Introduction
Lung cancer is one of the most prevalent cancer
types and the leading cause of cancer-related mortality worldwide,
accounting for an estimated 142,670 deaths in the USA in 2019
(1). Non-small cell lung cancer
(NSCLC) constitutes 80–85% of lung cancer cases, and the most
common histological subtype is lung adenocarcinoma (LUAD), followed
by lung squamous cell carcinoma (LUSC) (2). There has been much progress in the
development of molecular LUAD-targeted therapies (3). Conversely, there has been limited
progress in the development of treatments for LUSC, with the
exception of immunotherapy (4–6). It is
hypothesized that LUAD originates from epithelial secretory cells,
while LUSC originates from basal cells (2); furthermore, epidermal growth factor
receptor, KRAS proto-oncogene GTPase and EMAP-like 4-ALK receptor
tyrosine kinase mutations tend to occur more frequently in LUAD
(7). By contrast, LUSC exhibits more
molecular abnormalities in the fibroblast growth factor receptor 1,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha-keratin 3 and discoidin domain receptor tyrosine kinase 2
genes (8). In summary, advances in
the treatment, and the elucidation of the molecular mechanisms
involved in LUAD far outweigh those made in LUSC. Due to the high
incidence and mortality rate of LUSC, the need to reveal the
pathogenesis, explore novel biomarkers and develop effective
therapeutic strategies is imperative.
Genomics, gene microarrays and high-throughput
sequencing have been widely utilized in oncology research.
Moreover, dysregulated gene expression plays a significant role in
cancer development. Several studies have identified hub genes from
groups of differentially expressed genes (DEGs) based on integrated
bioinformatics methods; using integrated bioinformatics analysis,
Xia et al (9) identified
anillin actin binding protein as a key gene in cervical cancer
progression. Cui et al (10)
also used bioinformatics analysis to demonstrate that maternally
expressed 3 could function as a biomarker and predict the prognosis
of breast cancer. Studies on NSCLC do exist (11,12), but
most of these involve LUAD (13,14), and
few are related to LUSC.
In the present study, three gene expression profiles
for LUSC were downloaded from the Gene Expression Omnibus (GEO)
database and GEO2R was utilized to screen DEGs between LUSC tissue
samples and normal lung tissue samples. Subsequently, enrichment
analysis, protein-protein interaction (PPI) network construction
and module identification were performed to illustrate the
significant associations between DEGs. Furthermore, hub genes from
these DEGs were identified, validated and analyzed, revealing the
prognostic and clinical values of the DEGS and ncRNA regulatory
networks of hub genes involved in LUSC.
Materials and methods
Microarray data retrieval
The GEO (https://www.ncbi.nlm.nih.gov/gds/) database is a
public, functional genomics repository of array and sequence-based,
high-throughput gene expression data (15). In the present study, three gene
expression profiles (GSE19188, GSE21933 and GSE74706) were
downloaded from the GEO; GSE19188 contained 27 LUSC tissue samples
and 65 normal lung tissue samples; GSE21933 contained 10 LUSC
tissue samples and 10 matched normal lung tissue samples, and
GSE74706 contained 8 LUSC tissue samples and 8 matched normal lung
tissue samples. All probes were converted into their corresponding
official gene symbols according to the annotation information
provided each platform.
DEG screening
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) is an online
web tool that allows for the comparison between two groups of
samples in different experimental conditions (16). It uses Bioconductor R packages to
analyze selected datasets. In the present study, GEO2R was applied
to screen for DEGs between LUSC tissue samples and normal lung
tissue samples. The cut-off criteria were set as adjusted P-value
<0.01 and |logFC|<2. DEGs common to the three datasets were
selected for further analysis.
Gene ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analysis of DEGs
The Database for Annotation, Visualization and
Integrated Discovery (DAVID; http://david.ncifcrf.gov/) is a functional annotation
tool that integrates biological data and analysis for multiple
genes and proteins (17). GO and
KEGG pathway enrichment analyses of DEGs were performed using
DAVID. GO annotation includes biological process (BP), cellular
component (CC) and molecular function (MF). P<0.05 was
considered statistically significant.
PPI network construction and module
identification
The PPI network of DEGs was constructed using the
Search Tool for the Retrieval of Interacting Genes (STRING;
http://string-db.org/cgi/input.pl?UserId=LUbvQCh21nYS&sessionId=XJVXrbqj7wkb&input_page_show_search=on)
database (18). A combined score
≥0.4 was defined as the cut-off point. Cytoscape software (version
3.7.1; http://cytoscape.org/) was then employed
to visualize the PPI network (19).
The most significant module was identified using Molecular Complex
Detection (MCODE) (20), a plug-in
of Cytoscape. The screening options were set as degree cut-off=2,
node score cut-off=0.2, k-core=2 and Max. depth=100. GO and KEGG
pathway enrichment analyses of genes in the most significant mode
were subsequently performed using DAVID.
Hub gene selection and analysis
CytoHubba (21), a
plugin of Cytoscape, was used to select hub genes of DEGs by
identifying the intersection of the top 100 genes with 12
topological analysis methods. The biological process of hub genes
was then analyzed and visualized by the Biological Networks Gene
Oncology tool (BiNGO; http://apps.cytoscape.org/apps/bingo) plugin (22). The network of hub genes and their
co-expression genes was constructed on the cBioportal (v3.0.6)
platform (23).
Validation of hub genes
To validate the hub genes, RNA expression data from
LUSC samples stored in The Cancer Genome Atlas database (TCGA) were
visualized using USCS Xena (24).
Next, the immunohistochemistry (IHC) results of the hub genes were
verified on the Human Protein Atlas (HPA, http://www.proteinatlas.org/). The URLs that directly
link to the images in the HPA are provided in Appendix S1. Hub
genes with both higher RNA and protein expression levels in tumor
tissue compared with normal lung tissue were selected for further
analysis.
Survival analysis and clinical
comparison of hub genes
The Kaplan-Meier (KM) plotter is an online tool that
predicts the prognostic values of cancer-associated genes according
to their expression levels (25). In
the present study, analysis was restricted to squamous cell
carcinomas, and patients were divided into two groups according to
the expression levels of hub genes. Hazard ratio with 95% CI and
log-rank P-value were calculated, and the hub genes with
significant prognostic value were selected for further clinical
comparison. Gene Expression Profiling Interactive Analysis (GEPIA;
http://gepia.cancer-pku.cn/) is an
interactive web server for analyzing RNA sequencing expression data
from TCGA and the Genotype-Tissue Expression (GTEx) project
(26). The relevance between the
expression of selected hub genes and the clinical TNM stage in LUSC
was evaluated using data from the GEPIA database. P<0.05 was
considered as the threshold.
Non-coding (nc)RNA regulatory network
construction
Gene-Cloud Biotechnology information (GCBI;
http://www.gcbi.com.cn/gclib/html/index) is a web tool
that predicts the regulation of genes and ncRNAs, transcription
factors and gene expression levels in disease. In the present
study, GCBI was used to construct ncRNA regulatory networks of hub
genes.
