Introduction
Esophageal carcinomas (ECs) are classified as either
squamous cell carcinomas or adenocarcinomas. Esophageal squamous
cell carcinomas (ESCCs) comprise over 90% of ECs (1). ESCC is one of the most common
malignancies and has a poor outcome in China; there were an
estimated 298,500 deaths in 2012. Furthermore, among the patients
aged 0–74 years, the mortality rate was 9.65%. In males, the
mortality rate was 10.80%, while in female the mortality rate was
7.68% (2). China had a 20-fold
higher incidence rate than low-risk western Africa in 2002
(3). The 5-year survival rate of
patients with esophageal cancer with localized disease is less than
20% (4). Therefore, to improve
patient outcome, the molecular mechanism of ESCC requires further
investigation.
Approximately 5, 70 and 80% of the genomic regions
of bacteria, unicellular eukaryotes and invertebrates,
respectively, are annotated as noncoding (5). Noncoding RNAs include long noncoding
RNAs (lncRNAs), circular RNAs (circRNAs) and microRNAs (miRNAs). An
increasing number of studies reported that lncRNAs, circRNAs and
miRNAs may serve crucial roles in tumorigenesis and tumour
progression (6–11).
LncRNAs are a class of longer transcripts that range
from 200 nt to 100 kb and have little protein-coding potential
(7). Previous studies have reported
that lncRNAs can regulate multiple cellular and disease processes,
including cell differentiation, stem cell pluripotency, cell
proliferation and apoptosis, and cancer metastasis (8–11). To
the best of our knowledge, the majority of the lncRNAs functions
remain unclear. Regulation via lncRNAs does not occur
independently; rather, regulation occurs through a large and
complex network that involves mRNAs, miRNAs and proteins (6). LncRNAs may regulate gene expression by
both cis- and trans-regulation (12). Based on this knowledge, a systematic
study was performed, in order to understand the function of lncRNAs
and mRNAs.
CircRNAs are another novel type of endogenous
non-coding RNA, widespread in mammalian cells (13). Unlike linear RNA, circRNA consists of
RNA molecules with covalently joined 3′- and 5′- ends formed by a
back-splice event; this type of RNA therefore presents as a
covalently closed continuous loop structure and is considered to be
a major subtype of gene transcription (14). The characteristics of circRNA include
the following: Exists in nearly all types of species, is expressed
in a tissue- and disease-dependent manner; and is more stable in
tissue and circulation compared with linear RNA, since it resists
RNase (15). Recently, progress has
been made on the study of the formation and biogenesis of circRNAs,
adding to the evidence and possibilities for its biological value
(16). Similar to lncRNAs, abundant
evidence indicates that circRNAs may serve important roles in
non-alcoholic steatohepatitis, bladder cancer, hepatocellular
carcinoma, Hirschsprung's disease, colorectal cancer and lung
cancer (17–22). A number of studies have revealed that
circRNAs are differentially expressed in various cancerous tissues
or cell lines (such as HEK293 and HeLa) and can function as ‘miRNA
sponges’, regulators of splicing and transcription, and modifiers
of parental gene expression (23–25).
CircRNAs may negatively regulate the activity of miRNAs by
competing endogenous RNA (ceRNA) network that is composed of
lncRNAs, circRNAs and mRNAs (26).
If the dynamic balance of a given ceRNA network is disturbed,
tumorigenesis may occur (27).
To the best of our knowledge, the role of noncoding
RNAs in tumorigenesis and progression of ESCC remain unclear. In
the present study, using a microarray, the differential expression
patterns of lncRNAs, circRNAs and mRNAs were investigated in ESCC.
Subsequently, bioinformatics methods were used to identify the
associated pathways and gene ontology items, based on the
association between the different RNA species.
Furthermore, the lncRNA and circRNA profiles were
investigated and a number of lncRNA-mRNA and circRNA-miRNA-mRNA
networks were constructed. Finally, the functions of the lncRNAs
and circRNAs were predicted by constructing a co-expression
network. These findings may provide novel data for the diagnosis
and pathogenesis of ESCC, in addition to novel targets for ESCC
treatment.
Materials and methods
Patients and samples
A total of 6 ESCC tissue samples and para-carcinoma
tissues were obtained from 3 male patients (mean, 45 years), who
underwent surgical treatment without preoperative chemotherapy or
radiotherapy at the Affiliated Cancer Hospital of Xinjiang Medical
University (Urumqi, China) between July 2014 and April 2017
(Table I). The Union for
International Cancer Control TNM classification (7th edition, 2009)
was used to assess the anatomic extent of ESCC (28). The cancerous and paracancerous
tissues were matched samples. The samples were stored at the
Regional Bank of Tumour Resources of the Xinjiang Uygur Autonomous
Region at −80°C until use.
 | Table I.Details of ESCC and para-carcinoma
specimen used for microarray analysis. |
Table I.
Details of ESCC and para-carcinoma
specimen used for microarray analysis.
| Sample ID | Sex | Age, years | Tumor size, cm | TNM |
|---|
| ESCC-1 | Male | 70 | 5.5 | T3N0M0 |
| ESCC-2 | Male | 55 | 5 | T3N0M0 |
| ESCC-3 | Male | 44 | 4.5 | T3N0M0 |
Written informed consent was obtained from all
subjects. This experimental study was approved and supervised by
the Ethics Committee of The Third Affiliated Teaching Hospital of
Xinjiang Medical University, Affiliated Cancer Hospital.
RNA extraction
Total RNA was extracted from ESCC and para-carcinoma
tissues using Takara RNAiso (Takara Bio), according to the
manufacturer's protocols and was evaluated for a RIN number to
inspect the RNA integrity by an Agilent Bioanalyzer 2100 (Agilent
Technologies). Characterized total RNA was further purified using
an RNeasy mini kit (Qiagen GmBH) and an RNase-Free DNase Set,
according to the manufacturer's protocols. RNA quantity and quality
were measured by NanoDrop ND-2000 (Thermo Fisher Scientific, Inc.),
and RNA integrity was assessed using standard denaturing 10%
agarose gel electrophoresis following RNA extraction and prior to
sample labelling.
Microarray assay
Six tissue samples, including 3 ESCC samples and 3
control samples, were sent to Shanghai BioChip Co., Ltd. for ceRNA
microarray analysis to identify differentially expressed lncRNAs,
circRNAs and mRNAs.
Ribosomal RNA was removed from total RNA using a
Ribo-Zero rRNA Removal kit (Qiagen GmBH), and the RNA(-rRNA) was
subsequently amplified and labelled using a Low Input Quick Amp WT
Labeling kit (Agilent Technologies), according to the
manufacturer's protocols. Labelled cRNA was purified using an
RNeasy mini kit (Qiagen GmBH). Each slide was hybridized with 1.65
µg Cy3-labelled cRNA using a Gene Expression Hybridization kit
(Agilent Technologies) in a hybridization oven (Agilent
Technologies), according to the manufacturer's instructions, at
65°C for 17 h. Following which the slides were washed in staining
dishes (Thermo Fisher Scientific, Inc.) with a Gene Expression Wash
Buffer kit (Agilent Technologies), according to the manufacturer's
protocols.
Slides were scanned by Agilent Microarray Scanner
(cat. no. G2565CA, Agilent Technologies) with default settings, dye
channel: Green, scan resolution=3 µm, and photomultiplier tube
100%, 20bit. Data were extracted with Feature Extraction software
10.7 (Agilent Technologies). Raw data were normalized by Quantile
algorithm, using the limma packages of the R software (version
3.5.1) (18). Following quantile
normalization of the raw data, low intensity filtering was
performed.
The lncRNAs, circRNAs and mRNAs with 3 samples
flagged as ‘P’ or ‘A’ (all target values) were retained for further
analysis. When the profile differences were compared between two
groups, including the disease group vs. the control group, the
fold-change between the groups for each lncRNA, circRNA and mRNA
was computed. A scatter plot and volcano plots were prepared to
visualize the association between the fold-change and the
statistical significance of lncRNA, circRNA and mRNA expression
patterns among samples. The statistical significance of
differentially regulated lncRNAs, circRNAs and mRNAs between the
ESCC group and the para-carcinoma control group was evaluated based
on the P-value. Significantly differentially expressed transcripts
were calculated by screening for a fold-change ≥2.0 and
P<0.05.
