Introduction
Cervical cancer is one of the most common cancers
among women. According to statistics reported in 2018, ~528,000
patients worldwide are diagnosed with cervical cancer each year
(1). Global tumor statistics show
that cervical cancer ranks fourth in morbidity and mortality among
female malignancies worldwide; approximately 85% of cervical cancer
deaths worldwide occur in less-developed or developing countries,
and cervical cancer mortality in low- and middle-income countries
is 18 times higher than in high-income countries (2). The incidence of cervical cancer in
China is ranked second worldwide only to Chile in 2015, and its
prevalence in younger women is notable; ~93 women die of cervical
cancer every day (3). Efforts to
prevent cervical cancer, in areas of China with low economy, and to
improve morbidity and mortality in patients with cervical cancer
requires improvement in global cancer treatment regimens. Despite
the increasing popularity of and diagnostic techniques for early
screening of cervical cancer, the number of new cases in China is
13.15 million per year, accounting for 28.8% of new cases worldwide
in 2019, and shows an incidence trend in younger age groups (35–39
years) (4). The occurrence and
development of cervical cancer is affected by various regulatory
factors. A study in China and worldwide revealed that persistent
infection with high-risk human papillomavirus (HPV) was closely
related to the pathogenesis of cervical cancer (5). Mutations in some genes, such as p53,
p16, and Nm23, may also cause malignant transformation of cells
(6). Biomarkers are biochemical
indicators that can label changes in systems, organs, tissues, and
cells (6). To the best of our
knowledge, few studies on cervical cancer genes have been
conducted. One of the main limiting factors of better diagnosis and
treatment of cervical cancer is the lack of knowledge and
determination of effective molecular markers (7). Therefore, the identification of
specific molecules has become a research hotspot. The present study
explores cervical cancer-related genes based on the original data
in public databases and provides novel ideas for early cervical
cancer research.
In recent years, as gene chips and sequencing
technologies continue to evolve, high-throughput data have
burgeoned. The Cancer Genome Atlas (TCGA) and the human
tumor-associated Gene Expression Omnibus databases are two large
public databases that provide high-throughput data for a variety of
diseases, such as lung and breast cancer, for analysis (8). The introduction of TCGA database in
2005 through a new genomic analysis technology deepened
researchers' comprehensive understanding of cancer genetics and
provides an open dataset to assist with generating new cancer
treatments, diagnostic methods and prevention strategies (9). As a publicly funded project, TCGA
database aims to distinguish approximate genomic variations in
cancer to create a comprehensive ‘cancer genomic profile’ map.
Currently, TCGA database researchers have studied a total of 14,551
cumulative cases of >30 types of cancer in humans (such as lung
cancer, cervical cancer and colorectal cancer) through large-scale
genome sequencing and comprehensive multidimensional analysis.
Based on TCGA database, the present study used the R language to
mine 654 differentially expressed genes (DEGs) in cervical cancer
tissues and combined this approach with bioinformatics software and
literature mining methods to define the functions and pathways
enriched in the DEGs. Combining 304 cervical cancer clinical
samples from the TCGA database and analysing the prognosis of the
patients, important information for studying the molecular
mechanism of genes in the occurrence and development of cervical
cancer was assembled and provided novel ideas and targets for the
future treatment of cervical cancer.
Materials and methods
Screening of DEGs
TCGA database (https://cancergenome.nih.gov/) was used to download
raw sequencing data and clinical information. The standardized
datasets were obtained by annotation and integration. Perl
(http://www.perl.org/) scripting tools were used
to perform integration processing of datasets. Analysis of DEGs
between cervical cancer and normal tissues was conducted with the R
language package (version 3.5.1; Shengxin Self-study Network);
differential expression was defined as a Log2 fold
change (FC) of >4 and P adj=0.001). The data for clinical
survival times and the expression of DEGs were merged using Perl
scripting tools, and the prognostic value of each DEG was
subsequently evaluated using the log-rank method.
Gene ontology (GO) and Kyoto
encyclopedia of genes and genomes (KEGG) pathway enrichment
analysis
In the present study, a total of 654 DEGs were
screened from cervical cancer samples. The 654 DEGs were subjected
to KEGG analysis (https://www.kegg.jp/) and GO enrichment analysis
(http://geneontology.org). As a result, 195 genes
were enriched into the KEGG signaling pathway, and 246 genes were
enriched in GO and subsequently visualized to obtain KEGG map and
GO map, respectively. KEGG is a database resource for understanding
the advanced functions and activities of biological systems,
especially from large-scale molecular datasets generated by genome
sequencing and other high-throughput experimental techniques. The
KEGG database was used to perform pathway enrichment analysis on
the identified DEGs. The RSQLite program in the R package was used
to assist the transformation of DEGs into Entrez IDs. Then, the
enrichment data were visualized using Cytoscape software
(https://cytoscape.org/). GO was used to describe
gene functions and relationships between these concepts; GO
annotations are statements that use the concept of gene ontology to
describe the function of a particular gene (10,11).
DEGs were uploaded to the Database for Annotation, Visualization
and Integrated Discovery website (https://david.ncifcrf.gov/) to obtain the gene
enrichment results, and the results were then visualized with the R
language package as the GO analysis results.
Three-gene signature and risk score
assessment
DEGs were screened using single-factor Cox
regression analysis using the Survival package to screen out DEGs
associated the overall survival (OS) of patients. Multivariate Cox
regression analysis was used to establish a multigene prognosis
prediction model to derive three-gene signatures and calculate risk
scores as follows: Risk score=Σ βgenei ×
Expgenei (i=1-N)=βgene1 × Expgene1
+ βgene2 × Expgene2 +…+ βgeneN ×
ExpgeneN (12); where β
represents the regression coefficient, Exp represents the risk
ratio, i represents the running formula, and N represents the total
number of samples. Patients were divided into a high-risk group and
a low-risk group according to the median score. The effect of each
gene on patient survival was then assessed using the Kaplan-Meier
method. The receiver operating characteristic (ROC) curve was
plotted, and the survival ROC package in the R language was used to
estimate the area under the curve (AUC).
Statistical analysis
All the data in the present study were analyzed
using R software (version 3.5.1) and Perl scripting tools.