Results
Screening of DEGs in LUSC
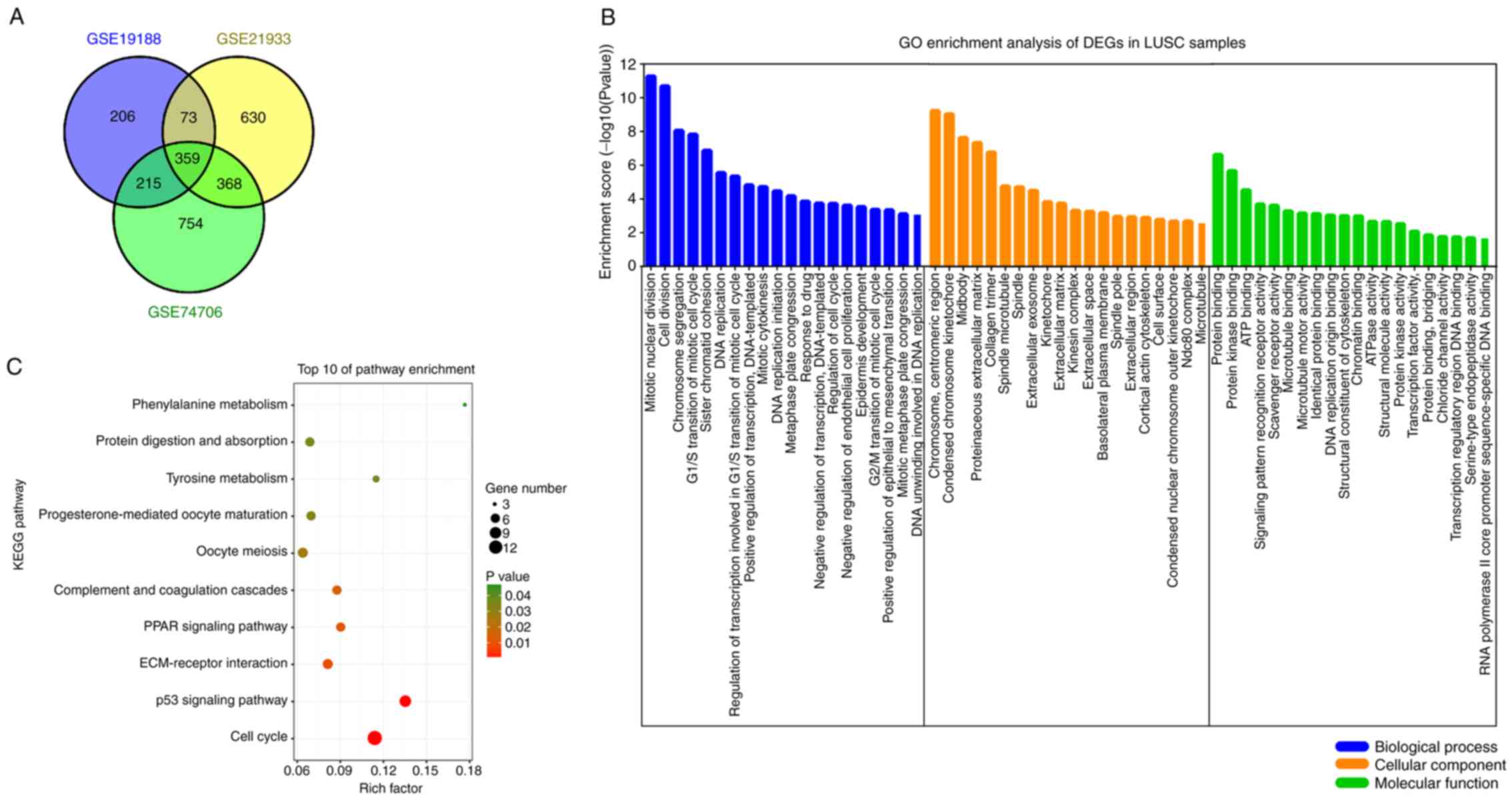
A total of 3 gene expression profiles (GSE19188,
GSE21933 and GSE74706) were analyzed using GEO2R. Collectively, 359
common DEGs were identified (Fig.
1A), including 155 upregulated and 204 downregulated genes
between normal lung tissue samples and tumor tissue samples in
LUSC.
GO and KEGG pathway enrichment
analysis of DEGs
To reveal the functions of the identified DEGs, GO
and KEGG pathway enrichment analysis was performed using DAVID. The
top 20 GO terms and the top 10 KEGG pathways are shown (Fig. 1B and C). GO annotation consisted of
three groups: BP, CC and MF. The DEGs related to BP terms were
predominantly enriched in ‘mitotic nuclear division’, ‘cell
division’, ‘chromosome segregation’, ‘G1/S transition of
mitotic cell cycle’ and ‘sister chromatid cohesion’. The DEGs
related to CC terms were mainly enriched in ‘chromosome’,
‘centromeric region’, ‘condensed chromosome kinetochore’,
‘midbody’, ‘proteinaceous extracellular matrix’ and ‘collagen
trimer’. The DEGs related to MF terms were mainly enriched in
‘protein binding’, ‘protein kinase binding’, ‘ATP binding’ and
‘signaling pattern recognition receptor activity’ and ‘scavenger
receptor activity’ (Fig. 1B). The
most enriched KEGG pathways were ‘cell cycle’, ‘p53 signaling
pathway’, ‘ECM-receptor interaction’, ‘PPAR signaling pathway’ and
‘complement and coagulation cascades’ (Fig. 1C). These enriched terms indicate the
pathogenic mechanisms of LUSC and provide direction for further
research.
PPI network construction, module
identification and analysis
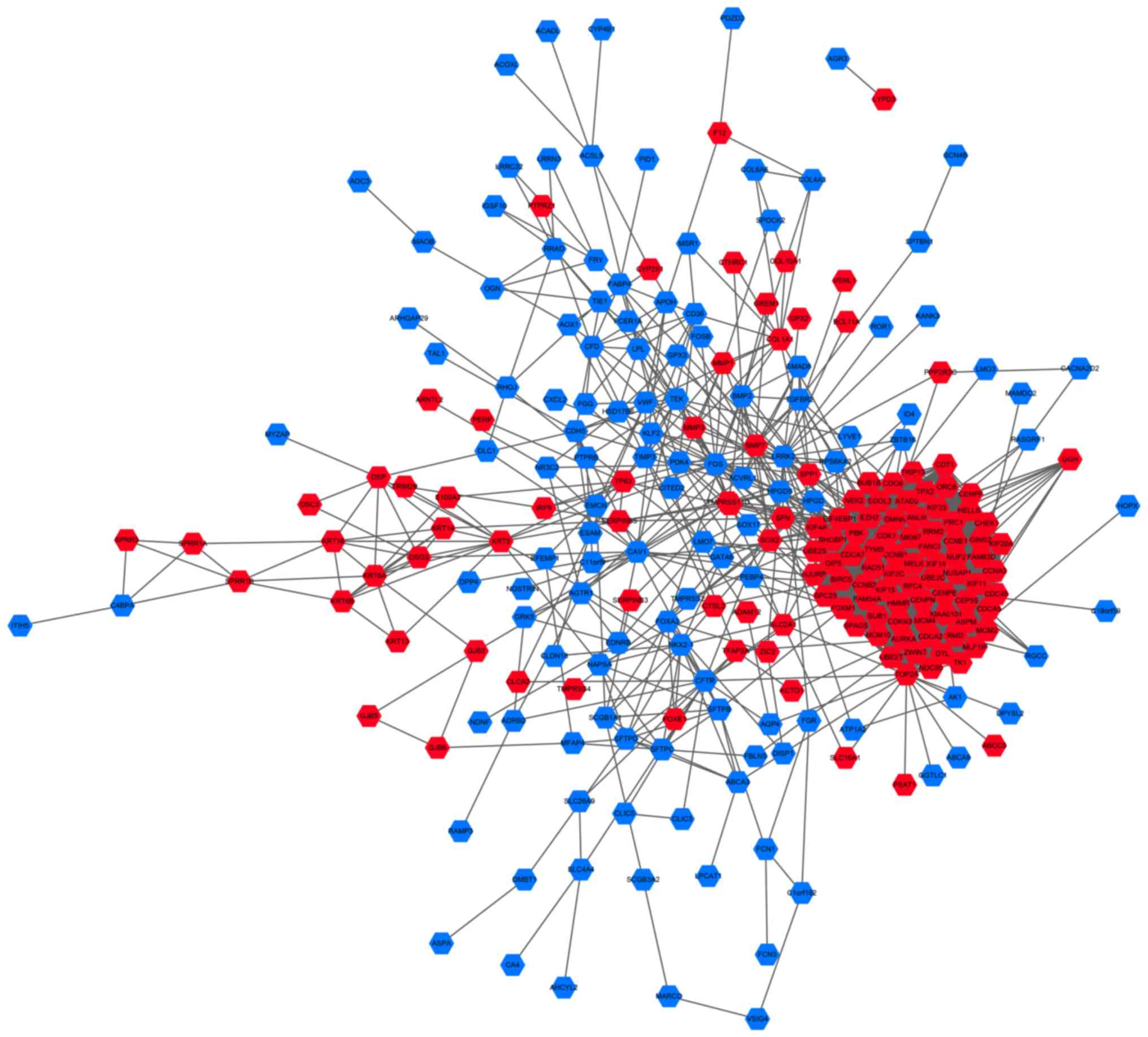
To investigate the interactions between DEGs, the
