Introduction
Triple-negative breast cancers (TNBCs) are
characterized by the lack of HER2/neu, progesterone receptor (PR)
and estrogen receptor (1) (ER) gene
expression. The TNBC subtype constitutes 10–15% of total breast
tumors and 80% of basal-like breast cancers (1). Triple-negative and basal-like tumors
commonly have a high histological grade (1). TNBCs also have a poor prognosis and
tend to result in earlier relapse compared with other subtypes of
breast cancer (1). Additionally,
TBNCs exhibit increased chemosensitivity compared with other
genotypes of breast cancer (2),
thus, chemotherapeutic methods are currently the most prevalently
used medical treatments (3). These
include epidermal growth factor receptor (EGFR)-targeted therapies,
multi-tyrosine kinase inhibitors, poly-ADP ribose polymerase (PARP)
inhibitors and anti-angiogenic agents (4). However, patients with TNBCs usually
exhibit poorer outcomes following chemotherapy, compared with
patients with breast cancers of other subtypes (3). Hence, it would be beneficial to
identify novel biomarkers associated with the progression of TNBCs,
and identify new targets to improve their precise diagnosis and
treatment.
Weighted-correlation network analysis (WGCNA) has
previously been performed to construct non-scale co-expressed gene
networks (5–8). In the present study, WGCNA was
performed to identify the hub-module that contained genes showing a
strong correlation with the pathological stage of TNBC.
Subsequently, differentially expressed gene (DEG) analysis was
performed on RNAsequencing (RNAseq) data of TNBC. The hub-genes
were defined as all genes contained in the overlap between the DEGs
and the hub-module. Gene ontology (GO) and gene enrichment analyses
were then employed, and identified several important terms in
biological process, molecular function and cellular components.
Survival analysis was also conducted, and 5 genes were selected
from the hub-genes. Receiver operating characteristic (ROC) and
Kaplan-Meier (KM) curves were plotted to indicate the capacity of
these genes to differentiate tumor and para-tumor tissues, and to
confirm the influence of these genes on the overall survival (OS)
of patients with TNBC.
Materials and methods
Data processing and co-expression gene
network construction using RNAseq data
A total of 1,240 RNAseq datasets from The Cancer
Genome Atlas (TCGA) and clinical data for patients with breast
cancer were downloaded from the University of California, Santa
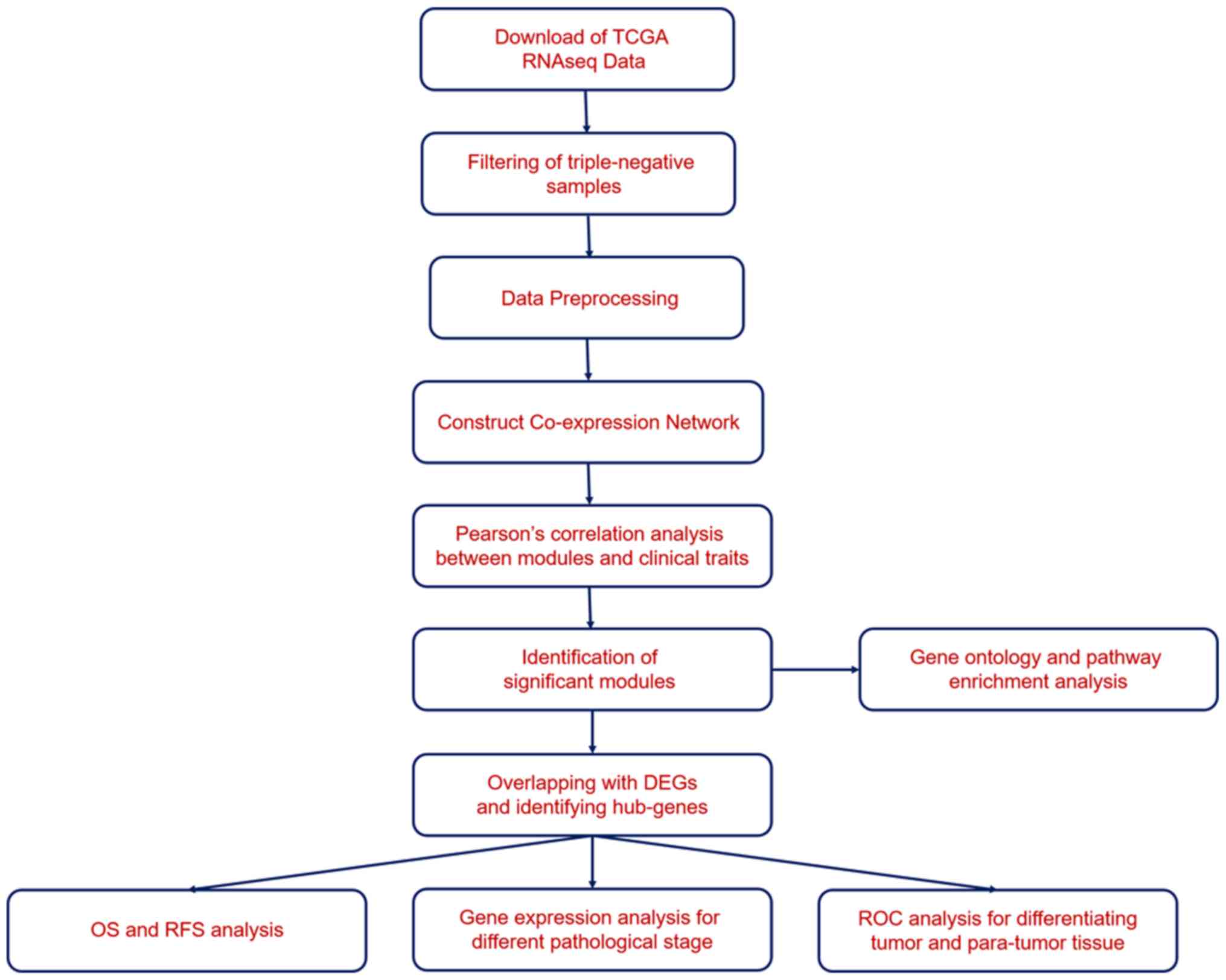
Cruz (UCSC) database using the Xena browser (https://xenabrowser.net/). The workflow is displayed
in Fig. 1. The TNBC samples were
filtered using the criteria of ‘not expressing genes for ER, PR and
HER2/neu’. R software (version 3.5.2; http://www.r-project.org) was applied to perform WGCNA
analysis. WGCNA was used to construct the gene co-expression
network; co-expression similarity (Si,j) was
defined as the absolute value of the correlation coefficient
between the mRNA expression profile of nodes i and
j:
si,j=|cor(xi,xj)|
Where Xi and Xj are mRNA
expression values for genes i and j, Si,j
was calculated using the Pearson's correlation coefficient between
genes i and j. Weighted-network adjacency was defined
by raising the co-expression similarity to a power:
αi,j=si,jβ
β≥1. The power of β=4 and scale free
R2=0.95 were selected as the soft-thresholding
parameters to ensure a signed scale-free gene network.
By evaluating the correlation between the
pathological stage of TNBC and the module membership with the ‘p.
weighted’, a high-correlated module was identified. The tan modules
which had the most significant adjusted P-values were selected.
Genes involved in the tan modules were presented using Cytoscape
v3.4.0 (https://cytoscape.org). The genes in the
tan module were selected as the input for GO and KEGG analysis,
which was performed using Metascape (http://metascape.org/gp/index.html).
