciferase plasmid
(pRL-TK vector; Nanjing Genebay Biotech Co., Ltd.) was transfected
as the internal control. The lysate was prepared by adding cell
lysis buffer 48 h after co-transfection. A Dual-Luciferase Reporter
Assay kit (Promega Corporation) was used to measure activity, which
was normalized to Renilla.
Western blot analysis
miR-16 mimic and inhibitor were transfected into
MCF-7/A and MCF-7/S cells, respectively, in a 60-mm dish.
Additionally, mi-mNC and mi-iNC were transfected into MCF-7/A and
MCF-7/S cells, respectively, as corresponding negative controls.
After incubation for 48 h, cell lysates were prepared by adding
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) containing 1 mM PMSF. Protein concentration was
determined using a bicinchoninic acid kit (Nanjing KeyGen Biotech
Co., Ltd.). A total of 50 µg protein were loaded and separated by
10% SDS-PAGE, followed by transfer onto polyvinylidene difluoride
membranes. The membranes were blocked in 5% skimmed milk in
Tris-buffered saline containing 0.05% Tween-20 (TBST) for 2 h at
room temperature, and then washed three times using TBST buffer.
Membranes were subsequently incubated overnight at 4°C with primary
antibodies (dilution, 1:1,000) against Wip1 (cat. no. 11901), Bcl-2
(cat. no. 4223) and β-actin (cat. no. 4970), followed by incubation
with a horseradish peroxidase-conjugated secondary antibody (cat.
no. 7074; dilution, 1:2,000; all Cell Signaling Technology, Inc.)
at room temperature for 2 h. Bands were visualized with enhanced
chemiluminescence on a ChemiDoc™ XRS+ imaging system (Bio-Rad
Laboratories, Inc.). Quantity One software v4.6.6 (Bio-Rad
Laboratories, Inc.) was used to quantify the blot intensities,
which were normalized to that of β-actin.
Immunofluorescent staining
MCF-7/A and MCF-7/S cells (2×103; no
prior treatment) were grown on a 15-mm confocal dish for 2 days and
fixed in 4% paraformaldehyde at room temperature for 15 min.
Subsequently, cells were washed with PBS and permeabilized with
ice-cold PBS containing 0.5% Triton X-100 for 20 min at room
temperature. The samples were incubated with the appropriate
primary antibodies (Bcl-2, cat. no. 15071; and Wip1, cat. no.
11901; Cell Signaling Technology, Inc.) at 4°C overnight, and
probed with Alexa Fluor® 647 Conjugated (cat. no. 4410;
Cell Signaling Technology, Inc.) or Alexa Fluor® 488
conjugated (cat. no. ab150077; Abcam) secondary antibodies (all
dilutions, 1:1,000) for 20 min at room temperature. The nuclei were
counterstained with DAPI (Invitrogen; Thermo Fisher Scientific,
Inc.) for 10 min at room temperature. Fluorescence was visualized
and captured using a confocal microscope (magnification, ×600;
Leica TCS SP5 MP; Leica Microsystems, Inc.).
RNA isolation and RT-qPCR
TRIzol® reagent (Thermo Fisher
Scientific, Inc.) was used to extract total RNA from the tissues or
MCF-7/S and MCF-7/A cells treated with miR-16 inhibitor or miR-16
mimic, respectively, in 6-well plates (2×105
cells/well). Then RNA was reverse transcribed into cDNA for 1 h at
37°C and 85°C for 5 min using a Mir-X miRNA First-Strand Synthesis
kit (cat. no. 638315; Takara Bio, Inc.). qPCR was performed using a
SYBR Premix Ex Taq II kit (Takara Bio, Inc.) on a LightCycler
system (Roche Molecular Diagnostics). The thermocycling conditions
were as follows: 95°C for 10 sec, 95°C for 5 sec and 60°C for 20
sec for 40 cycles, followed by 95°C for 60 sec, 55°C for 30 sec and
95°C for 30 sec. miR-16 mRNA expression was quantified by
normalizing to U6. Primer sequences were as follows: miR-16
forward, 5′-TAGCAGCACGTAAATATTGGCG-3′. The U6 forward and reverse
primers along with miR-16 reverse primers were included in the
Mir-X miRNA First-Strand Synthesis kit. The 2−ΔΔCq
method was used to analyze the RT-qPCR data (25).
Apoptosis assay
MCF-7/S cells were transfected with miR-16 inhibitor
or mi-iNC, and MCF-7/A cells were transfected with miR-16 mimics or
mi-mNC, as aforementioned. After 48 h, cells were treated with 10
µM ADM for 48 h to induce apoptosis. The attached and floating
cells were harvested, and flow cytometry analysis was performed
using an Annexin V-FITC/propidium iodide staining kit (Dojindo
Molecular Technologies, Inc.) according to the manufacturer's
protocol. Apoptotic cells were detected using a BD Accuri C6 Plus
flow cytometer (BD Biosciences) and the data were analyzed by BD
CSampler analysis software (cat. no. 653123; version 1.0.23.1; BD
Biosciences). The percentage of apoptotic cells was calculated by
dividing the number of proapoptotic and apoptotic cells by the
total number of cells.
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three replicates. Unpaired two-tailed Student's t-test
was used to analyze the significance between two groups. For
multiple comparisons, one-way analysis of variance followed by
Dunnett's or Bonferroni's post-hoc test was performed using
GraphPad Prism v5.01 (GraphPad Software, Inc.). Pearson's
correlation analysis and calculation of the IC50 for ADM
were also performed using GraphPad Prism. P<0.05 was considered
to indicate a statistically significant difference.