PPI dataset was downloaded from STRING and displayed using
Cytoscape (Fig. 2). The PPI network
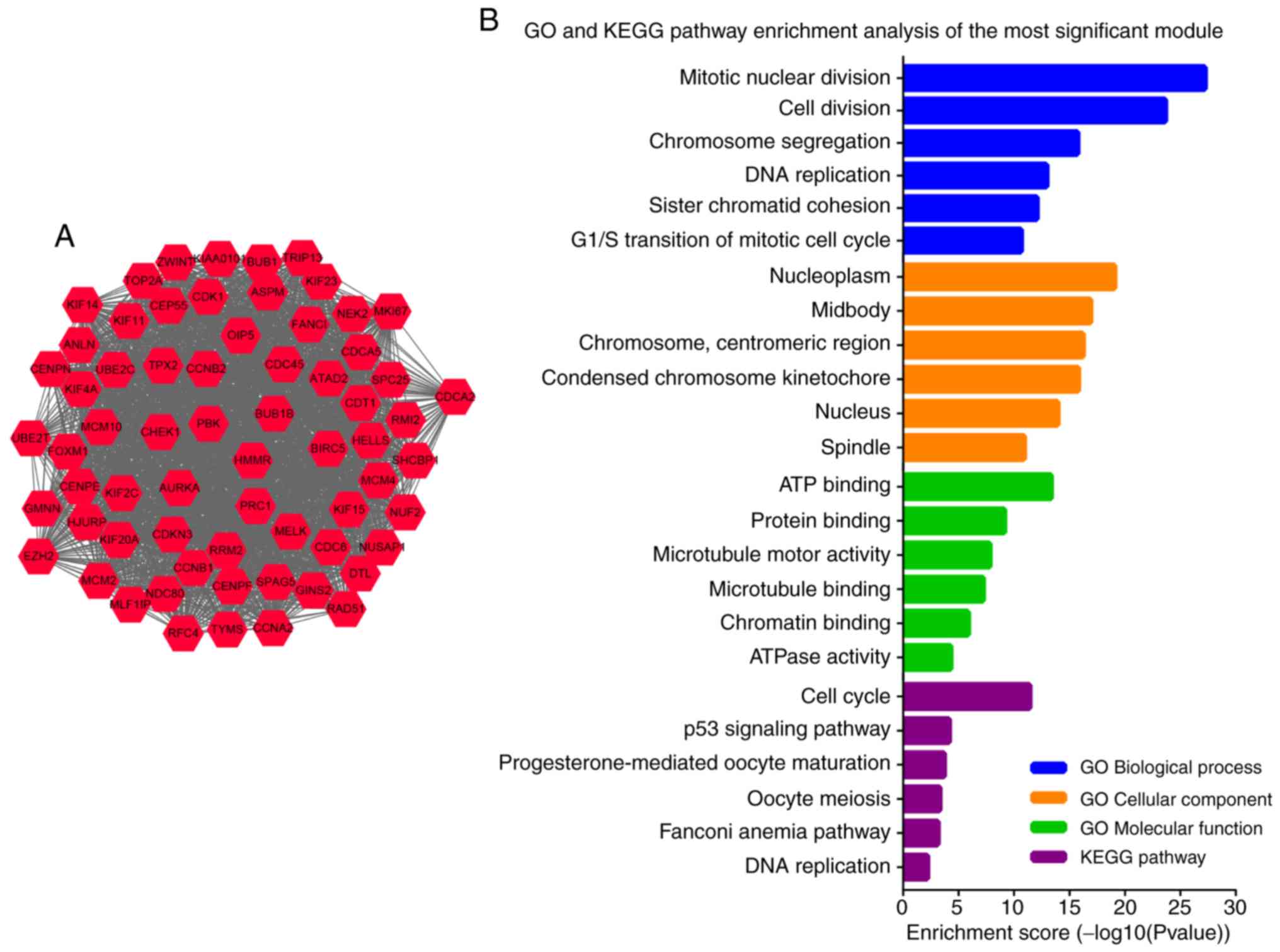
consisted of 257 nodes and 2,772 edges. The most significant module
was identified from the PPI network using the MCODE plugin, and
comprised 66 nodes and 2,050 edges (Fig.
3A). The genes in the module were all upregulated in LUSC.
Furthermore, GO and KEGG pathway enrichment analysis showed that
these genes were primarily involved in ‘mitotic nuclear division’,
‘cell division’, ‘cell cycle’ and ‘p53 signaling pathway’ (Fig. 3B).
Hub gene selection, validation and
analysis
A total of 19 hub genes were selected from the
intersection between the top 100 genes, using 12 topological
analysis methods in CytoHubba; the genes included AURKA, BIRC5,
BUB1, CCNB1, CDK1, CDKN3, CEP55, EZH2, FOXM1, HJURP, HMMR, MELK,
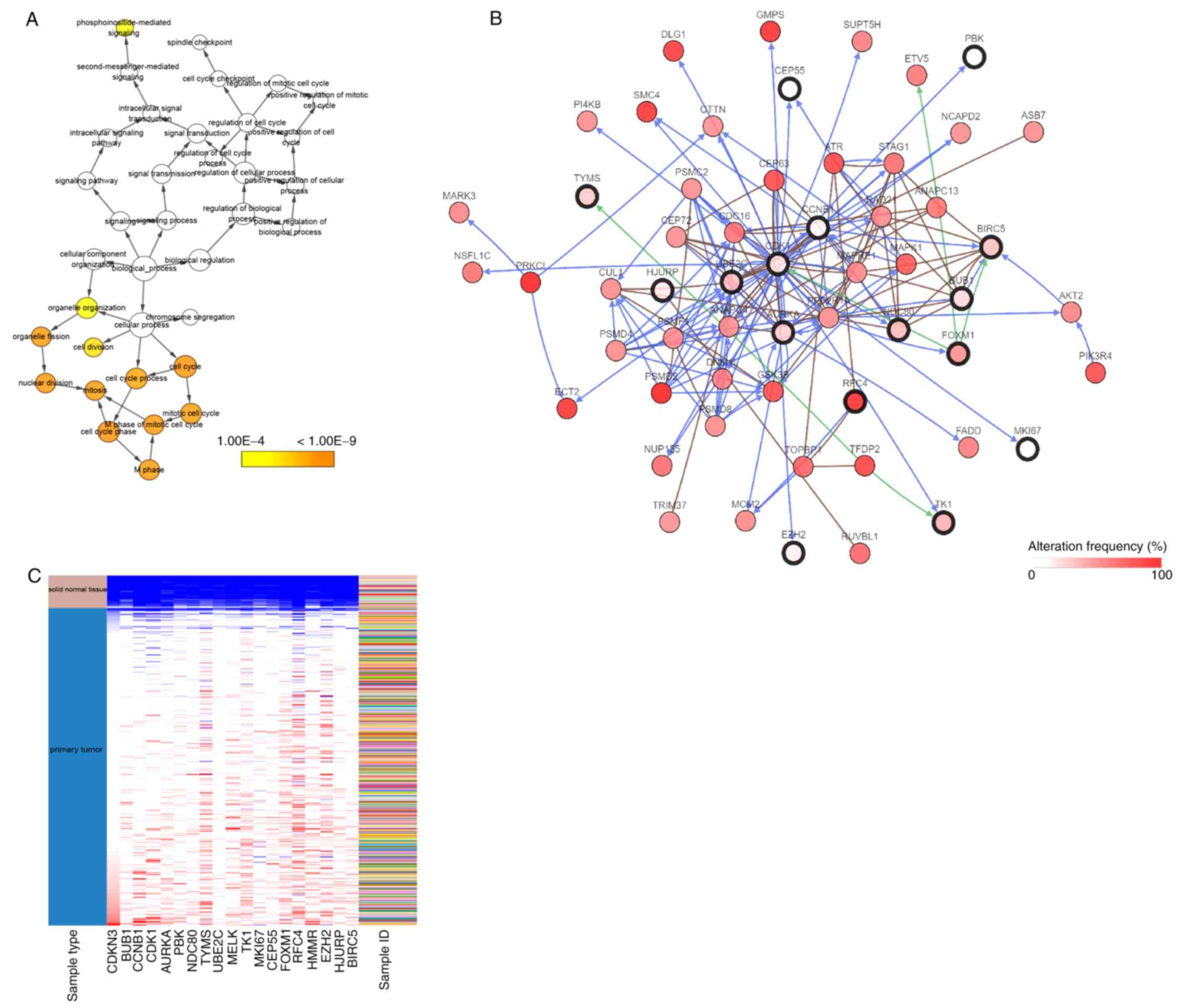
MKI67, NDC80, PBK, RFC4, TK1, TYMS and UBE2C (Table I). The biological process network of
hub genes was constructed using BiNGO (Fig. 4A). The network of hub genes and their
co-expression genes was constructed using cBioPortal (Fig. 4B). A heatmap of TCGA LUSC samples
revealed that these hub genes could differentiate LUSC tissues from
normal lung tissues (Fig. 4C), which
was also consistent with the former result (Fig. 1A). For further validation, the
protein levels of these hub genes appeared higher in LUSC tissue
samples than in normal lung tissue samples, based on the data
extracted from the HPA (Fig. 5);
namely, AURKA, BIRC5, CCNB1, CDK1, CEP55, EZH2, FOXM1, MKI67, RFC4
and TYMS. The protein expression levels of these genes were also
quantitatively analyzed using IHC (Fig.
