Introduction
Endometrial cancer (EC) is the most common
gynecologic malignancy in the western world and the fourth most
common cancer in women worldwide, with >280,000 cases per year
worldwide in 2017 (1). The number of
estimated deaths caused by EC in 2016 was 10,470, which was 1.8% of
all cancer deaths; the five-year survival rate was 81.7% (2). Two histologic categories have been
described among adenocarcinomas of the endometrium: Type 1 and type
2. Type 1 adenocarcinomas are estrogen-mediated, have an
endometrioid histology and are mostly lower grade. They account for
70–80% of new cases. Type 2 tumors occur more frequently in leaner,
older women, and this type consists of higher-grade tumors and
nonendometrioid histologies (usually serous or clear cells)
(3,4). Most patients who are diagnosed at an
early stage have a relatively better prognosis compared with those
who are diagnosed at an advanced stage or with recurrent tumor
(5). Thus, further investigation was
conducted aiming to reveal the possible mechanisms in the
occurrence and development of EC at the molecular level, to explore
potential candidate biomarkers as targets for more accurate and
early diagnosis, and treatment, in order to promote the overall
survival rate and prognosis of EC. The biological processes of EC
were explored; however the gene interactions and biological
pathways of EC were not accurately verified. In recent decades,
with the rapid development and wide application of microarray
technology and bioinformatics analysis, studies of diseases have
advanced to the genetic level. Increasing evidence has shown that
the abnormal expression and mutation of genes, including p53,
K-ras, PTEN (6,7), and mismatch repair (MMR) genes
(8,9), were associated with the carcinogenesis
and progression of EC. Thus, certain genes have the potential to
become biomarkers of EC. The identification of the differentially
expressed genes (DEGs) and pathways involved in EC can be achieved
by using bioinformatics methods. In the present study, three mRNA
microarray datasets were downloaded from the Gene Expression
Omnibus database (GEO) to avoid false-positive rates in any single
dataset. A total of 118 DEGs between EC and noncancerous tissues
were screened from the datasets and 11 hub genes were selected as
candidate biomarkers for the diagnosis, treatment and prognosis of
EC.
Materials and methods
Data sources
The GEO (http://www.ncbi.nlm.nih.gov/geo) database is an
international public repository that archives and freely
distributes high-throughput gene expression and other functional
genomics datasets (10). Three mRNA
datasets [GSE63678 (GPL571 platform, Affymetrix Human Genome U133A
2.0 Array) (11), GSE17025 (GPL570
platform, Affymetrix Human Genome U133 Plus 2.0 Array) (12) and GSE3013 (GPL8300 platform,
Affymetrix Human Genome U95 Version 2 Array) (13)] were downloaded from GEO. The GSE63678
dataset comprised 18 patients with gynecological cancer and 17
women as the control group. The clinicopathological data are listed
in Table SI. Seven samples of EC
and five samples of normal tissue were selected for the study.
GSE17025 contained 91 samples of EC (79 endometrioid cancer and 12
serous cancer) with a heterogeneous distribution of grade and depth
of myometrial invasion (i.e., 9 IAG1, 14 IAG2, 7 IAG3, 14 IBG1, 12
IBG2, 13 IBG3, 7 ICG1, 10 ICG2, and 6 ICG3) and 12 age-matched
normal endometrial samples from post-menopausal women as control.
GSE3013 contained one sample of endometrial epithelial cells (EECs)
from stage I endometrioid carcinomas, one sample of EECs from stage
I endometrioid carcinomas treated with oestrogen (E2), one sample
of EECs from stage I endometrioid carcinomas treated with tamoxifen
(TAM), one sample of EECs from stage II endometrioid carcinomas,
two samples of EECs from stage II endometrioid carcinomas treated
with E2, one sample of EECs from stage II endometrioid carcinomas
treated with TAM, and samples of normal endometrial epithelium in
each group (six samples total) as control. Two samples of EC
without any treatment and two samples of normal endometrial
epithelium were selected for the study.
Identification of DEGs
The DEGs were screened by using GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r).
Thus interactive web utility can be used to compare the datasets in
a GEO series and identify DEGs across experimental conditions.
Probe sets without corresponding gene symbols or with >1 gene
symbol were removed. Genes with >1 probe set were maximized. The
cut-off criteria were set as follows: Adj. P-value, <0.05 and
LogFC (fold change), >1.
Functional annotation and pathway
enrichment
The purpose of this step is to explain gene function
and find relevant pathways. Kyoto Encyclopedia of Genes and Genomes
(KEGG) and Gene ontology (GO) enrichment analyses were accomplished
in the Database for Annotation, Visualization and Integrated
Discovery (DAVID; https://david.ncifcrf.gov/; version 6.8). DAVID is a
bioinformatics data resource consisting of an integrated biological
knowledge base and analytic tools aimed at systematically
extracting the biological significance of genes and proteins from
large lists. It provides a comprehensive set of functional
annotation tools for investigating the biological mechanisms
underlying a list of genes (14).
KEGG (https://www.kegg.jp/) is a database
resource for understanding high-level functions and biological
systems from large-scale molecular datasets generated by
high-throughput experimental technologies (15). The GO knowledge base (http://geneontology.org/) is the world's largest
source of information on the functions of genes. Three independent
ontologies, including the biological process (BP), molecular
function (MF) and cellular component (CC) categories were
constructed to describe gene product attributes (16,17).
P<0.05 was considered to indicate a statistically significant
difference.
Protein-protein interaction (PPI)
network construction and module analysis
The DEGs were mapped using the Search Tool for the
Retrieval of Interacting Genes/Proteins (STRING; http://string-db.org; version 10.0) online database,
which is a database designed to provide a critical assessment and
integration of PPI (18). An
interaction with a combined score >0.4 was considered
statistically significant. Cytoscape 3.7.1 (19) was used to visualize the PPI network.
The most significant module was identified using the Molecular
Complex Detection (MCODE) plug-in (20). The selection criteria were: MCODE
scores >5; degree cut-off=2; node score cut-off=0.2; max
depth=100; and k-score=2. GO term and KEGG pathway enrichment were
assessed for the functional analysis of the 11 hub genes with
degrees ≥45 in the significant modules.
Hub gene analysis
A network of the 11 hub genes and their coexpression
genes was analyzed by the cBioPortal (http://www.cbioportal.Org; version 3.1.0) online
platform (21). The biological
process analysis was performed using the Biological Networks Gene
Ontology tool (BiNGO) plugin of Cytoscape (version 3.0.3) (22). Hierarchical clustering of the hub
genes was visualized by the University of California Santa Cruz
(UCSC) Xena Functional Genomics Explorer (https://xenabrowser.net/) (23), which showed the differential
expression of the hub genes between EC and normal tissue. The
overall survival (OS) rate and disease-free survival (DFS) of mRNA
expression was assessed using Kaplan-Meier curves in the cBioPortal
online platform. The expression profiles of cyclin B1 (CCNB1),
ubiquitin conjugating enzyme E2 C (UBE2C) and cell division cycle
20 (CDC20) were analyzed and displayed using Gene Expression
Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/index.html) (24). The association between expression
patterns and tumor grades were analyzed using the online database
UALCAN (http://ualcan.path.uab.edu/index.html) (25). These analyses were all based on data
from The Cancer Genome Atlas (TCGA) (26).