Hierarchical clustering analysis
To generate an overview of lncRNA, circRNA and mRNA
expression profiles between the two groups, hierarchical clustering
analysis was performed based on the expression values of all
expressed transcripts and significantly differentially expressed
transcripts using the Cluster and Tree View program (R software)
(18).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was used to confirm the lncRNA and circRNA
expression profiles obtained from the microarray data. All the
samples were normalized to the signal generated by GAPDH (Sangon
Biotech, Co., Ltd.) (25). Data are
shown as the fold-change (2−ΔΔCq) (29). Student's t-tests were used and
P<0.05 was considered to indicate a statistically significant
difference. The cDNA was used as the template in an SYBR-Green
Real-Time PCR Master Mixes (Invitrogen; Thermo Fisher Scientific,
Inc.) and in triplicate subjected to denaturation at 94°C for 4 min
and 35 cycles of 94°C for 30 sec, and 60°C for 30 sec, followed by
extension at 72°C for 10 min using the specific primers. The primer
sequences were as follows: Circ-0025580: F,
5′-CACGAGGGGCTTGTAGGATA-3′; R, 5′-AGGAAACCAAGCCACCAAG-3′.
Circ-0024108: F, 5′-AGGCAAGGGATAACTCTTCTAACAC-3′; R,
5′-TTGGCAAATCTGGCGTGTAA-3′. Circ-0025933: F,
5′-GGAATGGAACGACATGCAAA-3′; R, 5′-GACACACATTGTATTTTCACGACAGT-3′.
lnc-KLHDC7A-6:2: F, 5′-GGGCGTGAGGTGTGTGTTTA-3′; R,
5′-CGCTTACAAGCAGCAGGTAG-3′. LOC440173: F,
5′-GAGGTACCAAGAGAAGTGCTGATG-3′; R, 5′-GTTAATGCTTTCGGCCAAGATC-3′.
EPB41L4A-AS1: F, 5′-GTCATCTATGGAGAGGAAAGGTACAAA-3′; R,
5′-TGTCACCCCAAACCTCAAATG-3′. SMAD5-AS1_3: F,
5′-GTTCTGGTGGTGATGGCATTG-3′; R, 5′-CATCTGGCTCAGGGTGTTCA-3′. GAPDH:
F, 5′-TGACTTCAACAGCGACACCCA-3′; R, 5′-CACCCTGTTGCTGTAGCCAAA-3′.
Correlation and co-expression
analysis
The co-expression analysis was performed by
calculating the Pearson's correlation coefficient (PCC) between
coding genes and noncoding transcripts, according to their
expression levels. The absolute value of parameters PCC ≥0.90,
P<0.05 and false discovery rate <0.01 was recommended and
retained for further analysis.
Gene Ontology (GO) and Kyoto
Encyclopaedia of Genes and Genomes (KEGG) pathway enrichment
analysis
The DAVID 6.7 functional annotation clustering tool
was used to analyse the potential functions of lncRNAs, circRNAs
and co-expressed genes (18). The
permutated P-value cut-off was set at <0.05. GO is used to
describe biological genes and perform expression analyses of
product attributes and includes the following three subgroups:
Biological process (BP), cellular component (CC) and molecular
function (MF) (30). GO
classifications are calculated for selected genes using a
particular branch of the hypergeometric distribution relationship
for each gene to obtain a P-value <0.05 for the enrichment
items. GO suggests a role based on the differences in the gene
analysis. It can also indicate enriched genes that are
differentially categorized entries based on changes in different
samples (18). GO enrichment
analysis of significantly differentially expressed mRNAs can reveal
the role of obviously differentially regulated lncRNAs and
circRNAs. Therefore, lncRNAs and circRNAs have functions that may
be embodied in associated mRNA genes. The top 10 enriched GO terms
of the two groups, ranked according to fold enrichment and
enrichment score, are presented. The KEGG pathway analysis was
performed to determine the involvement of co-expressed genes in
different biological pathways. In addition, the -log10
(P-value) denotes the enrichment score, indicating the significance
of the pathway correlations (31).
LncRNA target gene prediction and KEGG
analysis
To identify the neighbouring target genes of
differentially expressed lncRNAs that may interact via cis- or
trans-regulatory effects, differentially expressed lncRNAs
(fold-change >5) were selected for potential target gene
prediction. LncRNAs have been found to regulate gene expression in
both cis- and trans-manners using cis- and trans-regulatory
mechanism-based algorithms (32).
The co-expressed protein-coding genes were defined as cis-regulated
genes when one differentially expressed lncRNA was indicated within
10 kb on the same chromosome or as trans-regulated genes if the
aforementioned criterion was not met.
Using gene annotations from UCSC (http://genome.ucsc.edu/), lncRNAs and potential target
genes were paired and visualized using the UCSC Genome Browser. The
genes transcribed within a 10-kb region upstream or downstream of
lncRNAs were considered cis target genes. The RNA plex software was
used to choose trans-acting target genes as described previously
(33). Biological pathways were
defined by KEGG.
Correlation analysis of circRNAs and
mRNAs in ESCC
An Agilent circRNA and mRNA expression profile
microarray was used to screen the differentially expressed circRNA
and mRNA gene expression. The regulation of the mRNA target
expression of circRNAs was evaluated to investigate whether
circRNAs could act as ‘miRNA sponges’. CircRNA-miRNA interaction
analysis was conducted by Cytoscape 3.2.1 software (Cytoscape
Consortium). The size of each node represents the number of
putative miRNAs that were functionally connected to each
circRNA.
LncRNA-miRNA-circRNA-mRNA ceRNA
networks
LncRNA-mRNA and circRNA-miRNA co-expression networks
were constructed based on the correlation analysis between the
differentially expressed lncRNA and mRNA, and circRNAs and miRNAs.
The expression of differentially expressed circRNA-miRNA,
lncRNA-mRNA and miRNA-mRNA pairs was analysed by Pearson's
correlation coefficient.
Statistical analysis
The results are reported as the mean ± standard
deviation for triplicate measurements. Statistically significant
differences between groups were estimated by two-tailed Student's
t-test using SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA).
Pearson's coefficient was used to compare the microarray data and
qPCR results. P<0.05 was considered to indicate a statistically
significant difference.
Results
Evaluation of extracted RNA
Three clear bands were observed, namely, 28S, 18S
and 5S, and RNA integrity of the 28S/18S band was ~2. The inclusion
criteria for RNA were set at an optical density (OD) A260/280 ratio
between 1.8 and 2.1 and an OD A260/230 ratio >1.8. All quality
criteria that were predefined by the manufacturer for successful
microarray analysis were fulfilled for each array.