Continuous variables are presented as mean ± SD, while categorical
variables are presented as frequencies and percentages. The
relationship between gene expression and clinical features was
assessed using χ2 and Student's t-test. Each risk gene
and prognosis gene marker were analyzed using Kaplan-Meier survival
analysis and univariate/multivariate Cox proportional hazards
regression. P<0.05 (two-tailed) were considered to indicate a
statistically significant difference.
Results
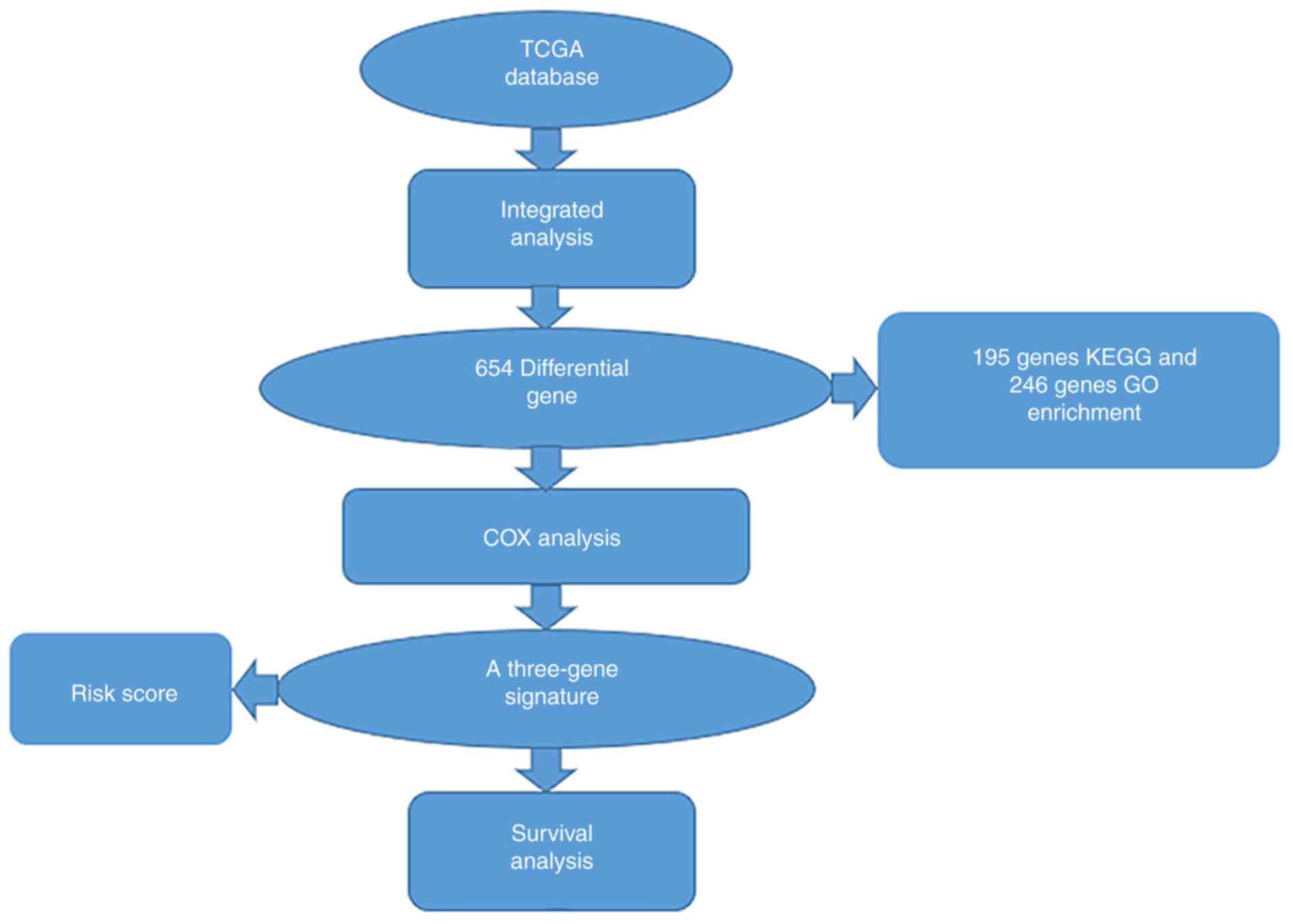
Workflow presentation
A series of combinatorial analysis were used to
identify DEGs in cervical cancer and to screen for three-gene
signatures associated with cervical cancer prognosis through a
variety of methods (Fig. 1).
Screening of DEGs in cervical cancer
samples
Data for 304 patients with cervical cancer and for
309 cervical cancer tissue samples were downloaded from TCGA
database, however only 301 cases were matched between the clinical
data and tissue sample data. A total of three of the tissue samples
did not have associated clinical data, and 3 patients with clinical
data did not have associated tissue samples; these samples were
excluded. The 309 cervical cancer tissue samples included 306
cervical cancer tumor tissues and 3 normal tissues from healthy
controls. The detailed characteristics of the clinical data were
stratified according to age, grade, T stage, lymph node status and
metastasis, as shown in Table I. The
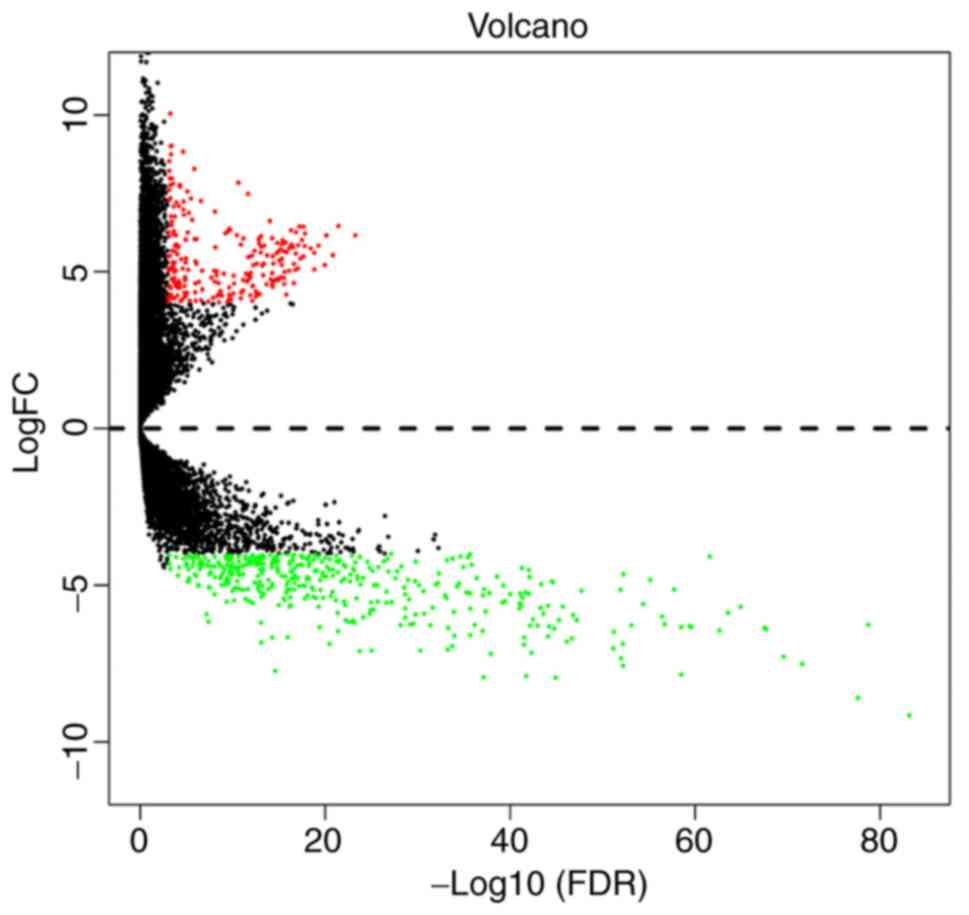
results of the differential gene expression in cervical cancer are
displayed as a volcano plot and show whether the P- and
|log2FC| values satisfied the logic of different tests
(Fig. 2). A total of 301 cervical
cancer samples downloaded from TCGA database were analyzed for
differential gene expression, resulting in the identification of