Results
Identification of DEGs in EC
Based on the cut-off criteria of adj. P-value
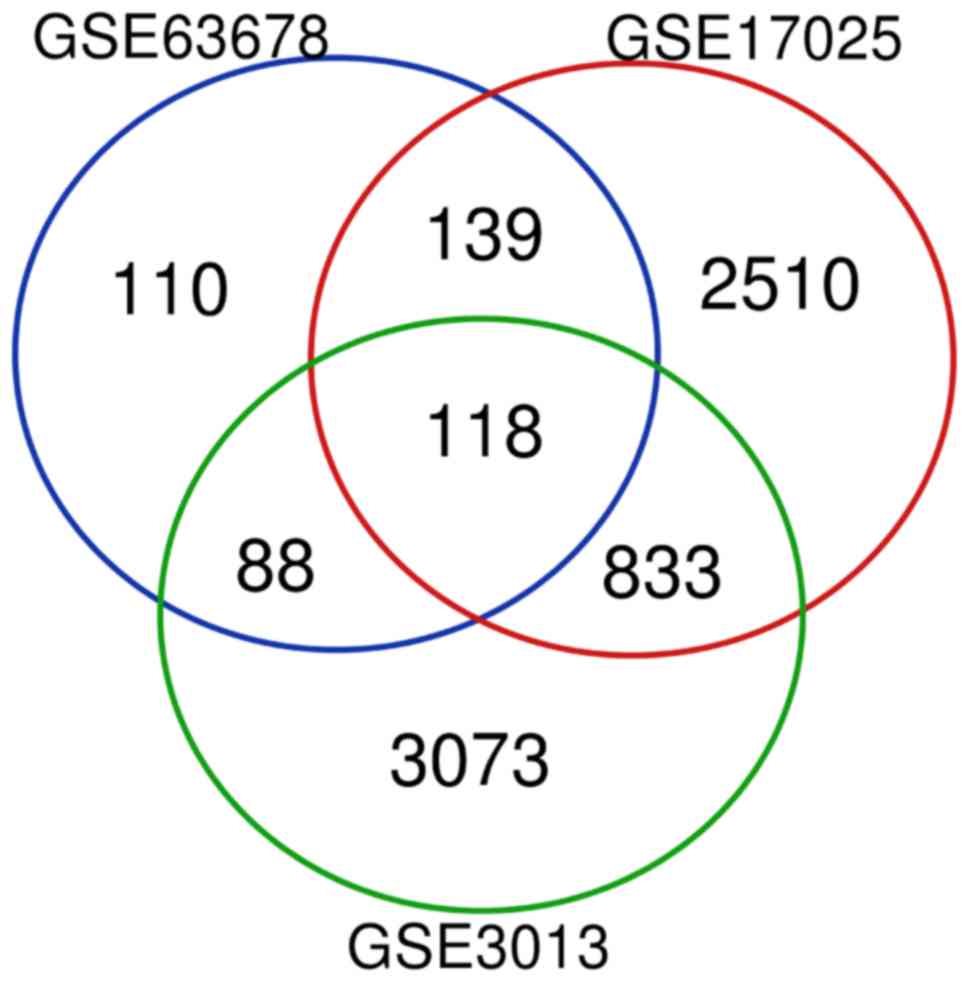
<0.05 and logFC>1, DEGs (455 in GSE63678; 3600 in GSE17025;
and 4740 in GSE3013) were identified in the EC samples. There were
118 genes that were differentially expressed among the three
datasets (Fig. 1), consisting of 27
downregulated genes and 91 upregulated genes.
Functional annotation and pathway
enrichment
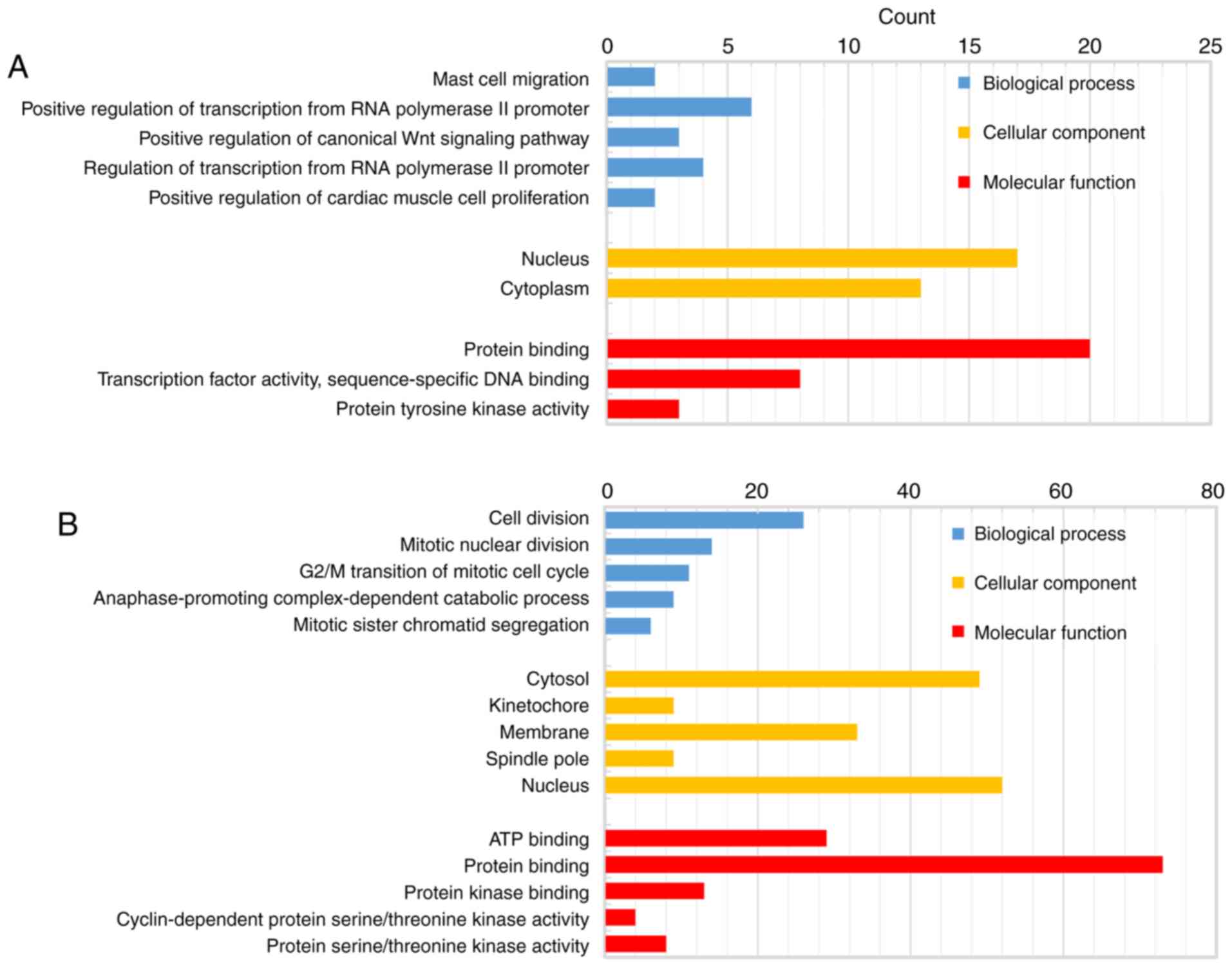
The biological classification analysis of 118 DEGs
was performed using DAVID, including functional and pathway
enrichment analyses. Sorting by P-value, the top five GO terms of
the BP, MF and CC categories are shown in Fig. 2. The downregulated genes were mainly
involved in the positive regulation of transcription from RNA
polymerase II promoter in the BP category, in protein binding in
the MF category, and mainly constituted the nucleus in the CC
category. The upregulated genes were mainly associated with cell
division in the BP category, protein binding in the MF category,
and mainly constituted the nucleus in the CC category. The KEGG
pathway analysis indicated that the downregulated DEGs were
primarily enriched in pathways associated with cancer, and the
upregulated DEGs were mainly enriched in the cell cycle (Fig. 3).