Statistical analysis
Statistical analysis was performed using R (R
Foundation for Statistical Computing; http://www.R-project.org/). The fold-change and
Q-value (adjusted P-value) for para-tumor and tumor samples were
calculated using the Limma package (9). A Q-value <0.05 was considered to be
statistically significant. The overall survival analysis was
conducted using the Survminer package (10), and the P-values in the KM curve were
obtained using the log-rank test. The false discovery rate was set
as 0.05 for analysis.
Results
WGCNA on RNAseq dataset of TNBC
In order to determine the co-expression network most
highly associated with the progress and prognosis of TNBC, TNBCTCGA
RNAseq datadownloaded from UCSC, was analyzed using WGCNA. The
analysis showed TNBC clustering using the average linkage and
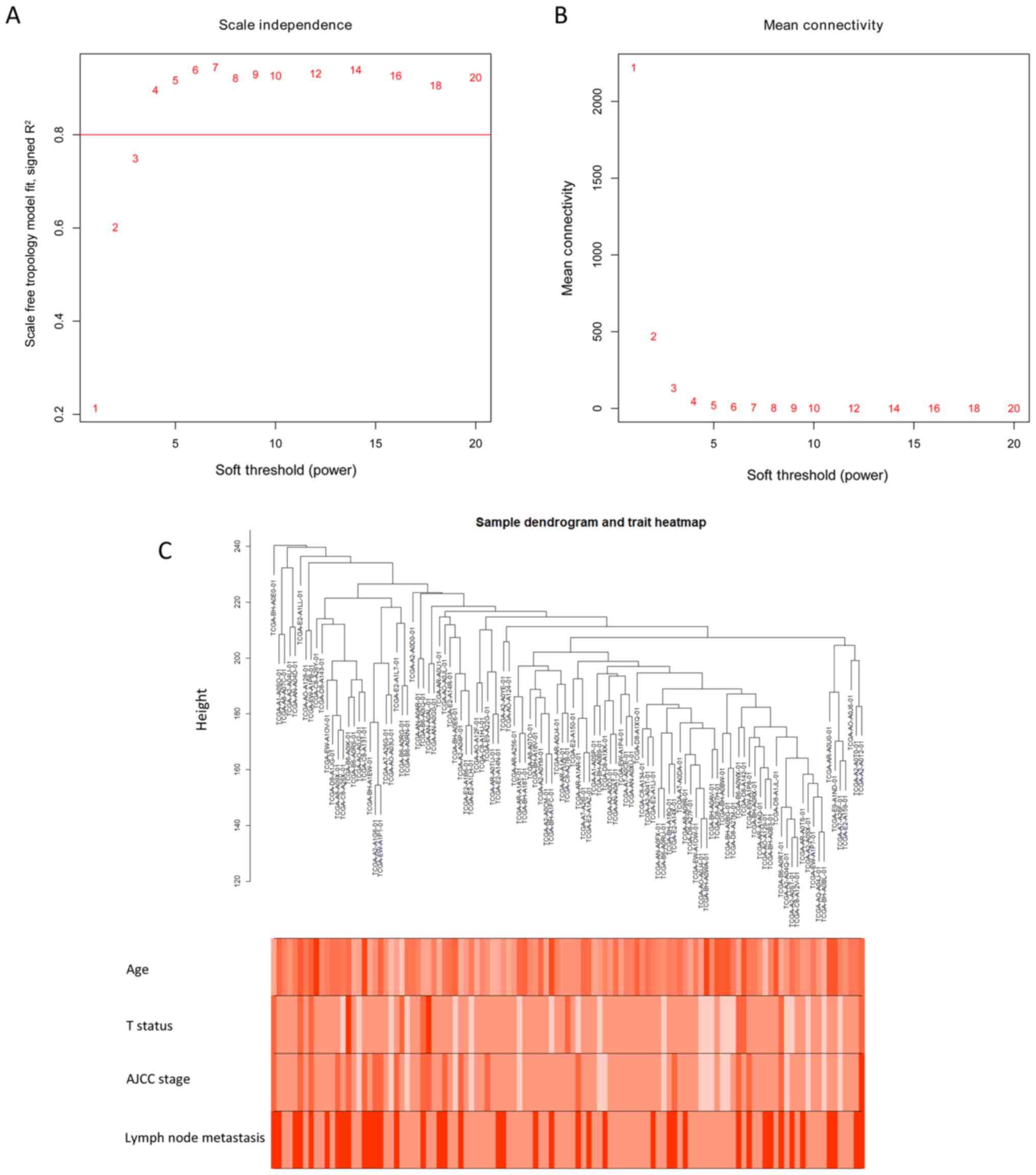
Pearson's correlation methods. The scale-free network was
constructed by raising the power of β to 4 and by ensuring that the
scale-free R2 reached 0.95 (Fig. 2A and B). The clustering dendrogram of
TNBC tissues is shown in Fig.
2C.
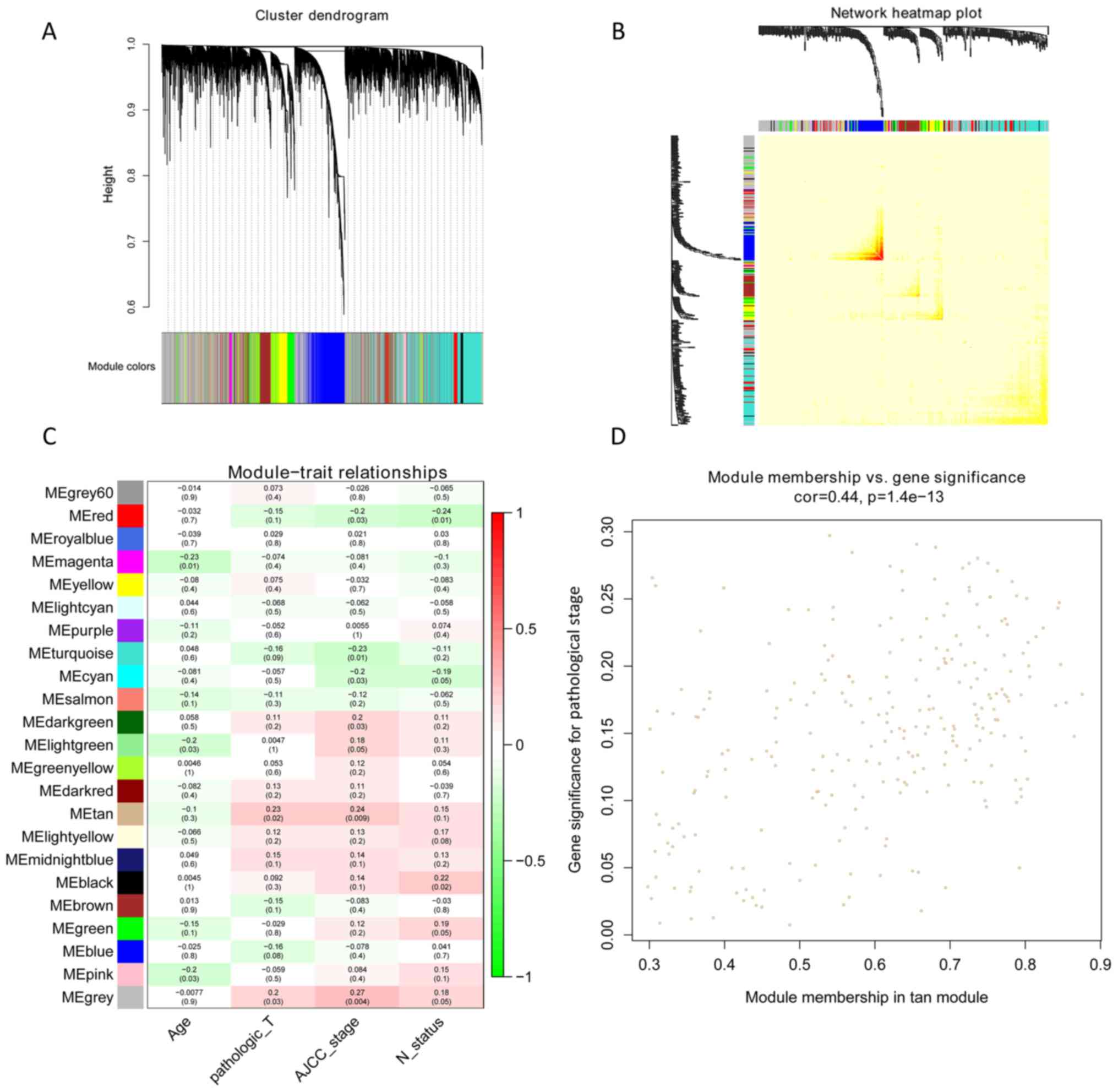
A total of 23 modules were found to be clustered,
and this gene clustering is displayed as a dendrogram in Fig. 3A. The weighted network of all genesis
exhibited in a heat map, depicting the topological overlap matrix
amongst the mRNA expression profiles (Fig. 3B). The tan module was determined
using a trait-heat map to be the module with the strongest
correlation with the pathological stage of TNBC (Fig. 3C). Fig.