S1).
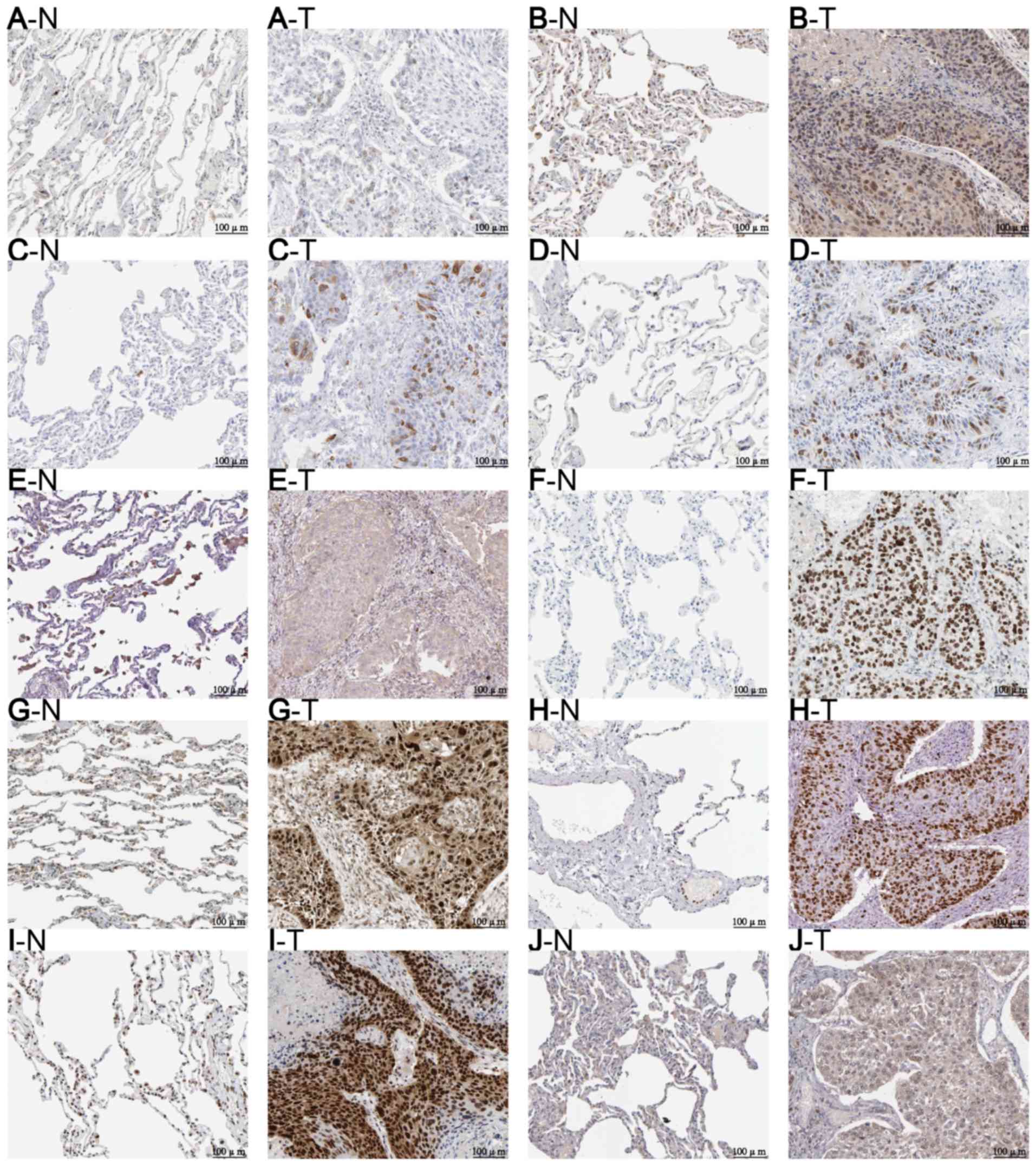 | Figure 5.Protein levels of the 10 hub genes
were higher in tumor, compared with normal tissues.
Immunohistochemistry of (A) AURKA (N: staining, not detected;
intensity, negative; quantity, negative. T: staining, low;
intensity, moderate; quantity, <25%) (B) BIRC5 (N: staining, not
detected; intensity, weak; quantity, <25%. T: staining, low;
intensity, moderate; quantity, 75–25%) (C) CCNB1 (N: staining, not
detected; intensity, negative; quantity, negative. T: staining,
high; intensity, strong; quantity, 75–25%) (D) CDK1 (N: staining,
not detected; intensity, negative; quantity, negative. T: staining,
high; intensity, strong; quantity, 75–25%) (E) CEP55 (N: staining,
not detected; intensity, negative; quantity, negative. T: staining,
low; intensity, weak; quantity, 75–25%) (F) EZH2 (N: staining, not
detected; intensity, negative; quantity, negative. T: staining,
high; intensity, strong; quantity, >75%) (G) FOXM1 (N: staining,
medium; intensity, moderate; quantity, 75–25%. T: staining, high;
intensity, strong; quantity, >75%) (H) MKI67 (N: staining, not
detected; intensity, negative; quantity, negative. T: staining,
high; intensity, strong; quantity, >75%) (I) RFC4 (N: staining,
low; intensity, weak; quantity, >75%. T: staining, high;
intensity, strong; quantity, >75%) (J) TYMS (N: staining, not
detected; intensity, negative; quantity, negative. T: staining,
