Introduction
Acute myeloid leukemia (AML), a type of aggressive
hematopoietic stem cell tumor, remains a considerable challenge in
the clinical setting due to its high relapse rate (1,2). Lethal
infections, bleeding and organ invasion are the main complications
of AML (3). In the past decades,
various studies have suggested that both environmental and genetic
factors are important in the occurrence of AML (1,4–6). However, the current knowledge of the
molecular mechanisms involved in the development of AML is limited,
and early diagnosis remains difficult, which may result in delayed
therapy. Thus, the identification of the key mechanisms regulating
AML management and patient survival may aid in the development of
AML-specific targeted therapies (3).
Microarray technology is a high-throughput and
powerful tool to generate large quantities of data, including mRNA
expression and DNA methylation (7).
The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO)
databases are two common public platforms obtaining such data.
These microarray results provide an opportunity to use
bioinformatics to identify novel targets (8,9).
Numerous public databases, including Oncomine (10), Gene Expression Profiling Interactive
Analysis (GEPIA) (11) and UALCAN
(12), provide several bioinformatic
analysis tools, including differential expression, co-expression
and comparative analyses, to identify novel tumor biomarkers and
potential therapeutic targets through the use of the stored
microarray data. Therefore, bioinformatics aid in the investigation
of the underlying regulatory networks involved in different types
of cancer, and constitute a powerful method of cancer research,
including the early diagnosis of cancer, grading and prognostic
prediction (8).
In the present study, microarray data were
downloaded to investigate the hub genes affecting the development
of AML from GEO in order to identify AML-associated genes via
bioinformatics analysis. The present study further investigated
those dysregulated genes at the molecular level and explored the
potential candidate genes for prognosis in AML.
Materials and methods
Microarray data preprocessing and
differentially expressed genes (DEGs) analysis
The original dataset GSE9476, provided by Stirewalt
et al (13), was downloaded,
which was based on the GPL96 Affymetrix Human Genome U133A Array
platform (Affymetrix; Thermo Fisher Scientific, Inc.). The
profiling dataset comprising 26 patients with AML and 10 normal
peripheral blood samples was selected for further analysis
(13). The raw data files were
processed using the Affy package in R version 3.3.2 (https://www.r-project.org/). The limma package version
3.40.2.2 (http://www.bioconductor.org/packages/release/bioc/html/limma.html)
was applied to identify the DEGs between AML samples and normal
peripheral blood mononuclear cells (12). |Fold-change (FC)| ≥2 and false
discovery rate (FDR) <0.05 were considered to indicate a
statistically significant difference.
GO and KEGG pathway enrichment
analyses
To obtain an insight into the function of the DEGs
identified, these DEGs were divided into an upregulated (FC≥2 and
FDR<0.05) and a downregulated group (FC≤-2 and FDR<0.05).
Then, Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathway analyses were performed using the
Database for Annotation, Visualization and Integrated Discovery
(DAVID; david.ncifcrf.gov/) online tool.
FDR<0.05 was considered to indicate a statistically significant
difference.
Oncomine analysis
The differences in mRNA levels of serine protease
inhibitor Kazal-type 2 (SPINK2) between the AML and control samples
were evaluated using gene expression array datasets from Oncomine,
a public cancer microarray database that is accessible online
(10,14,15). The
threshold was established at P<10−4 and FC>2. In
addition, the data type was restricted to mRNA levels only.
GEPIA analysis
GEPIA possesses key customizable and interactive
functions, including profiling plotting, differential expression
analysis, patient survival analysis, similar gene detection and
dimensionality reduction analysis (11). Comprehensive expression analyses
using GEPIA greatly facilitate data mining in numerous areas of
research, thus contributing to scientific discussions and the
identification of novel therapies. The present study employed
SPINK2 data into the GEPIA dataset to explore its differential
expression and effect on the prognosis of patients with AML.
UALCAN dataset analysis
UALCAN provides easy access to publicly available
cancer transcriptome data (namely, TCGA and MET500 transcriptome
sequencing). It allows users to identify biomarkers and to perform
in silico validations of potential genes of interest. In
addition, it can depict gene expression and patient survival
information based on gene expression (12). In the present study, SPINK2 was
included into the UALCAN dataset to explore its effect on the
prognosis of patients with AML.