3D illustrates the correlation of genes with pathological
stage, as well as module membership (the correlation of genes with
clusters) in the tan module. The results revealed that genes, which
had high a correlation with tan modules were also strongly
associated with the pathological stage of TNBC. Based on the
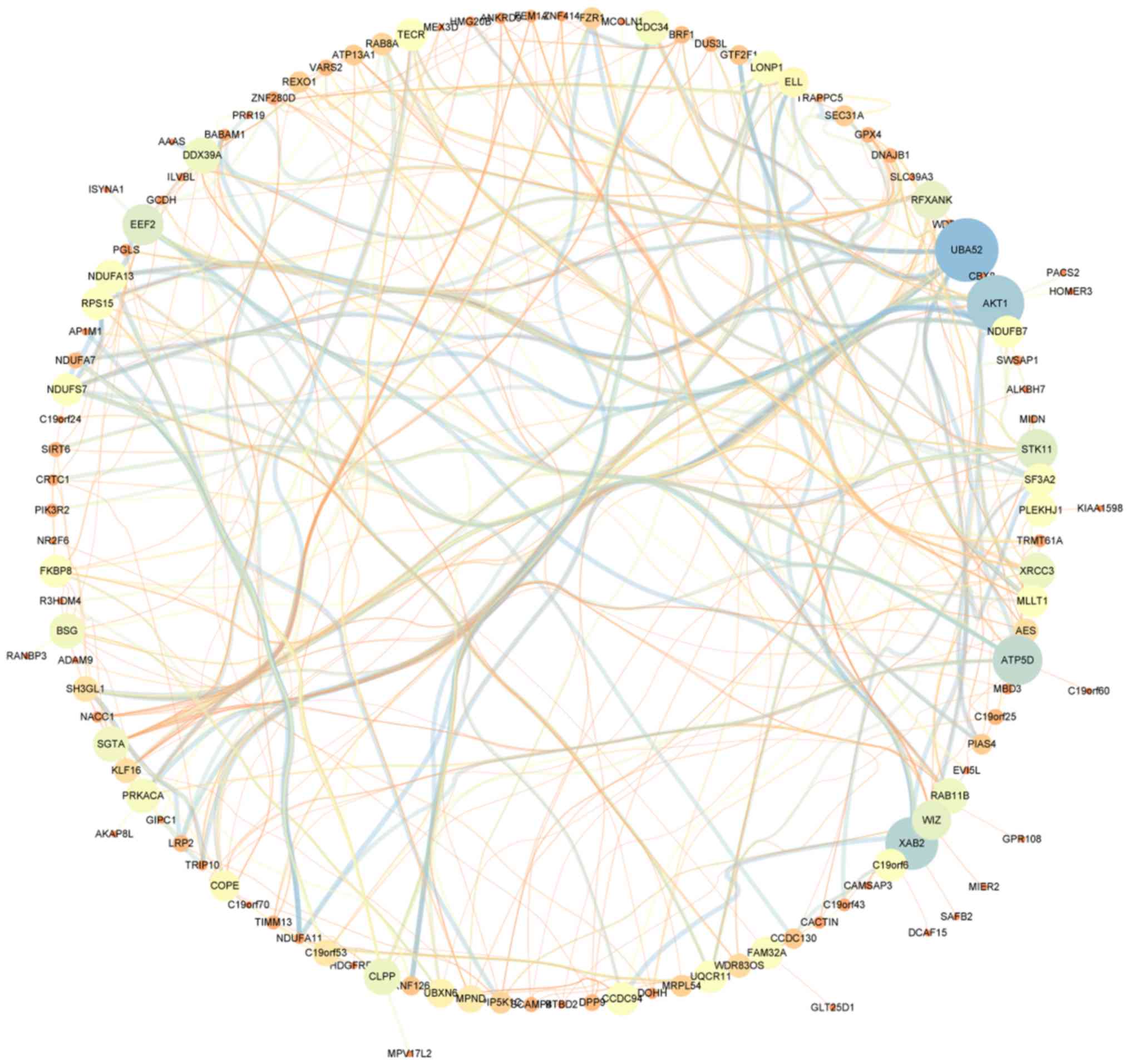
cut-off criteria (|GS|>0.4), 129 genes with high connectivity
were selected for the construction of the co-expression network.
The inner connectivity in the tan module with the threshold
(|GS|>0.4) was plotted. This showed strong co-expression
relationships in the tan module (Fig.
4).