Differentially expressed lncRNA, mRNA
and circRNA profiles by microarray
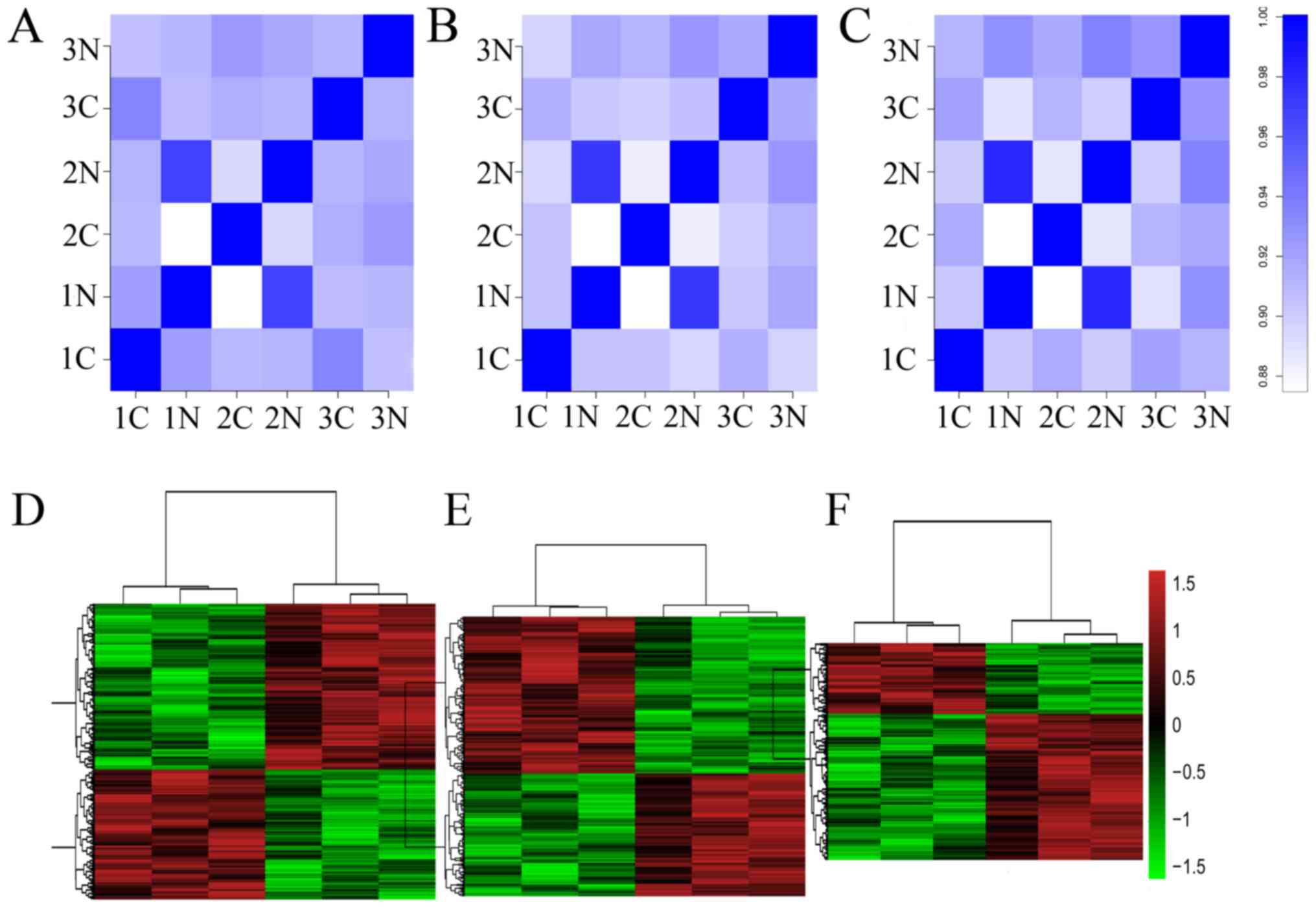
As indicated in the top row of Fig. 1, a correlation plot was used to
detect the correlation between samples and to verify the
homogeneity between biological replicates. In addition, as
presented in the hierarchical clustering in Fig. 1, 1,384 lncRNAs, 2,046 circRNAs and
936 mRNAs were differentially expressed, with fold-changes ≥2.0 and
P<0.05 in ESCC and para-carcinoma tissues. Among them, 608 and
776 lncRNAs were upregulated and downregulated, respectively; 1,148
and 898 circRNAs were upregulated and downregulated, respectively;
and 306 and 630 mRNAs were upregulated and downregulated
(fold-change ≥2.0 and P<0.05) in three ESCC tissue samples
compared with those of controls.
The scatter plots and volcano plots data suggested
that the expression of lncRNAs, circRNAs and mRNAs in ESCC tissues
differed from that of matched para-carcinoma tissues (Fig. 2).
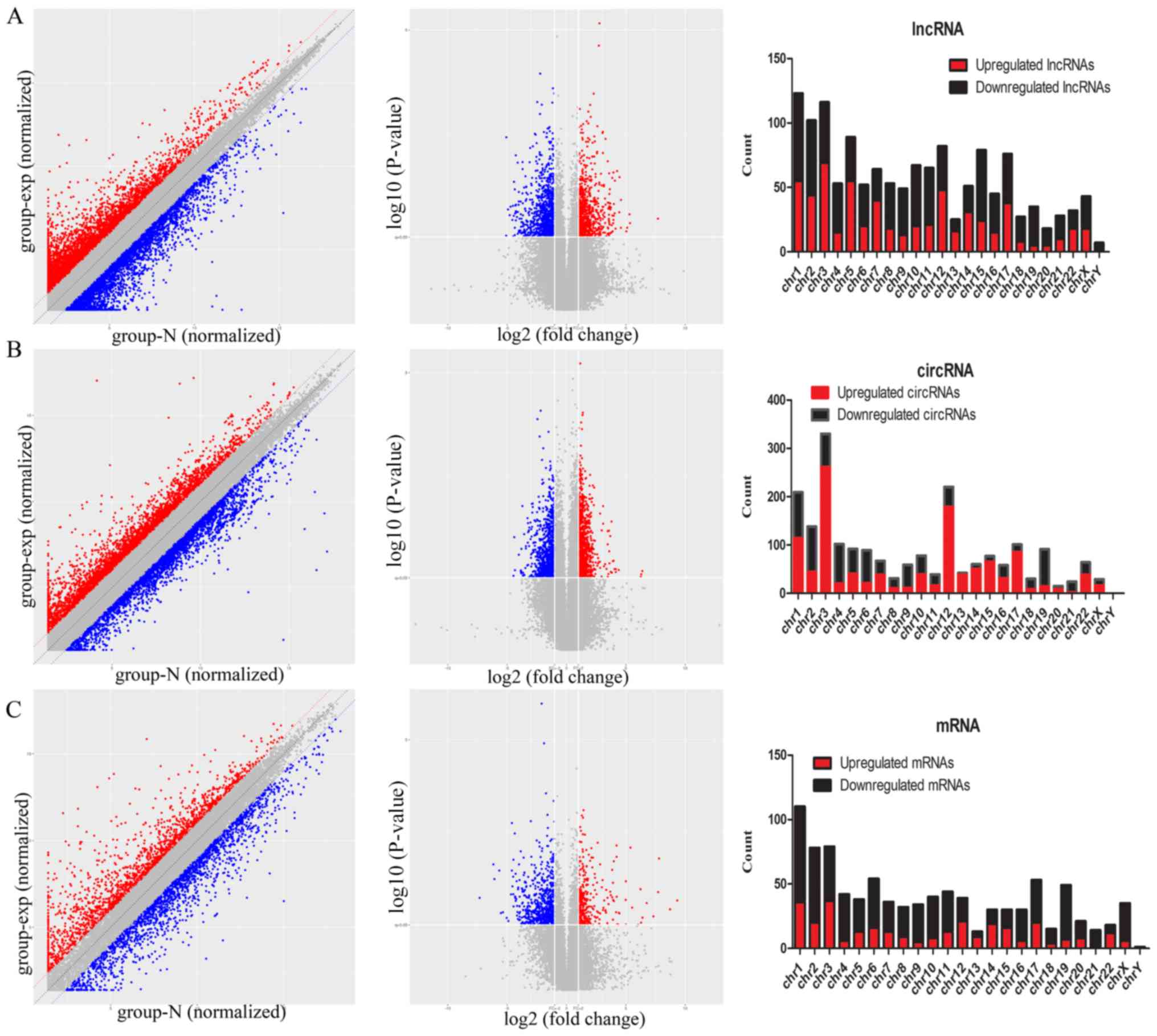 | Figure 2.Detected results for all lncRNAs,
circRNAs and mRNAs. Scatter plot and volcano plots based on the
expression values of significantly altered (A) lncRNAs, (B)
circRNAs and (C) mRNAs in ESCC. The upregulated and downregulated
lncRNAs, circRNAs and mRNAs are marked by red or blue bars,
respectively. The right vertical line corresponds to 2-fold
upregulation, the left vertical line corresponds to 2-fold
downregulation, and the horizontal line indicates a P=0.05. The
numbers of dysregulated lncRNAs, circRNAs and mRNAs identified in
ESCC tissues and matched para-carcinoma tissues are presented.
lncRNAs, long noncoding RNAs; circRNAs, circularRNAs; ESCC,
esophageal squamous carcinoma. |
The differentially regulated lncRNAs, circRNAs and
mRNAs were widely distributed among all chromosomes (Fig. 2). The dysregulated lncRNAs were
classified into six categories according to their association with
protein-coding genes: 34% were intergenic, 29% were exonic sense,
13% were exonic antisense, 11% were intronic sense, 7% were
bidirectional and 6% were intronic antisense. A total of 120
lncRNAs had a fold-change ≥10, including 46 upregulated lncRNAs and
74 downregulated lncRNAs. Lnc-SNX10-1:1 (fold-change: Approximately
208) was the most upregulated lncRNA. An overview of the coding
gene profile indicated that 40 circRNAs had a fold-change ≥10 (up:
20; down: 20) and 118 mRNAs had a fold-change ≥10 (up: 36; down:
82).
