Introduction
Retinoblastoma (RB) is the most prevalent
intraocular cancer that affects children (1). It occurs worldwide, with an incidence
of one case per 15,000-20,000 live births (2) and an estimated mortality rate of 50%
(3). Therefore, elucidation of the
characteristics of RB is necessary in order to identify more
effective therapeutic strategies. Although the most significant
event contributing to the oncogenesis of RB is the inactivation of
the two RB alleles on chromosome 13 (4), other oncogenes may also contribute to
RB tumorigenesis.
The c-myc proto-oncogene belongs to the MYC
family (5). Expression of
c-myc or its protein product c-Myc is upregulated in the
majority of malignant tumour types, including lymphoma,
neuroblastoma, melanoma, breast, ovarian, prostate and liver cancer
(6–9). c-Myc upregulation in tumours may result
from gene amplification, increased c-myc transcription, or
an increase in c-Myc protein stability and activity via
post-translational regulation (10).
Thus, it has been hypothesized that the oncogenicity of
c-myc is dependent on elevated expression levels. However,
the expression level of c-Myc in human cancer types ranges from
lower than average to greatly elevated (11), and it is differentially expressed
depending on the cell type. The expression level of c-Myc in RB is
yet to be identified, to the best of our knowledge.
Additionally, it has been determined that c-Myc is
regulated via different pathways in different cell lines. Histone
acylation and DNA methylation are involved in the transcriptional
regulation of c-myc. For example, c-myc is
downregulated by the demethylating reagent 5-azacytidine in human
prostate cancer cells (12,13), whereas 5-aza-deoxycytidine induces
the upregulation of c-myc in lung cancer cells (14). Moreover, c-myc expression is
regulated via histone deacetylation in human cervical cancer cells
(15). Nonetheless, whether
c-myc is regulated via histone acylation or DNA methylation
in RB cells has not yet been elucidated.
Furthermore, c-Myc is a pleiotropic transcription
factor that binds to the promoters, and regulates the expression,
of a large number of genes regulating metabolic processes,
macromolecular synthesis, the cell cycle and apoptosis (16). In a similar manner to the majority of
oncoproteins, c-Myc enhances cell proliferation and regulates cell
cycle (17). In both healthy and
tumorous cells, MYC-dependent signalling is an important regulator
of cell cycle progression from the G1 to S phases
(18), and inactivation of c-Myc
expression results in tumour regression accompanied by apoptosis,
differentiation or tumour dormancy (19). However, unlike most oncoproteins,
c-Myc also significantly enhances certain mechanisms of programmed
cell death (PCD), including senescence and apoptosis (20). Therefore, under conditions of limited
energy sources, downregulation of c-Myc may represent a survival
strategy enabling cancer cell proliferation (21). The conflicting roles discovered
indicate a complex role served by c-Myc, which varies depending on
cancer cell type. Thus, investigation of the bioactivity of c-Myc
may greatly improve the present understanding of RB
pathophysiology.
Based on the aforementioned findings, the present
study sought to determine the expression profile and bioactivity of
c-Myc in RB cells. It was discovered that c-Myc was downregulated
in the RB cell lines WERI-Rb1 and Y79. Moreover, the expression of
c-Myc was significantly upregulated following cell treatment with
HDAC inhibitors, such as trichostatin A (TSA), vorinostat (SAHA)
and entinostat (MS-275). The activity of the c-myc promoter
was significantly increased following TSA treatment in WERI-Rb1
cells. However, the low level of c-Myc expression in Y79 cells was
not upregulated by the HDAC inhibitors. Furthermore, exogenous
c-myc significantly reduced the viability of both WERI-Rb1
and Y79 cells. Therefore, the present data provide new insights
into the c-Myc expression mechanism and its bioactivity in RB
cells.
Materials and methods
Cell culture and transfection
Human retinoblastoma cell lines WERI-Rb1 and Y79
[both American Type Culture Collection (ATCC)], and the human colon
cancer cell line RKO (ATCC), were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) and
supplemented with 10% fetal bovine serum (FBS) and 1%
penicillin/streptomycin (both Gibco; Thermo Fisher Scientific,
Inc.), in a humidified 5% CO2 incubator at 37°C. The
cells selected for the assays were collected during the exponential
growth phase. TSA was obtained from Sigma-Aldrich; Merck KGaA, and
SAHA, MS-275 and VPA were obtained from Selleck Chemicals. WERI-Rb1
cells and Y79 cells were seeded at a density of 1×106
cells per well in a 6 well plate and were stably transfected with a
plasmid expressing c-Myc or an empty vector control (pMXs-c-Myc or
vector; Addgene, Inc.), using Lipofectamine®
(Invitrogen; Thermo Fisher Scientific Inc.) in Opti-MEM (Gibco;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The plasmid or vector was used at a concentration of
0.8 µg/well. Medium was changed to complete growth medium (10% FBS)
after 4 h.
Reverse-Transcription (RT) PCR
Total RNA was extracted from WERI-Rb1 and Y79 cells
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). One microgram of total RNA was subjected to
reverse transcription with the PrimeScriptTM RT-PCR kit (Takara
Bio, Inc.) according to the manufacture's protocol. Reverse
transcription was performed at 37°C for 15 min and 85°C for 5 sec.
The PCR reaction was carried out using a My Cycler thermal cycler
(Bio-Rad Laboratories, Inc.) with TaKaRa Premix Taq®
Version 2.0 (Takara Bio, Inc.). For c-Myc, PCR reaction was
performed for 35 cycles each at 98°C for 10 sec, 60°C for 30 sec,
72°C for 30 sec and a final extension at 72°C for 10 min. PCR
reaction for β-actin was performed for 20 cycles, with the same
temperature and time parameters as for c-Myc. The primer sequences
are presented in Table SI.