Results
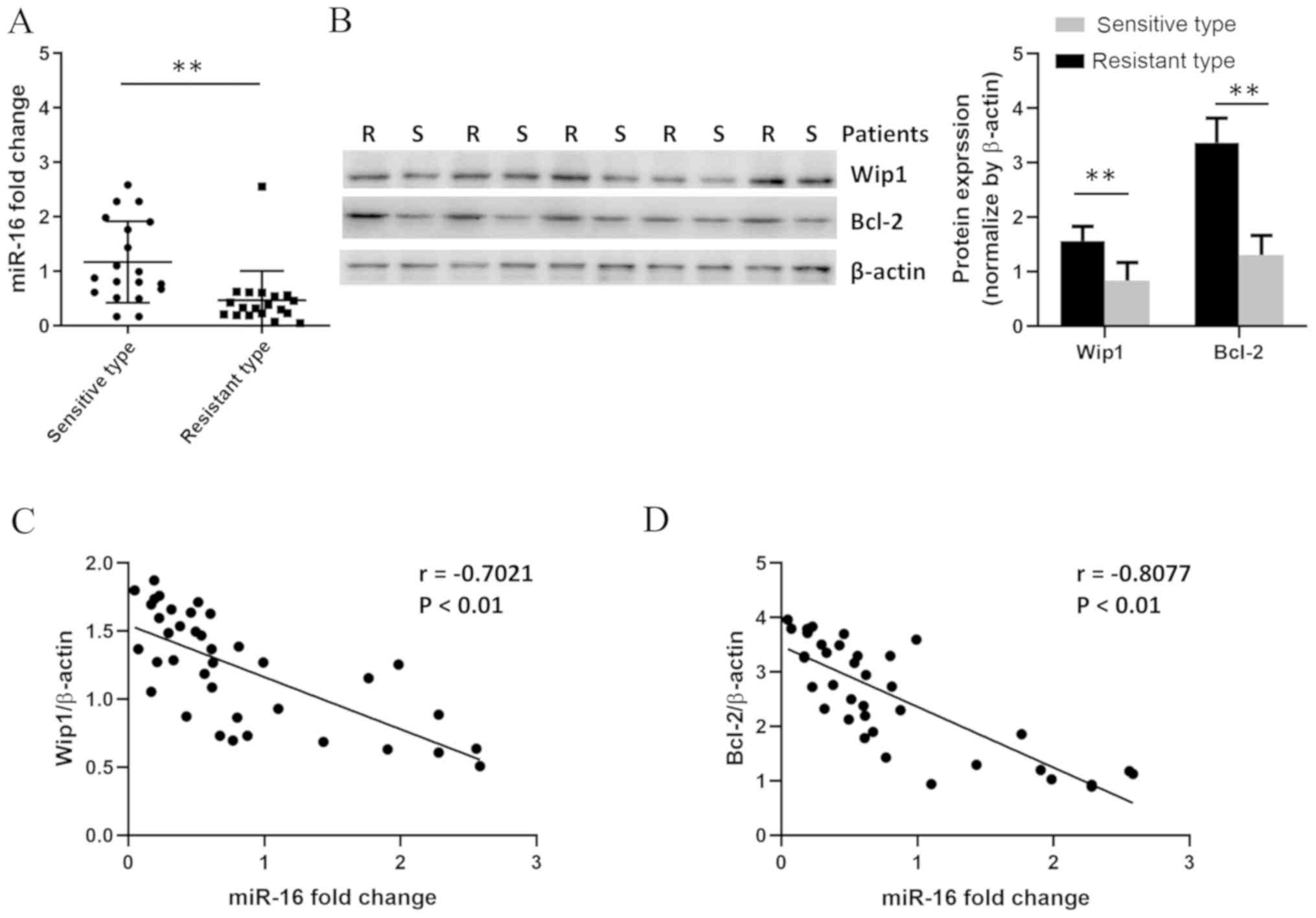
miR-16 is decreased in tumor tissues
from patients with drug-resistant BC
It has been reported that miR-16 is overexpressed in
multiple types of cancer, including BC (26). To determine whether miR-16 was
associated with BC drug resistance, tumor tissues were collected
from patients with BC, and miR-16 expression was quantified by
RT-qPCR. The clinicopathological characteristics of the patients
are presented in Table I. As shown
in Fig. 1A, miR-16 expression was
decreased in drug-resistant tumor samples compared with that in
drug-sensitive samples. To clarify the potential mechanism of the
association between miR-16 and BC drug resistance, the expression
of Wip1 and Bcl-2 was determined by western blotting. The
expression of these two proteins in ADM-resistant tumors was higher
compared with that in drug-sensitive tumors (Fig. 1B). Pearson's correlation analysis
showed that miR-16 expression was negatively correlated with the
expression of Wip1 and Bcl-2 (Fig. 1C
and D), suggesting that miR-16 may contribute to BC ADM
resistance.
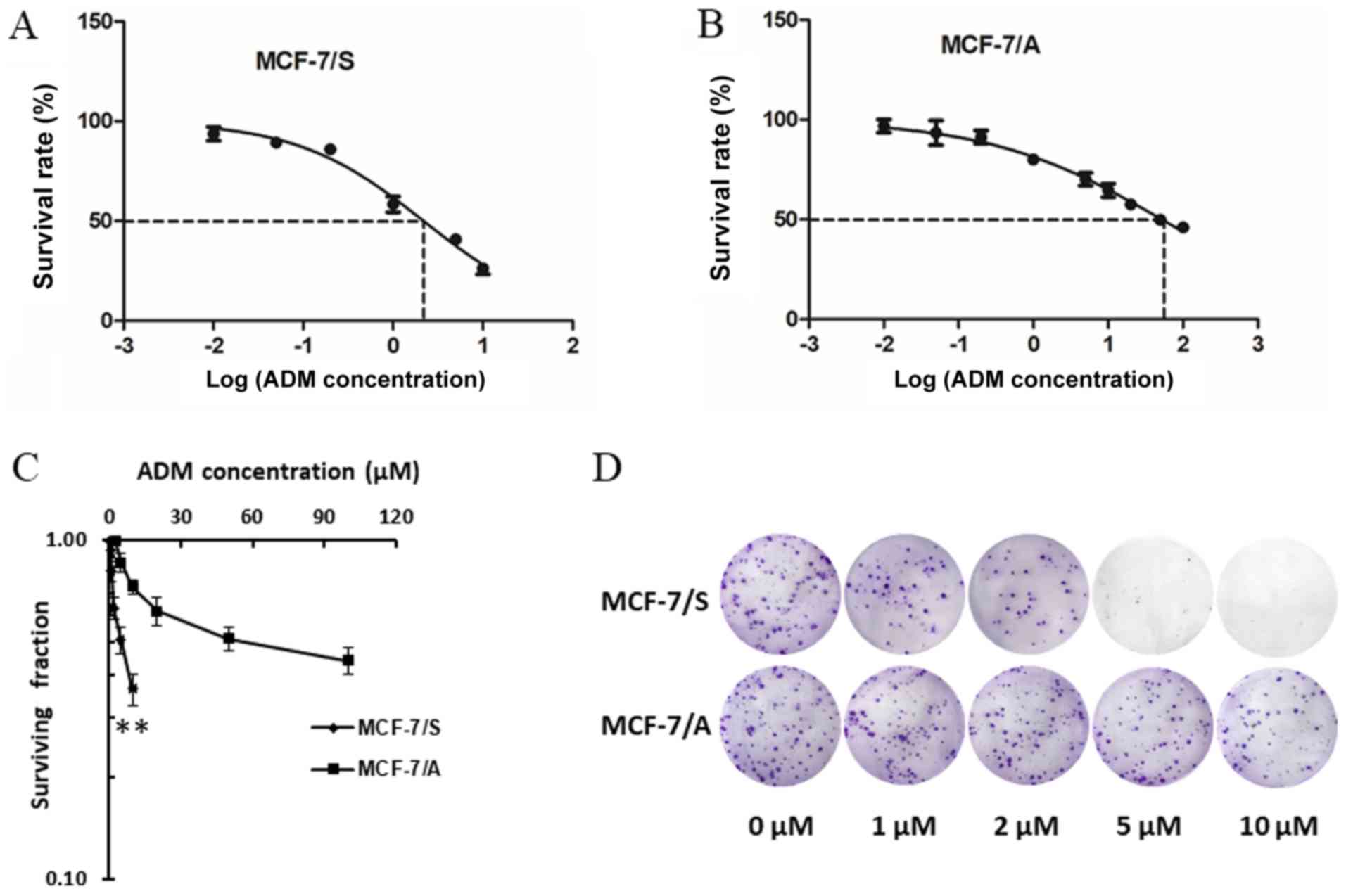
miR-16 expression is decreased in
ADM-resistant cells
To evaluate miR-16 expression in BC cells, a cell
line sensitive to ADM (MCF-7/S) and a resistant line (MCF-7/A) were
used. The IC50 values for ADM were determined in the