Validation of deregulated lncRNAs and
circRNAs
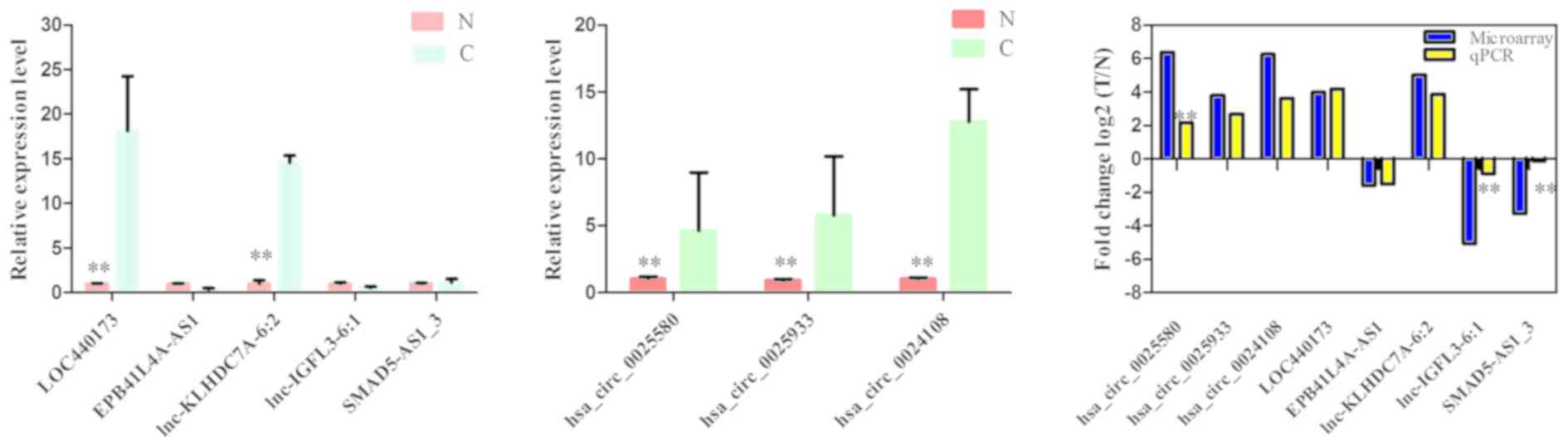
To validate the microarray profiling expression
data, RT-qPCR was performed and revealed four deregulated expressed
lncRNAs and three circRNAs. RT-qPCR assays indicated that the
expression of lncRNAs lnc-KLHDC7A-6:2 and LOC440173 was
upregulated, whereas the expression of EPB41L4A-AS1 and SMAD5-AS1_3
was downregulated (Fig. 3A). In
addition, circRNAs hsa_circ_0025580, hsa_circ_0024108 and
hsa_circ_0025933 were upregulated in ESCC compared with those of
the control (Fig. 3B). The
expression levels that were detected by the two methods were
consistent with each other, demonstrating the high reliability of
the microarray expression results (Fig.
3C). Therefore, the RT-qPCR data verified the validity of the
microarray results.
Delineation of GO and KEGG pathway
analysis
LncRNAs can regulate the expression of neighboring
and overlapping coding genes (10).
In addition, circRNAs can regulate their parental gene
transcription (14). Therefore, GO
analysis of the genes that produced differently expressed lncRNAs
and circRNAs was performed. The top 10 generally changed GO terms
in all comparison groups were classified as BP, CC, and MF and were
ranked by fold enrichment or enrichment score as listed.
Compared with those of para-carcinoma tissues, data
of the present study indicated that the mRNAs that were upregulated
by lncRNAs and were associated with biological processes were
associated to cell cycle and cell division (Fig. 4A). By contrast, the downregulated
transcripts were most relevant for endocytosis and intracellular
signal transduction (Fig. 4B). The
mRNAs that were upregulated by circRNAs and were associated with
biological processes were also associated with DNA repair and cell
division (Fig. 4C), whereas the
downregulated transcripts were most relevant for substrate
adhesion-dependent cell spreading and intracellular signal
transduction. The cell cycle and intracellular signal transduction
GO terms were correlated with cancer and served an important role
in the control of cell proliferation and gene expression (Fig. 4D).
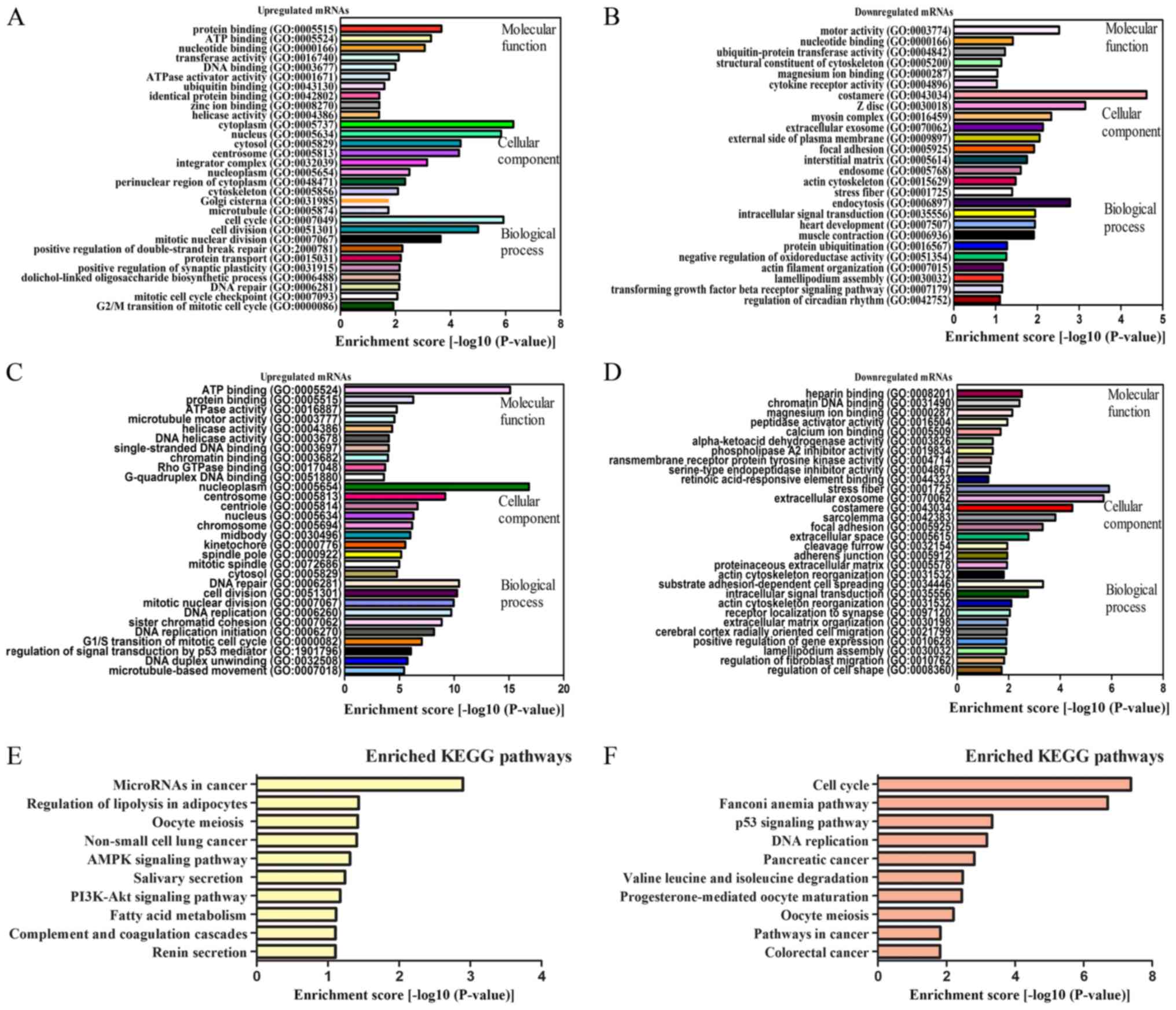 | Figure 4.GO and KEGG pathway analyses. GO
annotation of (A) upregulated lncRNAs and co-expressed mRNAs, (B)
downregulated lncRNAs and co-expressed mRNAs, (C) upregulated
circRNAs and co-expressed mRNAs, (D) downregulated circRNAs and
co-expressed mRNAs with top enrichment scores that fell in the
domains of BP, CC and MF. KEGG pathway enrichment analysis of (E)
dysregulated lncRNAs and co-expressed mRNAs and (F) dysregulated
circRNAs and co-expressed mRNAs with the top enrichment scores.