Reverse transcription-quantitative
(RT-q)PCR
The mRNA expression levels of histone deacetylases
(HDACs) in WERI-Rb1 cells were identified using a Roche 480 system
(Roche Diagnostics) and assays-on-demand primers (Invitrogen;
Thermo Fisher Scientific, Inc.) for HDACs. Total RNA was extracted
from WERI-Rb1 cells using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). One microgram of total RNA was
subjected to reverse transcription with the PrimeScriptTM RT-PCR
kit (Takara Bio, Inc.) according to the manufacture's protocol.
Reverse transcription was performed at 37°C for 15 min and 85°C for
5 sec. qPCR was then performed using the SYBR® Prime
Script TM RT-PCR kit (Takara Biotechnology Co., Ltd.), according to
the manufacturer's protocol. qPCR was conducted to determine the
expression levels of HDACs using the Roche 480 system (Roche
Diagnostics). PCR was performed as follows: 94°C for 5 min,
followed by 40 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C
for 30 sec. Relative target gene expression levels were quantified
using the 2−ΔΔCq method and normalized to the internal
reference gene, β-actin (22). The
data are presented as the inverse of the normalized Cq value
(InvCq), or as the relative fold change compared with the untreated
control. The primer sequences are presented in Table SI.
Western blot analysis
Total proteins were extracted following cell lysis
using RIPA buffer (Beyotime Institute of Biotechnology) at 4°C.
Protein concentration was determined using bicinchoninic acid
method (Beyotime Institute of Biotechnology). Western blotting was
performed according to standard protocols. Proteins (30 µg/well)
were separated by 8% SDS-PAGE and transferred onto polyvinylidene
difluoride membrane. The membranes were blocked with 5% non-fat
milk dissolved in TBS containing 0.1% Tween-20 (TBST) for 1 h at
room temperature, and incubated with primary antibodies diluted in
milk dissolved in TBST at 4°C overnight. The following primary
antibodies were used: Rabbit anti-c-Myc (1:500; cat. no. ab32072;
Abcam) and rabbit anti-GAPDH (1:20,000; cat. no. 10494-1-AP;
ProteinTech Group, Inc.). Proteins were visualized using
horseradish peroxidase-conjugated anti-rabbit secondary antibodies
(1:10,000; cat. no. 7074; Cell Signaling Technology, Inc.)
incubated for 1 h at room temperature followed by use of the ECL
system (Merck KGaA). Densitometric analysis was performed on the
western blotting data using computerized image analysis and
software (Gel-Pro Analyzer software v. 6.0; Media Cybernetics,
Inc.).
Immunofluorescence assay
WERI-Rb1 or RKO cells were fixed in ice-cold 95%
methanol at −20°C for 20 min. Then the fixed cells were incubated
with 0.1% Triton X-100 for 10 min, and blocked with 10% normal goat
serum (Beyotime Institute of Biotechnology) at room temperature for
30 min. Cells were stained with rabbit anti-c-Myc (1:100; cat. no.
ab32072; Abcam) at 4°C overnight. Secondary anti-rabbit antibody
(1:500; cat. no. 4413S; Cell Signaling Technology, Inc.) was added
at room temperature for 1 h, and the nuclei were stained using DAPI
at room temperature for 10 min. Images were acquired by
fluorescence microscopy (magnification, ×100; Leica Microsystems
GmbH).
Cell Counting Kit (CCK)-8 assay
The viability of WERI-Rb1 and Y79 cells was assessed
using a CCK-8 assay (Dojindo Molecular Technologies, Inc.)
according to the manufacture's protocol. The CCK-8 reagent was
added to each well and cells were incubated for 2 h at 37°C. The
absorbance (optical density) at 450 nm was measured.
Luciferase assay
A plasmid encoding the human c-Myc promoter upstream
of firefly luciferase (pDEL1) was obtained from Addgene, Inc., and
a pGL2-Control was purchased from Promega Corporation. WERI-Rb1
cells (1×106 cells in 60 mm dishes) were transfected
using Lipofectamine® (Invitrogen; Thermo Fisher
Scientific Inc.) according to the manufacturer's instructions. The
transfected plasmids contained 2 µg expression plasmid or
pGL2-Control, and 100 ng Renilla luciferase reporter
plasmid, pCMV-RL (Promega Corporation). The pCMV-RL plasmid
encoding Renilla luciferase was included in all samples to
monitor transfection efficiency. At 24 h following initial
transfection, WERI-Rb1 and Y79 cells were further treated with TSA,
SAHA, MS-275 or VPA for another 24 h. Subsequently, the cells were
harvested and a Dual-Luciferase reporter assay system (Promega
Corporation) was performed to identify the sequential measurements
of the firefly and Renilla luciferase activities. The
luciferase activities and calculation of the relative ratios were
quantified using a Turner Designs Luminometer Model TD-20/20
(TD-20/20; Turner Designs, Inc.). The levels of firefly luciferase
activity were normalized against Renilla luciferase activity.
Statistical analysis
The data shown are representative of three
independent experiments with each experiment performed in
triplicate. The data are expressed as the mean ± SD. Statistical
analyses were performed using the SPSS for Windows software
package, version 10.5 (SPSS, Inc.). The differences between mean
values were evaluated using either the Student's two-tailed t-test
(for comparisons between 2 groups) or ANOVA followed by Tamhane T2
post hoc test (for comparisons of mRNA expression levels between
different HDACs in WERI-Rb1 cells). P<0.05 was considered to
indicate a statistically significant difference.