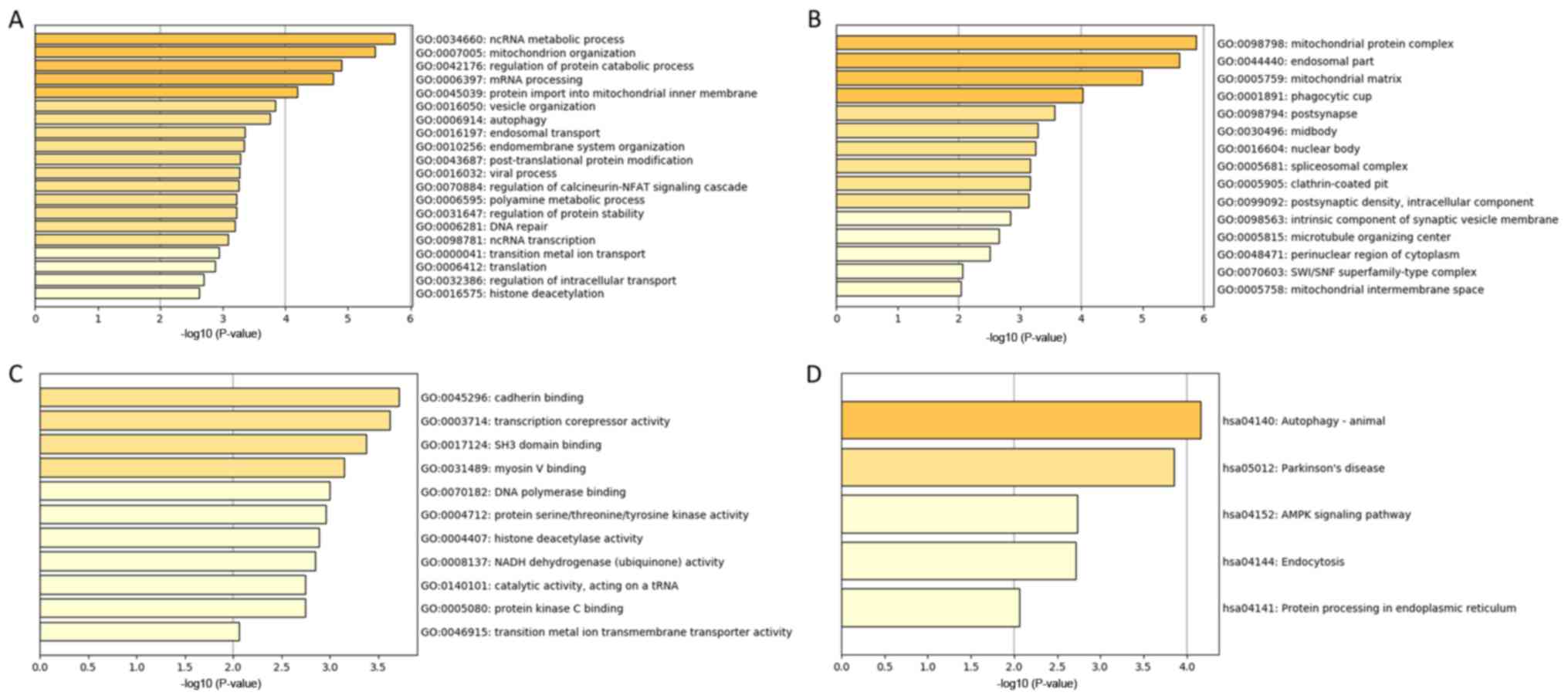
GO and Kyoto Encyclopedia of Genes and
Genomes (KEGG) Analysis
The genes in the tan module were divided into three
groups (biological process, cellular component, and molecular
function), which were then analyzed using GO and KEGG analysis. In
the biological process group, the most enriched genes concerned
epigenetic regulation, cell metabolism, DNA repair and mRNA
processing (Fig. 5A). The genes in
the cellular component group that were most enriched comprised
‘mitochondrial protein complex’, ‘endosomal part’ and ‘phagocytic
cup’ (Fig. 5B). The most enriched
genes in the molecular function group were in ‘cadherin binding’,
‘transcription corepressor activity’ and ‘SH3 domain binding’
(Fig. 5C). KEGG pathway analysis
indicated that ‘autophagy’ and ‘AMPK signaling pathway’ were
involved in the regulation of genes in the tan module (Fig. 5D).
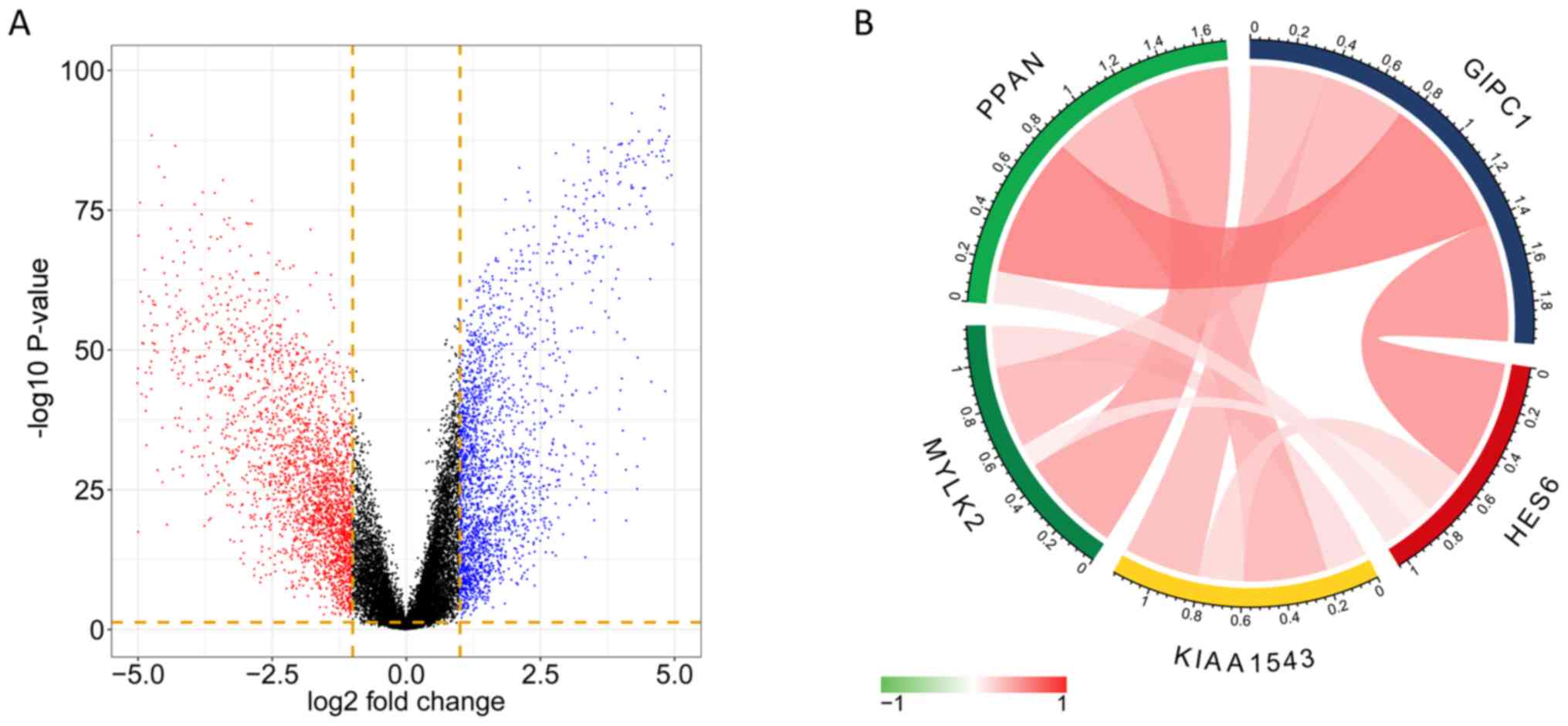
DEG analysis of TNBC tissues
The DEGs were obtained from the analysis of RNAseq
datasets, and a total of 2,169 significant genes were identified
(Q-value<0.05; fold change >2) from the comparison of TNBT
tissues and para-tumor tissues. The volcano plot indicates the
fold-change and P-value of DEGs (Fig.
6A). The overlap between the genes discovered in DEG analysis
and the genes in the tan module comprised 42 genes, which were
selected as hub-genes. Among the 42 genes, 5 of which showed high
correlation between expression level and survival probability, and
were used for further analysis.