lncRNAs, long noncoding RNAs; circRNAs, circularRNAs; GO, Gene
Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP,
biological processes; CC, cellular components; MF, molecular
functions. |
The KEGG pathway enrichment analysis was designed to
identify pathways and molecular interactions associated with mRNAs.
For lncRNAs, our data indicated that 7 pathways were associated
with upregulated mRNAs and that 10 were associated with
downregulated mRNAs. For all lncRNA studies, the microRNAs in
cancer were the most common pathway of dysregulated protein-coding
genes (Fig. 4E).
For genes that were co-expressed with circRNAs, the
top enriched KEGG pathway for upregulated mRNAs was associated with
the cell cycle, and for downregulated mRNAs, the top enriched
pathway was associated with valine, leucine and isoleucine
degradation (Fig. 4F). Cell cycle
signaling was the top pathway in dysregulated protein-coding genes.
Based on these results, these pathways may contribute significantly
to the pathogenesis and development of ESCC.
Cis- and trans-regulation function
prediction of lncRNAs
In order to understand the underlying function of
lncRNAs, cis- or trans-regulatory role of dysregulated lncRNAs in
nearby coding genes was investigated.
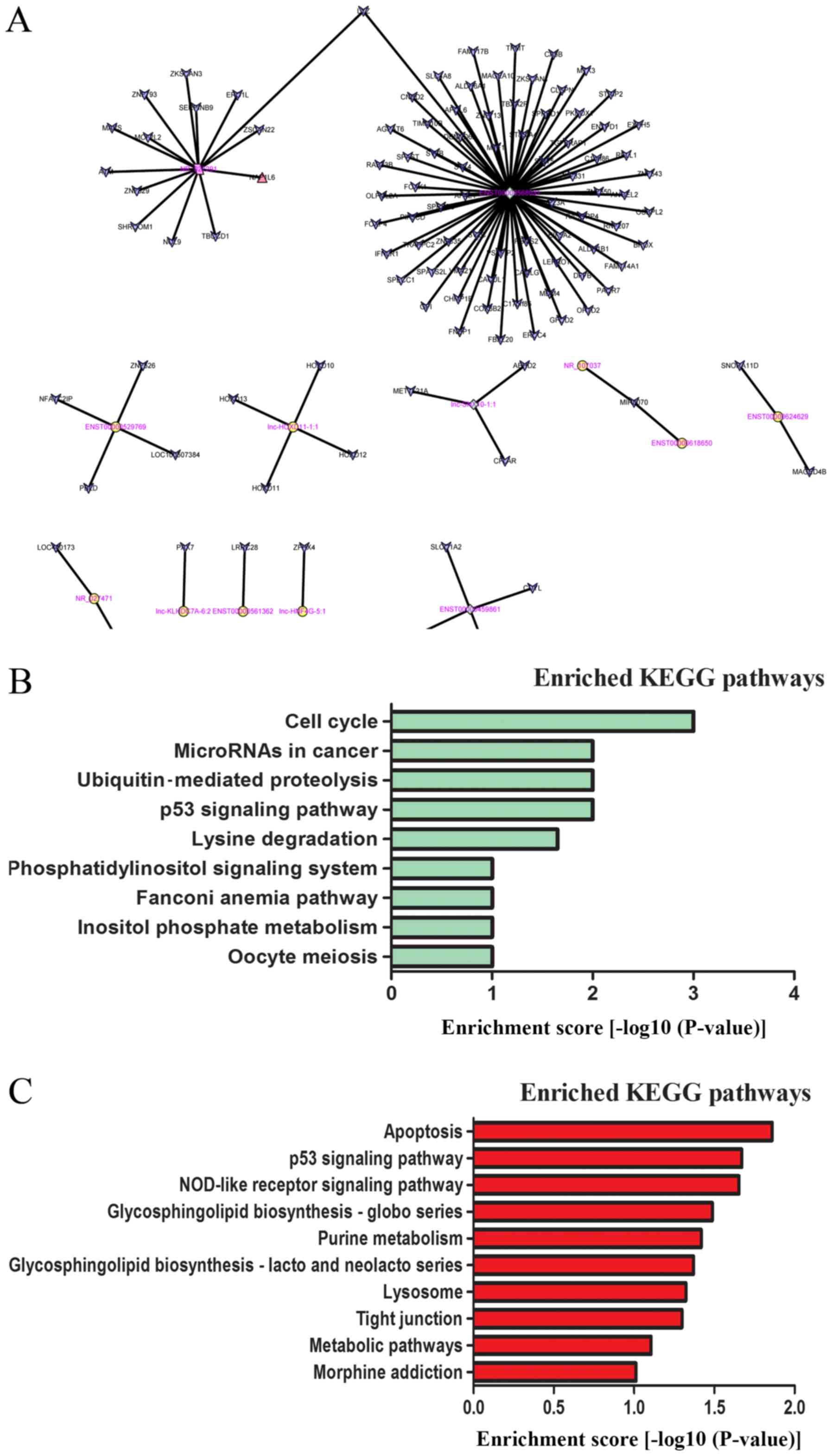
The networks were constructed to determine the
association of the differentially expressed lncRNAs and adjacent
coding genes. Each lncRNA had a different number of neighbouring
coding genes. For example, lnc-HOXD11-1:1 had a maximum number of 4
adjacent coding genes, whereas ENST00000529769 was indicated to
have only 1 nearby coding gene (Fig.
5A).
To identify lncRNA target genes and their
corresponding pathways, the DAVID 6.7 software was used to analyse
the potential functions of specific lncRNAs. KEGG pathway
enrichment analysis of 1,945 mRNAs was used, the genes for which
lncRNAs (fold-change >5) may serve a cis-regulatory role. The
results indicated that these mRNAs were implicated in a number of
biological processes, including the cell cycle, microRNA expression
in cancer, ubiquitin-mediated proteolysis and the p53 signalling
pathway (Fig. 5B).
The potential target genes of the 462 differentially
expressed lncRNAs (fold-change >5), which may serve a
trans-regulatory role, were predicted. In all, 2,893 target genes
were indicated. In addition, 282 differentially expressed genes
that had been identified by the mRNA arrays (fold-change >1.5)
were matched to these target genes. A number of well-known
tumour-associated pathways, including those that involve apoptosis
and the p53 signalling pathway, were indicated to be associated
with tumorigenesis (Fig. 5C).