MCF-7/S and MCF-7/A cells by MTT assay. As presented in Fig. 2A and B, the IC50 of ADM in
MCF-7/S and MCF-7/A cells was 2.192 µM and 52.25 µM, respectively.
The ratio of the IC50 in MCF-7/A cells to the
IC50 in MCF-7/S cells was 23.84 (52.25/2.192),
confirming that MCF-7/A cells were resistant to this drug. In
addition, colony-formation assays were used to further confirm
resistance to ADM in MCF-7/A cells. The results revealed that the
surviving fraction of MCF-7/S cells was significantly lower
compared with that in MCF-7/A cells at the same concentration of
ADM (Fig. 2C and D), consistent with
the MTT assay results.
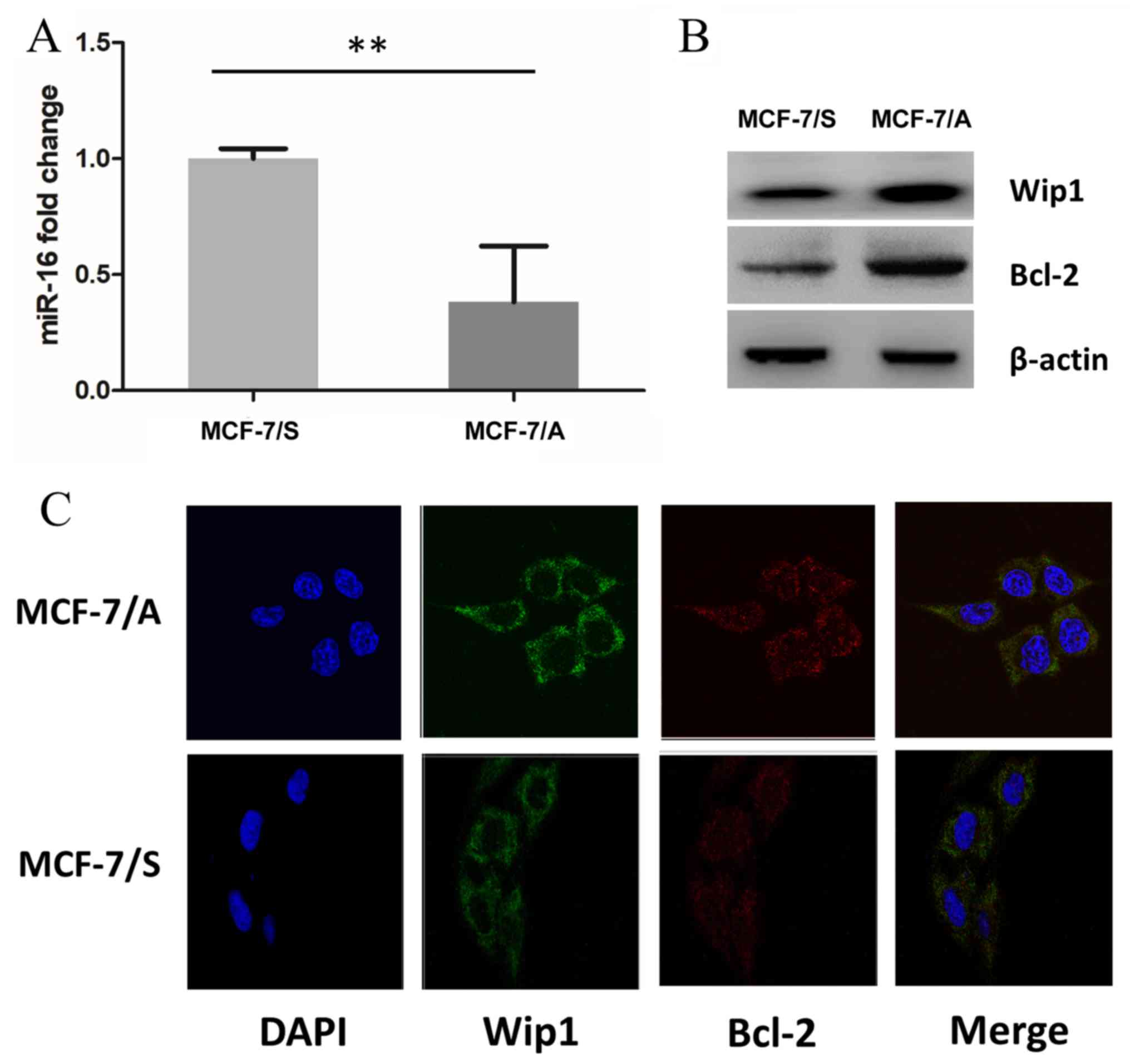
Wip1 and Bcl-2 protein expression is
higher in ADM-resistant MCF-7/A cells
miR-16 expression in ADM-sensitive and resistant BC
cells was examined to illustrate the potential mechanism of ADM
drug resistance. miR-16 expression was significantly decreased in
MCF-7/A cells, compared with that in MCF-7/S cells (Fig. 3A). Wip1 and Bcl-2 mRNAs are potential
targets of miR-16 as the 3′UTRs of these genes contain highly
conserved sites for miRNA binding. To assess whether miR-16
impacted these targets in BC, the expression of Wip1 and Bcl-2 was
determined in MCF-7/S and MCF-7/A cells. The results showed that
the expression of these proteins was increased in MCF-7/A cells, as
determined by western blotting (Fig.