Survival and expression of the 5
selected genes
From the aforementioned hub-genes, 5 genes [GIPC PDZ
domain containing family member 1 (GIPC1), hes family bHLH
transcription factor 6 (HES6), calmodulin-regulated
spectrin-associated protein family member 3 (KIAA1543), myosin
light chain kinase 2 (MYLK2) and peter pan homolog (PPAN)] were
selected to explore their association with the pathological
progress. The unsupervised clustering results illustrate the
co-expression of the 5 genes in the tan module and the extent of
correlation between the 5 genes in TNBC was determined. Fig. 6B indicates high correlation between
HES6 and GIPC1, and also between PPAN and GIPC1. The expression
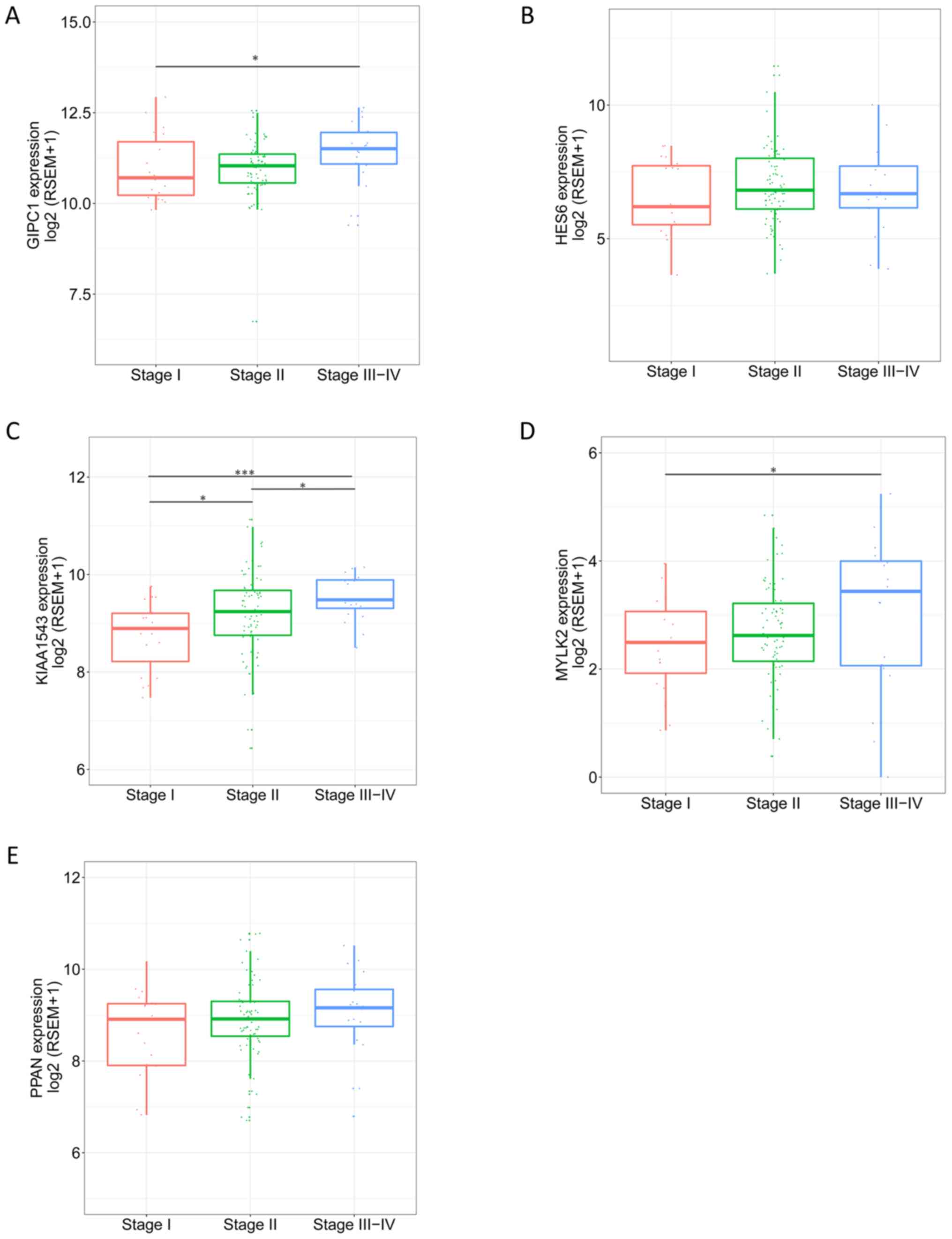
level of the genes in different pathological stages varied, and
tended to be higher in more progressed pathological stages
(Fig. 7). The expression levels of
the 5 genes in tumor and para-tumor tissues were also plotted in
Fig. 8, and showed significantly
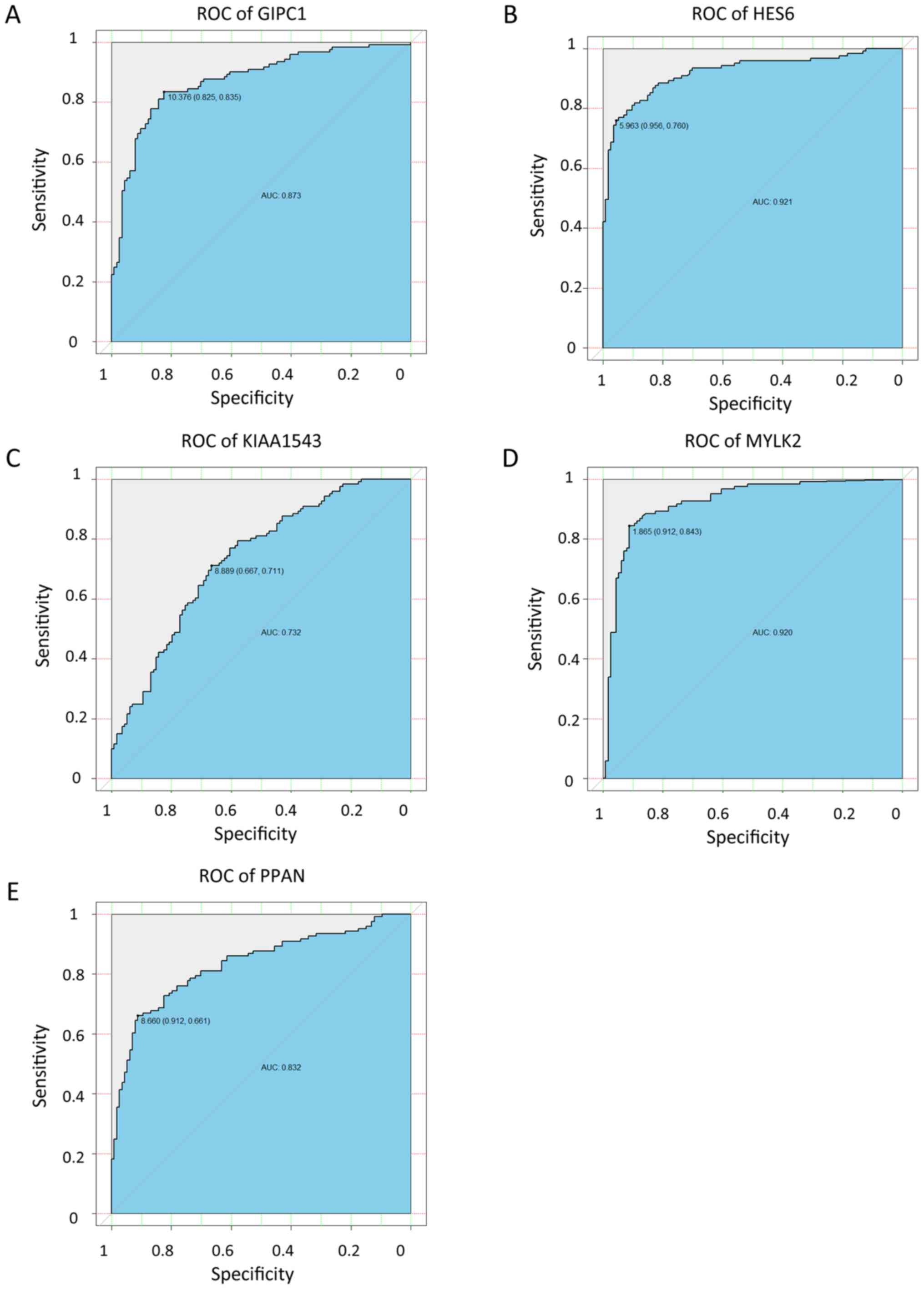
higher expression in tumor tissues (P<0.01). The ROC curves
indicate that GIPC1, HES6, KIAA1543, MYLK2 and PPAN all exhibited
high diagnostic accuracy for the identification of para-tumor and
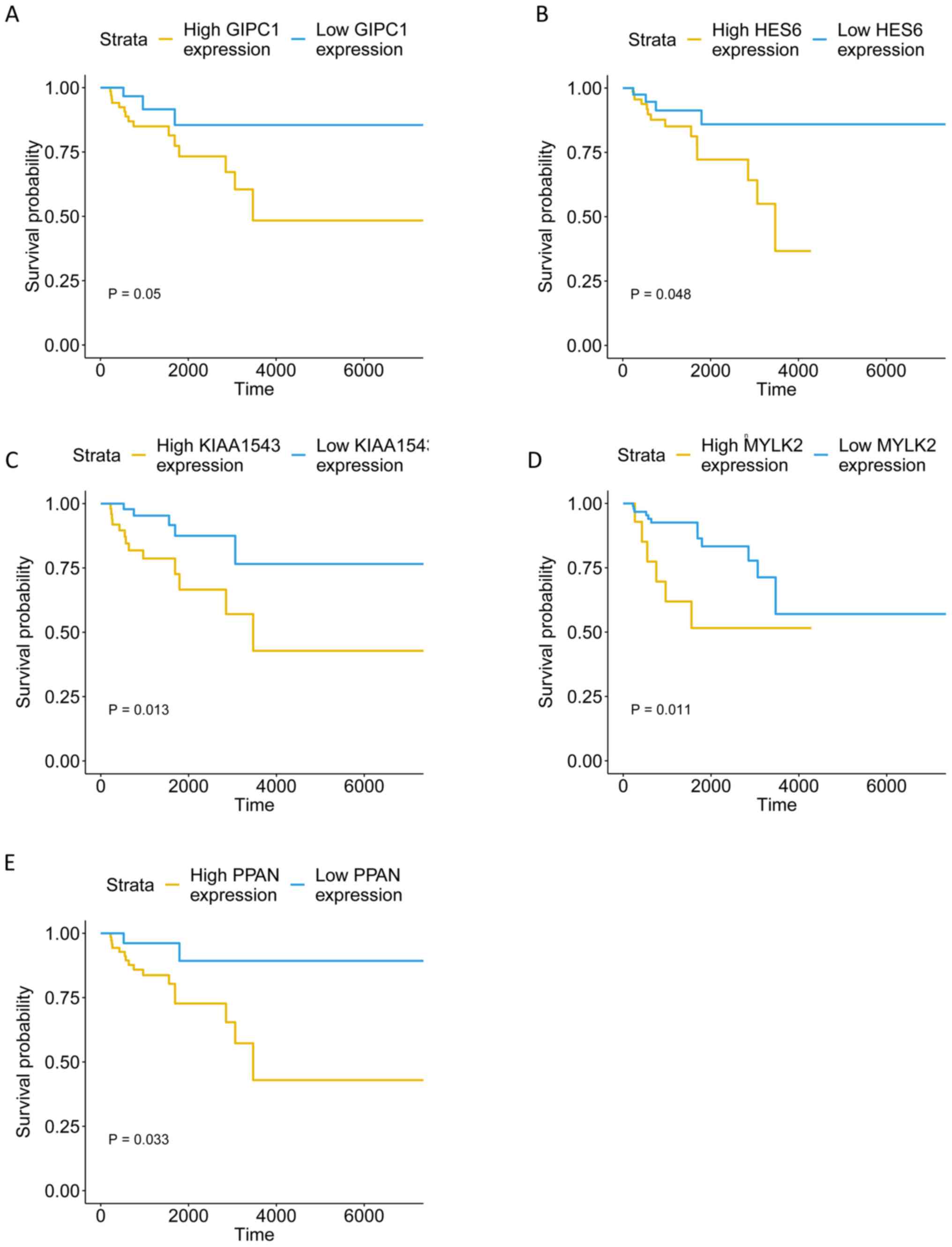
tumor tissues (Fig. 9). The KM
curves illustrate that the expression levels of the 5 genes were