PPI network construction and module
analysis
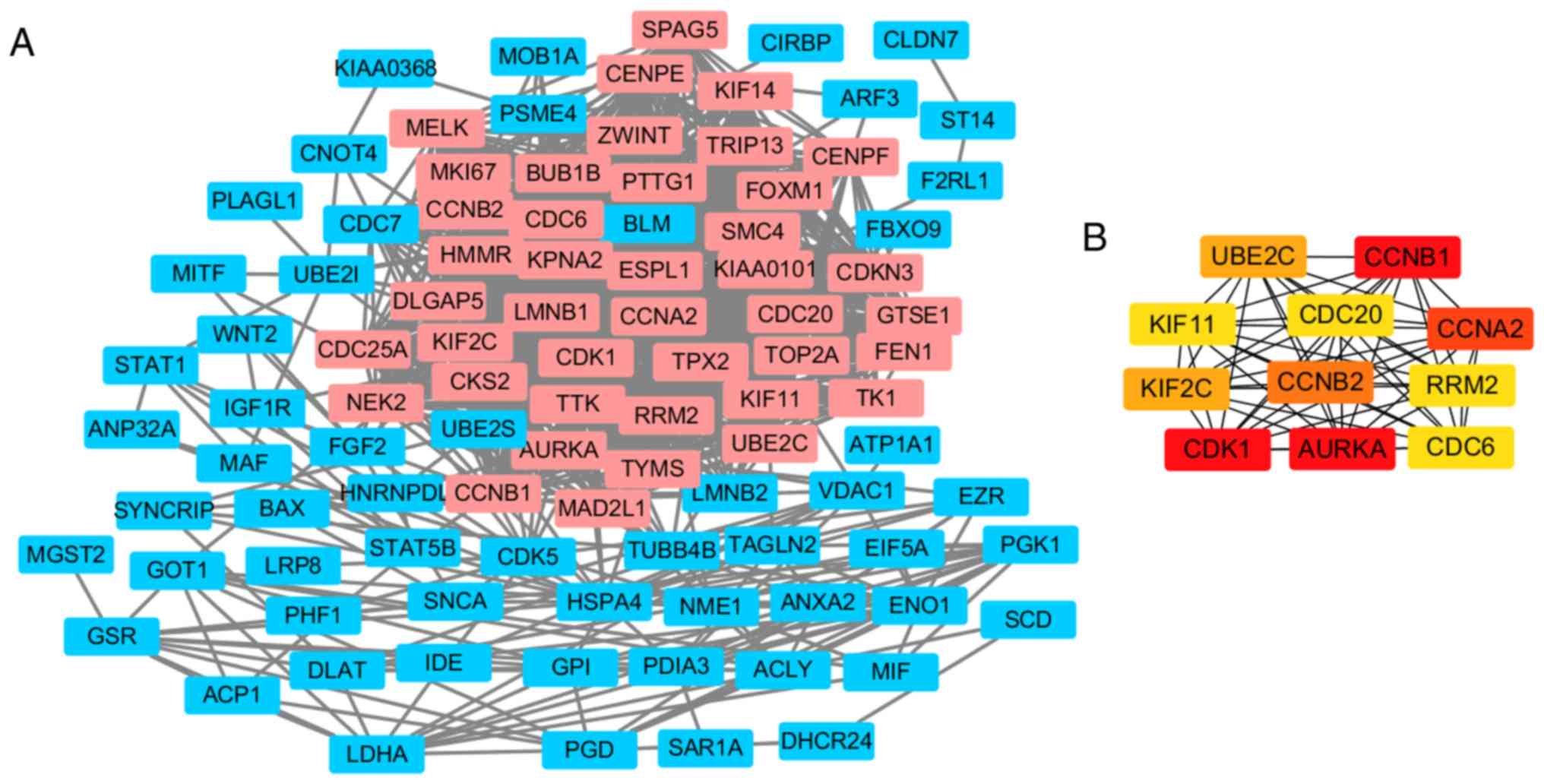
Following the prediction by STRING, the PPI network
of DEGs was constructed by using Cytoscape (Fig. 4A), which resulted in 98 nodes and
1078 edges. The most significant module including 41 nodes total
was obtained using MCODE (Fig. 4A),
in which 11 nodes of them with a degree of ≥45, were regarded as
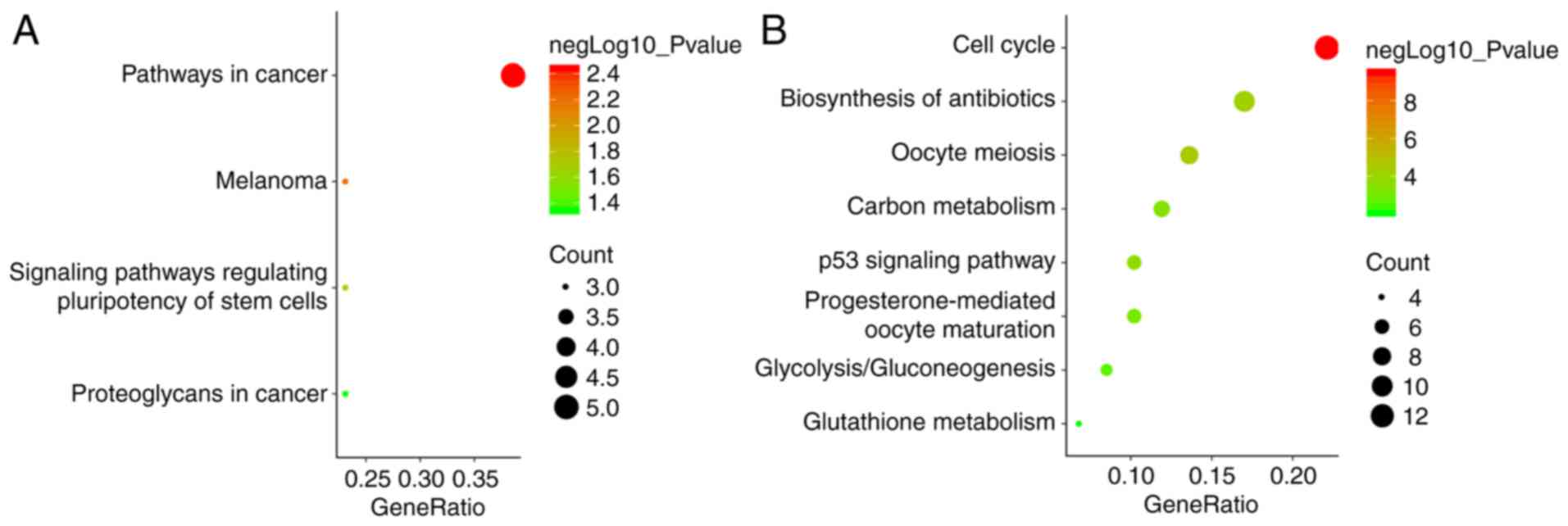
hub genes (Fig. 4B). The functional
analyses using DAVID showed that the hub genes were mainly enriched
in the cell cycle, oocyte meiosis, progesterone-mediated oocyte
maturation, the p53 signaling pathway and viral carcinogenesis
(Table I).
 | Table I.GO and KEGG pathway enrichment
analysis of hub genes. |
Table I.
GO and KEGG pathway enrichment
analysis of hub genes.
| Category | Term | Count | P-value |
|---|
| GOTERM_BP | GO:0051301-cell
division | 10 |
6.57×1015 |
| GOTERM_BP | GO:0007067-mitotic
nuclear division | 8 |
1.63×10−11 |
| GOTERM_BP |
GO:0031145-anaphase-promoting
complex-dependent catabolic process | 5 |
9.33×10−8 |
| GOTERM_BP | GO:
0051439-regulation of ubiquitin-protein ligase activity involved in
mitotic cell cycle | 4 |
2.68×10−7 |
| GOTERM_BP | GO:0042787-protein
ubiquitination involved in ubiquitin-dependent protein catabolic
process | 5 |
1.33×10−6 |
| GOTERM_CC |
GO:0005829-cytosol | 10 |
1.81×10−6 |
| GOTERM_CC |
GO:0005654-nucleoplasm | 9 |
9.89×10−6 |
| GOTERM_CC | GO:0000922-spindle
pole | 4 |
2.42×10−5 |
| GOTERM_CC |
GO:0005813-centrosome | 5 |
5.53×10−4 |
| GOTERM_CC |
GO:0005634-nucleus | 9 |
1.48×10−3 |
| GOTERM_MF | GO:
0004693-cyclin-dependent protein serine/threonine kinase
activity | 3 |
1.75×10−4 |
| GOTERM_MF | GO:0005524-ATP
binding | 6 |
9.33×10−4 |
| GOTERM_MF | GO:0019901-protein
kinase binding | 4 |
1.17×10−3 |
| GOTERM_MF | GO:0035173-histone
kinase activity | 2 |
2.36×10−3 |
| GOTERM_MF | GO:0005515-protein
binding | 10 |
1.49×10−2 |
| KEGG_PATHWAY | hsa04110:Cell
cycle | 6 |
9.42×10−8 |
| KEGG_PATHWAY | hsa04114:Oocyte
meiosis | 5 |
4.28×10−6 |
| KEGG_PATHWAY | hsa04115:p53
signaling pathway | 4 |
4.78×10−5 |
| KEGG_PATHWAY |
hsa04914:Progesterone-mediated oocyte
maturation | 4 |
1.05×10−4 |
| KEGG_PATHWAY | hsa05203:Viral
carcinogenesis | 3 |
2.20×10−2 |
Hub gene analysis
In the most significant module obtained using MCODE,
a total of 11 hub genes were identified with degrees ≥45 (Table II). The cBioPortal online platform
was used to analyze and to draw a network of the hub genes and
their coexpression genes (Fig. 5).