Construction of the circRNA-miRNA
co-expression network
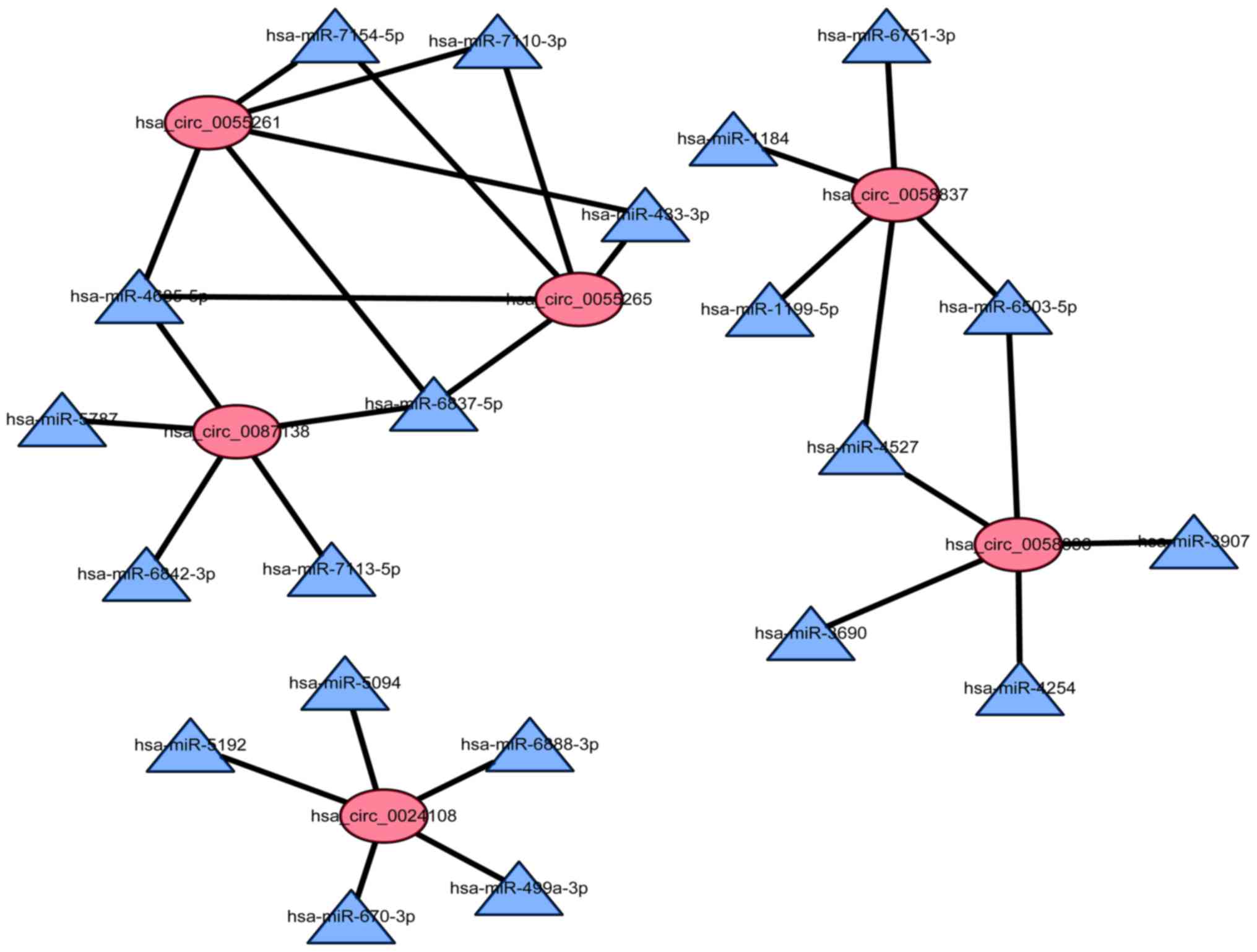
To determine the function of circRNAs, a
circRNA-miRNA co-expression network was subsequently constructed,
based on bioinformatics analysis. All of the differentially
expressed circRNA were predicted according to the complementary
miRNA sequence. An entire network of circRNA/miRNA interactions was
delineated using Cytoscape. Furthermore, the graph was enlarged to
display the top 6 up- and downregulated circRNAs and their
corresponding miRNAs (Fig. 6). In
the network, the ellipses represent circRNAs, and the triangles
represent miRNAs.
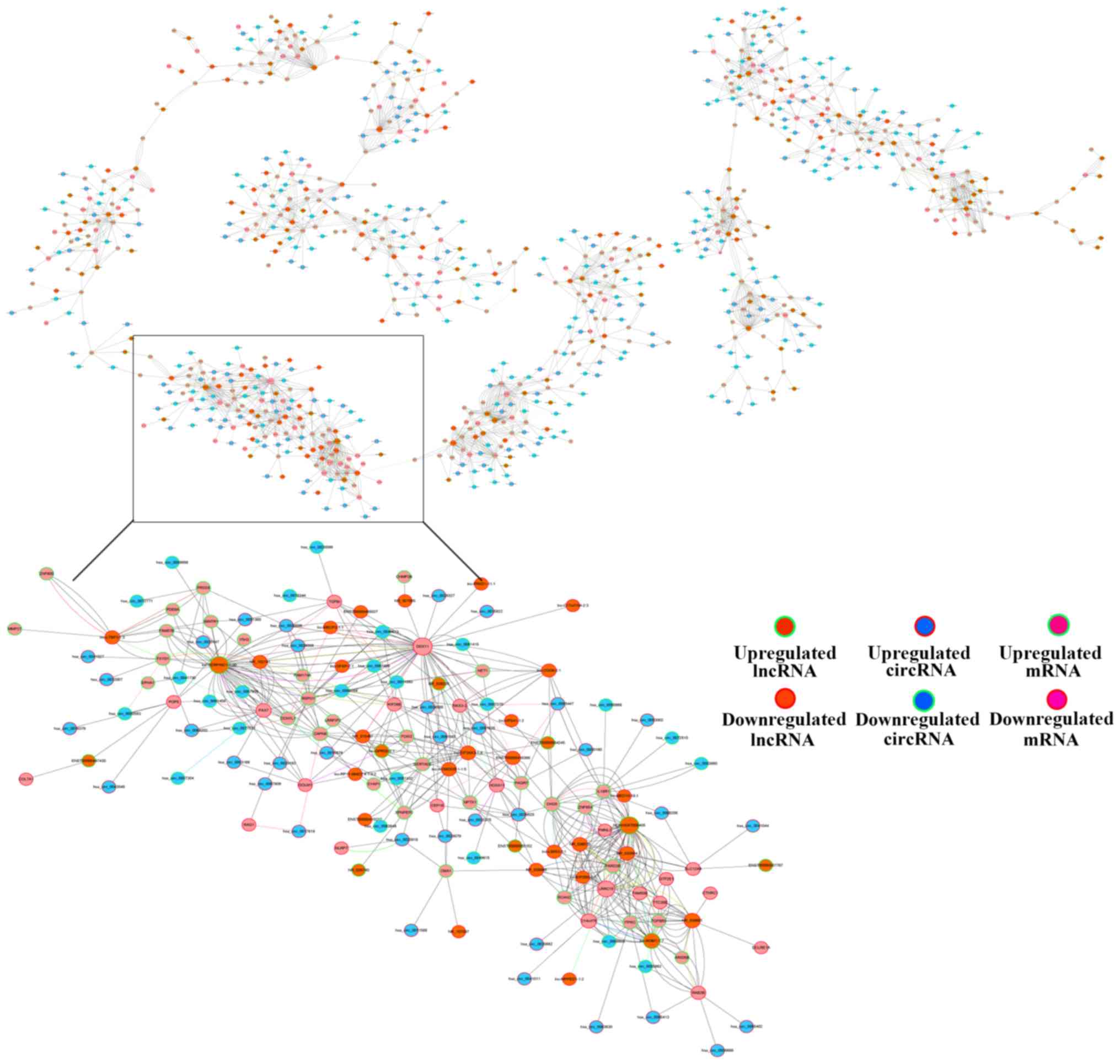
Construction of ceRNA networks
Based on the ceRNA hypothesis, ceRNAs can compete
for the same miRNA response elements (MREs) to regulate each other
(26). Bioinformatics methods to
establish a ceRNA network in ESCC were used according to our
microarray data, which included the circRNA, lncRNA and mRNA data
(Fig. 7). The association among
circRNA, lncRNA, mRNA and miRNA were established using the
expression values. The size of the circle represents the ability of
a given type of RNA to interact with other genes.