3B). Higher Wip1 and Bcl-2 protein expression was also observed
in MCF-7/A cells using immunofluorescence, suggesting that miR-16
may regulate the expression of these two endogenous proteins in BC
cells (Fig. 3C).
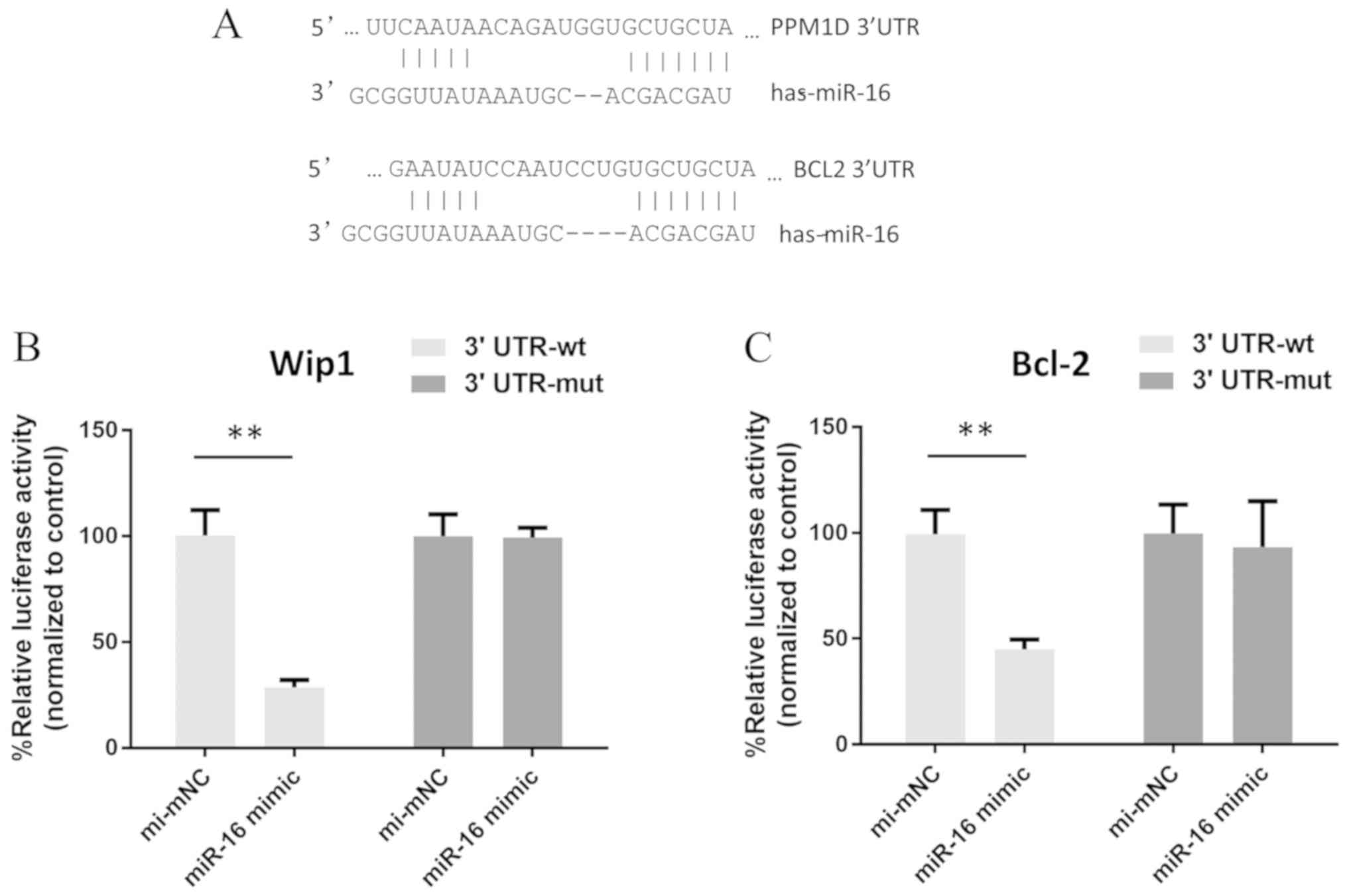
miR-16 inhibits Wip1 and Bcl-2
expression by targeting the 3′UTR of PPM1D and BCL-2
To confirm that miR-16 regulates the expression of
Wip1 and Bcl-2 in MCF-7 cells, the 3′UTR of the human PPM1D and
BCL-2 genes were predicted. It was found that 12 nucleotides from
miR-16 were complementary to target sequences in the 3′UTRs of
these two genes (Fig. 4A). To
further validate that PPM1D and BCL-2 were targets of miR-16, their
3′UTRs, containing the target sites of miR-16, were cloned into a
firefly luciferase reporter construct. Mutant versions of the
3′UTRs were transfected as controls. Luciferase activity was
detected following co-transfection of reporter constructs with
miR-16 into MCF-7/S cells. In the presence of miR-16, the
luciferase activity of constructs containing the wild-type PPM1D
and BCL-2 3′UTR was significantly decreased by 75 and 55%,
respectively (Fig. 4B and C).
However, luciferase activity was unaffected in cells transfected
with mutant PPM1D and BCL-2 3′UTRs and miR-16, indicating that the
PPM1D and BCL-2 genes are direct targets of miR-16.