RNA isolation, reverse transcription
and quantitative (RT-q) PCR
Blood samples of 6 healthy donors and 12 patients
with AML were collected from the left arm at the Linyi Central
Hospital between January 2017 and October 2017. The age range of
patients was 16–65 years and the male: Female ratio was 4:8. Blood
samples were centrifuged at 1,500 g for 5 min and at 4°C, and the
fraction containing blood cells was stored at −80°C for future
analyses. All blood cells were lysed using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.), and total RNAs were
extracted. First strand cDNA synthesis was conducted using 2 µg RNA
using the SYBR Premix Ex Taq™ II kit (Takara Biotechnology Co.)
complete with SYBR Green reagents (Bio-Rad Laboratories, Inc.). The
reverse transcriptase reaction was performed for 60 min at 37°C,
followed by 60 min at 42°C. qPCR was performed using a 7900HT
real-time PCR system (Thermo Fisher Scientific, Inc.), with the
Real-Time SYBR Green PCR Master Mix kit (Promega Corporation). The
primers used were as follows: SPINK2, forward
5′-GAGTGGCGCAGGTAACAGAC-3′, reverse, 5′-ACCAAATTGAGGGATCAGAGAG-3′;
and GAPDH, forward 5′-TGGTGAAGACGCCAGTGGA-3′ and reverse,
5′-GCACCGTAAGGCTGAGAAC-3′. The qPCR thermocycling conditions were
as follows: 95°C for 30 sec, 35 cycles at 95°C for 5 sec, then 60°C
for 30 sec. The relative mRNA expression was calculated using the
2−∆∆Cq method (16).
Statistical analysis
A paired t-test was used to determine the
statistical significance of the differences between healthy donors
and patients with AML. The Kaplan-Meier analysis and log rank were
used to assess patient survival rates. In the present study, the
cut-off of survival analysis was based on the median of expression
level in the GEPIA and UALCAN datasets. Data were presented as the
means ± standard deviation. Data were analyzed using SPSS 17.0
(SPSS, Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
DEGs in AML
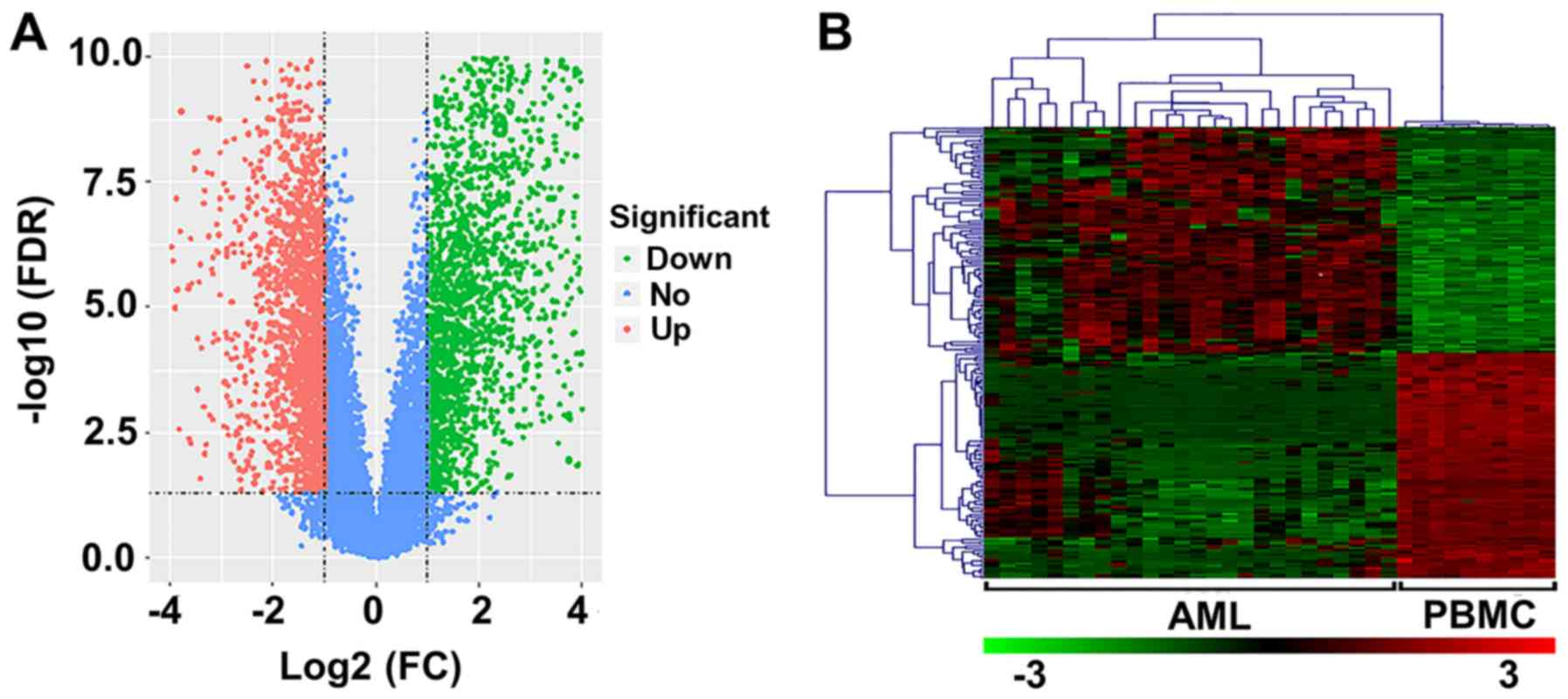
In the present study, 26 AML and 10 normal samples
were analyzed. Upon preprocessing, 3,700 DEGs (FDR<0.05;
log2 FC≥1) were identified, including 1,744 upregulated
and 1,956 downregulated genes (Fig.