In the present study, a total of 185 lncRNAs, 332
circRNAs and 318 mRNAs were selected to generate a network map with
Cytoscape. The red colour and green colour represent up- and
downregulation, respectively. The size of the circle represents the
size of the P-value, with a larger size resulting from a smaller
fold-change. The local graphic was enlarged to display the ceRNA
network. For instance, circ-0078344, circ-0026599, circ-0036698 and
NR-102741 are ceRNAs of different miRNAs that target transforming
grown factor β induced (TGFBI) (27). TGFBI is a type of protein that is
induced by the transforming growth factor beta secretory protein.
The analysis revealed that TGFBI is associated with cancer in the
general biological process subgroup, and may be a biological marker
of cancer. These RNA interactions provided a novel perspective on
the tumorigenic mechanisms of ESCC.
Discussion
Previous studies have focused on protein-coding
genes until the discovery of numerous non-coding RNAs, including
lncRNAs and circRNAs (12,14,16).
This study, to the best of our knowledge, presents the first
simultaneous investigation of lncRNAs, circRNAs and mRNAs in
clinical ESCC tissues.
A growing number of studies have confirmed that
lncRNAs may be one of the most important factors for the control of
gene expression (8–11,34).
LncRNA has been widely reported to participate in a wide range of
biological processes, and its dysregulated expression affects many
human disease phenotypes, including those of cancers (35–37).
Similar to lncRNAs, circRNAs are a large class of noncoding RNAs
(ncRNAs). CircRNAs were recently described as pivotal gene
regulators in humans, due to the different ways that they control
transcription and translation (38).
As increased attention is paid to the roles of circRNAs as
oncogenes and tumour suppressors in cancer, circRNAs have exhibited
great potential as diagnostic and prognostic biomarkers (19,39).
However, comprehensive analyses of differentially
expressed profiles of lncRNAs and circRNAs in ESCC, to the best of
our knowledge, have yet to be reported. To probe the functions of
lncRNAs and circRNAs in ESCC tumours, the genome-wide expression
profiles of lncRNAs and circRNAs were examined in the present study
in three ESCC specimens and matched adjacent tissues using a
microarray assay.
From the results of the present study, it was
determined that hundreds of lncRNAs, circRNAs and mRNAs were
differentially expressed in tumour tissues compared with those of
the control group. Overall, 1,384 lncRNAs, 2,046 circRNAs and 936
mRNAs were observed to be significantly differentially expressed in
ESCC. Four dysregulated lncRNAs and three circRNAs were further
verified by RT-qPCR. Therefore, the RT-qPCR data verified the
microarray data, demonstrating the reliability of the microarray
results. These differentially expressed lncRNAs, circRNAs and mRNAs
were subsequently integrated into hierarchical categories,
according to heat maps and hierarchical clustering.
To further study the function of these
differentially expressed lncRNAs and circRNAs, GO and KEGG pathway
analyses were performed. The annotation results of the most
significant GO terms of lncRNAs were cell cycle, cell division,
endocytosis and intracellular signal transduction. The notably
changed GO terms of circRNAs were DNA repair, cell division,
substrate adhesion-dependent cell spreading and intracellular
signal transduction. These biological processes identified coding
genes that were associated with the development of ESCC (40,41).
KEGG pathway analysis for the differentially
expressed lncRNAs and circRNAs revealed 10 pathways. These
pathways, including miRNAs in cancer, the 5′ AMP-activated protein
kinase signalling pathway, PI3K-Akt signalling pathway, p53
signalling pathway and cell cycle, may serve pivotal roles in the
tumorigenic mechanisms of ESCC, as they were more likely to be
identified in the ESCC group compared with in the normal control
group. This suggests that dysregulated lncRNAs and circRNAs may
affect these targets by regulating the associated pathways in
ESCC.
Increasing evidence indicates that lncRNAs and
circRNAs are extensively targeted by miRNAs and that they function
as ceRNAs (20,42). CeRNAs include lncRNAs, circRNAs and
mRNAs, as these transcripts can compete for the same MREs and
mutually regulate each other (23).
ceRNAs have been implicated in both physiological conditions and
cancer development. If the balance in the intricate ceRNA network
is disturbed, carcinogenesis may occur. To date, to the best of our
knowledge, no information on ceRNAs in ESCC has been reported. A
lncRNA-miRNA-circRNA-mRNA ceRNA network in ESCC was constructed
based on our microarray data. As indicated in Fig. 7, the association among these types of
RNA is extremely complex. Compared with those of the control group,
the lncRNAs and circRNAs that were dysregulated in the ESCC group
were identified. In the ceRNA network, 185 lncRNAs and 332 circRNAs
were included. In order to illustrate the ceRNA network, TGFBI was
selected, as it serves a role in the activation of morphogenesis,
cell proliferation, adhesion, migration, differentiation,
chemoresistance and inflammation (43). The RNAs circ-0078344, circ-0026599,
circ-0036698 and NR-102741 are ceRNAs of different miRNAs that
target TGFBI. Theoretically, lncRNAs and circRNAs may regulate the
tumorigenesis of ESCC by targeting TGFBI. However, the functions of
lncRNAs, circRNAs and their associated ceRNAs in ESCC remain
unclear. Further studies on these RNA interactions are required, in
order to provide a novel perspective of the tumorigenic mechanisms
of ESCC.
In conclusion, the present study presented a profile
of dysregulated lncRNAs, circRNAs and mRNAs in ESCC, as determined
by microarray analysis. GO and KEGG pathway analyses were performed
to annotate the potential functions of differentially expressed
lncRNAs and circRNAs. Co-expression networks were constructed for
lncRNA-miRNA-circRNA-mRNA. Our data may establish a foundation for
further functional research into lncRNAs and circRNAs in ESCC.
Therefore, these results suggest that ncRNAs may serve an important
diagnostic and therapeutic role in ESCC.
Acknowledgements
The authors would like to thank The Xinjiang Cancer
Biobank of Tumor Affiliated Hospital of Xinjiang Medical University
(Urumuqi, China) for providing samples of esophageal cancer.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81760498).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JS and YL conceived the study and JZ designed the
study. JS, YL and MH performed the experiments. WS, XD and YZ
collected clinical samples and analyzed the data. JS and YRL wrote
the paper. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Third
Affiliated Teaching Hospital of Xinjiang Medical University,
Affiliated Cancer Hospital's Protection of Human Subjects
Committee. Written informed consent was obtained from all
subjects.
Patient consent for publication
All patients signed written informed consent for the
publication.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BP
|
biological process
|
|
CC
|
cellular component
|
|
ceRNA
|
competing endogenous RNA
|
|
circRNAs
|
circularRNAs
|
|
EC
|
esophageal carcinomas
|
|
ESCC
|
esophageal squamous carcinoma
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
lncRNAs
|
long noncoding RNAs
|
|
miRNAs
|
microRNAs
|
|
MF
|
molecular function
|
|
MREs
|
miRNA response elements
|
|
ncRNAs
|
non-coding RNAs
|
|
PCC
|
Pearson's correlation coefficient
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
References
|
1
|
Forghanifard MM, Taleb Sh and Abbaszadegan
MR: Notch signaling target genes are directly correlated to
esophageal squamous cell carcinoma tumorigenesis. Pathol Oncol Res.
21:463–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Zuo T, Zeng H, Zhang S
and He J: National cancer incidence and mortality in China 2012.
Chin J Cancer Res. 28:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y: Epidemiology of esophageal
cancer. World J Gastroenterol. 19:5598–5606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lander ES, Linton LM, Birren B, Nusbaum C,
Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al:
Initial sequencing and analysis of the human genome. Nature.