associated with the OS of patients with TNBC (Fig. 10). Furthermore, high-expression
groups of KIAA1543, MYLK2 and PPAN were significantly associated
with lower OS times when compared with low-expression of the same
proteins (P<0.05).
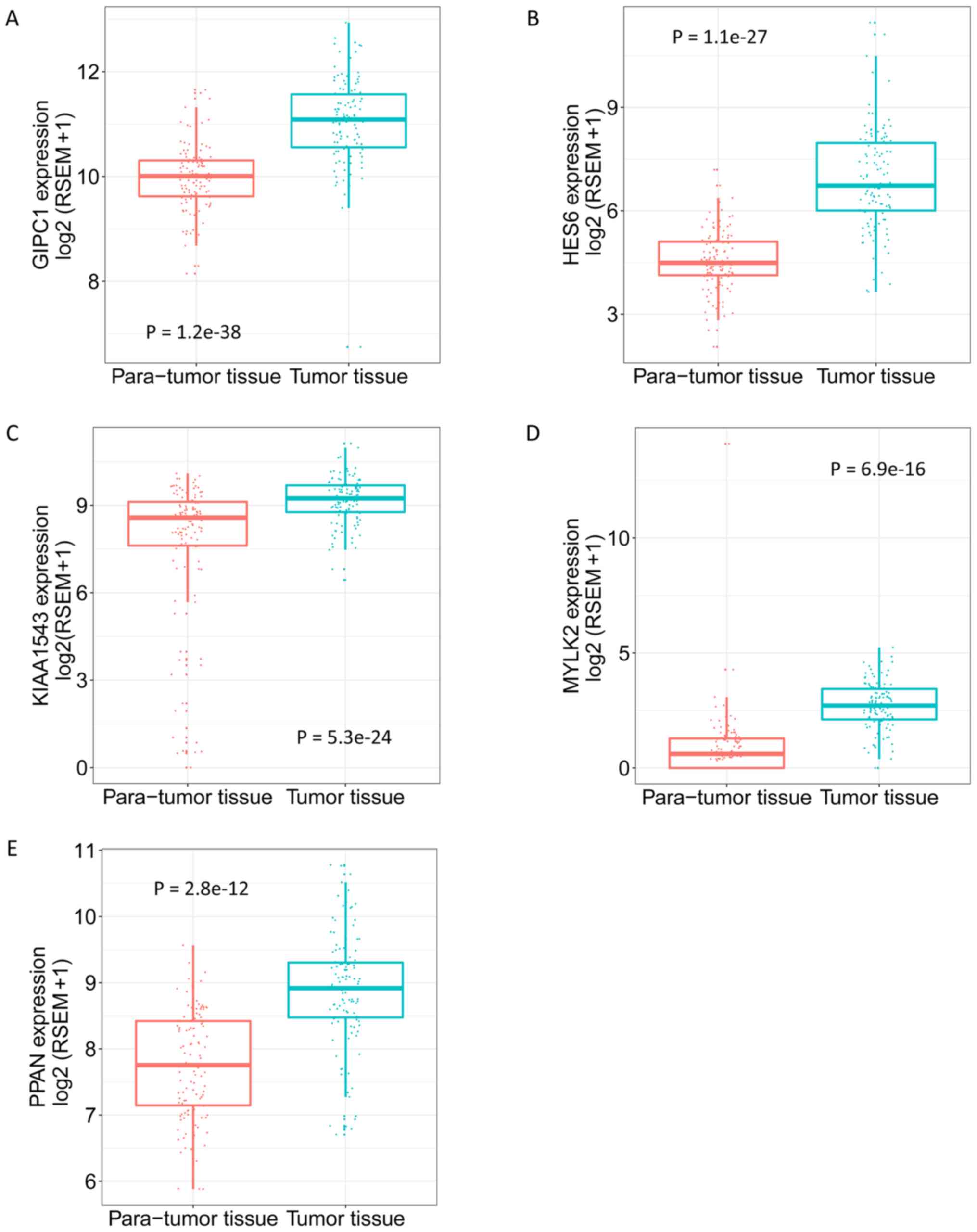 | Figure 8.Expression of GIPC1, HES6, KIAA1543,
MYLK2 and PPAN in para-tumor and tumor tissues. (A) GIPC1. (B)
HES6. (C) KIAA1543. (D) MYLK2. (E) PPAN. Adjusted Q value,
determined using the R Limma package, was used to evaluate the
statistical significance of differences. GIPC1, GIPC PDZ domain
containing family member 1; HES6, hes family bHLH transcription
factor 6; KIAA1543, calmodulin-regulated spectrin-associated
protein family member 3; MYLK2, myosin light chain kinase 2; PPAN,
peter pan homolog. |
Discussion
Breast cancer is an umbrella term summarizing
carcinomas originating from the breast, and is one of the most
prevalent cancer types worldwide. TNBCs are described as breast
cancers that simultaneously lack expression of the ER, PR and HER2
genes. Triple-negative tumors represent 80% of basal-like molecular
breast cancers (1). They have less
favorable outcomes compared with other molecular subtypes of breast
cancer, as they are commonly associated with a higher risk of early
relapse and poor survival (11).