Results
c-Myc is downregulated in WERI-Rb1
cells
c-myc is typically reported as an oncogene
with high expression levels in a variety of cancer types, including
glioma, colon and gastric cancer (6–9), though
its expression level in RB is yet to be reported. To investigate
the influence of c-Myc activity on RB progression, a quantitative
examination of c-myc expression in WERI-Rb1 cells was
performed; the RT-PCR results indicated that c-myc was
almost undetectable in WERI-Rb1 cells but significantly upregulated
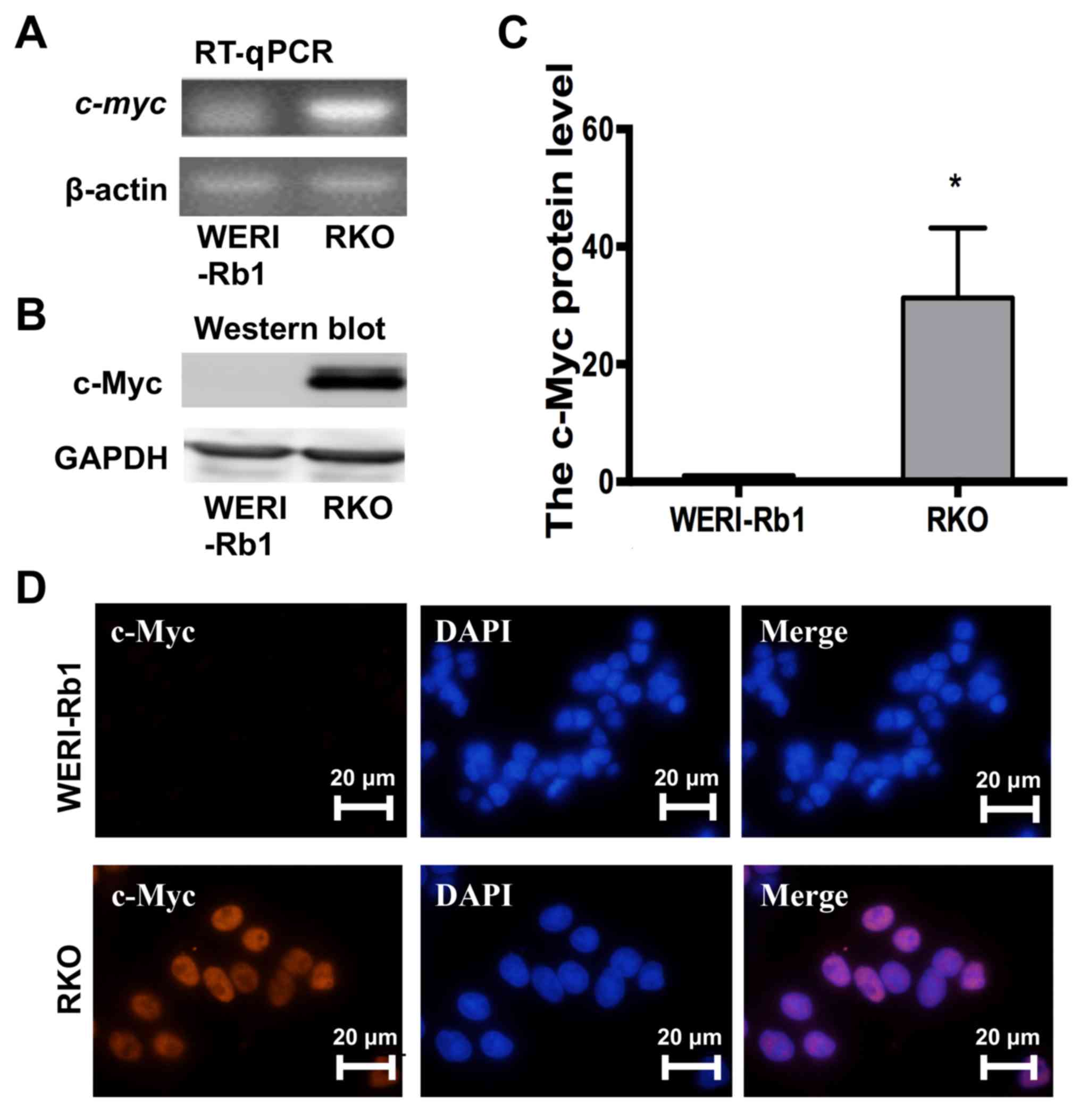
in RKO colon cancer cells (Fig. 1A).
Western blotting further supported this result (Fig. 1B and C; c-Myc protein expression;
WERI-Rb1 cells, 1; RKO cells, 31.26±11.92; P<0.05). Moreover, as
exhibited in Fig. 1D, nuclear
staining of c-Myc (red) was observed in RKO cells but not WERI-Rb1
cells. Thus, the current results indicated that c-myc was
not expressed in WERI-Rb1 cells.
c-Myc is upregulated in WERI-Rb1 cells
following treatment with the HDAC inhibitor TSA
To determine the influence of epigenetic
modifications on the silencing of the c-myc gene, WERI-Rb1
cells were treated with the HDAC inhibitor TSA. Fig. 2A indicates that c-myc
expression was increased in WERI-Rb1 cells treated with 250 or 500
nM TSA, according to the results of the RT-PCR analysis. Western
blotting was performed to further validate the aforementioned
results, and it was revealed that the level of c-Myc protein
expression was significantly increased in WERI-Rb1 cells treated
with 250 nM TSA for 24, 48 and 72 h, compared with the control
group (Fig. 2B and C; c-Myc relative
protein expression: Control, 1; 24 h, 3.84±1.06; 48 h, 7.44±3.43;
72 h, 8.12±2.69; P<0.05). Furthermore, nuclear staining of c-Myc
(red) was observed in WERI-Rb1 cells treated with TSA, but not in
the untreated cells (Fig. 2D).