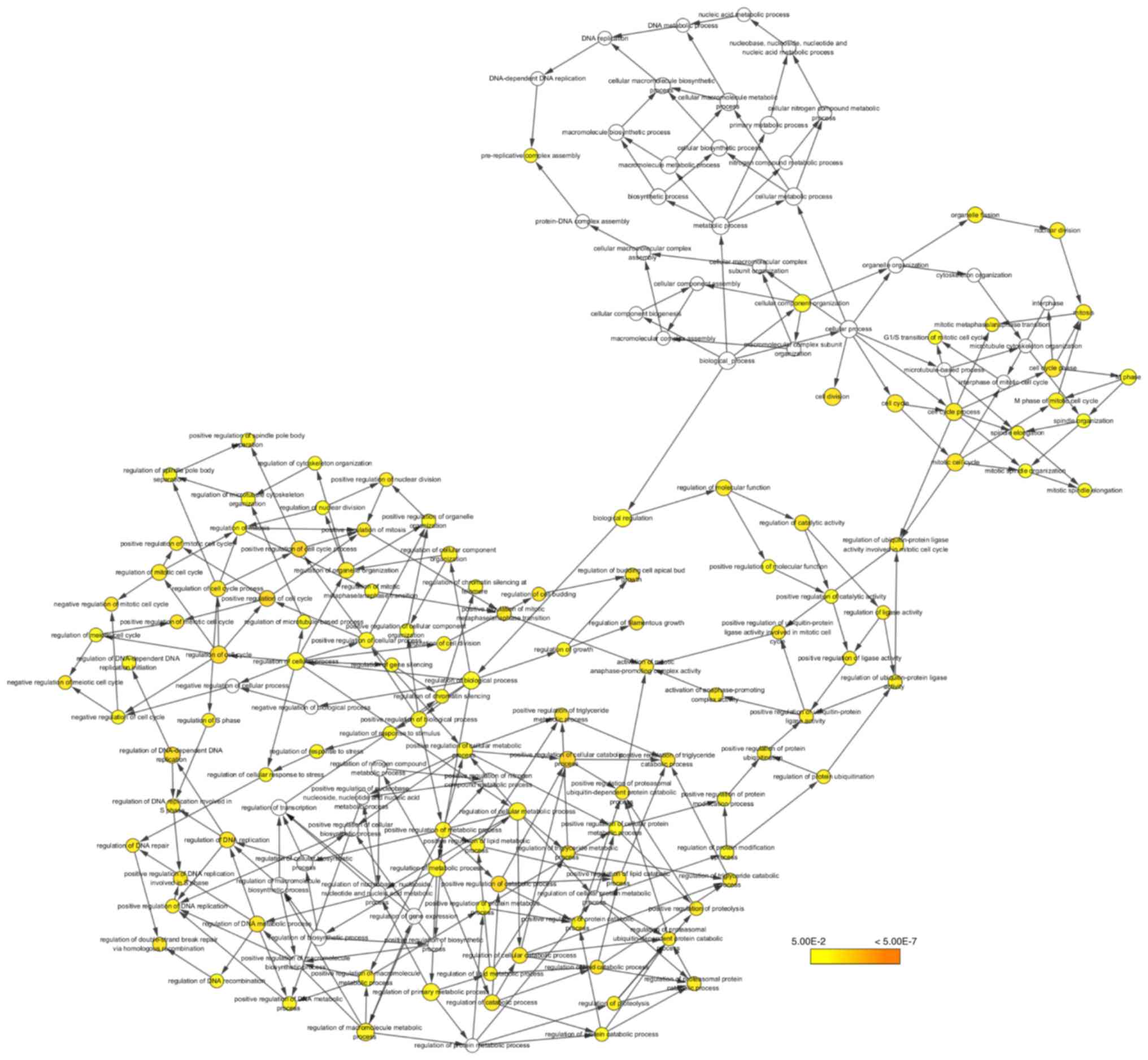
The biological process analysis of the hub genes is shown in
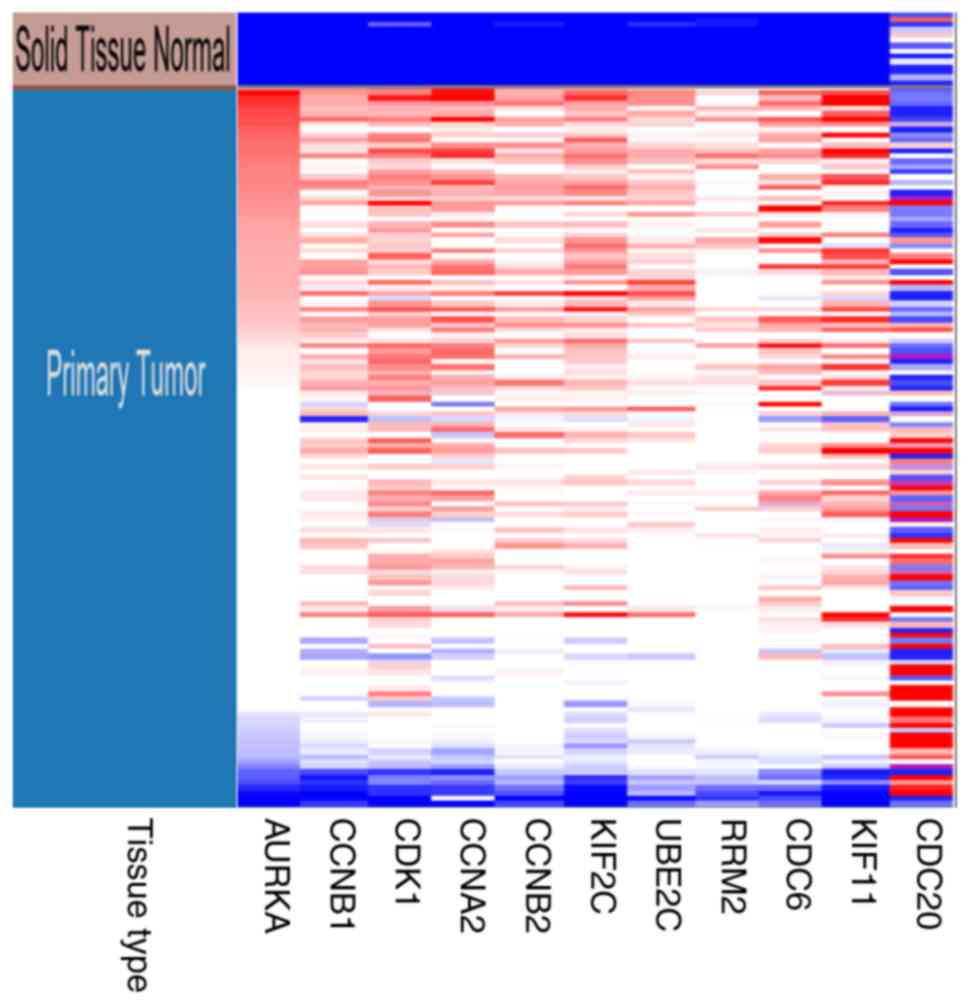
Fig. 6. Hierarchical clustering
showed that the hub genes could basically differentiate the EC
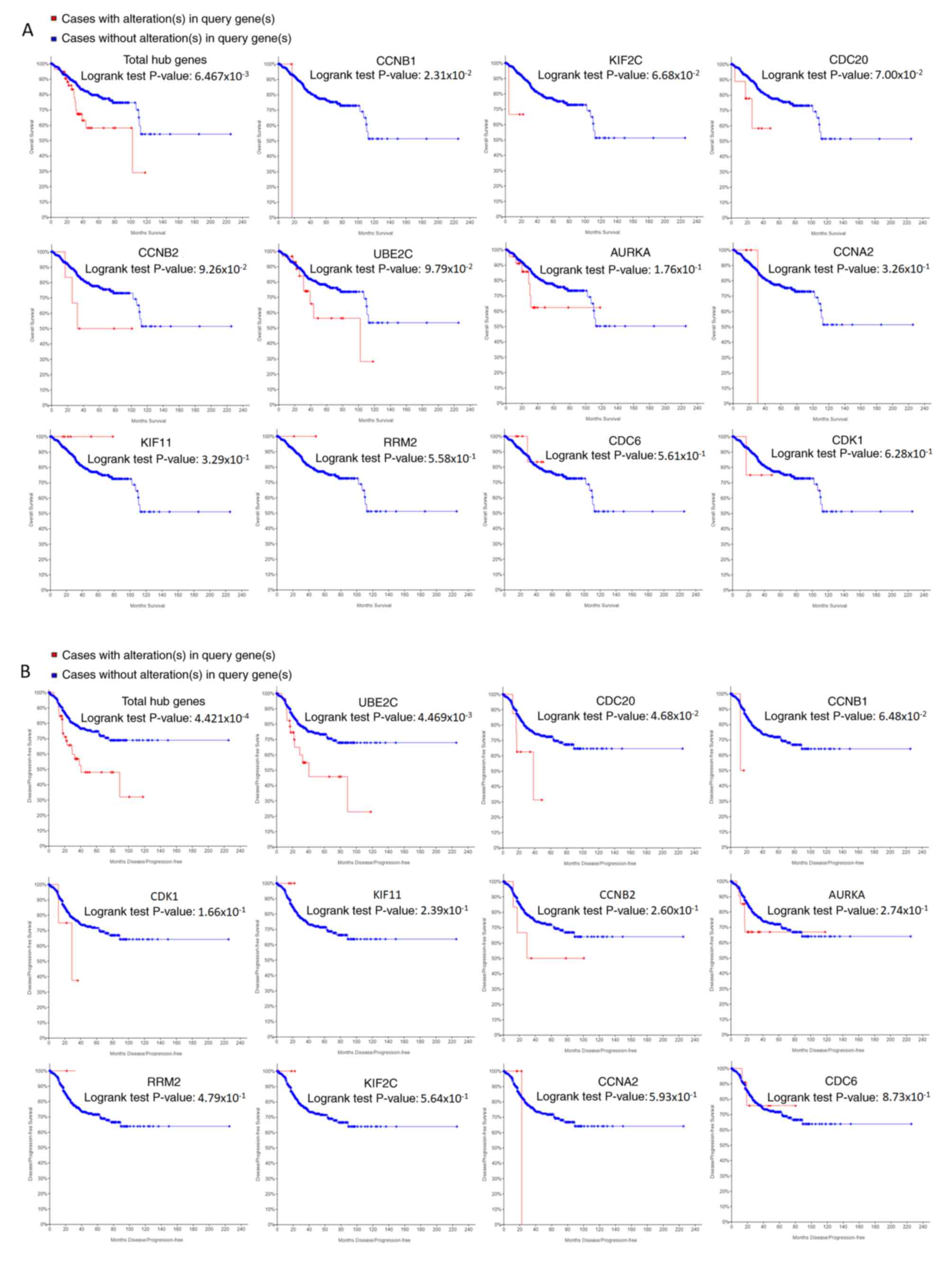
samples from the noncancerous samples, as is evident in Fig. 7. The OS and DFS analysis of the hub
genes was performed using Kaplan-Meier curves. Using the data from
cBioPortal, EC patients with hub gene alterations showed worse
overall survival and disease-free survival (P<0.05; Fig. 8A and B). Among these genes, cases
with a CCNB1 alteration showed worse overall survival (P<0.05;