1A). The heat map of the top 100 upregulated and downregulated
genes is presented in Fig. 1B.
GO term enrichment analysis for
DEGs
The upregulated and downregulated genes were
uploaded into the DAVID dataset to perform GO analysis. As
presented in Table I, the most
important biological processes (BPs) of overexpressed genes were
‘nitrogen compound metabolic process’, ‘cellular nitrogen compound
metabolic process’ and ‘cellular metabolic process’. For the
downregulated genes, BPs were significantly enriched in ‘immune
response’, ‘immune system process’ and ‘defense response’ (Table II). For molecular function (MF)
terms, upregulated DEGs were enriched in ‘RNA binding’, ‘poly (A)
RNA binding’ and ‘nucleic acid binding’ (Table I), while the downregulated DEGs were
enriched in ‘protein binding’, ‘binding’ and ‘anion binding’
(Table II). The most significant
component functions (CFs) for upregulated DEGs were ‘intracellular
organelle part’, ‘membrane-enclosed lumen’ and ‘organelle part’
(Table I), while the most
significant CF terms for downregulated DEGs were ‘cytosol’,
‘cytoplasm’ and ‘cell periphery’ (Table
II).
 | Table I.GO Enrichment analysis of upregulated
DEGs. |
Table I.
GO Enrichment analysis of upregulated
DEGs.
| A, Biological
processes |
|---|
|
|---|
| GO ID | Pathway
description | Gene count | FDR |
|---|
| GO.0006807 | Nitrogen compound
metabolic process | 562 |
5.32×10−57 |
| GO.0034641 | Cellular nitrogen
compound metabolic process | 537 |
4.68×10−56 |
| GO.0044237 | Cellular metabolic
process | 712 |
4.33×10−50 |
| GO.0044238 | Primary metabolic
process | 719 |
4.33×10−50 |
| GO.0071704 | Organic substance
metabolic process | 722 |
7.52×10−47 |
|
| B, Molecular
function |
|
| GO ID | Pathway
description | Gene
count | FDR |
|
| GO.000723 | RNA binding | 292 |
3.36×10−75 |
| GO.0044822 | Poly(A) RNA
binding | 249 |
1.00×10−73 |
| GO.0003676 | Nucleic acid
binding | 410 |
2.01×10−40 |
| GO.1901363 | Heterocyclic
compound binding | 520 |
1.27×10−36 |
| GO.0097159 | Organic cyclic
compound binding | 523 |
4.32×10−36 |
|
| C, Component
function |
|
| GO ID | Pathway
description | Gene
count | FDR |
|
| GO.0044446 | Intracellular
organelle part | 759 |
5.24×10−84 |
| GO.0031974 | Membrane-enclosed
lumen | 527 |
1.44×10−81 |
| GO.0044422 | Organelle part | 764 |
1.44×10−81 |
| GO.0070013 | Intracellular
organelle lumen | 516 |
9.85×10−81 |
| GO.0043233 | Organelle
lumen | 519 |
7.00×10−80 |
| Table II.GO Enrichment analysis of
downregulated DEGs. |
Table II.
GO Enrichment analysis of
downregulated DEGs.
| A, Biological
processes |
|---|
|
|---|
| GO ID | Pathway
description | Gene count | FDR |
|---|
| GO.0006955 | Immune
response | 264 |
1.30×10−72 |
| GO.0002376 | Immune system
process | 316 |
1.43×10−63 |
| GO.0006952 | Defense
response | 254 |
4.60×10−60 |
| GO.0007166 | Cell surface
receptor signaling pathway | 308 |
1.34×10−55 |
| GO.0045087 | Innate immune
response | 188 |
1.28×10−49 |
|
| B, Molecular
function |
|
| GO ID | Pathway
description | Gene
count | FDR |
|
| GO.0005515 | Protein
binding | 469 |
3.99×10−35 |
| GO.0005488 | Binding | 789 |
8.01×10−23 |
| GO.0043168 | Anion binding | 262 |
7.17×10−14 |
| GO.0019899 | Enzyme binding | 156 |
1.78×10−13 |
| GO.0060089 | Molecular
transducer activity | 184 |
5.74×10−13 |
|
| C, Component
function |
|
| GO ID | Pathway
description | Gene
count | FDR |
|
| GO.0005829 | Cytosol | 344 |
2.76×10−27 |
| GO.0005737 | Cytoplasm | 761 |
2.68×10−22 |
| GO.0071944 | Cell periphery | 413 |
7.57×10−19 |
| GO.0005886 | Plasma
membrane | 403 |
1.46×10−17 |
| GO.0005622 | Intracellular | 884 |
6.42×10−14 |
KEGG pathway analysis for DEGs
To gain further insight into the function of the
genes in the interaction network, the DAVID database was used to
identify the significant pathways associated with DEGs. According
to the results of KEGG pathway analysis, upregulated DEGs were
significantly enriched in ‘metabolic pathways’, ‘Huntington's
disease’ and ‘ribosome’ (Table
III). For the downregulated DEGs, the most significant pathways
were ‘osteoclast differentiation’, ‘tuberculosis’ and ‘chemokine
signaling pathway’ (Table
III).