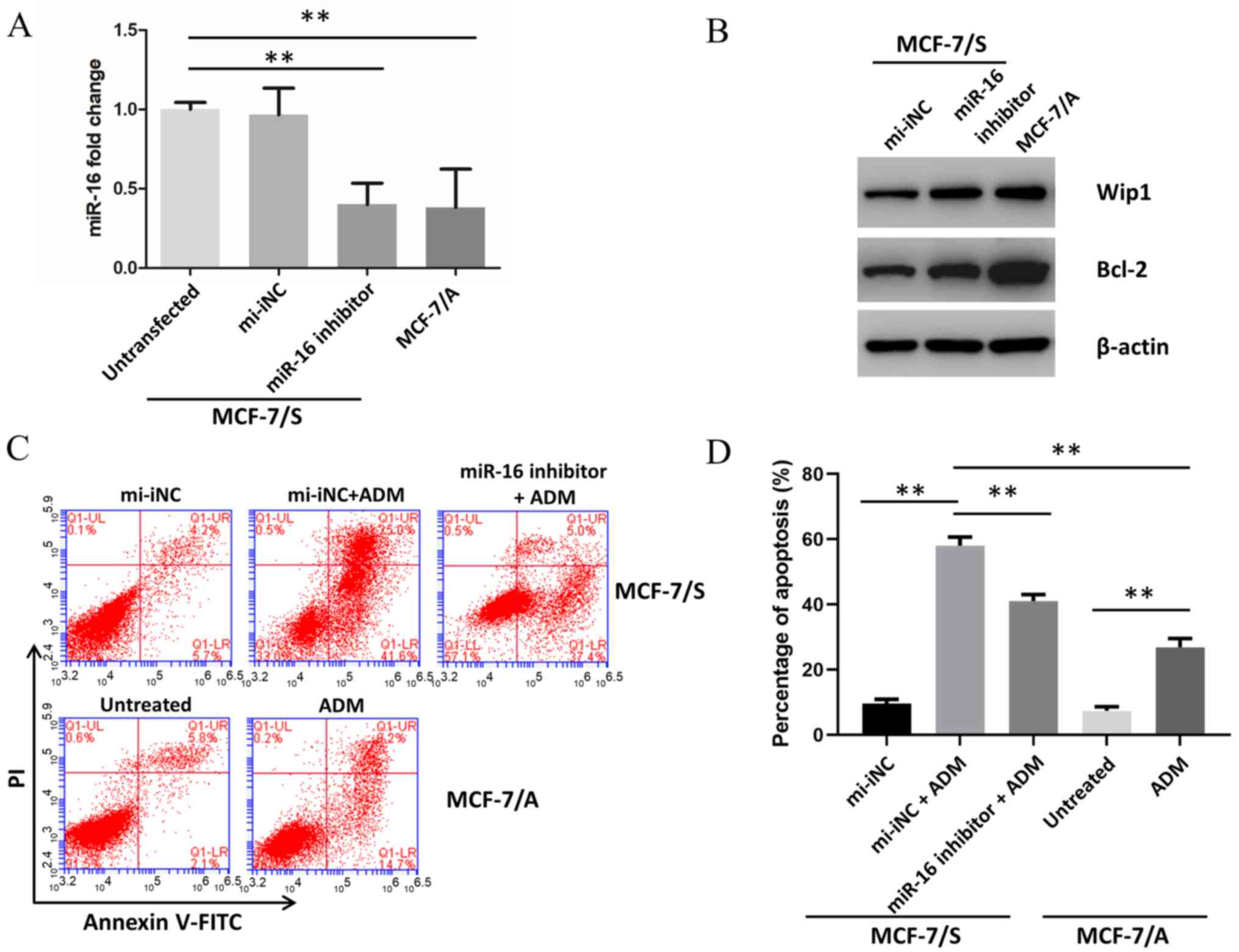
Downregulation of miR-16 in MCF-7/S
cells contributes to ADM resistance
To investigate the effects of miR-16 on BC drug
resistance, its expression in MCF-7/S cells was downregulated by
transfecting an inhibitor of miR-16. Transfection with the miR-16
inhibitor decreased expression of miR-16 compared to untransfected
MCF-7/S cells and those transfected with a non-targeting control
(Fig. 5A). Expression of miR-16 in
MCF-7/S cells after transfection with the inhibitor was comparable
to miR-16 expression in untransfected ADM-resistant MCF-7/A cells.
Downregulation of miR-16 in MCF-7/S cells resulted in increased
Wip1 and Bcl-2 protein expression compared with the negative
control (Fig. 5B). To investigate
the effects of downregulation of miR-16 on ADM resistance,
apoptosis following 10 µM ADM treatment was analyzed using flow
cytometry (Fig. 5C and D). The
apoptotic rate of MCF-7/S cells treated with ADM was significantly
higher compared with that of MCF-7/A cells. Apoptosis in MCF-7/S
cells transfected with miR-16 inhibitor and exposed to ADM was
markedly decreased compared with that in the ADM-treated mi-iNC
MCF-7/S group.