medium; intensity, moderate; quantity, >75%) based on data from
the Human Protein Atlas. N, normal tissue; T, tumor tissue. Gene
definitions are displayed in Table
I. |
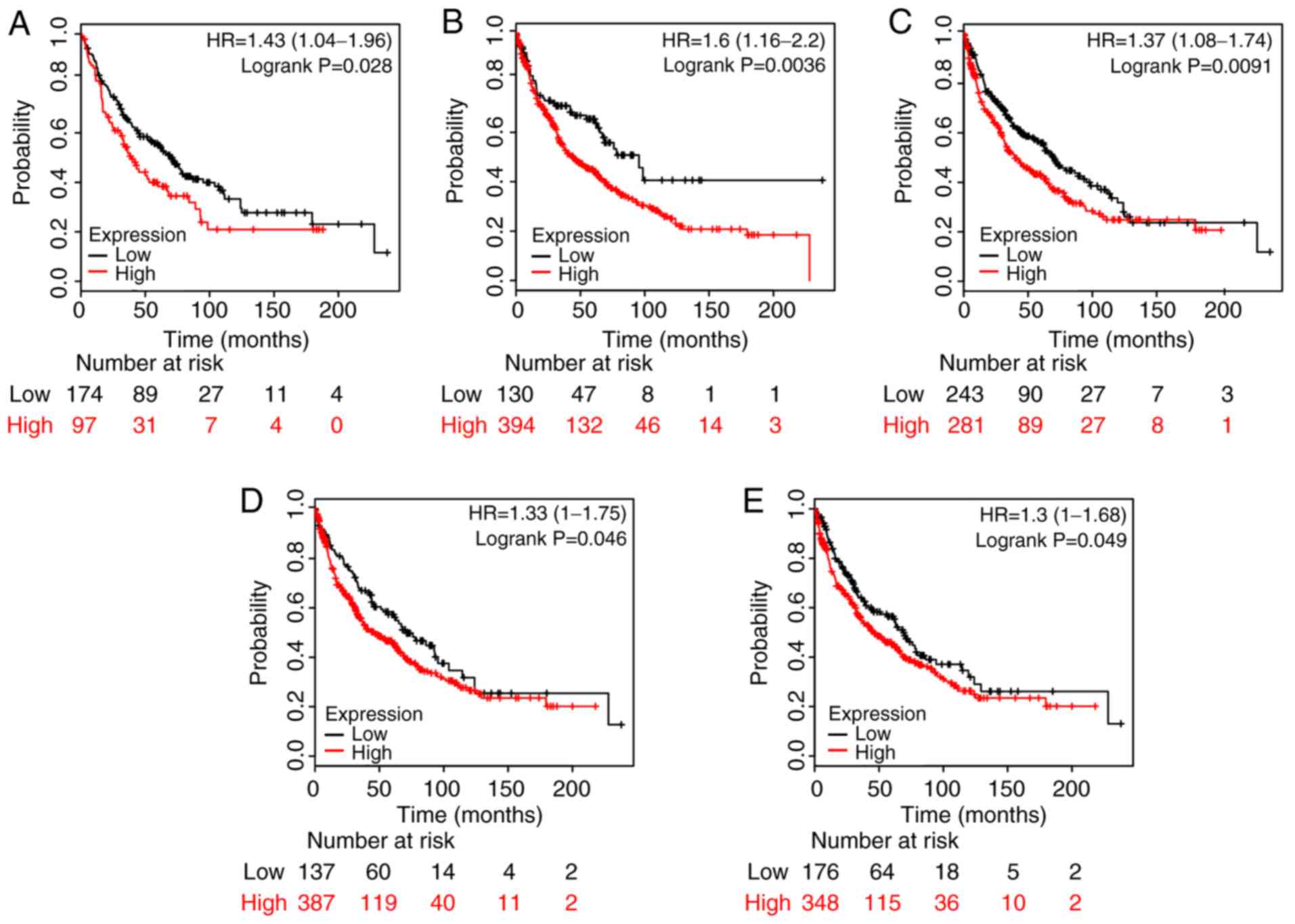
Survival analysis and clinical
comparison of hub genes
The aforementioned 10 hub genes were further
evaluated using the KM plotter to predict their prognostic values
in LUSC (according to their expression levels). The results showed
that higher mRNA expression levels of CCNB, CEP55, FOXM1, MKI67 and
TYMS 1, in tumor vs. normal tissues, were all significantly
associated with poor overall survival in LUSC patients (P<0.05;
Fig. 6), whereas the others genes
were not associated with prognosis. Multivariate analysis showed
that CEP55, MKI67 and TYMS together with stage and sex had a
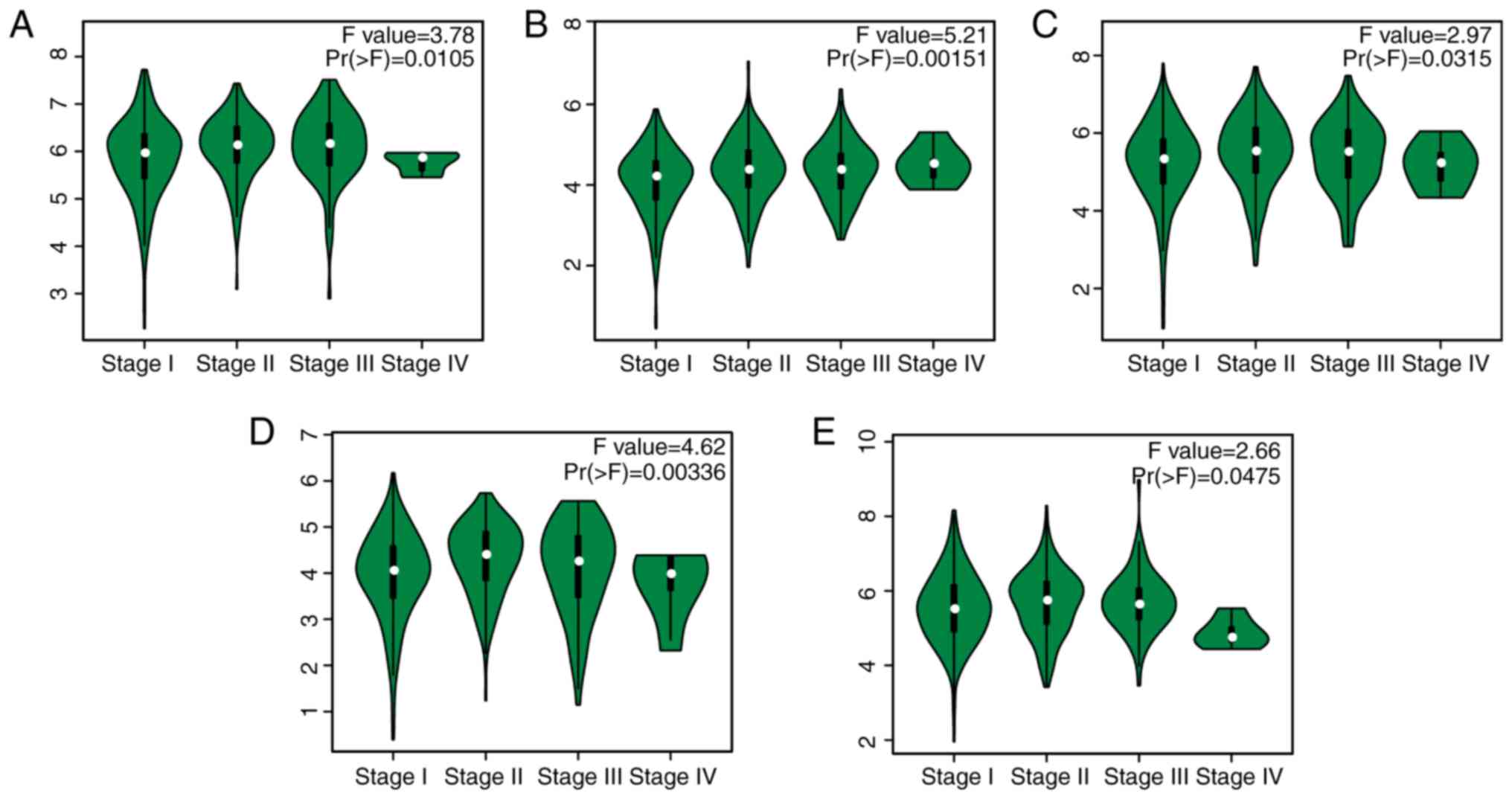
significant impact on patient survival time (Table SI). In addition, the expression
levels of the 5 hub genes were found to be associated with the
clinical pathological stage, displayed as violin plots (P<0.05;
Fig. 7). Higher expression levels of
5 hub genes tended to be associated with more advanced TNM stages,
except for CCNB1, FOXM1 and MKI67 in stage IV. Perhaps the small
sample size for stage IV disease could account for this finding.
Moreover, ROC curves of the five hub genes (Fig. S2) exhibited a significant effect in
distinguishing tumor tissues from normal tissues.