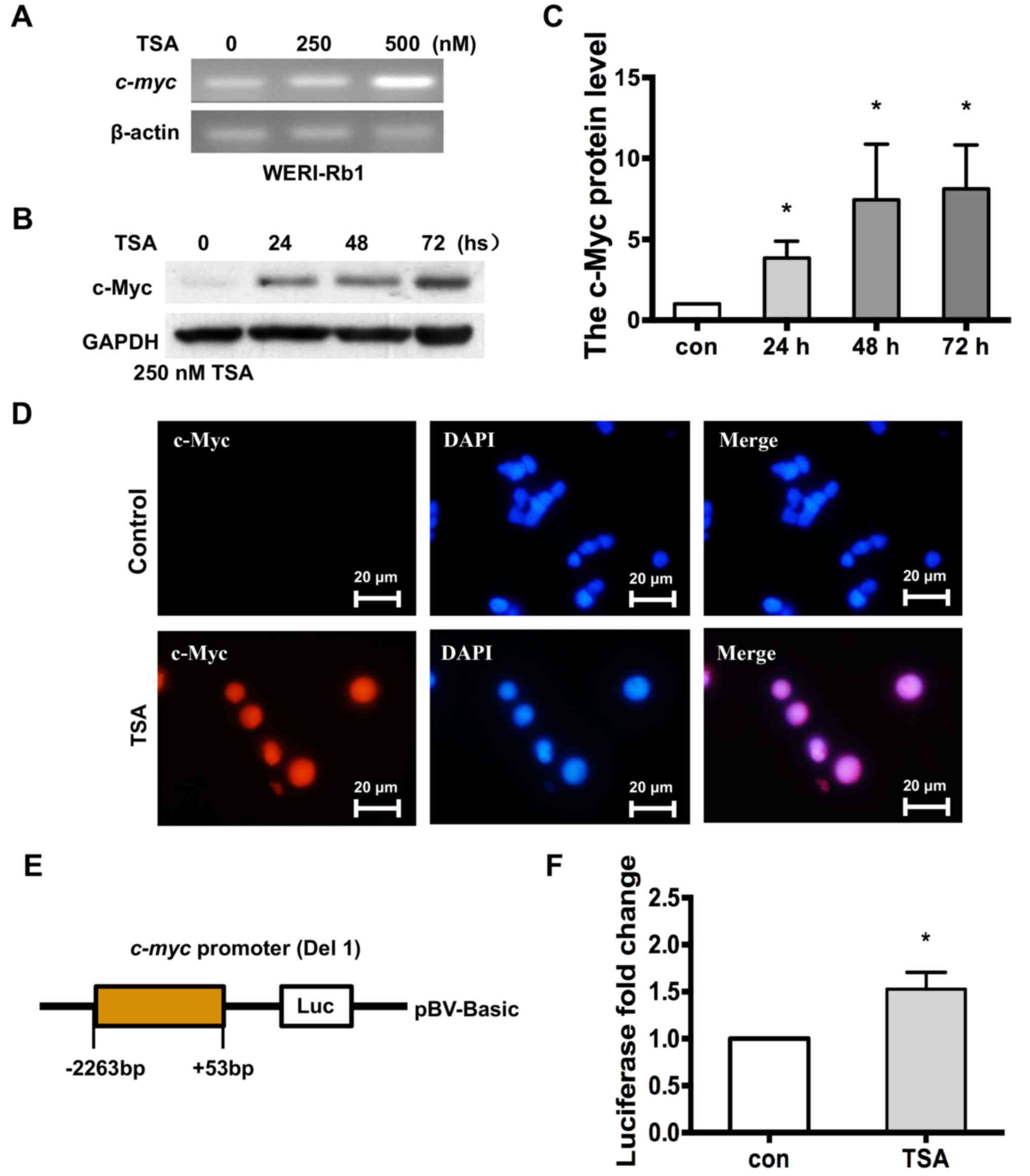 | Figure 2.c-myc is transcriptionally
upregulated in WERI-Rb1 cells following treatment with the histone
deacetylase inhibitor TSA. (A) RT-PCR analysis indicated that c-myc
was upregulated by TSA in a dose-dependent manner in WERI-Rb1
cells. (B) Western blot analysis of c-Myc in WERI-Rb1 cells
following TSA treatment. GAPDH is shown as an internal control. (C)
Relative expression of the c-Myc protein in WERI-Rb1 following TSA
treatment at different time points. Data are presented as
histograms. (D) Expression of c-Myc (red) was visualized in
WERI-Rb1 cells after TSA treatment, and compared with the control.
Magnification, ×100. (E) Luciferase plasmid structure, which
contains a c-myc promoter sequence from −2,263 to +53 bp.
(F) WERI-Rb1 cells were transfected with the reporter containing
the c-myc promoter and subsequently treated with 250 nM TSA for 24
h. Levels of luciferase activity were normalized to those of
Renilla luciferase (control, 1; TSA, 1.53±0.18; n=3 for each
group). All results were confirmed in triplicate. *P<0.05 vs.
respective control. RT-PCR, reverse transcription PCR; BP, base
pairs; TSA, trichostatin A; con, control. |
Moreover, to further investigate whether histone
deacetylation is specific to the expression of the endogenous
c-myc gene, a construct containing the human c-myc
promoter sequence between positions −2,263 and +53 upstream of the
luciferase reporter gene (Fig. 2E)
was utilised. WERI-Rb1 cells (transfected with a plasmid expressing
the aforementioned construct) were treated with 250 nM TSA and
subjected to luciferase assays. As exhibited in Fig. 2F, the basal activity of the
c-myc promoter was significantly higher in WERI-Rb1 cells
treated with TSA, compared to the control (control, 1; TSA,
1.53±0.18; P<0.05). Therefore, the current results indicated
that the expression of c-myc was significantly increased by
treatment with TSA in WERI-Rb1 cells.