654 DEGs, among which 239 were upregulated and 415 were
downregulated (fold change >4, P adj=0.001).
 | Table I.Number of cases and proportion of each
clinical feature of patients with cervical cancer. |
Table I.
Number of cases and proportion of each
clinical feature of patients with cervical cancer.
| Variable | Case, n (%) |
|---|
| Age |
|
| ≤46 | 154 (51.2) |
|
>46 | 147 (48.8) |
| Grade |
|
|
G1+G2 | 152 (50.5) |
|
G3+G4 | 117 (38.9) |
| GX | 24 (7.9) |
| NA | 8 (2.7) |
| T stage |
|
| Tis | 1 (0.3) |
|
T1+T2 | 209 (69.4) |
|
T3+T4 | 30 (10.0) |
| TX | 17 (5.6) |
| NA | 44 (14.6) |
| Lymph node
status |
|
| N0 | 132 (43.9) |
| N1 | 59 (19.6) |
| NX | 66 (21.9) |
| NA | 44 (14.6) |
| Metastasis |
|
| M0 | 115 (38.2) |
| M1 | 10 (3.3) |
| MX | 128 (42.5) |
| NA | 48 (15.9) |
KEGG enrichment analysis and signaling
pathway visualization
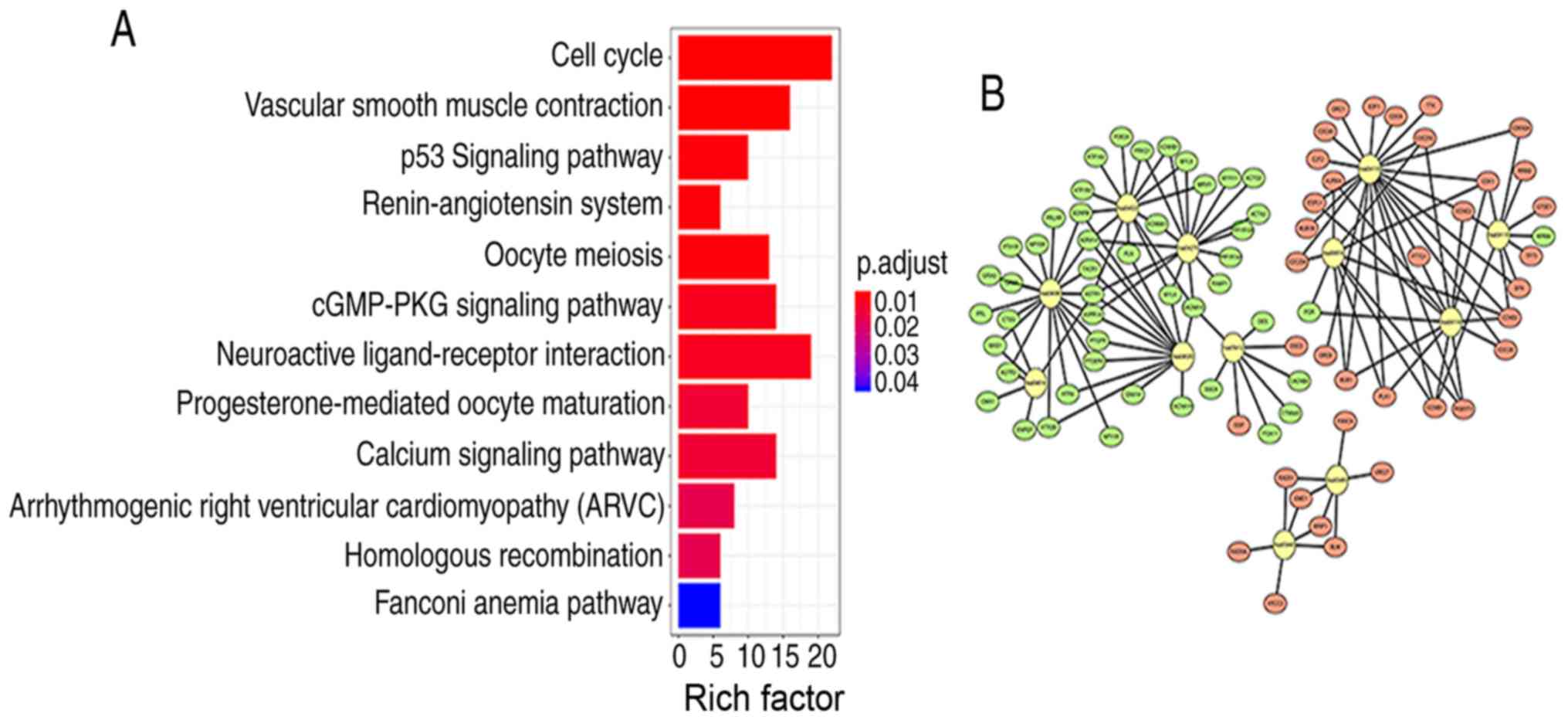
Signaling pathway enrichment analysis of the DEGs
was conducted using the KEGG database. The 654 differentially
expressed genes in cervical cancer samples were analyzed by KEGG
enrichment, and the results of 195 enriched genes were
statistically significant (P<0.05). The KEGG analysis results
showed that the 195 DEGs were significantly associated with 12
signaling pathways (P<0.05; Table
II). The top five signaling pathways with the strongest
enrichment in the KEGG analysis were as follows: hsa04110 (‘Cell
cycle’); hsa04270 (‘vascular smooth muscle contraction’); hsa04115
(‘p53 signaling pathway’); hsa04614 (‘renin-angiotensin system’)
and hsa04114 (‘oocyte meiosis’). Next, R package components were
used to construct KEGG signaling pathway diagrams (Fig. 3A). The larger the value of Rich
factor value, the greater the enrichment. Rich factor=number of
differentially expressed genes located under the pathway
term/number of all annotated genes located under the pathway term.