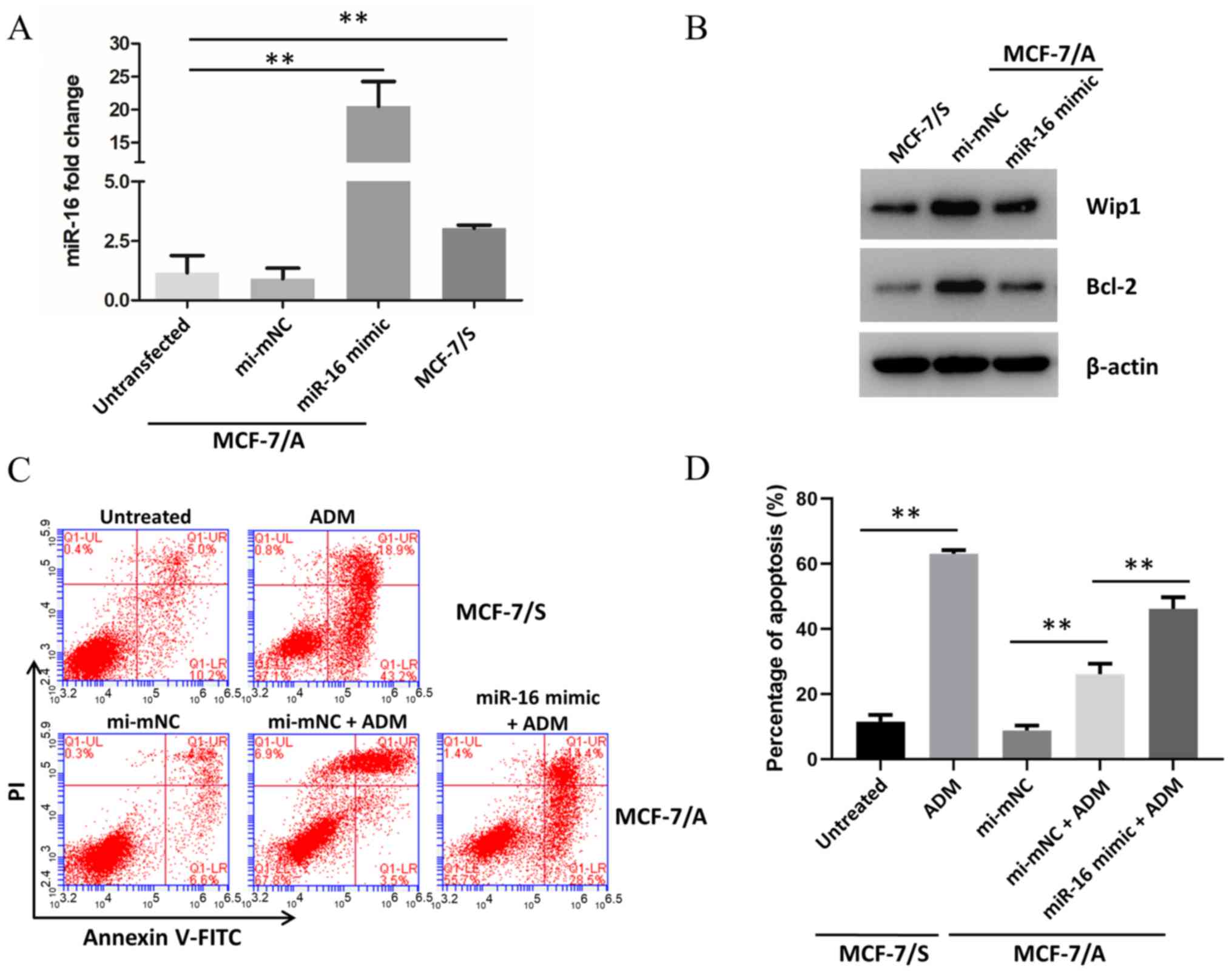
Overexpression of miR-16 sensitizes
MCF-7/A cells to ADM
To test the effect of miR-16 on chemotherapy
sensitization, MCF-7/A cells were transfected with an miR-16 mimic.
Transfection with the miR-16 mimic resulted in a 17-fold increase
in miR-16 expression compared with the mi-mNC group (Fig. 6A). Next, Wip1 and Bcl-2 protein
expression were examined by western blotting. As presented in
Fig. 6B, expression of these
proteins was decreased in MCF-7/A cells transfected with miR-16
mimic compared with mi-mNC MCF-7/A cells. Additionally, apoptosis
was assessed following exposure to ADM (Fig. 6C and D). It was found that the
apoptotic rate in the ADM-treated group was significantly
increased, compared with the untreated group, particularly in
MCF-7/S cells. However, in MCF-7/A cells transfected with miR-16
mimic, apoptosis was further elevated, compared with the
ADM-treated mi-mNC group.
Discussion
Conventional chemotherapy is regarded as the main
treatment option for cancer, but its effectiveness is often blocked
by intrinsic or acquired drug resistance (27). The repression of apoptosis or growth
signaling pathways may confer cancer cell resistance to
chemotherapy (28). In addition,
cancer cells activate DNA damage repair pathways to alleviate
cytotoxic stress induced by chemotherapeutics (28). Therefore, novel targets that can
enhance apoptosis are required to improve chemotherapy
efficacy.
Previous studies have demonstrated that alterations
in miRNA expression occur in numerous types of human cancer,
including BC, suggesting that miRNAs may be potential biomarkers
for cancer diagnosis, prognosis and pathogenesis (29,30).
miR-16 is a tumor-suppressor miRNA that has been found to suppress
proliferation, promote apoptosis and inhibit tumorigenicity in
vitro and in vivo (17).
miR-16 regulates the expression of a variety of proteins by
targeting multiple oncogenes, including BCL-2 (18) and PPM1D/Wip1 (9,31). Bcl-2
is highly conserved and is a vital regulator of apoptosis (32). Overexpression of miR-16 prevents cell
death induced by most chemotherapy agents and results in drug
resistance in multiple types of cancer (33,34). It
has been revealed that Wip1 directly phosphorylates the Ser15 site
of p53 to inactive it (31). As a
tumor-suppressor gene, TP53 blocks cell cycle progression to induce
apoptosis and promote DNA repair (35). Overexpression and mutations of TP53
are associated with enhanced drug resistance and are adverse
prognostic markers for patients diagnosed with BC (36,37). In
the present study, the MCF-7 cell line used expresses wild-type p53
(38). However, in p53-mutant BC
cells, the antitumor effect of miR-16 may depend on other targets
instead of p53. Therefore, gene sequencing is recommended in
clinical practice for selecting appropriate targets and strategies
for cancer treatment.
Decreased miR-16 expression contributes to
inhibition of apoptosis (18);
therefore, it was hypothesized that miR-16 expression in
ADM-resistant MCF-7/A cells may be lower than that in the more
sensitive cell line, MCF-7/S, and that this reduction in miR-16
could lead to ADM resistance by targeting Bcl-2 and Wip1. The
findings of the present study demonstrated that the expression of
miR-16 in drug-resistant tumor tissues was decreased compared with
drug-sensitive tissues, and was negatively correlated with Wip1 and
Bcl-2 expression. Additionally, it was found that MCF-7/A cells had
lower miR-16 expression and higher Wip1 and Bcl-2 expression
compared with MCF-7/S cells, which was consistent with the tumor
tissue results. Confirming that Wip1 and Bcl-2 expression was
regulated by miR-16, overexpression of miR-16 reduced luciferase
activity in cells transfected with PPM1D and BCL-2 3′UTRs. The
present study further demonstrated that the chemosensitivity of
MCF-7/A cells was enhanced by overexpressing miR-16 through
transfection with an miR-16 mimic. Expression of Wip1 and Bcl-2 was
decreased in response to increased miR-16, and apoptosis induced by
ADM was increased. These results demonstrated that the decreased
expression level of miR-16 in MCF-7/A cells may contribute to ADM
resistance in this cell line. Conversely, miR-16 knockdown in
ADM-sensitive MCF-7/S cells enhanced the expression of Wip1 and
Bcl-2 and reduced apoptosis after ADM treatment. Taken together,
these results suggest that targeting miR-16 may be a promising
approach to improve the effectiveness of chemotherapy in advanced
BC. Future research will focus on inducing normal BC cells to
become drug resistant, in order to further investigate the findings
of the present study.