c-Myc expression is differentially
regulated by three HDAC inhibitors in WERI-Rb1 cells
To further determine whether histone deacetylation
is implicated in c-myc silencing, three different HDAC
inhibitors (SAHA, VPA and MS-275) were used to investigate the
mechanism of c-myc silencing in WERI-Rb1 cells, via
treatment of the cells with various concentrations of the HDAC
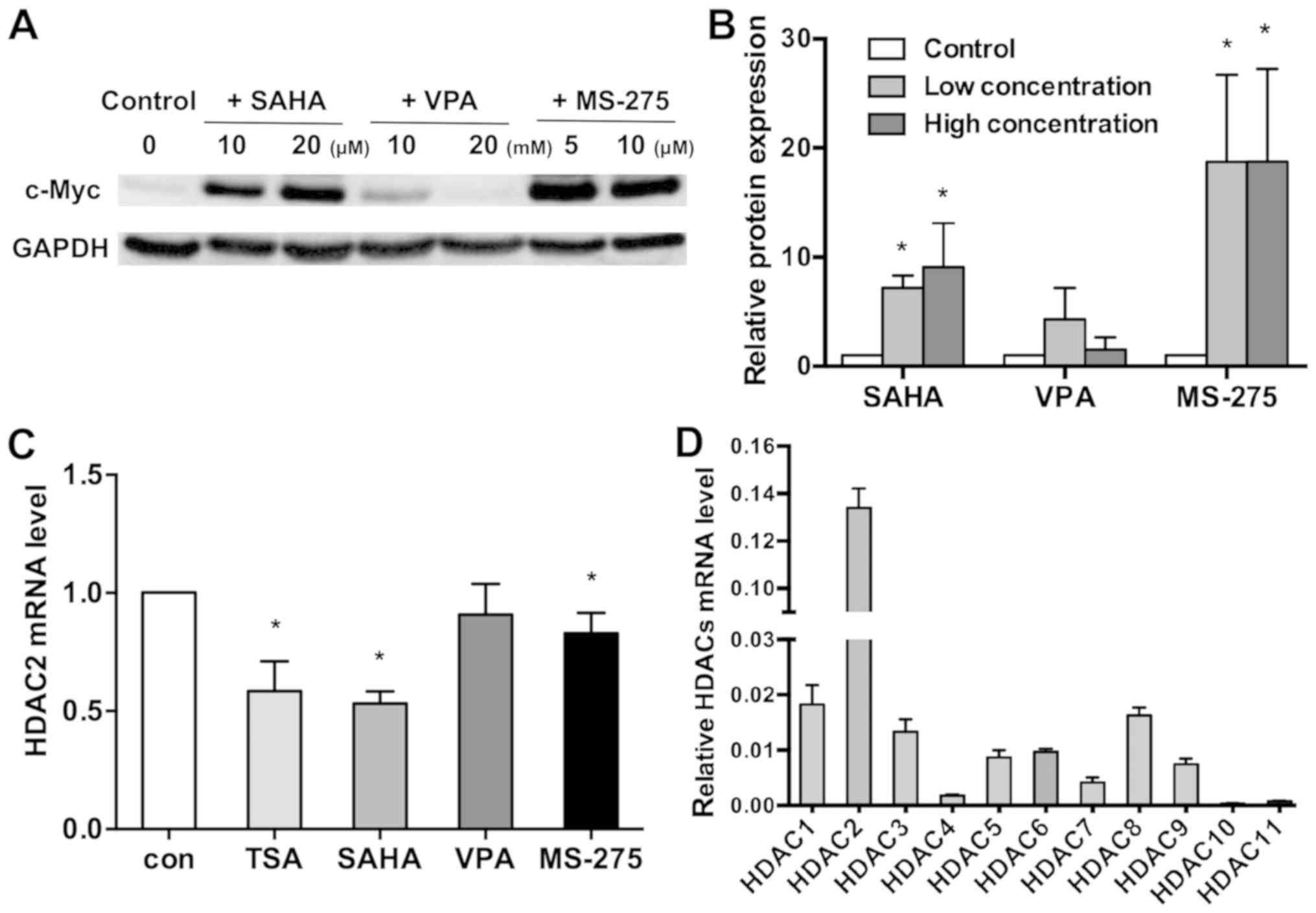
inhibitors SAHA, VPA and MS-275. As indicated in Fig. 3A and B, c-Myc expression was
significantly upregulated following treatment with 10 and 20 µM
SAHA, and 5 and 10 µM MS-275. However, 10 and 20 mM VPA did not
influence the expression of c-Myc (control, 1; 10 µM SAHA,
7.159±1.157; 20 µM SAHA, 9.027±4.066; 10 mM VPA, 4.301±2.856; 20 mM
VPA, 1.477±1.156; 5 µM MS-275, 18.750±7.971; 10 µM MS-275,
18.742±8.480; P<0.05).
To investigate the hypothesis that VPA does not
increase c-Myc expression in WERI-Rb1 cells, HDAC mRNA was assessed
in WERI-Rb1 cells treated with 500 nM TSA, 20 µM SAHA, 20 mM VPA
and 10 µM MS-275. The present results indicated that different HDAC
inhibitors differentially affected the mRNA expression of specific
HDAC family members in WERI-Rb1 cells, 6 h after treatment
(Fig. S1 and Table SII). All four HDAC inhibitors
downregulated HDAC7 and 8 in WERI-Rb1 cells. Notably, VPA did not
decrease HDAC2 mRNA expression like the other HDAC inhibitors did
(Fig. 3C; control, 1; TSA,
0.584±0.127; SAHA, 0.532±0.051; VPA, 0.909±0.130; MS-275,
0.829±0.087; P<0.05). The current data implied that HDAC2 may
influence HDAC inhibitor-mediated regulation of c-Myc.
Thus, the mRNA expression level of HDAC1-11 in
WERI-Rb1 cells was quantified using RT-qPCR. It was revealed that
HDAC2 expression was significantly upregulated in comparison with
the other members of the HDAC family (relative HDAC mRNA expression
levels: HDAC1, 0.0183±0.0035; HDAC2, 0.1339±0.0082; HDAC3,
0.0133±0.0023; HDAC4, 0.0018±0.0002; HDAC5, 0.0087±0.0013; HDAC6,
0.0097±0.0005; HDAC7, 0.0041±0.0009; HDAC8, 0.0163±0.0014; HDAC9,
0.0075±0.0010; HDAC10, 0.0003±0.0001; and HDAC11, 0.0007±0.0002;
Fig. 3D). Therefore, it is
speculated that HDAC2 may serve a key role in the regulation of
c-Myc in WERI-Rb1 cells.