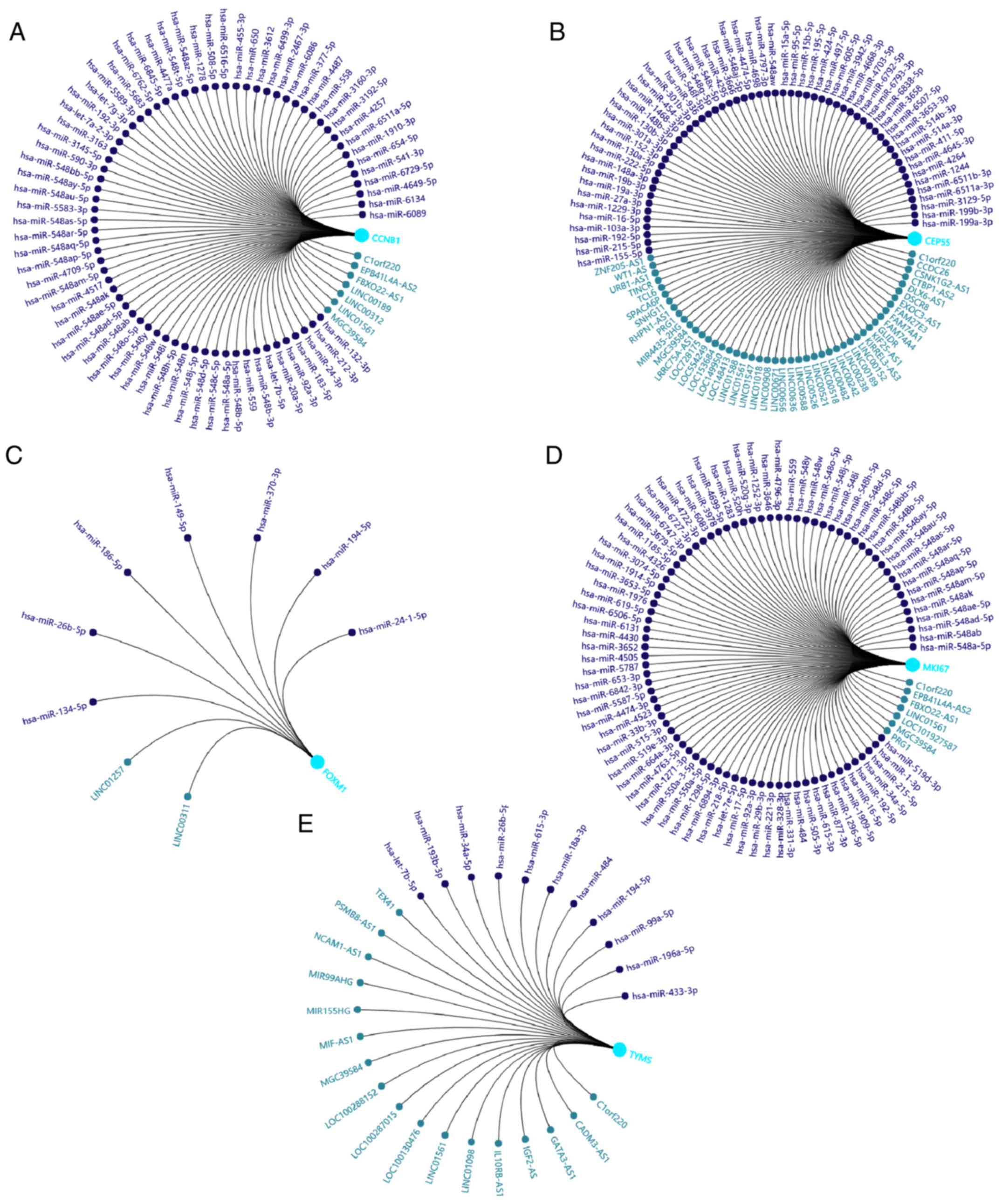
ncRNA regulatory network
construction
The aforementioned enrichment analysis results
revealed that the biological functions of the five selected hub
genes were related to the cell cycle; these included ‘mitotic
nuclear division’, ‘cell division’, ‘mitotic cytokinesis’,
‘regulation of cell cycle’, ‘G1/S transition of mitotic
cell cycle’ and ‘G2/M transition of mitotic cell cycle’.
Given the potent roles of ncRNAs in regulating biological
processes, the related ncRNAs of the five hub genes were predicted
using GCBI and exhibited as regulatory networks (Fig. 8). As shown, C1orf220, LINC01561 and
MGC39584 simultaneously regulated CCNB1, CEP55, MKI67 and TYMS,
indicating that these long non-coding RNAs (lncRNAs) play important
roles in LUSC pathogenesis. The target regions of CCNB1 and MKI67
possess the same micro RNA (miRNA/miR) binding sites, which include
miR-92a-3p, miR-559, miR-548a-5 and miR-548ab. CCNB1 and TYMS
shared let-7b-5p, and MKI67 and CEP55 shared miR-16-5p, miR-192-5p
and miR-215-5p. Moreover, the target regions of MKI67 and TYMS
shared the miR-34a-5p, miR-484 and miR-615-3p binding sites, and
TYMS and FOXM1 shared those of miR-194-5p and miR-26b-5p.
Discussion
Although the incidence of lung cancer is declining,
it is still responsible for the highest proportion of
cancer-related mortality (1). Until
now, there have been few specific therapeutics aimed at LUSC,
compared with LUAD. Hence, it is imperative to identify novel
biomarkers and effective therapeutic targets specific to LUSC.
In the present study, a total of 359 common DEGs
were selected from three datasets, including 155 upregulated and
204 downregulated genes. The relative biological functions were
primarily associated with ‘mitotic nuclear division’, ‘cell
division’, ‘protein binding’ and ‘protein kinase binding’. KEGG
pathways, including ‘cell cycle’, ‘p53 signaling pathway’, and
‘ECM-receptor interaction’, were dysregulated in LUSC. The PPI
network determined the interactions of DEGs and a significant
module was constructed and identified. GO and KEGG pathway
enrichment analysis indicated that 66 genes from the most
significant module in the PPI network were predominantly related to
‘mitotic nuclear division’, ‘cell division’, ‘cell cycle’ and ‘p53
signaling pathway’. Among the DEGs, 19 hub genes were selected. The
co-expression network further validated the relationship between
the hub genes, and revealed the pathways and potential therapeutic
targets in which they are involved. In total, 10 of the 19 hub
genes were validated as exhibiting elevated expressions levels of
both mRNA and protein. Survival analysis revealed that high mRNA
expression levels of CCNB1, CEP55, FOXM1, MKI67 and TYMS were
related to poor overall survival. These five hub genes were also
associated with advanced clinical pathological stages.
Cyclin B1 (CCNB1) is a pivotal member of the cyclin
family that complexes with CDC2, exerting control over the cell
cycle (27). As it gradually
accumulates in the S phase, CCNB1 reaches its maximum level before
mitosis and is then rapidly degraded in the M phase (27). Hence, CCNB1 is a key mediator of
G2-M phase checkpoint surveillance. Dysregulation of
CCNB1 leads to cell hyperplasia and tumorigenesis, which has been
reported in various cancers, including esophageal squamous cell
carcinoma (28), breast cancer
(29) and gastric cancer (30). Also, CCNB1 was highly expressed in
NSCLC tissues compared with normal lung tissues (31,32).
NSCLC patients with CCNB1 upregulation tend to have a poorer
prognosis compared with patients with normal CCNB1 expression
(32), particularly in LUSC
(31). Furthermore, CCNB1 is also
associated with long-term smokers and preneoplastic lesions
(33), which could partially account
for smoking being the leading cause of LUSC (34). Consistent with previous studies
(27,35), the results of the present study
revealed that CCNB1 was implicated in both the cell cycle and the
p53-signaling pathway. Additionally, CCNB1 overexpression was
associated with poor prognosis and advanced clinical pathological
stages in LUSC in the present study. Yoshida et al (36) revealed that the upregulation of CCNB1
correlated with higher Ki-67 and PCNA in NSCLC, while CCNB1 and
MKI67 (Ki-67) served as hub genes in the present study. Considering
these findings, CCNB1 shows promise as a biomarker for LUSC
diagnosis.
Centrosomal Protein 55 (CEP55), a highly coiled
centrosomal protein, is an indispensable regulator of cytokinesis
(37). During abscission, CEP55
recruits members of the endosomal-sorting complex to tear the
cytokinetic bridge and divide the cytoplasm into two daughter cells
(37,38). Cytokinetic disorders result in
cellular transformation and malignancy (39). Certain studies have demonstrated that
CEP55 upregulation contributes to different cancer types, such as
breast cancer, ovarian cancer, colon cancer (39,40).
CEP55 upregulation also promotes a number of events related to
neoplasia, such as cell migration, invasion and anchorage
independent growth (39,41). Kalimutho et al (39) revealed that CEP55 is implicated in
the MEK1/2-MYC axis, which mediates aneuploidy and genomic
instability in breast cancer. In NSCLC patients, elevated levels
CEP55 promote migration and invasion via activation of the PI3K/AKT
pathway (42), in addition to
predicting unfavorable clinical outcomes (43). However, few studies have been
conducted specifically on LUSC. GO enrichment analysis revealed
that CEP55 was engaged in mitotic cytokinesis and mitotic nuclear
division. CEP55 also exhibited a co-expression relationship with
mitogen-activated protein kinase (MAPK) 1, which could implicate
CEP55 in the MAPK-signaling pathway. In addition, CEP55 was highly
expressed in LUSC and indicated poor clinical outcome. Accordingly,
the findings of the present study indicate CEP55 as a potential
therapeutic target.