The enrichment analysis data were visualized using Cytoscape
software (Fig. 3B).
| Table II.Significantly enriched KEGG pathway
of differential expressed genes. |
Table II.
Significantly enriched KEGG pathway
of differential expressed genes.
| ID | Description | GeneRatio | BgRatio | P-value | Adjust P-value | Count |
|---|
| hsa04110 | Cell cycle | 22/195 | 124/7469 |
6.20×10−13 |
1.32×10−10 | 22 |
| hsa04270 | Vascular smooth
muscle contraction | 16/195 | 121/7469 |
8.12×10−8 |
8.65×10−6 | 16 |
| hsa04115 | p53 signaling
pathway | 10/195 | 72/7469 |
1.53×10−5 |
8.81×10−5 | 10 |
| hsa04614 | Renin-angiotensin
system | 6/195 | 23/7469 |
2.04×10−5 |
8.81×10−5 | 6 |
| hsa04114 | Oocyte meiosis | 13/195 | 125/7469 |
2.07×10−5 |
8.81×10−5 | 13 |
| hsa04022 | cGMP-PKG signaling
pathway | 14/195 | 163/7469 |
8.50×10−5 |
3.02×10−3 | 14 |
| hsa04080 | Neuroactive
ligand-receptor interaction | 19/195 | 277/7469 | 10.5
×10−5 |
3.19×10−3 | 19 |
| hsa04914 |
Progesterone-mediated oocyte
maturation | 10/195 | 99/7469 | 24.2
×10−5 |
6.43×10−3 | 10 |
| hsa04020 | Calcium signaling
pathway | 14/195 | 183/7469 | 2.9
×10−4 |
6.86.4×10−3 | 14 |
| hsa05412 | Arrhythmogenic
right ventricular cardiomyopathy | 8/195 | 72/7469 |
5.35×10−4 |
1.14×10−2 | 8 |
| hsa03440 | Homologous
recombination | 6/195 | 41/7469 |
6.15×10−4 |
1.19×10−2 | 6 |
| hsa03460 | Fanconi anemia
pathway | 6/195 | 54/7469 |
2.67×10−3 |
4.74×10−5 | 6 |
Functional analysis of DEGs via GO
enrichment analysis
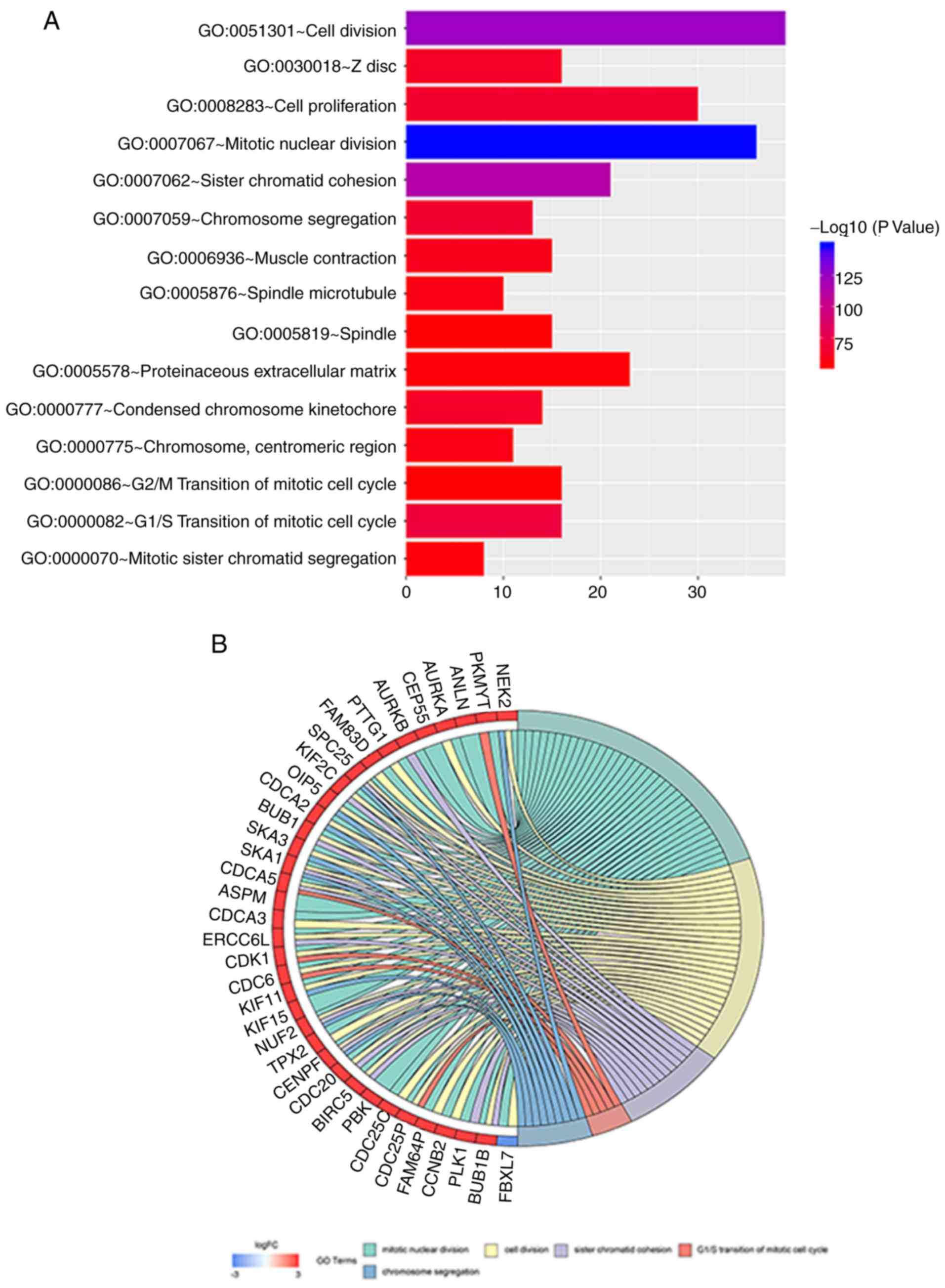
GO analysis was performed on all 654 differentially
expressed genes, of which 246 genes were enrichment and
statistically significant (P<0.05). The 246 DEGs were also
analyzed for GO term enrichment using the Gene Ontology Enrichment
Analysis Software Toolkit. The following GO biological process
terms were mainly enriched in the DEGs: Regulation of mitotic
nuclear division, regulation of cell division and regulation of
sister chromatid cohesion (Table
III). The main biological functions of the DEGs studied that
are enriched, the number of enriched genes and the degree of
enrichment of different GO terms arepresented in Fig. 4A. The DEGs in the top 5 GO terms with
the highest enrichment level (the top 5 with the lowest P-value)
demonstrated the highest degree of enrichment are shown; the genes
marked in red were upregulated, while those marked in blue were
downregulated (Fig. 4B).