Exogenous c-Myc influences the
viability of WERI-Rb1 cells
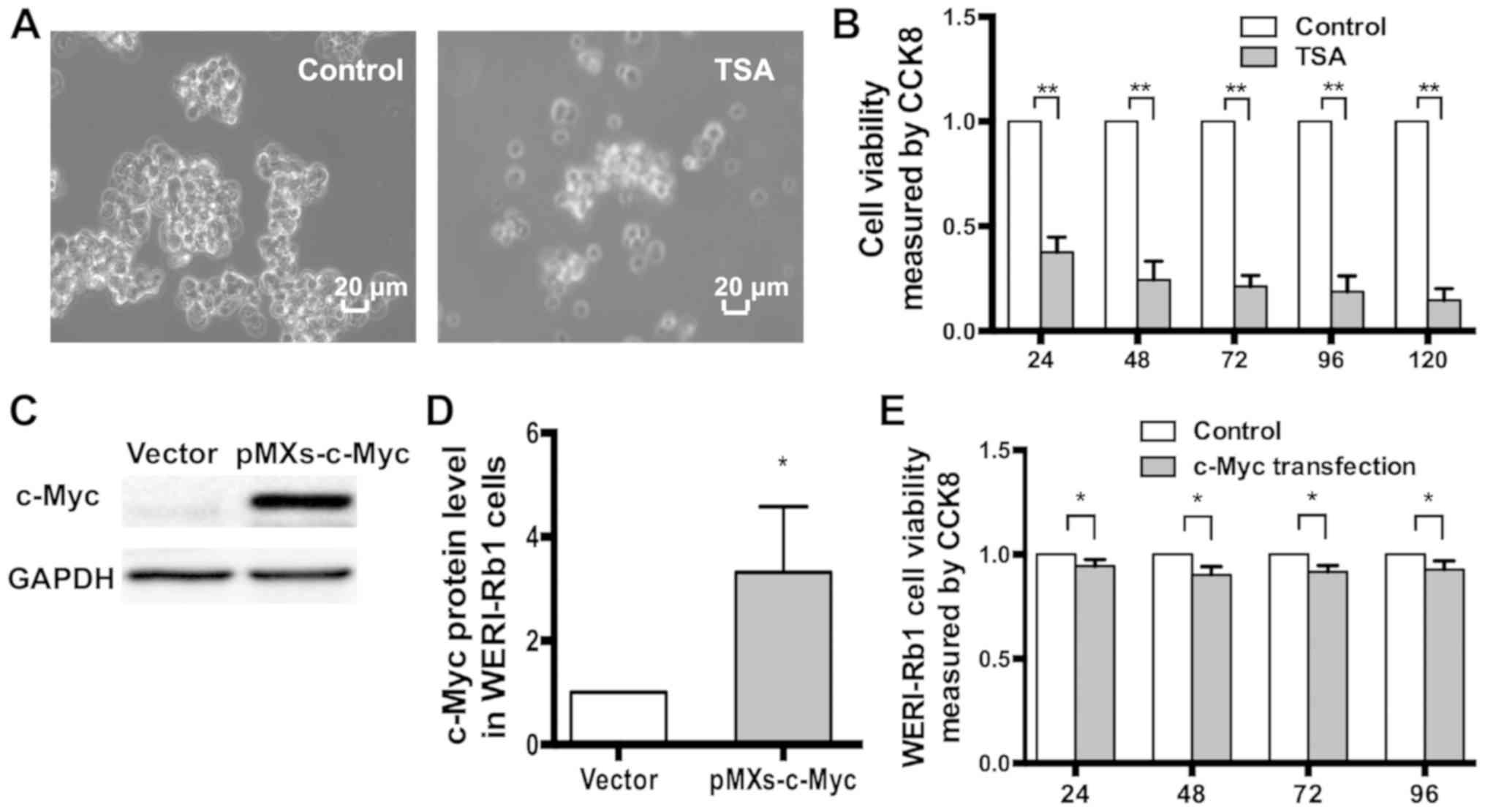
TSA treatment resulted in an increase in c-Myc
expression, but also affected the morphology of WERI-Rb1 cells.
Normally, WERI-Rb1 cells appear as small, round cells in large
clusters. However, following treatment with 250 nM TSA for 72 h,
the clusters reduced in size, and single cells were observed
(Fig. 4A). The CCK-8 assay indicated
that the viability of WERI-Rb1 cells was markedly reduced (in a
continuous manner), following treatment with 250 nM TSA (Fig. 4B; TSA, 24 h, 0.377±0.072; 48 h,
0.244±0.089; 72 h, 0.213±0.053; 96 h, 0.188±0.075; 120 h,
0.147±0.056) compared to the respective controls (control, 1;
P<0.01). Therefore, TSA significantly inhibited the viability of
WERI-Rb1 cells.
The c-Myc protein possesses versatile functions in
human cancer (16,20). To investigate whether the TSA-induced
inhibition of viability in WERI-Rb1 cells was associated with
c-Myc, the cells were transfected with either pMXs-c-Myc or an
empty control vector. The CCK-8 assay was then performed to
evaluate the effects of exogenous c-Myc on WERI-Rb1 proliferation.
As indicated in Fig. 4C and D,
western blot analysis revealed a higher level of c-Myc expression
in WERI-Rb1 cells transfected with pMXs-c-Myc compared with the
empty vector (c-Myc protein expression: Vector, 1; pMXs-c-Myc,
3.32±1.27; P<0.05). Notably, the viability of WERI-Rb1 cells was
significantly reduced following pMXs-c-Myc transfection (exogenous
c-Myc; 24 h, 0.929±0.014; 48 h, 0.886±0.028; 72 h, 0.901±0.012; and
96 h, 0.913±0.030) compared to vector transfection (control, 1)
(Fig. 4E; P<0.05). Therefore, the
present results demonstrated that exogenous c-myc may
significantly decrease the viability of WERI-Rb1 cells.
c-Myc downregulation results from
histone deacetylation in WERI-Rb1 cells, but not in Y79 cells
To examine whether the silencing mechanism was
conserved across other RB cell lines, Y79 cells were chosen for
comparison with WERI-Rb1 cells. Compared with RKO cells,
c-myc was also nearly silenced in Y79 cells, similar to
WERI-Rb1 cells (Fig. 5A). However,
the expression level of c-myc was not upregulated in Y79
cells after treatment with 250 nM and 500 nM TSA. Moreover, western
blot analysis demonstrated that the protein level of c-Myc was not
induced by 250 nM TSA at different time points (Fig. 5B).