Fig. 8A), and those without UBE2C
and CDC20 alterations showed better disease-free survival
(P<0.05; Fig. 8B). The survival
curves of cases with alterations in CDC20 and UBE2C showed that OS
was also decreased (0.05<P<0.1), indicating that the
difference was close to being significant. The same result was
found for CCNB1 in the DFS curve. These genes were all upregulated
in EC tissues in the three datasets from GEO and were considered to
take part in the carcinogenesis or progression of EC. The
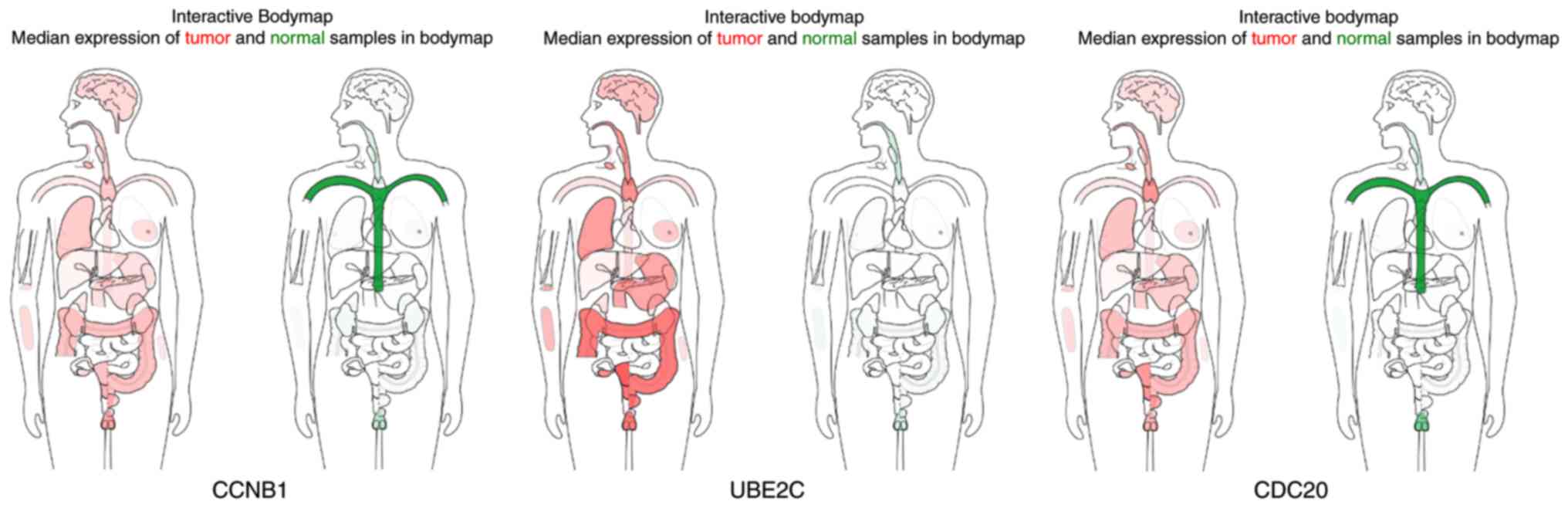
expression profiles of CCNB1, UBE2C and CDC20 in human tissue were
displayed using GEPIA (Fig. 9).