A study reported that the miR-15/16 cluster resides
at chromosome 13q14.3, and its overexpression led to increased
transcription of 265 genes and suppression of 3,300 genes (16). Among the suppressed genes, 27 coded
for proteins that were also found to be repressed by miR-15/16
using proteomics analysis (16). A
number of the downregulated proteins were associated with
tumorigenesis, cell growth or apoptosis, and eight were predicted
to be targets of miR-15/16 (16).
Furthermore, TP53-binding sites were identified upstream of the
miR-15/16 cluster, and p53 stimulates the transcription of the
miR-15/16 cluster (39). In
addition, the 3′UTR of TP53 contains binding sites for miR-16
(39), suggesting that this miRNA
may influence the regulation of intracellular homeostasis by p53
and the response to chemotherapeutics. Other proteins of
significance have been reported as targets of miR-16. For example,
it has been reported that miR-15/16 inhibits expression of the cell
cycle regulator, cyclin D1 in several malignancies (40,41). In
addition, the myeloid cell leukemia 1 oncogene in CLL, and the BMI1
polycomb ring finger oncogene in BC have also been identified to be
miR-16 targets (16,42). According to the findings of the
present study, miR-16 increases chemosensitivity in ADM-resistant
cells. However, drug resistance is a complex clinical obstacle and
targeting more than one site may achieve a more effective clinical
outcome. Therefore, further study is required to verify the effects
of miR-16 on proteins associated with drug resistance in BC. To
confirm the potential clinical application of miR-16 as a
chemosensitizing agent and investigate the mechanism, further
research is required using primary cell culture and animal models
of tumorigenesis.
Acknowledgements
Not applicable.
Funding
This study was funded by the National Key Research
and Development Program of China (grant no. 2016YFC0905900), the
National Natural Science Foundation of China (grant no. 81872365),
the ‘333’ Talent Project of Jiangsu Province [grant no. 4(2016)],
and the Innovation Team Construction Project of Nanjing Medical
University (grant no. JX102GSP201727).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XG, MW, YZ and JT conceived and designed the study.
XG, MW, YZ and ZX performed the experiments. JD and MW analyzed the
data. YZ and MW wrote the manuscript. JT revised the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The protocols for the use of human samples were
reviewed and approved by the Ethics Committee of Nanjing Medical
University (approval no. LCYJ20160306001; Nanjing, China). Patients
provided written informed consent for the use of their samples for
research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hong W and Dong E: The past, present and
future of breast cancer research in China. Cancer Lett. 351:1–5.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Turati F, Carioli G, Bravi F, Ferraroni M,
Serraino D, Montella M, Giacosa A, Toffolutti F, Negri E, Levi F
and La Vecchia C: Mediterranean diet and breast cancer risk.
Nutrients. 10:E3262018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ni W, Chen B, Zhou G, Lu C, Xiao M, Guan
C, Zhang Y, He S, Shen A and Ni R: Overexpressed nuclear BAG-1 in
human hepatocellular carcinoma is associated with poor prognosis
and resistance to doxorubicin. J Cell Biochem. 114:2120–2130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pelden S, Insawang T, Thuwajit C and
Thuwajit P: The trefoil factor 1 (TFF1) protein involved in
doxorubicininduced apoptosis resistance is upregulated by estrogen
in breast cancer cells. Oncol Rep. 30:1518–1526. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cunha NL, Teixeira GM, Martins TD, Souza
AR, Oliveira PF, Simaro GV, Rezende KC, Gonçalves Ndos S, Souza DG,
Tavares DC, et al: (−)-Hinokinin induces G2/M arrest and
contributes to the antiproliferative effects of doxorubicin in
breast cancer cells. Planta Medica. 82:530–538. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hermawan A, Wagner E and Roidl A:
Consecutive salinomycin treatment reduces doxorubicin resistance of
breast tumor cells by diminishing drug efflux pump expression and
activity. Oncol Rep. 35:1732–1740. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wijdeven RH, Pang B, Assaraf YG and
Neefjes J: Old drugs, novel ways out: Drug resistance toward
cytotoxic chemotherapeutics. Drug Resist Updat. 28:65–81. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Das CK, Linder B, Bonn F, Rothweiler F,
Dikic I, Michaelis M, Cinatl J, Mandal M and Kögel D: BAG3
overexpression and cytoprotective autophagy mediate apoptosis
resistance in chemoresistant breast cancer cells. Neoplasia.
20:263–279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang X, Wan G, Mlotshwa S, Vance V,
Berger FG, Chen H and Lu X: Oncogenic Wip1 phosphatase is inhibited
by miR-16 in the DNA damage signaling pathway. Cancer Res.
70:7176–7186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM
and Zhang GZ: Biological functions of microRNAs: A review. J
Physiol Biochem. 67:129–139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai Y, Yu X, Hu S and Yu J: A brief review
on the mechanisms of miRNA regulation. Genomics Proteomics
Bioinformatics. 7:147–154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wahid F, Shehzad A, Khan T and Kim YY:
MicroRNAs: Synthesis, mechanism, function, and recent clinical
trials. Biochim Biophys Acta. 1803:1231–1243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bushati N and Cohen SM: microRNA
functions. Annu Rev Cell Dev Biol. 23:175–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Calin GA, Cimmino A, Fabbri M, Ferracin M,
Wojcik SE, Shimizu M, Taccioli C, Zanesi N, Garzon R, Aqeilan RI,
et al: MiR-15a and miR-16-1 cluster functions in human leukemia.