Currently, no drug is available that specifically targets TNBC, and
the results of chemotherapy are unsatisfactory. The molecular
characteristics of TNBC include disruption of BRCA1 DNA repair
associated (BRCA)-1 function (12,13). The
low expression level of BRCA-1 and inhibitor of DNA binding 4, HLH
protein (ID4) lead to the change from homologous repair (HR) to
non-homologous end joining (NHEJ) or single-strand annealing (SSA)
pathways (14,15). HR is the most important DNA repair
mechanism in healthy tissues and maintains genomic stability.
However, the change to NHEJ (the alternative, more error-prone
DNA-repair mechanism) can lead to genetic instability in tumor
tissues. The inhibitor olaparib, which targets PARP-1, can block
the NHEJ of breast cancer types and ultimately inhibit DNA repair
(16). Moreover, epigenetic
dysregulation is well established as having a crucial role in
cancer pathology and progression (17,18), and
the current study identified enrichment of epigenetic regulation in
TNBC.
However, the molecular characteristics of TNBC are
not well understood. Exploring the mechanisms involved in the
progress and prognosis of TNBC would be helpful for improving
diagnosis and treatment, and new targets or biomarkers are
required. The present study determined a number of candidates using
TGCA RNAseq datasets downloaded from UCSC database to identify
promising biomarkers related to TNBC progression. WGCNA is a
commonly used bioinformatics analysis tool used to identify the key
modules and genes, which are associated with specific clinical
traits. A study applied WGCNA analysis in breast cancer,
identifying the association between cyclin B2 (CCNB2), F-box
protein 5 (FBXO5), kinesin family member 4A (KIF4A),
minichromosomal maintenance 10 replication initiation factor
(MCM10) and TPX2 microtubule nucleation factor (TPX2) expression,
and the survival of breast cancer patients (19). Another study used WGCNA to reveal the
association between ATRX chromatin remodeler (ATRX) and TNBC
(20). In the present study, WGCNA
was performed to determine the association between American Joint
Committee on Cancer-TNM stage and the gene co-expression module. By
overlapping the genes from the tan module and DEG analysis, 42 hub
genes were identified. Of these 42 genes, GIPC1, HES6, KIAA1543,
MYLK2 and PPAN were selected to validate the strength of
association with TNBC progression.
GIPC1 (also known as C19orf3) is a cytoplasmic
protein that acts as an adaptor protein, linking receptor
interactions to intracellular signaling pathways, including cell
cycle regulation (21). GIPC1
protein is highly expressed both in cultured human breast cancer
cells and in primary and metastatic tumor tissues (22). It is a cancer-associated immunogenic
antigen in breast cancer which is also associated with bone
metastasis development in breast cancer (23). The functions of GIPC1 include the
regulation of apoptotic cell death, G2 cell-cycle
arrest, modified cell adhesion and the migration of breast cancer
cells (24). In the present
analysis, GIPC1 was significantly upregulated in both
triple-negative breast tumor tissue and a progressed pathological
stage.
HES6 encodes a subfamily of basic helix-loop-helix
transcription repressors with homology to the Drosophila enhancer
of split genes (25). The
overexpression of HES6 is found in metastatic carcinomas of
different origins. Hes6 ectopic expression stimulates cell
proliferation, not only in breast cancer T47D cells, but also in
breast tumor growth in xenografts. The overexpression of HES6 also
led to the induction of E2F transcription factor 1, a crucial
target gene for the transcriptional repressor HES1 (26). Furthermore, the ER α-negative breast
cancer cell lines MDA-MB-231 and SKBR3express 4 to 10 times higher
levels of Hes-6 mRNA, compared with ERα-positive T47D and MCF-7
cells. The aforementioned studies complement the present findings
of an association between HES6 expression and estrogen in TNBC.
KIAA1543, also known as Nezha, was shown to bridge
the minus ends of non-centrosome anchored microtubules with p120,
which plays a crucial role in stabilizing cadherin-catenin mediated
cell-cell adhesion complexes (27,28).
KIAA1543 is also involved in epithelial-mesenchymal transition, and
mediates the interaction between cadherin and microtubules. It is
overexpressed in invasive lobular carcinomas but its role is poorly
characterized (29,30). The underlying mechanisms of KIAA1543
on TNBCs need to be further established. The current study found
KIAA1543 to be significantly upregulated in TNBC tissues and at
advanced pathological stages. Moreover, high KIAA1543 expression
was associated with poor OS in TNBC patients.
MYLK2 encodes a calcium/calmodulin-dependent
serine/threonine kinase (31). A
somatic mutation in, or amplification of, MYLK2 has been detected
in several cancer tissues, compared with normal tissues (31). Moreover, proteomic analyses conducted
on serum protein profiling results revealed the upregulation of
MYLK2 in pancreatic cancer patients (32). The present study showed that in TNBC
tissues, MYLK2 was overexpressed and was also associated with the
poor patient OS.
PPAN encodes an evolutionarily conserved protein
similar to the Drosophila gene peter pan. A signature inferred from
Drosophila mitotic genes revealed an association between PPAN
expression and the survival of breast cancer patients (33). Tumor patients with high expression
levels of PPAN have been shown to respond more favorably to
treatment with anti-EGFR antibodies such as cetuximab (34). The data presented reveal a
significant association between high PPAN expression and a poor OS
in patients with TNBC.
Taken together, WGCNA and DEG analysis on RNAseq
datasets from TNBC tissues revealed that the tan module was the
most significantly associated with AJCC-TNM stage. The genes in the
tan module showed high inter-connectivity with the co-expression
network. Furthermore, GO and KEGG analysis revealed the enrichment
of the genes in the related terms from GO. The overlap between the
module and DEG analysis identified 42 genes, 5 of which were
negatively associated with the OS of patients with TNBC. Therefore,
the current study provides 5 novel biomarkers for TNBC, which
exhibit potential as targets in the diagnosis and treatment of the
disease.
Acknowledgements
Not applicable.
Funding
This study was supported by the Jinhua technology
division fund (grant no. 2018-4-120).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available in the University of California, Santa
Cruz repository, (https://xenabrowser.net/datapages/).