CCNB1 mRNA displayed higher levels in tumors of the brain,
lymphonodus, lung, colon, uterus and cervix-uteri compared with the
matched normal tissues. UBE2C mRNA displayed higher levels in
tumors of the brain, lymphonodus, lung, breast, stomach, colon,
ovary, uterus, cervix uteri, bladder and testis compared with
normal tissues. Furthermore, CDC20 displayed higher levels in the
brain, lymphonodus, thymus, lung, colon, ovary, uterus, cervix
uteri and bladder compared with the matched normal tissues. The
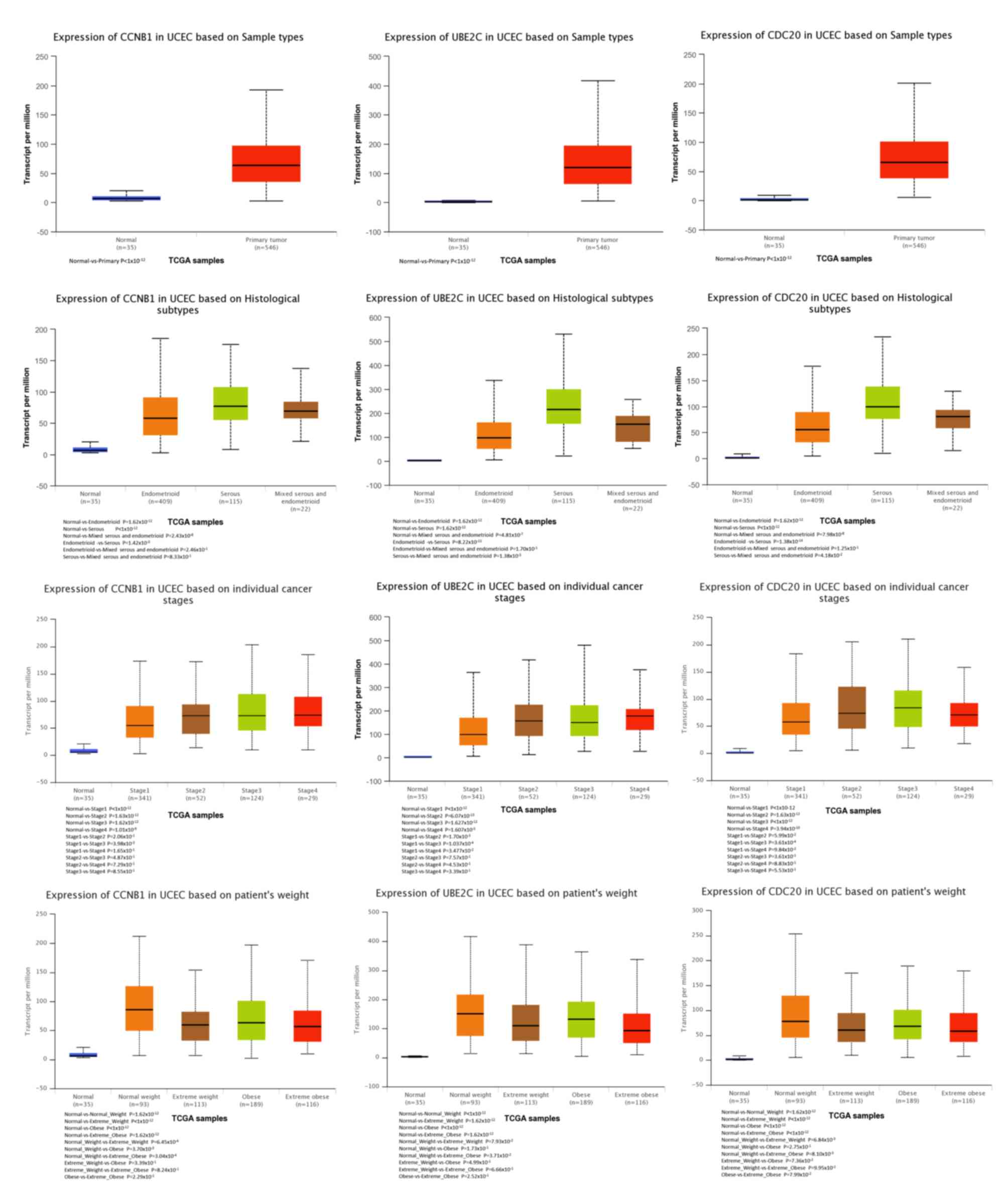
analysis of tumor vs. normal tissues by UALCAN demonstrated that
CCNB1, UBE2C and CDC20 had significantly increased expression in EC
in the different datasets (Fig.
10). All had increased expression in serous carcinoma compared
with endometrioid carcinoma. In addition, the three genes,
particularly UBE2C, showed a tendency toward higher expression in
the late stage. An association between the three genes and body
weight was identified, and patients with EC and normal weight had a
higher expression than those with extreme obese weight.
| Table II.Key nodes in the protein-protein
interaction network with a degree ≥45. |
Table II.
Key nodes in the protein-protein
interaction network with a degree ≥45.
| No. | Name | Degree | Gene title | Function |
|---|
| 1 | AURKA | 50 | Aurora kinase
A | The protein encoded
by AURKA is a cell cycle-regulated kinase that appears to be
involved in microtubule formation and/or stabilization at the
spindle pole during chromosome segregation. |
| 2 | CCNB1 | 50 | Cyclin B1 | The protein encoded
by CCNB1 is a regulatory protein involved in mitosis, which is
necessary for regulation of the G2/M transition phase of
the cell cycle. |
| 3 | CDK1 | 50 | Cyclin dependent
kinase 1 | The protein encoded
by CDK1 is a member of the Ser/Thr protein kinase family, which is
essential for G1/S and G2/M phase transitions
of eukaryotic cell cycle. |
| 4 | CCNA2 | 48 | Cyclin A2 | The protein encoded
by CCNA2 binds and activates cyclin-dependent kinase 2 and thus
promotes transition through G1/S and
G2/M. |
| 5 | CCNB2 | 47 | Cyclin B2 | Cyclin B2 is a
member of the B-type cyclins. The B-type cyclins, B1 and B2,
associate with p34cdc2 and are essential components of the cell
cycle regulatory machinery. Cyclin B2 is primarily associated with
the Golgi region. |
| 6 | KIF2C | 46 | Kinesin family
member 2C | KIF2C encodes a
kinesin-like protein that functions as a microtubule-dependent
molecular motor. The encoded protein can depolymerize micro tubules
at the plus end, thereby promoting mitotic chromosome
segregation. |
| 7 | UBE2C | 46 | Ubiquitin
conjugating enzyme E2 C | UBE2C encodes a
member of the E2 ubiquitin-conjugating enzyme family. The encoded
protein is required for the destruction of mitotic cyclins and for
cell cycle progression, and may be involved in cancer
progression. |
| 8 | RRM2 | 45 | Ribonucleotide
reductase regulatory subunit M2 | RRM2 encodes one of
two non-identical subunits for ribonucleotide reductase.
Transcription from this gene can initiate from alternative
promoters, which results in two isoforms that differ in the lengths
of their N-termini. |
| 9 | CDC6 | 45 | Cell division cycle
6 | The protein encoded
by CDC6 is highly similar to Saccharomyces cerevisiae Cdc6,
a protein essential for the initiation of DNA replication. |
| 10 | KIF11 | 45 | Kinesin family
member 11 | The function of
KIF11 product includes chromosome positioning, centro some
separation and establishing a bipolar spindle during cell
mitosis. |
| 11 | CDC20 | 45 | Cell division cycle
20 | CDC20 appears to
act as a regulatory protein interacting with several other proteins
at multiple points in the cell cycle. It is required for nuclear
movement prior to anaphase and chromosome separation. |
Discussion
EC is the fourth most common cancer in women
worldwide in recent years, and the number of estimated deaths due
to EC in 2016 was 10,470, which accounts for 1.8% of all cancer
deaths (2). Unfortunately, the
etiology of endometrial cancer remains poorly defined, hampering
the developments of early diagnostic and effective therapeutic
options for this disease. Using microarrays is a powerful technique
to monitor the expression of thousands of genes in a single
experiment. Key genes and pathways can be screened by analyzing
microarray datasets from different experiments in GEO or other
databases to identify the mechanisms in all biological processes
(27,28). In the field of obstetrics and
gynecology, microarrays have been used to study diseases such as
ovarian cancer, cervical cancer and preeclampsia. The technology is
applied to explore the occurrence and development of disease for
better detection and treatment (29–31).
Thus, the present study sought to apply microarray technology to
explore the genetic alterations in EC.
In the present study, three mRNA microarray datasets
were downloaded from GEO and were analyzed to acquire DEGs between
EC and noncancerous tissues. The selected 118 DEGs contained 27
downregulated genes and 91 upregulated genes. GO term enrichment
analyses revealed that the downregulated genes were mainly enriched
in the positive regulation of transcription from RNA polymerase II
promoter and in protein binding, and mainly constituted the
nucleus, whereas the upregulated DEGs were mainly enriched in cell
division and protein binding, and mainly constituted the nucleus.
Previous studies have reported that these processes, functions and
cellular components play important roles in the tumorigenesis or
progression of tumors (32–34). KEGG pathway analysis showed that the
DEGs were primarily enriched in pathways in cancer and the cell
cycle.