| Table III.Enrichment analysis of differentially
expressed genes in the Gene Onotology enrichment analysis. |
Table III.
Enrichment analysis of differentially
expressed genes in the Gene Onotology enrichment analysis.
| ID | Biological
function | Count | P-value |
|---|
| GO:0007067 | Mitotic nuclear
division | 36 |
1.30×10−15 |
| GO:0051301 | Cell division | 39 |
4.01×10−13 |
| GO:0007062 | Sister chromatid
cohesion | 21 |
4.58×10−12 |
| GO:0000082 | G1/S
transition of mitotic cell cycle | 16 |
1.12×10−7 |
| GO:0007059 | Chromosome
segregation | 13 |
2.74×10−7 |
| GO:0008283 | Cell
proliferation | 30 |
3.10×10−7 |
| GO:0000777 | Condensed
chromosome kinetochore | 14 |
5.72×10−7 |
| GO:0030018 | Z disc | 16 |
6.73×10−7 |
| GO:0006936 | Muscle
contraction | 15 |
1.27×10−6 |
| GO:0005876 | Spindle
microtubule | 10 |
2.17×10−6 |
| GO:0000775 | Chromosome,
centromeric region | 11 |
2.60×10−6 |
| GO:0000070 | Mitotic sister
chromatid segregation | 8 |
3.42×10−6 |
| GO:0005578 | Proteinaceous
extracellular matrix | 23 |
3.98×10−6 |
| GO:0005819 | Spindle | 15 |
4.90×10−6 |
| GO:0000086 | G2/M
transition of mitotic cell cycle | 16 |
5.19×10−6 |
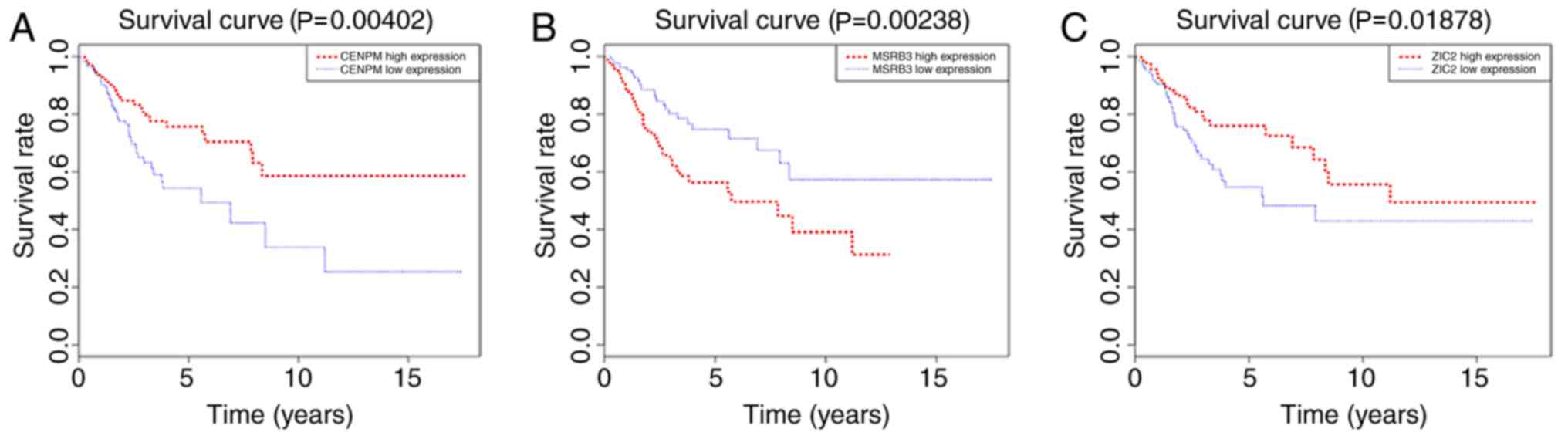
Detection of three-gene signatures and
risk scores
Three specific genes were screened using univariate
survival analysis and Cox regression analysis, and the significant
association of three genes as independent predictors of OS were
validated (P<0.05). This three-gene signature comprised
methionine sulfoxide reductase B3 (MSRB3), centromere protein M
(CENPM) and Zic family member 2 (ZIC2). The risk score for each
patient was calculated based on the sum of the weighted expressions
of the three genes (likelihood ratio test, 48.04;
P=3.484×10−9; n=304; Table
IV). The result of the analysis revealed that the higher the
risk score, the worse the clinical prognosis. To identify possible
genes associated with OS in patients with cervical cancer,
Kaplan-Meier curves and the log-rank test were used to evaluate the
association between gene expression and patient survival. As shown
in Fig. 5, the expression of CENPM
and ZIC2 was positively associated with the survival rate of
patients with cervical cancer, while the expression of MSRB3 was
negatively associated with the survival rate of patients with
cervical cancer.
| Table IV.Multivariate Cox regression analysis
in a three-gene signature |
Table IV.
