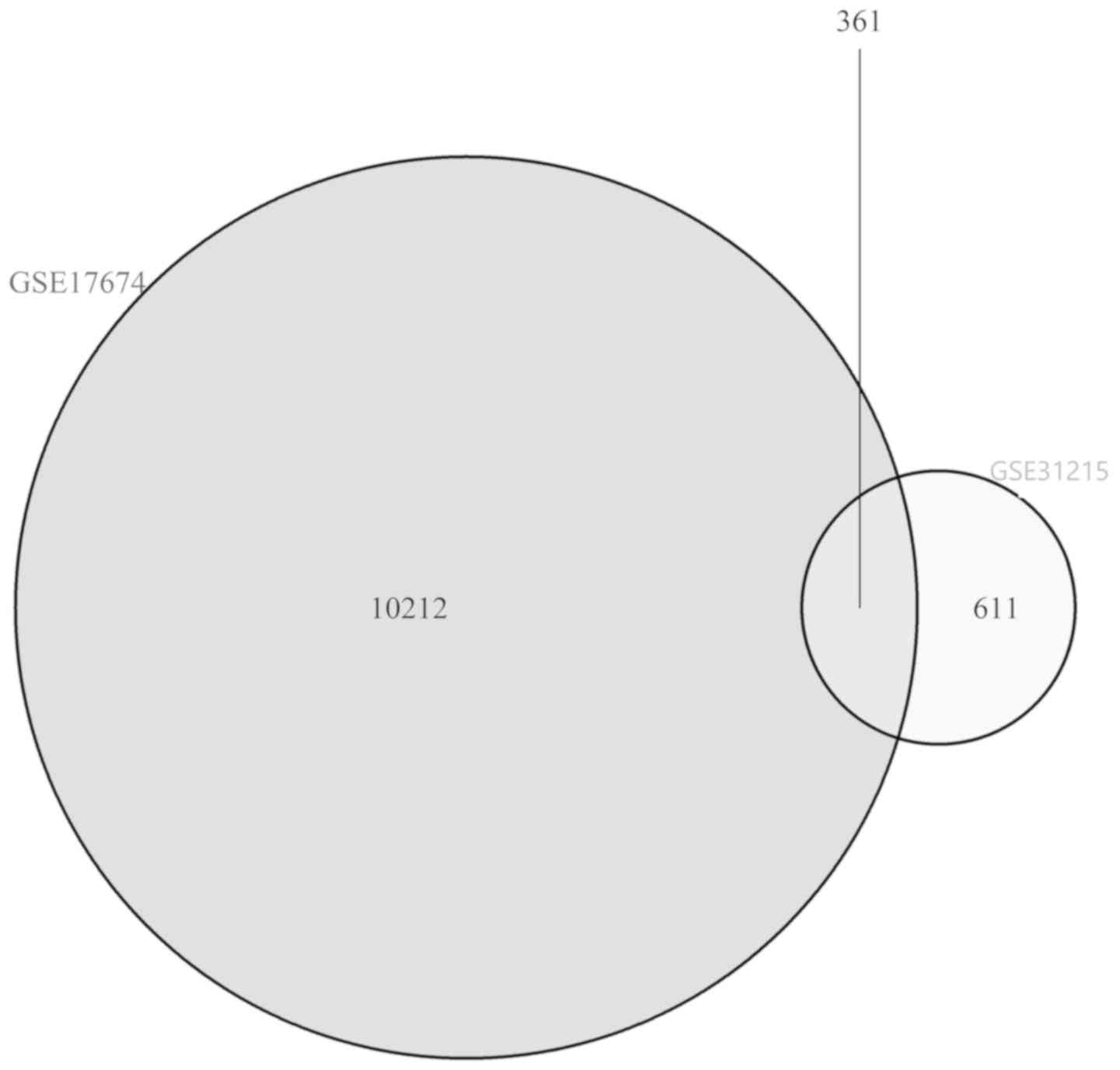
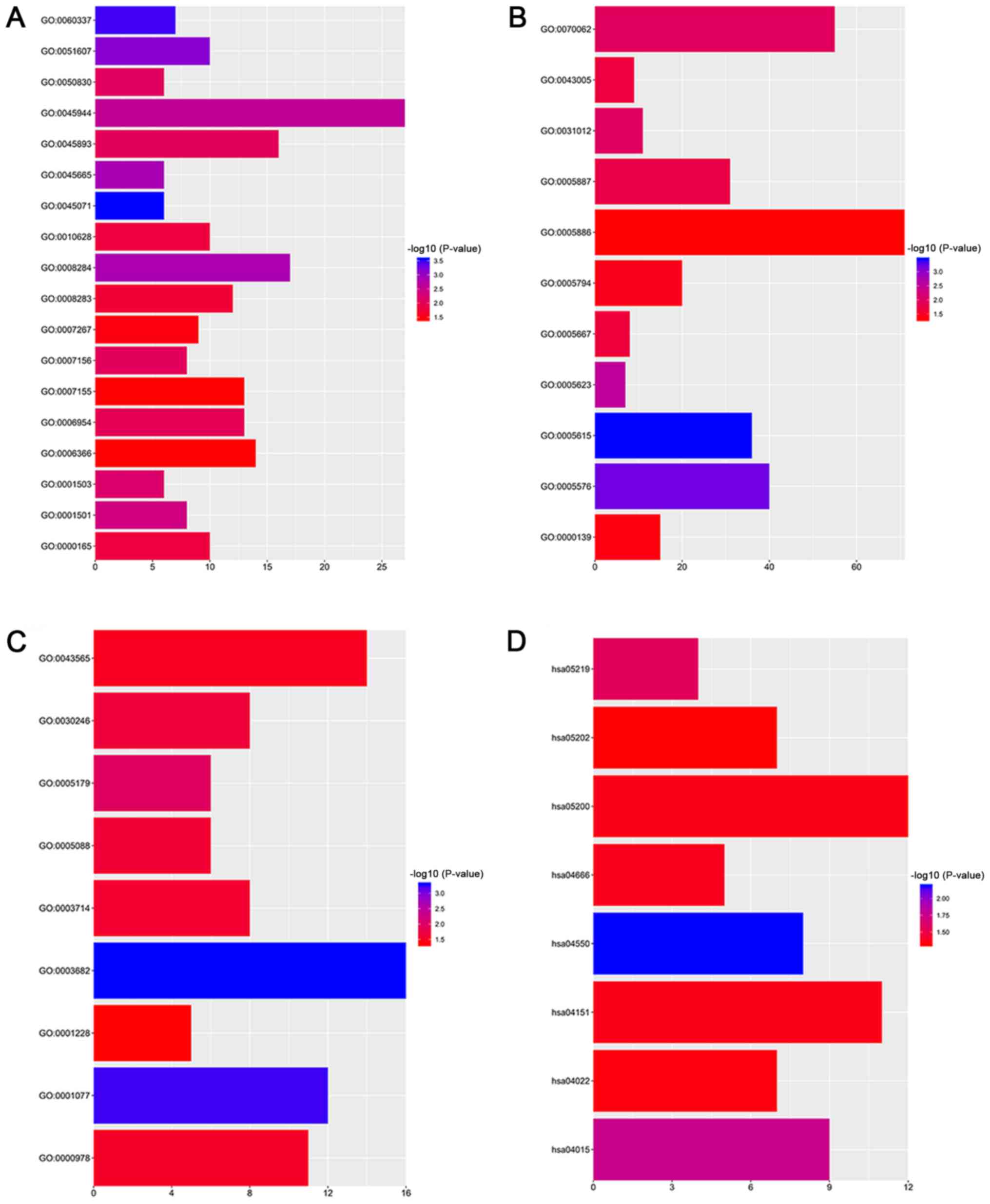
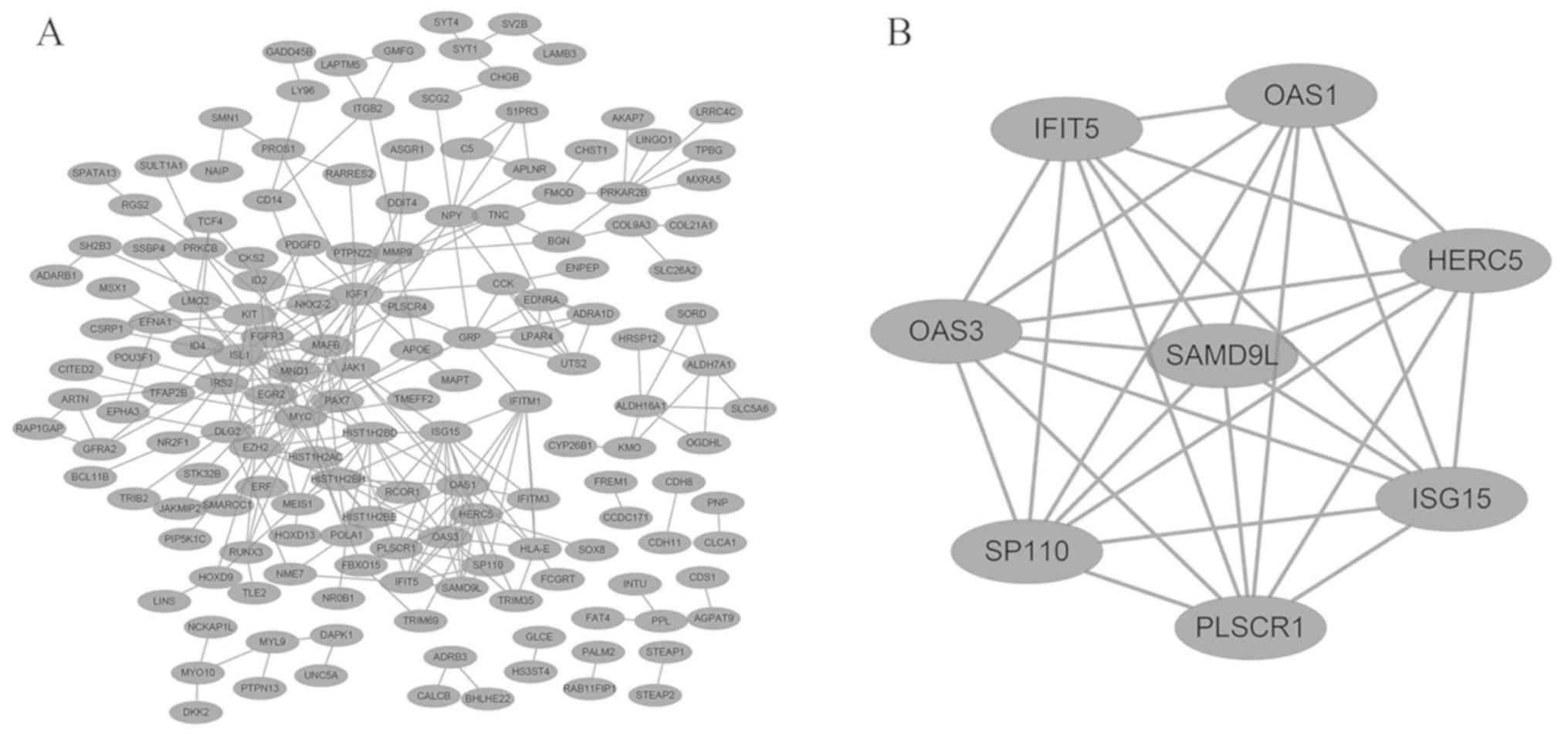
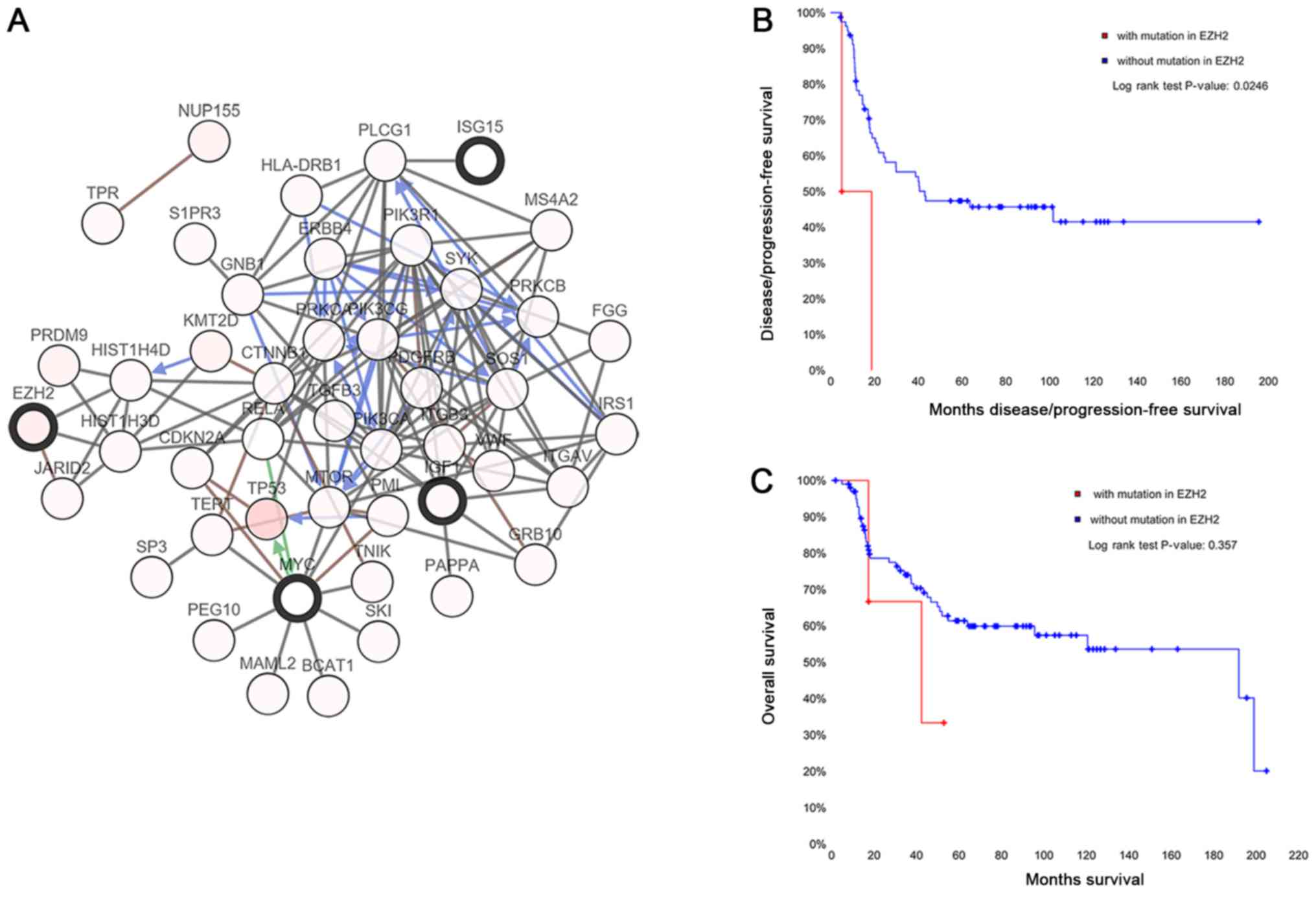
| Upregulated | RIDA, EWSAT1,
KHDRBS3, CDH8, SAMD9L, HOXD9, LPAR4, OGDHL, SV2B, SLC5A6, WBSCR17,
MYO10, CAPN15, GFRA2, GADD45B, NR2F1, CCDC155, LRRC4C, COL21A1,
ZNF57, CA11, DLX3, CD99P1, CDS1, HSD17B2, PARM1, RASSF2, TMEM176A,
SOX8, RPP25, POLA1, CHGBPIP5K1C, PCDHB5, ZNF711, CHST1, PDGFD,