A total of 11 hub genes were selected from the most
significant module obtained using MCODE with degrees ≥45. Among
these hub genes, CCNB1, UBE2C and CDC20 showed a correlation with
the prognosis of EC patients. Patients with an alternative
expression of CCNB1, UBE2C and CDC20 were more concentrated in type
2 (elderly patients with normal weight, late stages and serous
adenocarcinoma), which indicated that these genes are probably
involved in the progression and poor prognosis of EC. The CCNB1
gene is indispensable for the control of the cell cycle at the G2/M
(mitosis) transition. The gene product complexes with p34 (cdc2) to
form the maturation-promoting factor (MPF; provided by RefSeq, Aug
2017), which may promote the progression to mitosis and augment the
cellular growth rate. In various tumor types, the overexpression of
CCNB1 was reported to be related to increased mitotic activity
during malignant metastasis. CCNB1 was considered to be related to
increased proliferative potential in EC (35). Wild-type p53 was reported to mediate
the control of CCNB1 (36). As a
target gene of the CCAAT-binding factor NF-Y, CCNB1 is upregulated
by the complex of mutant p53 and NF-Y, which promotes cell cycle
progression and might play a role in the chemoresistance of
colorectal carcinoma by inducing DNA damage (37,38).
Alterations of TP53 result in an aberrant form of P53 with a longer
half-life that accumulates in the cell. The presence of TP53/P53
expression/accumulation and tp53 mutations was associated with an
aggressive type of EC (39).
Furthermore, a SNP (rs2069433) in the CCNB1 gene was associated
with a reduction in EC risk, but the role of SNPs in the CCNB1 gene
regarding the oncogenesis of EC remains to be further studied
(40). UBE2C encodes a member of the
E2 ubiquitin-conjugating enzyme family. It is a member of the
anaphase promoting complex/cyclosome, which promotes the
degradation of several target proteins during cell cycle
progression, particularly during the metaphase/anaphase transition.
UBE2C may present in several human neoplasia. For instance, in
esophageal squamous cell carcinoma, as a transcriptional target of
FOXM1, UBE2C contributes to the loss of G2/M checkpoint control due
to the deregulation of FOXM1 (41).
The upregulation of UBE2C in several distinct tumor types has been
associated with a highly malignant phenotype and poor survival,
suggesting its role in cancer progression (42–44). The
present study also showed that the expression of UBE2C was
significantly higher in stage III than in stage I and II EC
tissues. In malignant tissues including esophagus, colon and
prostate, the 20q13.1 locus amplification is one of the important
mechanisms underlying the aberrant expression of UBE2C (44–49).
Wild-type p53 was reported to mediate the suppression of UBE2C
expression, whereas mutant p53 acts in the opposite manner
(46). Nevertheless, the concrete
mechanisms of UBE2C in tumorigenesis and progression in EC should
be further investigated. CDC20 is required for two
microtubule-dependent processes, nuclear movement prior to anaphase
and chromosome separation (provided by RefSeq, Jul 2008). CDC20
possibly acts as a regulatory protein interacting with several
other proteins at multiple points in the cell cycle, and is
necessary for the degradation of an S-phase cyclin, which can
antagonize anaphase-promoting complex (APC) activity (50). The abnormal regulation of CDC20 may
lead to the accumulation of deleterious chromosomal alterations
promoting tumor development and progression. In breast cancer, the
increased expression of CDC20 is associated with increased
chromosomal instability (51). High
CDC20 expression was strongly associated with advanced tumor stage
in carcinoma of the breast, colon, endometrium and prostate
(52). Thus, CDC20 is probably a
biomarker for the diagnosis and prognosis of EC and may serve as a
therapeutic target. A number of similar studies have been
previously conducted and showed various results. For example, the
analysis of the GSE17025 dataset by Liu et al (53) reported that PCDH10, CCL20 and TOP2A
was associated with EC and may be potential molecular markers. The
main reason for the differences is that the data was obtained from
three datasets and some genes were filtered out by Wayne chart in
in the present study. In addition, although the analytical methods
were used correctly, the possibility of error cannot be completely
excluded, and using different datasets may lead to different
results. Thus, the findings of the present study require validation
in experiments by using samples of EC and normal tissues.
Furthermore, there are many other DEGs except the three key genes
selected, which may also be involved in the biological process in
EC.
Nevertheless, the present study provides a new
direction for further studies on EC. The DEGs identified in EC
tissues may be involved in carcinogenesis or progression. The 11
hub genes in the significant module, particularly CCNB1, UBE2C and
CDC20, may be important in the pathogenesis and progression of EC,
and may be regarded as biomarkers for the diagnosis or prognosis of
EC. However, further investigation is required to determine the
definite functions of these genes in EC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SL conceived and designed the study. SL, ZW and XX
analyzed the data. XX and ZW generated the figures. SL and ZW wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eritja N, Yeramian A, Chen BJ,
Llobet-Navas D, Ortega E, Colas E, Abal M, Dolcet X, Reventos J and
Matias-Guiu X: Endometrial carcinoma: Specific targeted pathways.
Adv Exp Med Biol. 943:149–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patni R: Endometrial carcinoma: Evolution
and overviewCurrent Concepts in Endometrial Cancer. Singapore:
Springer; pp. 1–9. 2017, View Article : Google Scholar
|
|
3
|
Bokhman JV: Two pathogenetic types of
endometrial carcinoma. Gynecol Oncol. 15:10–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tran AQ and Gehrig P: Recent advances in
endometrial cancer. F1000Res. 6:812017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sheets SSF: Endometrial cancer.
Surveillance, Epidemiology, and End Results Program. seer. cancer.
gov. 2015.
|
|
6
|
Lax SF, Kendall B, Tashiro H, Slebos RJ
and Ellenson L: The frequency of p53, k-ras mutations, and
microsatellite instability differs in uterine endometrioid and
serous carcinoma: Evidence of distinct molecular genetic pathways.
Cancer. 88:814–24. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng ZZ, Chen WJ, Yang ZR, Lu ZG and Cai
ZG: Expression of PTTG1 and PTEN in endometrial carcinoma:
Correlation with tumorigenesis and progression. Med Oncol.
29:304–310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Basil JB, Goodfellow PJ, Rader JS, Mutch
DG and Herzog TJ: Clinical significance of microsatellite
instability in endometrial carcinoma. Cancer. 89:1758–1764. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hecht JL and Mutter GL: Molecular and
pathologic aspects of endometrial carcinogenesis. J Clin Oncol.
24:4783–4791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Clough E and Barrett T: The gene
expression omnibus database//Statistical GenomicsNew York: Humana
Press; pp. 93–110. 2106
|
|
11
|
Pappa KI, Polyzos A, Jacob-Hirsch J,
Amariglio N, Vlachos GD, Loutradis D and Anagnou NP: Profiling of
discrete gynecological cancers reveals novel transcriptional
modules and common features shared by other cancer types and
embryonic stem cells. PLoS One. 10:e01422292015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Day RS, McDade KK, Chandran UR, Lisovich
A, Conrads TP, Hood BL, Kolli VK, Kirchner D, Litzi T and Maxwell
GL: Identifier mapping performance for integrating transcriptomics
and proteomics experimental results. BMC Bioinformatics.
12:2132011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu H, Chen Y, Liang J, Shi B, Wu G, Zhang
Y, Wang Y, Wang D, Li R, Yi X, et al: Hypomethylation-linked
activation of PAX2 mediates tamoxifen-stimulated endometrial
carcinogenesis. Nature. 438:981–987. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanehisa M: The KEGG database//‘In
Silico'Simulation of Biological Processes: Novartis Foundation
Symposium 247. Chichester, UK, John Wiley & Sons, Ltd,.
247:91–103. 2002.
|
|
16
|
Harris MA, Clark J, Ireland A, Lomax J,
Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C,
et al: The Gene Ontology (GO) database and informatics resource.