Multivariate Cox regression analysis
in a three-gene signature
| Gene | Coef | Exp (coef) | SE (coef) | z | P-value |
|---|
| MSRB3 | 0.18134 | 1.19882 | 0.08608 | 2.107 |
3.51×10−2 |
| CENPM | 0.45186 | 0.63644 | 0.16121 | −2.803 |
5.06×10−3 |
| ZIC2 | 0.11147 | 0.89452 | 0.05007 | −2.226 |
2.60×10−2 |
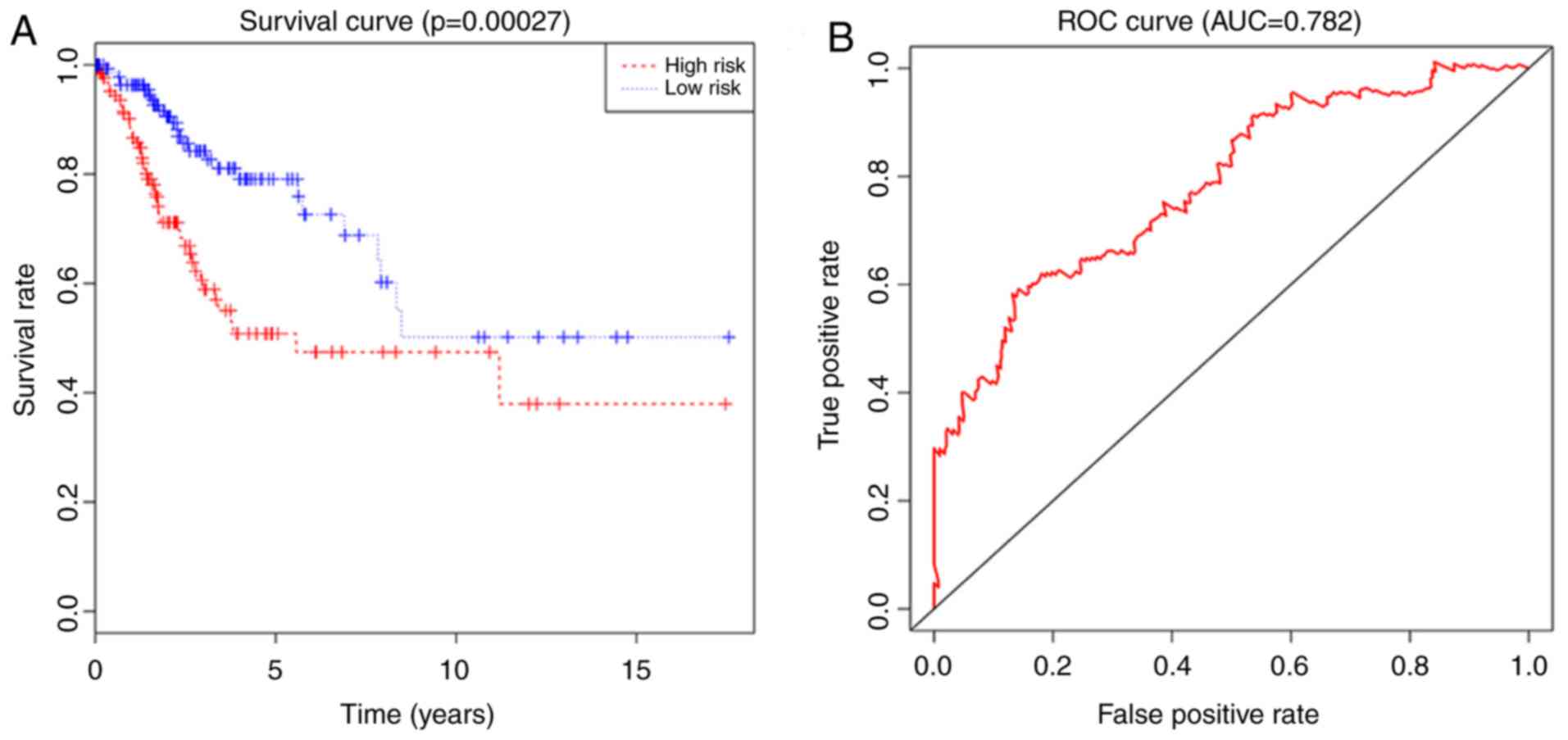
Verification of the regression model
and risk degree
Each patient from the dataset was assigned to the
high-risk group or the low-risk group based on their risk score. In
the present study, the median risk score was calculated to be 1.33
for the 304 patients based on the risk score formula, 152 patients
with the score <1.33 were included in the low-risk group, and
152 patients with the score ≥1.33 were included in the high-risk
group. The analysis results indicated that the risk score was
significantly associated with OS (P<0.05) as a prognostic factor
in univariate regression analysis. The results also showed that the
risk score was an independent prognostic factor for OS in
multivariate Cox regression analysis (P<0.05; Fig. 6A). In addition, the ROC curve and AUC
(0.782) analysis results showed high sensitivity and specificity
(P<0.05; Fig. 6B).