TGIF2, HERC5, TMEM91, ADGRL1, PLSCR1, CLCA1, LINC01503, C5,
ALDH16A1, ATP6V0B, ITGB2-AS1, CYTL1, TBCCD1, CLSTN2, PALM2, COL9A3,
ASAP1, PRKAR2B, SULT1A1, DDIT4, PNP, FGFR3, IFITM3, ISL1, HLA-E,
LAPTM5, ARTN, ADARB1, MYL9, TRIM69, MAPT, ADRA1D, SPATA13, MND1,
TLE2, GMFG, FBXO15, C1orf226, TDRD3, FAM213A, LAMB3, FMOD, ZNF195,
INTS6L, HIST1, H2BD, KCNE4, IFIT5, SP110, ASGR1, SLCO5A1, STK32B,
GALNT14, IFITM1, IGSF21, ZNF703, ITGB2, OAS3, ST6GAL1, NYNRIN,
ERFE, CLDN1, YPEL5, SYT4, OAS1, FREM1, CITED2, IL1RAP, MBD4, STMN4,
NR0B1, LMO2, ERF, PTPN22, NCKAP1L, CSRNP2, BHLHE22, LINS1,
PEG3-AS1, CAV2, RAP1GAP, ALG6, ADGRE5, INTU, L3MBTL3, HIST1H2BE,
TOX3, EFNA1, EPHA3, SORD, TRIM35, MEIS1, NME7, ODF2L, KIAA1462,
IGF1, ZNF468, ZNF432, TRHDE, NCKAP5, UNC5A, PROS1, TMEM71, PLXDC2,
EMILIN1, LBH, HIST1H2BH, CADPS2, ZNF426 TCF4, LOC102724275, SSBP4,