Nucleic Acids Res. 32((Database Issue)): D258–D261. 2004.PubMed/NCBI
|
|
17
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler HL, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huertacepas J, Simonovic M, Roth A, Santos A,
Tsafou K, et al: STRING v10: Protein-protein interaction networks,
integrated over the tree of life. Nucleic Acids Res. 43((Database
Issue)): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smoot ME, Ono K, Ruscheinski J, Wang P and
Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bandettini WP, Kellman P, Mancini C,
Booker OJ, Vasu S, Leung SW, Wilson JR, Shanbhag SM, Chen MY and
Arai AE: MultiContrast delayed enhancement (MCODE) improves
detection of subendocardial myocardial infarction by late
gadolinium enhancement cardiovascular magnetic resonance: A
clinical validation study. J Cardiovasc Magn Reson. 14:832012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne AJ, Heuer ML, Larsson EG, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of Gene Ontology
categories in Biological Networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goldman M, Craft B, Hastie M, Repečka K,
Kamath A, McDade F, Rogers D, Brooks AN, Zhu J and Haussler D: The
UCSC Xena Platform for cancer genomics data visualization and
interpretation. BioRxiv. 3264702019.
|
|
24
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tomczak K, Czerwinska P and Wiznerowicz M:
Review the cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
27
|
Werner T: Bioinformatics applications for
pathway analysis of microarray data. Curr Opin Biotechnol.
19:50–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moreau Y, De Smet F, Thijs G, Marchal K
and De Moor B: Functional bioinformatics of microarray data: From
expression to regulation. Proceedings IEEE. 90:1722–1743. 2002.
View Article : Google Scholar
|
|
29
|
Ye Q, Lei L and Aili AX: Identification of
potential targets for ovarian cancer treatment by systematic
bioinformatics analysis. Eur J Gynaecol Oncol. 36:283–289.
2015.PubMed/NCBI
|
|
30
|
Ai Z, Wang J, Xu Y and Teng Y:
Bioinformatics analysis reveals potential candidate drugs for
cervical cancer. J Obstet Gynaecol Res. 39:1052–1058. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tejera E, Bernardes J and Rebelo I:
Preeclampsia: A bioinformatics approach through protein-protein
interaction networks analysis. BMC Syst Biol. 6:972012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang V, Zheng J, Qi Z, Wang J, Place RF,
Yu J, Li H and Li LC: Ago1 Interacts with RNA polymerase II and
binds to the promoters of actively transcribed genes in human
cancer cells. PLoS Genet. 9:e10038212013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lacey JV, Potischman N, Madigan MP, Berman
ML, Mortel R, Twiggs LB, Barrett RJ, Wilbanks GD, Lurain JR,
Fillmore CM, et al: Insulin-like growth factors, insulin-like
growth factor-binding proteins, and endometrial cancer in
postmenopausal women: Results from a U.S. case-control study.
Cancer Epidemiol Biomarkers Prev. 13:607–612. 2004.PubMed/NCBI
|
|
34
|
Suman S and Mishra A: Network analysis
revealed aurora kinase dysregulation in five gynecological types of
cancer. Oncol Lett. 15:1125–1132. 2018.PubMed/NCBI
|
|
35
|
Ferguson SE, Olshen AB, Viale A, Awtrey
CS, Barakat RR and Boyd J: Gene expression profiling of
tamoxifen-associated uterine cancers: Evidence for two molecular
classes of endometrial carcinoma. Gynecol Oncol. 92:719–725. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Krause K, Wasner M, Reinhard W, Haugwitz
U, Dohna CL, Mössner J and Engeland K: The tumour suppressor
protein p53 can repress transcription of cyclin B. Nucleic Acids
Res. 28:4410–4418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Agostino SD, Strano S, Emiliozzi V,
Zerbini V, Mottolese M, Sacchi A, Blandino G and Piaggio G: Gain of
function of mutant p53: The mutant p53/NF-Y protein complex reveals
an aberrant transcriptional mechanism of cell cycle regulation.
Cancer Cell. 10:191–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Alam SK, Yadav VK, Bajaj S, Datta A, Dutta
SK, Bhattacharyya M, Bhattacharya S, Debnath S, Roy S, Boardman LA,
et al: DNA damage-induced ephrin-B2 reverse signaling promotes
chemoresistance and drives EMT in colorectal carcinoma harboring
mutant p53. Cell Death Differ. 23:707–722. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sisovsky V, Lasabova Z, Straka L, Telkova
M, Rychly B, Minarik G, Gemzova K, Petrovic R, Szemes T, Turna J,
et al: Tumour suppressor gene and protein TP53/P53 in normal
endometrium and endometrial carcinoma. Pathology. 46 (Suppl
2):S912014. View Article : Google Scholar
|
|
40
|
Cai H, Xiang Y, Qu S, Long J, Cai Q, Gao
J, Zheng W and Shu XO: Association of genetic polymorphisms in
cell-cycle control genes and susceptibility to endometrial cancer
among Chinese women. Am J Epidemiol. 173:1263–1271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nicolau-Neto P, Palumbo A, De Martino M,
Esposito F, de Almeida Simão T, Fusco A, Nasciutti LE, Meireles Da
Costa N and Ribeiro Pinto LF: UBE2C is a transcriptional target of
the cell cycle regulator FOXM1. Genes. 9(pii): E1882018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang H, Zhang C, Rorick A, Wu D, Chiu M,
Thomas-Ahner J, Chen Z, Chen H, Clinton SK, Chan KK and Wang Q:
CCI-779 inhibits cell-cycle G2/M progression and invasion of
castration resistant prostate cancer via attenuation of UBE2C
transcription and mRNA stability. Cancer Res. 71:4866–4876. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shen Z, Jiang X, Zeng C, Zheng S, Luo B,
Zeng Y, Ding R, Jiang H, He Q, Guo J and Jie W: High expression of
ubiquitin-conjugating enzyme 2C (UBE2C) correlates with
nasopharyngeal carcinoma progression. BMC Cancer. 13:1922013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fujita T, Ikeda H, Taira N, Hatoh S, Naito
M and Doihara H: Overexpression of UbcH10 alternates the cell cycle
profile and accelerate the tumor proliferation in colon cancer. BMC
Cancer. 9:872009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guo L, Ding Z, Huang N, Huang Z, Zhang N
and Xia Z: Forkhead Box M1 positively regulates UBE2C and protects
glioma cells from autophagic death. Cell Cycle. 16:1705–1718. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bajaj S, Alam SK, Roy KS, Datta A, Nath S
and Roychoudury S: E2 Ubiquitin-conjugating Enzyme, UBE2C Gene, is
reciprocally regulated by Wild-type and Gain-of-Function Mutant
p53. J Biol Chem. 291:14231–14247. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Takahashi Y, Ishii Y, Nishida Y, Ikarashi
M, Nagata T, Nakamura T, Yamamori S and Asai S: Detection of
aberrations of ubiquitin-conjugating enzyme E2C gene (UBE2C) in
advanced colon cancer with liver metastases by DNA microarray and
two-color FISH. Cancer Genet Cytogenet. 168:30–35. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tzelepi V, Zhang J, Lu JF, Kleb B, Wu G,
Wan X, Hoang A, Efstathiou E, Sircar K, Navone NM, et al: Modeling
a lethal prostate cancer variant with small-cell carcinoma
features. Clin Cancer Res. 18:666–677. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sakai N, Kajiyama Y, Iwanuma Y, Tomita N,
Amano T, Isayama F, Ouchi K and Tsurumaru M: Study of abnormal
chromosome regions in esophageal squamous cell carcinoma by
comparative genomic hybridization: Relationship of lymph node
metastasis and distant metastasis to selected abnormal regions. Dis
Esophagus. 23:415–421. 2010.PubMed/NCBI
|
|
50
|
Harper JW, Burton JL and Solomon MJ: The
anaphase-promoting complex: It's not just for mitosis any more.
Genes Dev. 16:2179–2206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yuan B, Xu Y, Woo JH, Wang Y, Bae YK, Yoon
DS, Wersto RP, Tully E, Wilsbach K and Gabrielson E: Increased
expression of mitotic checkpoint genes in breast cancer cells with
chromosomal instability. Clin Cancer Res. 12:405–410. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gayyed MF, Elmaqsoud NM, Tawfiek ER, El
Gelany SA and Rahman MF: A comprehensive analysis of CDC20
overexpression in common malignant tumors from multiple organs: Its
correlation with tumor grade and stage. Tumor Biol. 37:749–762.
2016. View Article : Google Scholar
|
|
53
|
Liu L, Chen F, Xiu A, Du B, Ai H and Xie
W: Identification of key candidate genes and pathways in
endometrial cancer by integrated bioinformatical analysis. Asian
Pac J Cancer Prev. 19:969–975. 2018.PubMed/NCBI
|