| Table III.KEGG pathway analysis of DEGs. |
Table III.
KEGG pathway analysis of DEGs.
| A, Upregulated |
|---|
|
|---|
| Pathway
description | Observed gene
count | FDR |
|---|
| Metabolic
pathways | 197 |
1.71×10−38 |
| Huntington's
disease | 47 |
1.58×10−15 |
| Ribosome | 37 |
7.29×10−14 |
| Oxidative
phosphorylation | 36 |
5.91×10−13 |
| Parkinson's
disease | 37 |
7.94×10−13 |
|
| B,
Downregulated |
|
| Pathway
description | Observed gene
count | FDR |
|
| Osteoclast
differentiation | 46 |
1.66×10−21 |
| Tuberculosis | 53 |
6.51×10−21 |
| Chemokine signaling
pathway | 53 |
4.78×10−20 |
| Natural killer
cell-mediated cytotoxicity | 43 |
2.71×10−19 |
| Influenza A | 47 |
6.17×10−17 |
SPINK2 expression is elevated in
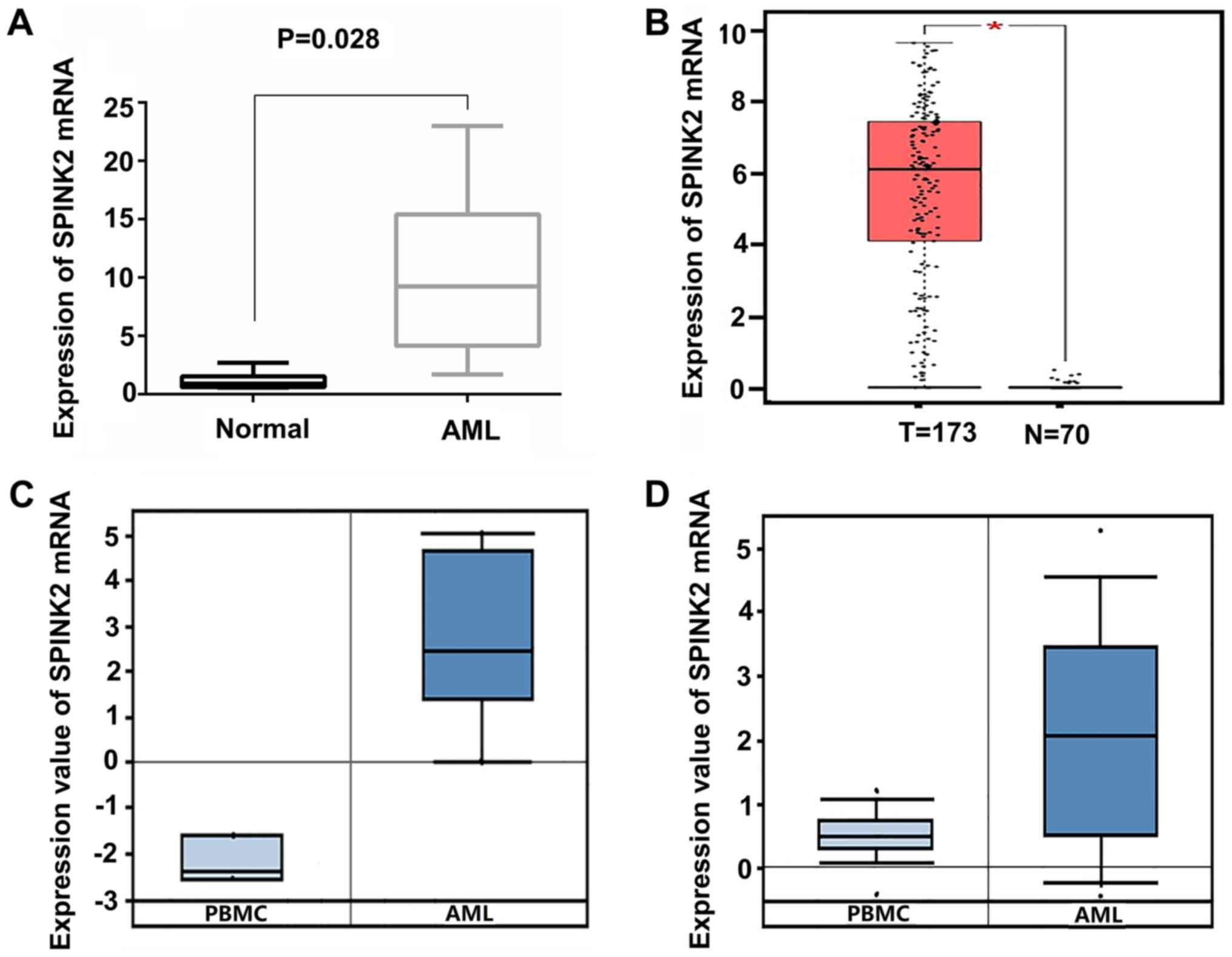
patients with AML
Among all the upregulated genes, SPINK2 ranked first
(log2 FC, 6.59; FDR, 9.86×10−8). To validate
the expression level of SPINK2 in AML, 6 healthy individuals and 12
patients with AML were recruited in the present study. qPCR results
indicated that the levels of SPINK2 mRNA were significantly
increased by 10.11-fold (P=0.028) in patients with AML compared
with the healthy controls (Fig. 2A).