FAM107B, TEAD2, MAFB, SMARCC1, SH2B3, CPNE4, LINGO1, GOT1L1, CRIP1,
RGS2, POU3F1, NPY, CRIP2, APELA, CARD16, ENPEP, ISG15, LECT1,
GDF10, ADRB3, FAT3, MSX1, HIST1H2AC, TRIB2, SYT1, OLFML3, AKAP7,
CD14, HS3ST4, PLSCR4, CLIP2, SEC11C, RAB11FIP1, S1PR3, LOXHD1,
OLFM3, HOXD13, RUNX3, PGLYRP2, UTS2, KIT, DNAJC12, LGALS8, H2BFS,
CPVL, CALCB, OLFM1, RCOR1, MFAP4, CSRP1, ZDHHC21, BGN, STEAP2,
DLG2, IRS2, DUSP6, TFAP2B, TRPM4, GPR137B, IVNS1ABP, GIMAP2, AMER2,
MXRA5, PCDH17, RARRES2, HOOK1, JAK1, TPBG, ITM2A, MYC, SMA4, APOE,
SLAIN1, CCK, ABHD6, JAKMIP2, MMP9, GRP, RGL1, SCG2, EGR2, GALNT7,
NPTXR, FCGRT, CDH11, NUDT11, NELL2, NAIP, MARCKSL1, ALDH7A1, LIPI,
TOX2, TNC, SLC26A2, GLCE, APCDD1, FAT4, EDNRA, KMO, TMEFF2,
TSPAN13, CCDC171, LY96, CYP26B1, ID4, DAPK1, PAX7, ATP1A1, RBM11,
DKK2, EZH2, KDSR, CKS2, PRSS35, ADGRG2, PCDH8, ID2, PTPN13, STEAP1,
HMCN1, FAM84B, BCL11B, PRKCB, NKX2-2 |