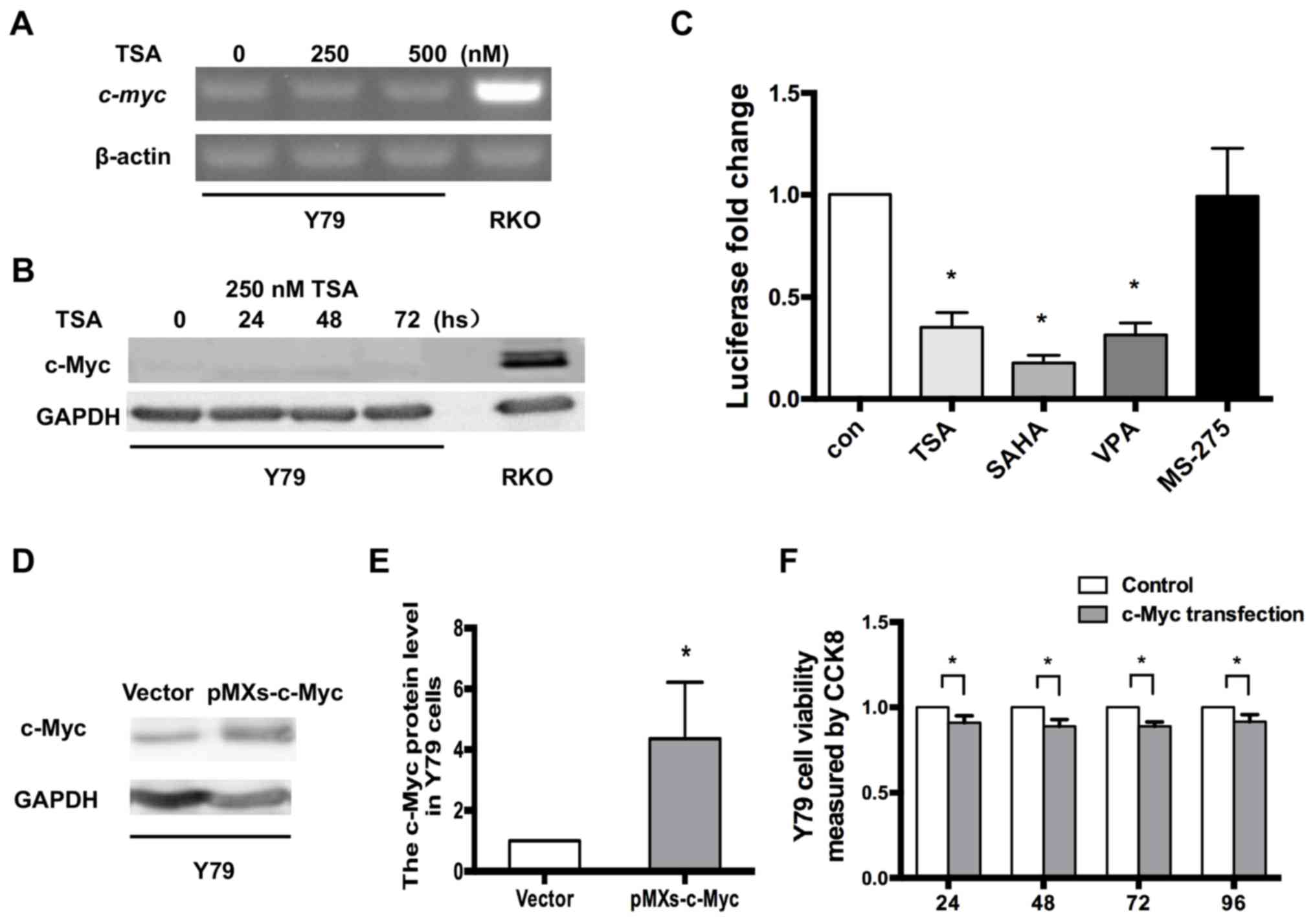 | Figure 5.c-Myc is not upregulated by HDAC
inhibitors in RB Y79 cells. (A) RT-PCR analysis indicated that
c-myc expression was not induced by treatment with 250 or 500 nM
TSA, in Y79 cells. (B) Western blot analysis of c-Myc in Y79 cells
following TSA treatment. GAPDH was used as a reference gene. (C)
Y79 cells were transfected with the c-myc promoter reporter
and then treated with 250 nM TSA, 10 µM SAHA, 10 mM VPA or 5 µM
MS-275 for 24 h. Levels of luciferase activity were normalized to
those of Renilla luciferase (control, 1; TSA, 0.35±0.07;
SAHA, 0.18±0.04; VPA, 0.31±0.59; MS-275, 0.99±0.24; n=3 for each
group) *P<0.05 vs. control. (D) Expression level of c-Myc in Y79
cells after c-Myc transfection. (E) Relative expression of c-Myc in
Y79 cells was quantified using densitometry, and the data are
presented as histograms. *P<0.05 vs. vector. (F) CCK-8 assays
indicated that the viability of Y79 cells may also be reduced by
exogenous c-Myc, similar to WERI-Rb1 cells. All the results were
confirmed in triplicate. *P<0.05. RT-PCR, reverse transcription
PCR; TSA, trichostatin A; SAHA, vorinostat; VPA, valproic acid
sodium salt; MS-275, entinostat; con, control. |
Y79 cells were also transfected with the plasmid
carrying the human c-myc promoter with the firefly
luciferase gene (Fig. 2E) and
treated with 250 nM TSA, 10 µM SAHA, 10 mM VPA or 5 µM MS-275.