Discussion
Cervical cancer was reported as the third most
common gynecological malignancy worldwide in 2012 (13). In China, the incidence of cervical
cancer ranks first among malignant tumors in the female
reproductive system (14), and
>99% of patients with cervical cancer are infected with HPV
(11,15,16). In
2011, the American Cancer Society, the American Society for
Colposcopy and Cervical Pathology, and the American Society for
Clinical Pathology updated their joint guidelines for cervical
cancer screening, as well as the US Preventive Services Task Force
(17–19). It can be seen that cervical cancer
screening for the early detection and prevention of cervical cancer
is crucial to reduce the incidence and mortality of cervical cancer
in China. In addition, understanding the pathological mechanisms
and finding potential survival-related genes involved in cervical
cancer development is a direction requiring diligent study. With
the latest developments in biotechnology, an increasing number of
technologies, such as gene sequencing, transcriptome analysis, DNA
methylation and epigenetic analysis, are being used to study tumor
genomes (20). In the present study,
TCGA database was used to analyze normal tissues from healthy
controls and tumor tissues of patients with cervical cancer to
determine the DEGs in tumor tissues. Then, the three-gene signature
and risk scores were verified by univariate and multivariate Cox
regression analysis, and the risk scores were found to be
significantly associated with OS. It can be concluded that our
three-gene signature can predict the pathogenesis of cervical
cancer at the genetic level. Therefore, our research results not
only provide an experimental background for future experiments on
cervical cancer but also provide strong evidence for prognostic
evaluation of cervical cancer in a clinical setting.
The three genes (MSRB3, CENPM and ZIC2) were found
to function both as risk factors and protective factors. The
molecular functions of these three genes in other tumors, will
provide a reference for further studies on these genes in cervical
cancer. MSRB3 is a member of the MSR family of proteins (21) and plays a vital role in cold
tolerance by eliminating methionine sulfoxide and reactive oxygen
species, which accumulate in the endoplasmic reticulum during cold
acclimation (22). Recent study
found that MSRB3 deficiency induces apoptosis in breast cancer
cells, lung cancer cells and colon cancer cells through
p53-independent and ER stress-dependent pathways (23,24).
CENPM, also known as proliferation-associated nuclear element 1, is
a protein encoded by the CENPM gene in humans, and is involved in
centromere assembly and immune responses (25). Human CENPM is mainly present in
immune cells of tumor tissues of leukemias and lymphomas and in
tumor-derived cell lines (26). In
addition, CENPM participates in the formation of complexes in the
nucleoplasm of viable human breast cancer cells outside centromeres
(27). ZIC2 is located on chromosome
13q32, and its amino acid sequence is highly conserved; a previous
study has found that loss of heterozygosity in the 13q32 region of
ZIC2 leads to anterior cerebral schizophrenia (28). It has been reported that ZIC2 is a
critical regulator of Kaposi's sarcoma-associated herpesvirus
latency maintenance (29). Marchini
et al (30) found that ZIC2
overexpression was closely related to proliferation and metastasis
of ovarian cancer cells. Additionally, ZIC2 plays an indispensable
role in cell proliferation and apoptosis in pancreatic ductal
adenocarcinoma (PDAC) (31) and
hepatocellular carcinoma, and ZIC2 is a potential therapeutic
target in PDAC (32). In summary,
these three genes play almost completely different biological roles
in different types of cancers, however these have not been
validated in further functional and mechanistic experiments in
cervical cancer.
In the present study, a three-gene signature was
established and identified as an independent prognostic factor for
patients with cervical cancer. The Cox coefficient based on
univariate Cox regression was obtained and established a risk
score. In addition, ROC analysis was used to determine the optimal
cutoff for classifying high- and low-risk patients. In the
univariate Cox regression model, patients with high-risk scores had
significantly shorter survival times. The expression of the MSRB3,
CENPM and ZIC2 genes was significantly associated with the OS rate
of patients with cervical cancer (P<0.05), revealing that these
three genes include both protective factors and risk factors. These
results indicate that genes play a key role in the progression and
prognosis of cervical cancer. The present study has the advantage
of using large databases, complete clinical data, and good sample
quality control procedures and provides novel ideas for
tumor-targeted therapy. The limitations of the present study are
that the gene expression level data obtained from TCGA database may
not fully represent the expression of MSRB3, CENPM and ZIC2 at the
protein level. Subsequent studies require further experiments to
validate these results and further analysis, including
immunohistochemistry assays. To make the present study more
clinically meaningful, it is necessary to study the three target
genes and their related signaling pathways via functional
experiments; in clinical terms, further association between these
genes and the prognosis of patients with cervical cancer needs to
be evaluated from clinical data. Therefore, cervical tumor
specimens and clinical prognosis information will be collected from
The Affiliated Hospital of Zunyi Medical University) and verify the
results through specific experiments in our hospital oncology
laboratory. In addition, the development of biomarkers usually
includes the establishment of models such as training sets,
validation sets and test sets. This model was not built in the
present study as the literature states it is not suitable for small
sample data. Since the sample is relatively small, it is not
suitable to randomly divide data into three parts for verification
when setting up grouping. Finally, statistical analysis showed that
the P-value of the survival curves of each group was not
statistically significant, and the AUC value of the ROC curve was
relatively small. When the sample is small, the criteria for
establishing the three models are not enough and the results are
less accurate. In conclusion, the present study used TCGA database
and found that MSRB3, CENPM and ZIC2 can be used as prognostic
biomarkers for cervical cancer, thus providing novel ideas and
targets for future research on cervical cancer. This research
belongs to the research field of biological information prediction.
The main purpose is to provide prima facie evidence for the future
assessment of patient risk and prognosis, but further verification
is required from further investigation and clinical experiments to
improve its accuracy.
In summary, our study shows that the identified
three-gene signature can be used not only as a prognostic indicator
for cervical cancer but also as a predictor of patient risk. In
addition, further validation by functional experiments combined
with clinical trials are required, for application of the
three-gene signature in clinical practice for the benefit of
patients with cervical cancer.
Acknowledgements
Not applicable.
Funding
This study was financially supported by the Natural
Science Foundation of China (grant nos. 81360351) and grants from
the National Natural Science Foundation of China (grant no.
81560407).
Availability of data and materials
Data sets used and/or analyzed during the current
study are available in the TCGA database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).
Authors' contributions
TTD, HM and JHF conceived and designed the study.
TTD and HM conducted the database mining. HM and JHF analyzed data
and compiled charts. TTD wrote the manuscript and approved the
final version.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liang B, Li Y and Wang T: A three miRNAs
signature predicts survival in cervical cancer using bioinformatics
analysis. Sci Rep. 7:56242017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pimple S, Mishra G and Shastri S: Global
strategies for cervical cancer prevention. Curr Opin Obstet
Gynecol. 28:4–10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li H, Wu X and Cheng X: Advances in
diagnosis and treatment of metastatic cervical cancer. J Gynecol
Oncol. 27:e432016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bahrami A, Hasanzadeh M, Shahidsales S,
Farazestanian M, Hassanian SM, Moetamani Ahmadi M, Maftouh M,
Gharib M, Yousefi Z, Kadkhodayan S, et al: Genetic susceptibility
in cervical cancer: From bench to bedside. J Cell Physiol.