Forkhead Box M1 (FOXM1) is a well-known
transcription factor from the Forkhead family of proteins, which is
upregulated in a broad range of tumors (44,45). As
such, the FOXM1 regulatory network has been identified as a major
predictor of unfavorable outcomes in several cancer types, such as
breast cancer, colon cancer, prostate cancer (46). Similar to NSCLC, elevated expression
levels of FOXM1 are significantly associated with poor prognosis
(47). In LUSC, the findings of the
present study resulted in similar conclusions. Expression of FOXM1
varies between different pathological stages; generally, FOXM1
mediates its pro-tumorigenic effect via transcriptional activation
of its target (44). Accordingly, it
has been revealed that FOXM1 regulates the expression levels of
CCNB1, CEP55 and TYMS (48–50), which were identified as hub genes in
the present study. Thus, it was demonstrated that FOXM1 plays a
crucial role in LUSC tumorigenesis.
Marker of Proliferation Ki-67 (MKI67), a protein
phosphatase 1-binding protein, is known as a cell proliferation
marker in both laboratory and clinical cancer applications
(51). MKI67 is degraded in the
G0 and G1 phases, and gradually accumulates
in the nucleoli following the onset of S phase (52). Moreover, MKI67 not only represents
proliferation status, but also distinguishes rapid-growing from
slow-growing tumors (51,53). In NSCLC, MKI67 has been defined as a
diagnostic and prognostic marker (54). The results of the present study
further validate that MKI67 upregulation indicates an advanced
pathological stage and adverse overall survival time in LUSC.
Consequently, the results of the present study support the crucial
value of MKI67 in clinical diagnosis and during treatment.
Thymidylate Synthetase (TYMS) functions as a
fundamental participant in thymidylate biosynthesis and de
novo DNA replication (55). TYMS
inhibition results in cell cycle arrest at the S phase (56), and high TYMS expression accelerates
cell proliferation and leads to malignant behaviors in various
types of solid tumor (56–58). In NSCLC, TYMS is reported to be
upregulated in tumor tissues and linked to adverse prognosis
(56). Further studies have revealed
that TYMS is more highly expressed in LUSC than in LUAD (56,59).
This is hypothesized to be responsible for the unsatisfactory
treatment response to pemetrexed-based chemotherapy in LUSC
(60). The present study revealed
that TYMS was associated with the GO terms ‘G1/S
transition of mitotic cell cycle’ and ‘regulation of
transcription’. Thus, the upregulation of TYMS may indicate poor
prognosis and be pathologically detrimental. As indicated, TYMS
disturbances contribute to LUSC development, and may therefore be a
promising therapeutic target for further research.
With advances in next-generation deep sequencing, an
increasing number of ncRNAs have been recognized in various
diseases, including cancer (61).
ncRNAs have the robust ability to regulate gene expression via
versatile mechanisms that orchestrate biological processes. The
present study assessed ncRNA regulatory networks to further
investigate the effect of the hub genes in LUSC. Using, a competing
endogenous RNA (ceRNA) network, Sui et al (62) reported that C1orf220 was upregulated
in LUSC and indicated poor prognosis. Jiang et al (63) determined that the
linc01561-miR-145-5p-MMP11 interaction contributed to the
progression of breast cancer. However, the role of linc01561 in
LUSC pathology is not fully understood, and MGC39584 (also known as
LINC01667) remains to be investigated. Hub gene-related ncRNAs were
revealed in the present study; C1orf220, LINC01561 and MGC39584 are
all lncRNAs that simultaneously regulate the hub genes CCNB1,
CEP55, MKI67 and TYMS and, hub genes sharing the same miRNAs may
form a ceRNA network. Based on these findings, the present study
suggests further research targets concerning the role of ncRNAs in
LUSC.
RNA-based bioinformatics analyses could be
considered a limitation of the present study. The verification of
direct protein interactions, as well as functional experiments,
would improve the validity of the results.
In conclusion, five hub genes (CCNB1, CEP55, FOXM1,
MKI67 and TYMS) have been identified to play a central role in LUSC
tumorigenesis, exhibiting ample diversity to the results derived
from LUAD studies (13,14). Furthermore, ncRNAs such as C1orf220,
LINC01561 and MGC39584, were shown to regulate these hub genes.
This is the first proposal of key genes specifically upregulated in
LUSC via integrated bioinformatics analysis. The findings of the
present study may help inform the development of targeted
therapeutics, though further experimental studies are required to
verify these findings.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by a grant from the
National Natural Scientific Foundation of China (grant no.
31627801).
Availability of data and materials
The datasets downloaded and analyzed during the
current study are available on the GEO database: GSE19188,
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19188;
GSE21933, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21933;
GSE74706, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74706.
Authors' contributions
YS conducted the study and wrote the manuscript. YL
applied statistical, computational and other techniques to
synthesize and interpret the data, and revised the manuscript. CY
and HS downloaded and analyzed the data. KY designed and directed
the study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Einhorn LH: First-line chemotherapy for
non-small-cell lung cancer: Is there a superior regimen based on
histology? J Clin Oncol. 26:3485–3486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brahmer J, Reckamp KL, Baas P, Crinò L,
Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE,
Holgado E, et al: Nivolumab versus docetaxel in advanced
squamous-cell non-small-cell lung cancer. N Engl J Med.
373:123–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Herbst RS, Baas P, Kim DW, Felip E,
Pérez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, et al:
Pembrolizumab versus docetaxel for previously treated,
PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010):
A randomised controlled trial. Lancet. 387:1540–1550. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zugazagoitia J, Ponce S and Paz-Ares L:
Necitumumab for first-line treatment of advanced, squamous,
non-small-cell lung cancer: A relevant step forward? Transl Lung
Cancer Res. 5:95–97. 2016.PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Research Network, .
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Drilon A, Rekhtman N, Ladanyi M and Paik
P: Squamous-cell carcinomas of the lung: Emerging biology,
controversies, and the promise of targeted therapy. Lancet Oncol.
13:e418–e426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xia L, Su X, Shen J, Meng Q, Yan J, Zhang
C, Chen Y, Wang H and Xu M: ANLN functions as a key candidate gene
in cervical cancer as determined by integrated bioinformatic
analysis. Cancer Manag Res. 10:663–670. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cui X, Yi Q, Jing X, Huang Y, Tian J, Long
C, Xiang Z, Liu J, Zhang C, Tan B, et al: Mining prognostic
significance of MEG3 in human breast cancer using bioinformatics
analysis. Cell Physiol Biochem. 50:41–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shao Y, Liang B, Long F and Jiang SJ:
Diagnostic MicroRNA biomarker discovery for non-small-cell lung
cancer adenocarcinoma by integrative bioinformatics analysis.
Biomed Res Int. 2017:25630852017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Piao J, Sun J, Yang Y, Jin T, Chen L and
Lin Z: Target gene screening and evaluation of prognostic values in
non-small cell lung cancers by bioinformatics analysis. Gene.
647:306–311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song YJ, Tan J, Gao XH and Wang LX:
Integrated analysis reveals key genes with prognostic value in lung
adenocarcinoma. Cancer Manag Res. 10:6097–6108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou LN, Li SC, Li XY, Ge H and Li HM:
Identification of differential protein-coding gene expressions in
early phase lung adenocarcinoma. Thorac Cancer. 9:234–240. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Davis S and Meltzer PS: GEOquery: A bridge
between the Gene Expression Omnibus (GEO) and BioConductor.
Bioinformatics. 23:1846–1847. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang da DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45:D362–D368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: cytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 8 (Suppl 4):S112014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goldman M, Craft B, Zhu J and Haussler D:
Abstract 2584: The UCSC Xena system for cancer genomics data
visualization and interpretation. Cancer Res. 77 (Suppl
13):S25842017.
|
|
25
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Smits VA and Medema RH: Checking out the
G(2)/M transition. Biochim Biophys Acta. 1519:1–12. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song Y, Zhao C, Dong L, Fu M, Xue L, Huang
Z, Tong T, Zhou Z, Chen A, Yang Z, et al: Overexpression of cyclin
B1 in human esophageal squamous cell carcinoma cells induces tumor
cell invasive growth and metastasis. Carcinogenesis. 29:307–315.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aaltonen K, Amini RM, Heikkilä P,
Aittomäki K, Tamminen A, Nevanlinna H and Blomqvist C: High cyclin
B1 expression is associated with poor survival in breast cancer. Br
J Cancer. 100:1055–1060. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Begnami MD, Fregnani JH, Nonogaki S and
Soares FA: Evaluation of cell cycle protein expression in gastric
cancer: Cyclin B1 expression and its prognostic implication. Hum
Pathol. 41:1120–1127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soria JC, Jang SJ, Khuri FR, Hassan K, Liu
D, Hong WK and Mao L: Overexpression of cyclin B1 in early-stage
non-small cell lung cancer and its clinical implication. Cancer
Res. 60:4000–4004. 2000.PubMed/NCBI
|
|
32
|
Cooper WA, Kohonen-Corish MR, McCaughan B,
Kennedy C, Sutherland RL and Lee CS: Expression and prognostic
significance of cyclin B1 and cyclin A in non-small cell lung
cancer. Histopathology. 55:28–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Suzuki H, Graziano DF, McKolanis J and
Finn OJ: T cell-dependent antibody responses against aberrantly
expressed cyclin B1 protein in patients with cancer and
premalignant disease. Clin Cancer Res. 11:1521–1526. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Egawa H, Furukawa K, Preston D, Funamoto
S, Yonehara S, Matsuo T, Tokuoka S, Suyama A, Ozasa K, Kodama K and
Mabuchi K: Radiation and smoking effects on lung cancer incidence
by histological types among atomic bomb survivors. Radiat Res.