The expression level of SPINK2 in the AML samples was validated in
the Oncomine and GEPIA datasets. In the GEPIA database, 70 normal
individuals and 173 patients with AML were included. The results
suggested that the mRNA level of SPINK2 was elevated in patients
with AML (P<0.01; Fig. 2B). From
the Oncomine leukemia dataset of Stegmaier et al (14), SPINK2 expression was determined to be
elevated by 20.21-fold in 9 AML samples compared with the PMBCs of
3 normal samples (P=2.12×10−5; Fig. 2C). The study on leukemia conducted by
Haferlach et al (15)
revealed that SPINK2 levels were elevated by 2.86-fold
(P=8.15×10−42) in 542 AML samples compared with 74
normal samples (Fig. 2D). Therefore,
the high expression of SPINK2 in AML was validated in larger
cohorts.
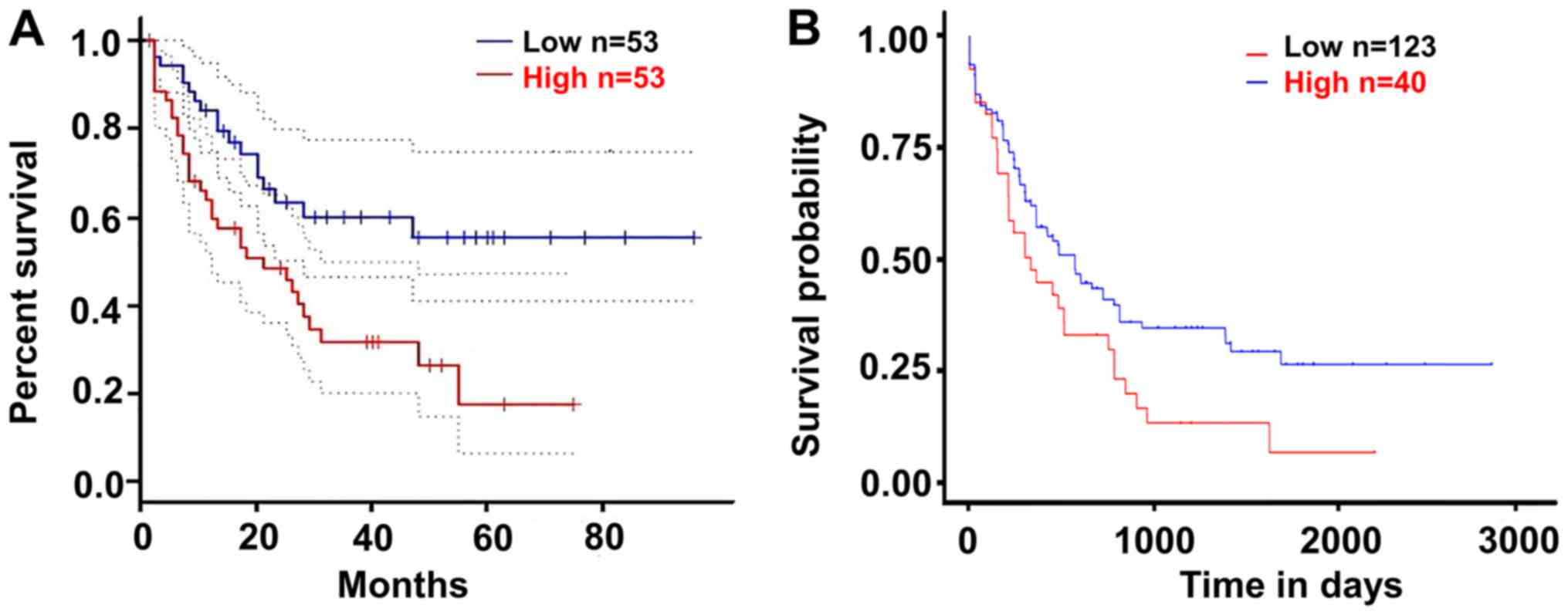
Upregulated SPINK2 is associated with
poor outcomes in patients with AML
The present study validated the effect of SPINK2 on
the prognosis of patients with AML using the GEPIA and UALCAN
datasets. According to the median expression level of SPINK2, the
patients were divided into a high-expression and a low-expression
group. The results suggested that patients with high SPINK2
expression had shorter survival time than those with low SPINK2
levels in the GEPIA dataset (hazard ratio, 2.3; P=0.0039; Fig. 3A). In the UALCAN dataset, 163
patients with AML were analyzed for their expression levels of
SPINK2. The Kaplan-Meier analysis demonstrated that patients with
high SPINK2 expression had shorter survival time than patients with
lower SPINK2 levels (P=0.024; Fig.