Proc Natl Acad Sci USA. 105:5166–5171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rivas MA, Venturutti L, Huang YW,
Schillaci R, Huang TH and Elizalde PV: Downregulation of the
tumor-suppressor miR-16 via progestin-mediated oncogenic signaling
contributes to breast cancer development. Breast Cancer Res.
14:R772012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mobarra N, Shafiee A, Rad SM, Tasharrofi
N, Soufi-Zomorod M, Hafizi M, Movahed M, Kouhkan F and Soleimani M:
Overexpression of microRNA-16 declines cellular growth,
proliferation and induces apoptosis in human breast cancer cells.
In vitro Cell Dev Biol Anim. 51:604–611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pekarsky Y, Balatti V and Croce CM: BCL2
and miR-15/16: From gene discovery to treatment. Cell Death Differ.
25:21–26. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Booser DJ, Esteva FJ, Rivera E, Valero V,
Esparza-Guerra L, Priebe W and Hortobagyi GN: Phase II study of
liposomal annamycin in the treatment of doxorubicin-resistant
breast cancer. Cancer Chemother Pharmacol. 50:6–8. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giuliano AE, Connolly JL, Edge SB,
Mittendorf EA, Rugo HS, Solin LJ, Weaver DL, Winchester DJ and
Hortobagyi GN: Breast cancer-major changes in the American joint
committee on cancer eighth edition cancer staging manual. CA Cancer
J Clin. 67:290–303. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Côme C, Laine A, Chanrion M, Edgren H,
Mattila E, Liu X, Jonkers J, Ivaska J, Isola J, Darbon JM, et al:
CIP2A is associated with human breast cancer aggressivity. Clin
Cancer Res. 15:5092–5100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shah MV, Wiktor AE, Meyer RG, Tenner KS,
Ballman KV, Green SJ, Sukov WR, Ketterling RP, Perez EA and Jenkins
RB: Change in pattern of HER2 fluorescent in situ hybridization
(FISH) results in breast cancers submitted for FISH testing:
Experience of a reference laboratory using US food and drug
administration criteria and American society of clinical oncology
and college of American pathologists guidelines. J Clin Oncol.
34:3502–3510. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wei T, Ye P, Peng X, Wu LL and Yu GY:
Prognostic value of miR-222 in various cancers: A systematic review
and meta-analysis. Clin Lab. 62:1387–1395. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gonen N and Assaraf YG: Antifolates in
cancer therapy: Structure, activity and mechanisms of drug
resistance. Drug Resist Updat. 15:183–210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Borst P, Jonkers J and Rottenberg S: What
makes tumors multidrug resistant? Cell Cycle. 6:2782–2787. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo L, Zhao Y, Yang S, Cai M, Wu Q and
Chen F: Genome-wide screen for aberrantly expressed miRNAs reveals
miRNA profile signature in breast cancer. Mol Biol Rep.
40:2175–2186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harquail J, Benzina S and Robichaud GA:
MicroRNAs and breast cancer malignancy: An overview of
miRNA-regulated cancer processes leading to metastasis. Cancer
Biomark. 11:269–280. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhan XH, Xu QY, Tian R, Yan H, Zhang M, Wu
J, Wang W and He J: MicroRNA16 regulates glioma cell proliferation,
apoptosis and invasion by targeting Wip1-ATM-p53 feedback loop.
Oncotarget. 8:54788–54798. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liang X, Xu K, Xu Y, Liu J and Qian X:
B1-induced caspase-independent apoptosis in MCF-7 cells is mediated
by down-regulation of Bcl-2 via p53 binding to P2 promoter TATA
box. Toxicol Appl Pharmacol. 256:52–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Srivastava RK, Sasaki CY, Hardwick JM and
Longo DL: Bcl-2-mediated drug resistance: Inhibition of apoptosis
by blocking nuclear factor of activated T lymphocytes
(NFAT)-induced Fas ligand transcription. J Exp Med. 190:253–265.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang T, Xu F, Sheng Y, Zhang W and Chen Y:
A targeted proteomics approach to the quantitative analysis of
ERK/Bcl-2-mediated anti-apoptosis and multi-drug resistance in
breast cancer. Anal Bioanal Chem. 408:7491–7503. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Marcel V, Catez F and Diaz JJ: p53, a
translational regulator: Contribution to its tumour-suppressor
activity. Oncogene. 34:5513–5523. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamamoto M, Hosoda M, Nakano K, Jia S,
Hatanaka KC, Takakuwa E, Hatanaka Y, Matsuno Y and Yamashita H: p53
accumulation is a strong predictor of recurrence in estrogen
receptor-positive breast cancer patients treated with aromatase
inhibitors. Cancer Sci. 105:81–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Varna M, Bousquet G, Plassa LF, Bertheau P
and Janin A: TP53 status and response to treatment in breast
cancers. J Biomed Biotechnol. 2011:2845842011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Casey G, Lo-Hsueh M, Lopez ME, Vogelstein
B and Stanbridge EJ: Growth suppression of human breast cancer
cells by the introduction of a wild-type p53 gene. Oncogene.
6:1791–1797. 1991.PubMed/NCBI
|
|
39
|
Fabbri M, Bottoni A, Shimizu M, Spizzo R,
Nicoloso MS, Rossi S, Barbarotto E, Cimmino A, Adair B, Wojcik SE,
et al: Association of a microRNA/TP53 feedback circuitry with
pathogenesis and outcome of B-cell chronic lymphocytic leukemia.
JAMA. 305:59–67. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cai CK, Zhao GY, Tian LY, Liu L, Yan K, Ma
YL, Ji ZW, Li XX, Han K, Gao J, et al: miR-15a and miR-16-1
downregulate CCND1 and induce apoptosis and cell cycle arrest in
osteosarcoma. Oncol Rep. 28:1764–1770. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang QQ, Liu B and Yuan T: MicroRNA-16
inhibits bladder cancer proliferation by targeting cyclin D1. Asian
Pac J Cancer Prev. 14:4127–4130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Patel N, Garikapati KR, Pandita RK, Singh
DK, Pandita TK, Bhadra U and Bhadra MP: Erratum: MiR-15a/miR-16
down-regulates BMI1, impacting Ub-H2A mediated DNA repair and
breast cancer cell sensitivity to doxorubicin. Sci Rep.
7:129322017. View Article : Google Scholar : PubMed/NCBI
|