409:860–921. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rong D, Sun H, Li Z, Liu S, Dong C, Fu K,
Tang W and Cao H: An emerging function of circRNA-miRNAs-mRNA axis
in human diseases. Oncotarget. 8:73271–73281. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guttman M, Donaghey J, Carey BW, Garber M,
Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al:
lincRNAs act in the circuitry controlling pluripotency and
differentiation. Nature. 477:295–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han X, Wang L, Ning Y, Li S and Wang Z:
Long non-coding RNA AFAP1-AS1 facilitates tumor growth and promotes
metastasis in colorectal cancer. Biol Res. 49:362016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou M, Hou Y, Yang G, Zhang H, Tu G, Du
YE, Wen S, Xu L, Tang X, Tang S, et al: LncRNA-Hh strengthen cancer
stem cells generationin Twist-positive breast cancer via activation
of Hedgehog signaling pathway. Stem Cells. 34:55–66. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu S, Wang P, You Z, Meng H, Mu G, Bai X,
Zhang G, Zhang J and Pang D: The longnon-coding RNAEPB41L4A-AS2
inhibits tumor proliferation and is associated with favorable
prognoses in breast cancer and other solid tumors. Oncotarget.
7:20704–20717. 2016.PubMed/NCBI
|
|
12
|
Lee S, Kopp F, Chang TC, Sataluri A, Chen
B, Sivakumar S, Yu H, Xie Y and Mendell JT: Noncoding RNA NORAD
regulates genomic stability by sequestering PUMILIO proteins. Cell.
164:69–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kristensen LS, Hansen TB, Venø MT and
Kjems J: Circular RNAs in cancer: Opportunities and challenges in
the field. Oncogene. 37:555–565. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salzman J, Gawad C, Wang PL, Lacayo N and
Brown PO: Circular RNAs are the predominant transcript isoform from
hundreds of human genes in diverse cell types. PLoS One.
7:e307332012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qu S, Yang X, Li X, Wang J, Gao Y, Shang
R, Sun W, Dou K and Li H: Circular RNA: A new star of noncoding
RNAs. Cancer Lett. 365:141–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Geng Y, Jiang J and Wu C: Function and
clinical significance of circRNAs in solid tumors. J Hematol Oncol.
11:982018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin X, Feng CY, Xiang Z, Chen YP and Li
YM: CircRNA expression pattern and circRNA-miRNA-mRNA network in
the pathogenesis of nonalcoholic steatohepatitis. Oncotarget.
7:66455–66467. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang M, Zhong Z, Lv M, Shu J, Tian Q and
Chen J: Comprehensive analysis of differentially expressed profiles
of lncRNAs and circRNAs with associated co-expression and ceRNA
networks in bladder carcinoma. Oncotarget. 7:47186–47200.
2016.PubMed/NCBI
|
|
19
|
Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z,
Yang J, Fan J, Liu L and Qin W: Hsa_circ_0001649: A circularRNA and
potential novel biomarker for hepatocellular carcinoma. Cancer
Biomark. 16:161–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peng L, Chen G, Zhu Z, Shen Z, Du C, Zang
R, Su Y, Xie H, Li H, Xu X, et al: CircularRNA ZNF609 functions as
a competitive endogenous RNA to regulate AKT3 expression by
sponging miR-150-5p in Hirschsprung's disease. Oncotarget.
8:808–818. 2017.PubMed/NCBI
|
|
21
|
Xie H, Ren X, Xin S, Lan X, Lu G, Lin Y,
Yang S, Zeng Z, Liao W, Ding YQ and Liang L: Emerging roles of
circRNA_001569 targeting miR-145 in the proliferation and invasion
of colorectal cancer. Oncotarget. 7:26680–26691. 2016.PubMed/NCBI
|
|
22
|
Yao JT, Zhao SH, Liu QP, Lv MQ, Zhou DX,
Liao ZJ and Nan KJ: Over-expression of CircRNA_100876 in non-small
cell lung cancer and its prognostic value. Pathol Res Pract.
213:453–456. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han D, Li J, Wang H, Su X, Hou J, Gu Y,
Qian C, Lin Y, Liu X, Huang M, et al: Circular RNA circMTO1 acts as
the sponge of microRNA-9 to suppress hepatocellular carcinoma
progression. Hepatology. 66:1151–1164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen L, Zhang S, Wu J, Cui J, Zhong L,
Zeng L and Ge S: CircRNA_100290 plays a role in oral cancer by
functioning as a sponge of the miR-29 family. Oncogene.
36:4551–4561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolf PP: A ceRNA hypothesis: The rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou B and Yu JW: A novel identified
circular RNA, circRNA_010567, promotes myocardial fibrosis via
suppressing miR-141 by targeting TGF-β1. Biochem Biophys Res
Commun. 487:769–775. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sobin LH, Gospodarowicz MK and Wittekind
C: Oesophagus including oesophagogastric junctionTNM classification
of malignant tumours. 7th. Oxford: Wiley-Blackwell; pp. 66–72.
2009
|
|
29
|
Coates DE, Zafar S and Milne TJ:
Quantitative real-time gene profiling of human alveolar
osteoblasts. Methods Mol Biol. 1537:447–459. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
The Gene Ontology Consortium, . Gene
ontology consortium: Going forward. Nucleic Acids Res. 43((Database
Issue)): D1049–D1056. 2015.PubMed/NCBI
|
|
31
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Petruk S, Sedkov Y, Riley KM, Hodgson J,
Schweisguth F, Hirose S, Jaynes JB, Brock HW and Mazo A:
Transcription of bxd noncoding RNAs promoted by trithorax represses
Ubx in cis by transcriptional interference. Cell. 127:1209–1221.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tafer H and Hofacker IL: RNAplex: A fast
tool for RNA-RNA interaction search. Bioinformatics. 24:2657–2663.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Annu Rev Biochem. 81:145–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou M, Wang X, Li J, Hao D, Wang Z, Shi
H, Han L, Zhou H and Sun J: Prioritizing candidate disease-related
long non-coding RNAs by walking on the heterogeneous lncRNA and
disease network. Mol Biosyst. 11:760–7569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun J, Shi H, Wang Z, Zhang C, Liu L, Wang
L, He W, Hao D, Liu S and Zhou M: Inferring novel lncRNA-disease
associations based on a random walk model of a lncRNA functional
similarity network. Mol Biosyst. 10:2074–2081. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sahu A, Singhal U and Chinnaiyan AM: Long
noncoding RNAs in cancer: From function to translation. Trends
Cancer. 1:93–109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li J, Yang J, Zhou P, Le Y, Zhou C, Wang
S, Xu D, Lin HK and Gong Z: Circular RNAs in cancer: Novel insights
into origins, properties, functions and implications. Am J Cancer
Res. 5:472–480. 2015.PubMed/NCBI
|
|
39
|
Wang X, Zhang Y, Huang L, Zhang J, Pan F,
Li B, Yan Y, Jia B, Liu H, Li S and Zheng W: Decreased expression
of hsa_circ_001988 in colorectal cancer and its clinical
significances. Int J Clin Exp Pathol. 8:16020–16025.
2015.PubMed/NCBI
|
|
40
|
Cai X, Hu X, Tan X, Cheng W, Wang Q, Chen
X, Guan Y, Chen C and Jing X: Metformin induced AMPK activation,
G0/G1 phase cell cycle arrest and the inhibition of growth of
esophageal squamous cell carcinomas in vitro and in vivo. PLoS One.
10:e01333492015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Peng HH, Zhang X and Cao PG: MMP-1/PAR-1
signal transduction axis and its prognostic impact in esophageal
squamous cell carcinoma. Braz J Med Biol Res. 45:86–92. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou M, Diao Z, Yue X, Chen Y, Zhao H,
Cheng L and Sun J: Construction and analysis of dysregulated
lncRNA-associated ceRNA network identified novel lncRNA biomarkers
for early diagnosis of human pancreatic cancer. Oncotarget.
7:56383–56394. 2016.PubMed/NCBI
|
|
43
|
Zhu J, Chen X, Liao Z, He C and Hu X:
TGFBI protein high expression predicts poor prognosis in colorectal
cancer patients. Int J Clin Exp Pathol. 8:702–710. 2015.PubMed/NCBI
|