3B).
Discussion
The pathogenesis of AML is heterogeneric and complex
(17). To improve the outcome of AML
treatment, it is important to understand the mechanism of AML
(5,18). To detect DEGs in patients with AML,
high-throughput platforms for the analysis of gene expression, such
as microarrays, are increasingly used to identify novel molecular
biomarkers and drug targets for clinical applications. Currently,
microarray technology that combines bioinformatics analysis allows
the wide exploration of the molecular mechanisms involved in the
development and progression of AML. Using this method, Gal et
al (19) compared DEGs of
cluster of differentiation (CD) 34+ CD38−
cells and CD34+ CD38+ cells using
microarrays, and observed that 409 genes were 2-fold overexpressed
or downregulated between the two cell populations. Previous focus
on the Notch signaling pathway revealed that regulated leukemic
stem cell self-renewal could be investigated to identify novel
targets for therapy (19). Based on
microarray data from 163 patients, Metzeler et al (20) developed a gene signature including an
86-probe set to predict the overall survival rate of patients with
AML. When the authors applied the prognostic score in an
independent cohort, this continuous score remained a significant
predictor for the outcomes of patients with AML (20). Therefore, bioinformatics analysis
could accelerate our understanding of AML (21,22).
The present study analyzed the gene expression
patterns of patients with AML and healthy individuals by
computational methods, and identified 1,744 upregulated and 1,956
downregulated genes in patients with AML. GO and KEGG functional
enrichment analyses demonstrated that the upregulated genes were
mainly associated with metabolic processes, which was in agreement
with the results of a previous study (23). Furthermore, the genes involved in the
immune response and cell surface signaling were downregulated
according to our results. Cancer cells are able to avoid the immune
response via a number of mechanisms (24). Tumor cells escape the immune response
either by employing mechanisms to suppress the immune response, or
by downregulating the expression of immunogenic molecules (25). Controlling the immune system in AML
may facilitate the treatment of AML. Notably, the expression level
of SPINK2 was the most altered among all the upregulated genes.
Further analysis validated the upregulation of SPINK2 in patients
with AML from the Oncomine and GEPIA datasets. Analysis further
suggested that patients with AML and high expression levels of
SPINK2 would have poor outcomes. Therefore, the findings from the
present study indicated that SPINK2 may serve an important role in
the development of AML.
SPINK2, also known as human seminal plasma inhibitor
II, belongs to the SPINK family, which consists of members
containing ≥1 conserved Kazal domain composed of 6 cysteine
residues (26–28). The expression of SPINK2 is closely
associated with cancer development, and high transcript levels of
SPINK2 can be detected in patients with primary cutaneous follicle
center cell lymphoma (29). SPINK2
serves as a classification marker for lymphoma, as well as a
predictive marker of response to cancer therapy. Chen et al
(30) reported that SPINK2 was
significantly elevated in the majority of the leukemia cell lines
investigated, and served an important role in tumor progression and
response to treatment. Hoefnagel et al (29) proposed a possible interaction between
SPINK2 and its currently unknown cancer-associated target
proteinase, which appeared to be essential for tumor progression
and metastasis. In the present study, patients with AML and high
levels of SPINK2 had reduced survival times. Thus, we hypothesize
that SPINK2 may serve an important role in the process of AML.
However, the molecular mechanism of SPINK2 affecting the
tumorigenic process remains unclear. Thus, further functional
studies are required to deeply investigate how SPINK2 affects the
development of AML.
There are some limitations to the present study.