Authors' contributions
MB, JW, LB and KZ conceived the experiment design.
LB, JW, KZ and TG performed the data analysis. JW and KZ revised
the manuscript. LB, TG, JW, KZ and MB wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dawood S: Triple-negative breast cancer:
Epidemiology and management options. Drugs. 70:2247–2258. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liedtke C, Mazouni C, Hess KR, André F,
Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B,
Green M, et al: Response to neoadjuvant therapy and long-term
survival in patients with triple-negative breast cancer. J Clin
Oncol. 26:1275–1281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Zhang Q, Gao Z, Xin S, Zhao Y,
Zhang K, Shi R and Bao X: A novel 4-gene signature for overall
survival prediction in lung adenocarcinoma patients with lymph node
metastasis. Cancer Cell Int. 19:1002019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang K, Maruyama T and Fan G: The naive
state of human pluripotent stem cells: A synthesis of stem cell and
preimplantation embryo transcriptome analyses. Cell Stem Cell.
15:410–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Xin S, Zhang K, Shi R and Bao X:
Low GAS5 levels as a predictor of poor survival in patients with
lower-grade gliomas. J Oncol. 2019:17850422019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smyth GK: Limma: linear models for
microarray data. Bioinformatics and computational biology solutions
using R and BioconductorStatistics for Biology and Health.
Gentleman R..Carey V.J..Huber W..Irizarry R.A..Dudoit S.: Springer;
New York, NY: pp. 397–420. 2005, View Article : Google Scholar
|
|
10
|
Kassambara A, Kosinski M and Biecek P:
Survminer: Drawing Survival Curves using ‘ggplot2’. R package
version 03. 2017 1;
|
|
11
|
Haffty BG, Yang Q, Reiss M, Kearney T,
Higgins SA, Weidhaas J, Harris L, Hait W and Toppmeyer D:
Locoregional relapse and distant metastasis in conservatively
managed triple negative early-stage breast cancer. J Clin Oncol.
24:5652–5657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Atchley DP, Albarracin CT, Lopez A, Valero
V, Amos CI, Gonzalez-Angulo AM, Hortobagyi GN and Arun BK: Clinical
and pathologic characteristics of patients with BRCA-positive and
BRCA-negative breast cancer. J Clin Oncol. 26:4282–4288. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cleator S, Heller W and Coombes RC:
Triple-negative breast cancer: Therapeutic options. Lancet Oncol.
8:235–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Powell SN and Kachnic LA: Roles of BRCA1
and BRCA2 in homologous recombination, DNA replication fidelity and
the cellular response to ionizing radiation. Oncogene.
22:5784–5791. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Evers B, Helleday T and Jonkers J:
Targeting homologous recombination repair defects in cancer. Trends
Pharmacol Sci. 31:372–380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang M, Wu W, Wu W, Rosidi B, Zhang L,
Wang H and Iliakis G: PARP-1 and Ku compete for repair of DNA
double strand breaks by distinct NHEJ pathways. Nucleic Acids Res.
34:6170–6182. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Deng H, Xin S, Zhang K, Shi R and
Bao X: Prognostic and predictive value of three DNA methylation
signatures in lung adenocarcinoma. Front Genet. 10:3492019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baylin SB and Jones PA: Epigenetic
determinants of cancer. Cold Spring Harb Perspect Biol.
8:a0195052016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang J, Kong D, Cui Q, Wang K, Zhang D,
Gong Y and Wu G: Prognostic genes of breast cancer identified by
gene co-expression network analysis. Front Oncol. 8:3742018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lan L, Xu B, Chen Q, Jiang J and Shen Y:
Weighted correlation network analysis of triple-negative breast
cancer progression: Identifying specific modules and hub genes
based on the GEO and TCGA database. Oncol Lett. 18:1207–1217.
2019.PubMed/NCBI
|
|
21
|
Valdembri D, Caswell PT, Anderson KI,
Schwarz JP, König I, Astanina E, Caccavari F, Norman JC, Humphries
MJ, Bussolino F and Serini G: Neuropilin-1/GIPC1 signaling
regulates alpha5beta1 integrin traffic and function in endothelial
cells. PLoS Biol. 7:e252009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rudchenko S, Scanlan M, Kalantarov G,
Yavelsky V, Levy C, Estabrook A, Old L, Chan GL, Lobel L and Trakht
I: A human monoclonal autoantibody to breast cancer identifies the
PDZ domain containing protein GIPC1 as a novel breast
cancer-associated antigen. BMC Cancer. 8:2482008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Westbrook JA, Cairns DA, Peng J, Speirs V,
Hanby AM, Holen I, Wood SL, Ottewell PD, Marshall H, Banks RE, et
al: CAPG and GIPC1: Breast cancer biomarkers for bone metastasis
development and treatment. J Natl Cancer Inst. 108:2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chittenden TW, Pak J, Rubio R, Cheng H,
Holton K, Prendergast N, Glinskii V, Cai Y, Culhane A, Bentink S,
et al: Therapeutic implications of GIPC1 silencing in cancer. PLoS
One. 5:e155812010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bae S, Bessho Y, Hojo M and Kageyama R:
The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal
differentiation. Development. 127:2933–2943. 2000.PubMed/NCBI
|
|
26
|
Hartman J, Lam EW, Gustafsson JA and Ström
A: Hes-6, an inhibitor of Hes-1, is regulated by 17beta-estradiol
and promotes breast cancer cell proliferation. Breast Cancer Res.
11:R792009. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Akhmanova A and Hoogenraad CC: Microtubule
minus-end-targeting proteins. Curr Biol. 25:R162–R171. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fu Y, Zheng S, An N, Athanasopoulos T,
Popplewell L, Liang A, Li K, Hu C and Zhu Y: β-catenin as a
potential key target for tumor suppression. Int J Cancer.
129:1541–1551. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Castellana B, Escuin D, Pérez-Olabarria M,
Vázquez T, Muñoz J, Peiró G, Barnadas A and Lerma E: Genetic
up-regulation and overexpression of PLEKHA7 differentiates invasive
lobular carcinomas from invasive ductal carcinomas. Hum Pathol.
43:1902–1909. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanaka N, Meng W, Nagae S and Takeichi M:
Nezha/CAMSAP3 and CAMSAP2 cooperate in epithelial-specific
organization of noncentrosomal microtubules. Proc Natl Acad Sci
USA. 109:20029–20034. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soung YH, Lee JW, Kim SY, Nam SW, Park WS,
Lee JY, Yoo NJ and Lee SH: Mutational analysis of the kinase domain
of MYLK2 gene in common human cancers. Pathol Res Pract.
202:137–140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rong Y, Jin D, Hou C, Hu J, Wu W, Ni X,
Wang D and Lou W: Proteomics analysis of serum protein profiling in
pancreatic cancer patients by DIGE: Up-regulation of
mannose-binding lectin 2 and myosin light chain kinase 2. BMC
Gastroenterol. 10:682010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Damasco C, Lembo A, Somma MP, Gatti M, Di
Cunto F and Provero P: A signature inferred from drosophila mitotic
genes predicts survival of breast cancer patients. PLoS One.
6:e147372011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tabernero J, Cervantes A, Rivera F,
Martinelli E, Rojo F, von Heydebreck A, Macarulla T,
Rodriguez-Braun E, Eugenia Vega-Villegas M, Senger S, et al:
Pharmacogenomic and pharmacoproteomic studies of cetuximab in
metastatic colorectal cancer: Biomarker analysis of a phase I
dose-escalation study. J Clin Oncol. 28:1181–1189. 2010. View Article : Google Scholar : PubMed/NCBI
|