178:191–201. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pines J and Hunter T: Isolation of a human
cyclin cDNA: Evidence for cyclin mRNA and protein regulation in the
cell cycle and for interaction with p34cdc2. Cell. 58:833–846.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yoshida T, Tanaka S, Mogi A, Shitara Y and
Kuwano H: The clinical significance of Cyclin B1 and Wee1
expression in non-small-cell lung cancer. Ann Oncol. 15:252–256.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee HH, Elia N, Ghirlando R,
Lippincott-Schwartz J and Hurley JH: Midbody targeting of the ESCRT
machinery by a noncanonical coiled coil in CEP55. Science.
322:576–580. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fabbro M, Zhou BB, Takahashi M, Sarcevic
B, Lal P, Graham ME, Gabrielli BG, Robinson PJ, Nigg EA, Ono Y and
Khanna KK: Cdk1/Erk2- and Plk1-dependent phosphorylation of a
centrosome protein, Cep55, is required for its recruitment to
midbody and cytokinesis. Dev Cell. 9:477–488. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kalimutho M, Sinha D, Jeffery J, Nones K,
Srihari S, Fernando WC, Duijf PH, Vennin C, Raninga P, Nanayakkara
D, et al: CEP55 is a determinant of cell fate during perturbed
mitosis in breast cancer. EMBO Mol Med. 10:e85662018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jeffery J, Sinha D, Srihari S, Kalimutho M
and Khanna KK: Beyond cytokinesis: The emerging roles of CEP55 in
tumorigenesis. Oncogene. 35:683–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen CH, Shiu LY, Su LJ, Huang CY, Huang
SC, Huang CC, Yin YF, Wang WS, Tsai HT, Fang FM, et al: FLJ10540 is
associated with tumor progression in nasopharyngeal carcinomas and
contributes to nasopharyngeal cell proliferation, and metastasis
via osteopontin/CD44 pathway. J Transl Med. 10:932012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen CH, Lai JM, Chou TY, Chen CY, Su LJ,
Lee YC, Cheng TS, Hong YR, Chou CK, Whang-Peng J, et al: VEGFA
upregulates FLJ10540 and modulates migration and invasion of lung
cancer via PI3K/AKT pathway. PLoS One. 4:e50522009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma XP, Zhang W, Wu BQ and Qin J:
Correlations between mRNA levels of centrosomal protein 55 (CEP55)
and clinical features of patients with lung cancer. Med Sci Monit.
24:3093–3097. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Halasi M and Gartel AL: FOX(M1) news-it is
cancer. Mol Cancer Ther. 12:245–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gentles AJ, Newman AM, Liu CL, Bratman SV,
Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al: The
prognostic landscape of genes and infiltrating immune cells across
human cancers. Nat Med. 21:938–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang IC, Ustiyan V, Zhang Y, Cai Y, Kalin
TV and Kalinichenko VV: Foxm1 transcription factor is required for
the initiation of lung tumorigenesis by oncogenic Kras(G12D.).
Oncogene. 33:5391–5396. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Leung TW, Lin SS, Tsang AC, Tong CS, Ching
JC, Leung WY, Gimlich R, Wong GG and Yao KM: Over-expression of
FoxM1 stimulates cyclin B1 expression. FEBS Lett. 507:59–66. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gemenetzidis E, Bose A, Riaz AM, Chaplin
T, Young BD, Ali M, Sugden D, Thurlow JK, Cheong SC, Teo SH, et al:
FOXM1 upregulation is an early event in human squamous cell
carcinoma and it is enhanced by nicotine during malignant
transformation. PLoS One. 4:e48492009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Intuyod K, Saavedra-García P, Zona S, Lai
CF, Jiramongkol Y, Vaeteewoottacharn K, Pairojkul C, Yao S, Yong
JS, Trakansuebkul S, et al: FOXM1 modulates 5-fluorouracil
sensitivity in cholangiocarcinoma through thymidylate synthase
(TYMS): Implications of FOXM1-TYMS axis uncoupling in 5-FU
resistance. Cell Death Dis. 9:11852018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sales GR and Vagnarelli P: Ki-67: More
hidden behind a ‘classic proliferation marker’. Trends Biochem Sci.
43:747–748. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sobecki M, Mrouj K, Colinge J, Gerbe F,
Jay P, Krasinska L, Dulic V and Fisher D: Cell-cycle regulation
accounts for variability in Ki-67 expression levels. Cancer Res.
77:2722–2734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Miller I, Min M, Yang C, Tian C, Gookin S,
Carter D and Spencer SL: Ki67 is a graded rather than a binary
marker of proliferation versus quiescence. Cell Rep.
24:1105–1112.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Warth A, Cortis J, Soltermann A, Meister
M, Budczies J, Stenzinger A, Goeppert B, Thomas M, Herth FJ,
Schirmacher P, et al: Tumour cell proliferation (Ki-67) in
non-small cell lung cancer: A critical reappraisal of its
prognostic role. Br J Cancer. 111:1222–1229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Carreras CW and Santi DV: The catalytic
mechanism and structure of thymidylate synthase. Annu Rev Biochem.
64:721–762. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Takezawa K, Okamoto I, Tsukioka S, Uchida
J, Kiniwa M, Fukuoka M and Nakagawa K: Identification of
thymidylate synthase as a potential therapeutic target for lung
cancer. Br J Cancer. 103:354–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Foekens JA, Romain S, Look MP, Martin PM
and Klijn JG: Thymidine kinase and thymidylate synthase in advanced
breast cancer: Response to tamoxifen and chemotherapy. Cancer Res.
61:1421–1425. 2001.PubMed/NCBI
|
|
58
|
Squires MR III, Fisher SB, Fisher KE,
Patel SH, Kooby DA, El-Rayes BF, Staley CR III, Farris AR III and
Maithel SK: Differential expression and prognostic value of ERCC1
and thymidylate synthase in resected gastric adenocarcinoma.
Cancer. 119:3242–3250. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sun JM, Han J, Ahn JS, Park K and Ahn MJ:
Significance of thymidylate synthase and thyroid transcription
factor 1 expression in patients with nonsquamous non-small cell
lung cancer treated with pemetrexed-based chemotherapy. J Thorac
Oncol. 6:1392–1399. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sun JM, Ahn JS, Jung SH, Sun J, Ha SY, Han
J, Park K and Ahn MJ: Pemetrexed plus cisplatin versus gemcitabine
plus cisplatin according to thymidylate synthase expression in
nonsquamous non-small-cell lung cancer: A biomarker-stratified
randomized phase II trial. J Clin Oncol. 33:2450–2456. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Beermann J, Piccoli MT, Viereck J and Thum
T: Non-coding RNAs in development and disease: Background,
mechanisms, and therapeutic approaches. Physiol Rev. 96:1297–1325.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sui J, Xu SY, Han J, Yang SR, Li CY, Yin
LH, Pu YP and Liang GY: Integrated analysis of competing endogenous
RNA network revealing lncRNAs as potential prognostic biomarkers in
human lung squamous cell carcinoma. Oncotarget. 8:65997–66018.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jiang R, Zhao C, Gao B, Xu J, Song W and
Shi P: Mixomics analysis of breast cancer: Long non-coding RNA
linc01561 acts as ceRNA involved in the progression of breast
cancer. Int J Biochem Cell Biol. 102:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|