First, the GSE9476 dataset provides the complete data of AML and
healthy people, but a larger dataset, including more patients,
would be more robust. Of note, the GSE9476 dataset was only used to
investigate the DEGs, and so the results from the present study
require the analysis of additional datasets in order to be
validated. The different expression levels of SPINK2 were validated
in the present study by analyzing 70 healthy individuals and 173
patients with AML from the GEPIA database, and by analyzing 542
patients with AML and 74 healthy individuals in the report of
leukemia by Haferlach et al (15). Furthermore, the prognosis of patients
with AML were validated by analyzing 106 patients with AML from the
GEPIA dataset (11) and 163 patients
with AML from the UALCAN dataset (12). Therefore, despite the limited sample
size, the results are credible. Secondly, AML has several
subgroups, and the DEGs in patients with different subtypes were
not analyzed in the present study. Thirdly, although the
upregulated levels of SPINK2 mRNA in the Oncomine dataset were
investigated in the present study, the protein levels of SPINK2
were not. Additional AML samples are required to validate these
results. Finally, the present study only determined that SPINK2 may
contribute to the development of AML; further studies are required
that investigate the underlying molecular mechanisms of SPINK2 in
the progression of AML.
To conclude, the present study has provided a
comprehensive investigation of dysregulated genes involved in the
progression of AML. SPINK2 could be regarded as a hub gene in
patients with AML, and elevated levels of SPINK2 were associated
with the poor prognosis in these patients. However, further
molecular biology investigations are needed to confirm the
mechanisms of SPINK2 in AML.
Acknowledgements
The authors would like to thank Dr Jun Zhang
(Hematology Department, Linyi Central Hospital) for providing
critical review and elaborated revision of this manuscript. In
addition, the authors would like to thank Dr Guoming Deng and Dr
Ebun Omoyinmi (Hematology Department, Linyi Central Hospital) for
their helpful discussion regarding this report.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
CX, JZ, YX and GZ curated the data. CX and GZ
performed the investigations. CX and XW designed the study. JZ
performed the statistical analysis. CX and XW wrote the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Academic
Committee of Linyi Central Hospital. Written informed consent was
obtained from all patients included within the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AML
|
acute myeloid leukemia
|
|
DEGs
|
differentially expressed genes
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
DAVID
|
Database for Annotation, Visualization
and Integrated Discovery
|
|
SPINK2
|
serine protease inhibitor Kazal-type
2
|
|
GEO
|
Gene Expression Omnibus
|
|
TCGA
|
The Cancer Genome Atlas
|
|
FC
|
fold change
|
|
FDR
|
false discovery rate
|
|
BP
|
biological process
|
|
MF
|
molecular function
|
|
CF
|
component function
|
References
|
1
|
Zhu B, Zhang J, Wang X, Chen J and Li C:
Correlation between acute myeloid leukemia and IL-17A, IL-17F, and
IL-23R gene polymorphism. Int J Clin Exp Pathol. 8:5739–5743.
2015.PubMed/NCBI
|
|
2
|
Quotti Tubi L, Canovas Nunes S, Brancalion
A, Doriguzzi Breatta E, Manni S, Mandato E, Zaffino F, Macaccaro P,
Carrino M, Gianesin K, et al: Protein kinase CK2 regulates AKT,
NF-κB and STAT3 activation, stem cell viability and proliferation
in acute myeloid leukemia. Leukemia. 31:292–300. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Godwin CD, Gale RP and Walter RB:
Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia.
31:1855–1868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Belson M, Kingsley B and Holmes A: Risk
factors for acute leukemia in children: A review. Environ Health
Perspect. 115:138–145. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Estey E and Dohner H: Acute myeloid
leukaemia. Lancet. 368:1894–1907. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schuz J and Erdmann F: Environmental
exposure and risk of childhood leukemia: An overview. Arch Med Res.