233:1929–1939. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guido R: Cervical cancer screening. Clin
Obstet Gynecol. 61:40–51. 2018.PubMed/NCBI
|
|
8
|
Clough E and Barrett T: The gene
expression omnibus database. Methods Mol Biol. 1418:93–110. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chandran UR, Medvedeva OP, Barmada MM,
Blood PD, Chakka A, Luthra S, Ferreira A, Wong KF, Lee AV, Zhang Z,
et al: TCGA expedition: A data acquisition and management system
for TCGA data. PLoS One. 11:e01653952016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park S, Eom K, Kim J, Bang H, Wang HY, Ahn
S, Kim G, Jang H, Kim S, Lee D, et al: MiR-9, miR-21, and miR-155
as potential biomarkers for HPV positive and negative cervical
cancer. BMC Cancer. 17:6582017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oliveira CA, Russomano FB, Gomes Júnior SC
and Corrêa Fde M: Risk of persistent high-grade squamous
intraepithelial lesion after electrosurgical excisional treatment
with positive margins: A meta-analysis. Sao Paulo Med J.
130:119–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Q, Su YL and Shen WX: A novel
prognostic signature of seven genes for the prediction in patients
with thymoma. J Cancer Res Clin Oncol. 145:109–116. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hou C, Zhuang Z, Deng X, Xu Y, Zhang P and
Zhu L: Knockdown of Trio by CRISPR/Cas9 suppresses migration and
invasion of cervical cancer cells. Oncol Rep. 39:795–801.
2018.PubMed/NCBI
|
|
14
|
Li W, Liang J, Zhang Z, Lou H, Zhao L, Xu
Y and Ou R: MicroRNA-329-3p targets MAPK1 to suppress cell
proliferation, migration and invasion in cervical cancer. Oncol
Rep. 37:2743–2750. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Di J, Rutherford S and Chu C: Review of
the cervical cancer burden and population-based cervical cancer
screening in China. Asian Pac J Cancer Prev. 16:7401–7407. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Farzaneh E, Heydari H, Shekarchi AA and
Kamran A: Breast and cervical cancer-screening uptake among females
in Ardabil, northwest Iran: A community-based study. Onco Targets
Ther. 10:985–992. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moyer VA; U.S. Preventive Services Task
Force, : Screening for cervical cancer: U.S. Preventive Services
Task Force recommendation statement. Ann Intern Med. 156:880–891.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huh WK, Ault KA, Chelmow D, Davey DD,
Goulart RA, Garcia FA, Kinney WK, Massad LS, Mayeaux EJ, Saslow D,
et al: Use of primary high-risk human papillomavirus testing for
cervical cancer screening: Interim clinical guidance. Obstet
Gynecol. 125:330–337. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakada T, Akiba T, Yabe M, Tanaka K,
Nakano M, Suzuki M and Morikawa T: Clinicopathological features of
thymoma with ring calcification: Case reports. Ann Thorac
Cardiovasc Surg. 23:256–261. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim HY: The methionine sulfoxide reduction
system: Selenium utilization and methionine sulfoxide reductase
enzymes and their functions. Antioxid Redox Signal. 19:958–969.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee E, Kwak GH, Kamble K and Kim H:
Methionine sulfoxide reductase B3 deficiency inhibits cell growth
through the activation of p53-p21 and p27 pathways. Arch Biochem
Biophys. 547:1–5. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kwak GH and Kim HY: MsrB3 deficiency
induces cancer cell apoptosis through p53-independent and ER
stress-dependent pathways. Arch Biochem Biophys. 621:1–5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kwak GH, Kim TH and Kim HY:
Down-regulation of MsrB3 induces cancer cell apoptosis through
reactive oxygen species production and intrinsic mitochondrial
pathway activation. Biochem Biophys Res Commun. 483:468–474. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu C, Zhang W, Yang D and Liu Y:
Molecular characterization, polymorphism, and association of
porcine GADD45G Gene. Anim Biotechnol. 26:230–236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oliveras-Ferraros C, Vazquez-Martin A,
Cuyàs E, Corominas-Faja B, Rodríguez-Gallego E, Fernández-Arroyo S,
Martin-Castillo B, Joven J and Menendez JA: Acquired resistance to
metformin in breast cancer cells triggers transcriptome
reprogramming toward a degradome-related metastatic stem-like
profile. Cell Cycle. 13:1132–1144. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hoischen C, Yavas S, Wohland T and
Diekmann S: CENP-C/H/I/K/M/T/W/N/L and hMis12 but not CENP-S/X
participate in complex formation in the nucleoplasm of living human
interphase cells outside centromeres. PLoS One. 13:e01925722018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sutherland MJ, Wang S, Quinn ME, Haaning A
and Ware SM: Zic3 is required in the migrating primitive streak for
node morphogenesis and left-right patterning. Hum Mol Genet.
22:1913–1923. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lyu Y, Nakano K, Davis RR, Tepper CG,
Campbell M and Izumiya Y: ZIC2 is essential for maintenance of
latency and is a target of an immediate early protein during
Kaposi's sarcoma-associated Herpesvirus lytic reactivation. J
Virol. 91(pii): e00980–17. 2017.PubMed/NCBI
|
|
30
|
Marchini S, Poynor E, Barakat RR, Clivio
L, Cinquini M, Fruscio R, Porcu L, Bussani C, D'Incalci M, Erba E,
et al: The zinc finger gene ZIC2 has features of an oncogene and
its overexpression correlates strongly with the clinical course of
epithelial ovarian cancer. Clin Cancer Res. 18:4313–4324. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Inaguma S, Ito H, Riku M, Ikeda H and
Kasai K: Addiction of pancreatic cancer cells to zinc-finger
transcription factor ZIC2. Oncotarget. 6:28257–28268. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu P, Wang Y, He L, Huang G, Du Y, Zhang
G, Yan X, Xia P, Ye B, Wang S, et al: ZIC2-dependent OCT4
activation drives self-renewal of human liver cancer stem cells. J
Clin Invest. 125:3795–808. 2015. View
Article : Google Scholar : PubMed/NCBI
|