47:607–614. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yin F, Shu L, Liu X, Li T, Peng T, Nan Y,
Li S, Zeng X and Qiu X: Microarray-based identification of genes
associated with cancer progression and prognosis in hepatocellular
carcinoma. J Exp Clin Cancer Res. 35:1272016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yin F, Yi S, Wei L, Zhao B, Li J, Cai X,
Dong C and Liu X: Microarray-based identification of genes
associated with prognosis and drug resistance in ovarian cancer. J
Cell Biochem. 120:6057–6070. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Raich T and Powell S: Identification of
bacterial and fungal pathogens from positive blood culture bottles:
A microarray-based approach. Methods Mol Biol. 1237:73–90. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stirewalt DL, Meshinchi S, Kopecky KJ, Fan
W, Pogosova-Agadjanyan EL, Engel JH, Cronk MR, Dorcy KS, McQuary
AR, Hockenbery D, et al: Identification of genes with abnormal
expression changes in acute myeloid leukemia. Genes Chromosomes
Cancer. 47:8–20. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stegmaier K, Ross KN, Colavito SA,
O'Malley S, Stockwell BR and Golub TR: Gene expression-based
high-throughput screening (GE-HTS) and application to leukemia
differentiation. Nat Genet. 36:257–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haferlach T, Kohlmann A, Wieczorek L,
Basso G, Kronnie GT, Béné MC, De Vos J, Hernández JM, Hofmann WK,
Mills KI, et al: Clinical utility of microarray-based gene
expression profiling in the diagnosis and subclassification of
leukemia: Report from the International Microarray Innovations in
Leukemia Study Group. J Clin Oncol. 28:2529–2537. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang YX, Zhang TJ, Yang DQ, Yao DM, Yang
L, Zhou JD, Deng ZQ, Ma JC, Guo H, Wen XM, et al: Reduced miR-215
expression predicts poor prognosis in patients with acute myeloid
leukemia. Jpn J Clin Oncol. 46:350–356. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ryotokuji T, Yamaguchi H, Ueki T, Usuki K,
Kurosawa S, Kobayashi Y, Kawata E, Tajika K, Gomi S, Kanda J, et
al: Clinical characteristics and prognosis of acute myeloid
leukemia associated with DNA-methylation regulatory gene mutations.
Haematologica. 101:1074–1081. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gal H, Amariglio N, Trakhtenbrot L,
Jacob-Hirsh J, Margalit O, Avigdor A, Nagler A, Tavor S, Ein-Dor L,
Lapidot T, et al: Gene expression profiles of AML derived stem
cells; similarity to hematopoietic stem cells. Leukemia.
20:2147–2154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Metzeler KH, Hummel M, Bloomfield CD,
Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M,
Marcucci G, Whitman SP, et al: An 86-probe-set gene-expression
signature predicts survival in cytogenetically normal acute myeloid
leukemia. Blood. 112:4193–4201. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Delas MJ, Sabin LR, Dolzhenko E, Knott SR,
Munera Maravilla E, Jackson BT, Wild SA, Kovacevic T, Stork EM,
Zhou M, et al: lncRNA requirements for mouse acute myeloid leukemia
and normal differentiation. ELife. 6(pii): e256072017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meyer SE, Qin T, Muench DE, Masuda K,
Venkatasubramanian M, Orr E, Suarez L, Gore SD, Delwel R, Paietta
E, et al: DNMT3A haploinsufficiency transforms FLT3ITD
myeloproliferative disease into a rapid, spontaneous, and fully
penetrant acute myeloid leukemia. Cancer Ddiscov. 6:501–515. 2016.
View Article : Google Scholar
|
|
23
|
Salvestrini V, Zini R, Rossi L, Gulinelli
S, Manfredini R, Bianchi E, Piacibello W, Caione L, Migliardi G,
Ricciardi MR, et al: Purinergic signaling inhibits human acute
myeloblastic leukemia cell proliferation, migration, and
engraftment in immunodeficient mice. Blood. 119:217–226. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Austin R, Smyth MJ and Lane SW: Harnessing
the immune system in acute myeloid leukaemia. Crit Rev Oncol
Hematol. 103:62–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Davidson-Moncada J, Viboch E, Church SE,
Warren SE and Rutella S: Dissecting the immune landscape of acute
myeloid leukemia. Biomedicines. 6(pii): E1102018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dietrich MA, Slowinska M, Karol H, Adamek
M, Steinhagen D, Hejmej A, Bilińska B and Ciereszko A: Serine
protease inhibitor Kazal-type 2 is expressed in the male
reproductive tract of carp with a possible role in antimicrobial
protection. Fish Shellfish Immunol. 60:150–163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kherraf ZE, Christou-Kent M, Karaouzene T,
Amiri-Yekta A, Martinez G, Vargas AS, Lambert E, Borel C, Dorphin
B, Aknin-Seifer I, et al: SPINK2 deficiency causes infertility by
inducing sperm defects in heterozygotes and azoospermia in
homozygotes. EMBO Mol Med. 9:1132–1149. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee B, Park I, Jin S, Choi H, Kwon JT, Kim
J, Jeong J, Cho BN, Eddy EM and Cho C: Impaired spermatogenesis and
fertility in mice carrying a mutation in the Spink2 gene expressed
predominantly in testes. J Biol Chem. 286:29108–29117. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hoefnagel JJ, Dijkman R, Basso K, Jansen
PM, Hallermann C, Willemze R, Tensen CP and Vermeer MH: Distinct
types of primary cutaneous large B-cell lymphoma identified by gene
expression profiling. Blood. 105:3671–3678. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen T, Lee TR, Liang WG, Chang WS and Lyu
PC: Identification of trypsin-inhibitory site and structure
determination of human SPINK2 serine proteinase inhibitor.
Proteins. 77:209–219. 2009. View Article : Google Scholar : PubMed/NCBI
|