After 24 h, the cells were harvested and subjected to luciferase
assays. As detailed in Fig. 5C,
unlike in WERI-Rb1 cells, TSA, SAHA, VPA and MS-275 did not
upregulate the basal activity of the c-myc promoter in Y79
cells. By contrast, c-myc promoter activity was decreased in
Y79 cells treated with TSA, SAHA and VPA. In addition, c-myc
promoter activity was not influenced by MS-275 compared to the
controls (control, 1; TSA, 0.35±0.07; SAHA, 0.18±0.04; VPA,
0.31±0.59; MS-275, 0.99±0.24; P<0.05). Therefore, the present
results indicated that the mechanism of c-myc silencing is
specific to WERI-Rb1 cells compared with Y79 cells.
Moreover, to determine whether exogenous
c-myc is able to inhibit the viability of Y79 cells, the
cells were transfected with either pMXs-c-Myc or a control vector,
and analysed using a CCK-8 assay. As revealed in Fig. 5D and E, western blot analysis
confirmed the change in the c-Myc protein expression level in Y79
cells when transfected with pMXs-c-Myc, compared with the vector
(c-Myc protein expression: Vector, 1; pMXs-c-Myc: 4.36±1.85;
P<0.05). Consistent with WERI-Rb1 cells, Fig. 5F shows that the viability of Y79
cells was also significantly reduced by exogenous c-myc at
24 h (0.923±0.033), 48 h (0.898±0.037), 72 h (0.903±0.004) and 96 h
(0.92±0.040) (all P<0.05). Thus, according to the current
results, exogenous c-myc also significantly inhibits the
proliferation of Y79 cells.
Discussion
It has been revealed that c-Myc is highly expressed
in a variety of tumour types (6–9), though
the expression level of c-Myc in RB cells is yet to be reported, to
the best of our knowledge. In the present study, it was revealed
that the expression level of c-Myc was very low in the RB cell line
WERI-Rb1. It was confirmed that c-Myc expression in WERI-Rb1 cells
was increased at the transcriptional level by the HDAC inhibitor
TSA, and was also differentially regulated by three other HDAC
inhibitors. However, none of the HDAC inhibitors induced the
upregulation of c-Myc expression in Y79 cells. Moreover, exogenous
c-Myc significantly inhibited the proliferation of WERI-Rb1 and Y79
cells. Thus, the results of the current study revealed the
expression mechanism of c-Myc and the role it serves in RB
cells.
c-myc has been reported to be regulated by
histone acylation or DNA methylation in numerous types of cancer
cell (12–15). The present data revealed that
expression levels of c-Myc were significantly upregulated in
WERI-Rb1 cells following treatment with TSA, SAHA and MS-275. This
result is consistent with a previous study, which revealed that TSA
significantly increased the expression of c-myc mRNA
resulting from nerve growth factor (NGF), and blocked both
oncogenic ras- and NGF-induced neurite outgrowth from PC12 cells
(23). By contrast, previous studies
have indicated that c-myc may also be regulated by certain
DNA demethylating reagents, such as 5-azacytidine (12–14).
Certain previous studies on different cancer cells have reported
that c-myc expression was downregulated or unaffected by
treatment with HDAC inhibitors. For example, Kretzner et al
(24) demonstrated that both TSA and
SAHA decreased c-Myc mRNA and protein expression, as well as
c-Myc-regulated microRNA expression. Nebbioso et al
(25) reported that treatment with
SAHA and MS-275 resulted in both c-Myc acetylation at lysine
residue 323, and c-Myc downregulation in acute myeloid leukaemia
cell lines. Furthermore, silencing of c-Myc was not influenced by
TSA in prostate cancer PC3 cells (26). Thus, the present data indicated that
histone deacetylation was implicated in the silencing mechanism of
c-Myc in WERI-Rb1 cells.
Moreover, in the present study, the expression of
c-Myc in WERI-Rb1 cells was discovered not to be increased by VPA,
by contrast to TSA, SAHA and MS-275. In addition, HDAC2 was reduced
by all the other HDAC inhibitors except VPA. HDAC2 might be
involved in HDAC inhibitor-medicated regulation of c-Myc. HDAC2 has
a pivotal role in the modulation chromatin architecture leading to
transcriptional changes (27).
Previous studies reported that HDAC2 is upregulated in numerous
cancer types, including breast and prostate cancer (28–30).
Additionally, upregulation of HDAC2 has been revealed to
significantly enhance cancer cell proliferation, migration and
invasion (28,30–33).
These findings support the results of the present study which
indicated that the HDAC2 mRNA level was the higher than that of any
other HDACs in WERI-Rb1 cells. Additionally, several studies have
demonstrated that VPA treatment may decrease or have no effect on
HDAC2 expression (34–37). This may be partially attributable to
the fact that VPA is tumour-selective, similar to the other HDAC
inhibitors (38). However, HDAC2 has
been reported to enhance c-Myc expression in numerous cell types
(39,40), by contrast to the present study which
discovered that c-Myc expression increased when HDAC2 was
downregulated. Further research should investigate the association
between HDAC2 and c-Myc expression in RB cells. Taken together, the
current data suggest that HDAC2 may serve an important role in the
regulation of c-Myc expression in WERI-Rb1 cells treated with TSA,
SAHA and MS-275.
Notably, none of the HDAC inhibitors significantly
altered c-Myc expression in Y79 cells. This result is partially
consistent with the different effects on gene expression caused by
SAHA in human breast carcinoma cell line MDA 468, compared with MDA
435 (41). Moreover, the RB mutation
in WERI-Rb1 cells is a complete deletion of the gene, whereas a
partial deletion is present in Y79 cells (42). The two RB cell lines possess
differing growth characteristics (43), which may contribute to the variation
in their gene expression patterns (44) and result in contrasting
c-myc-silencing mechanisms. Overall, the current results
demonstrated that histone acetylation of the c-myc gene was
specific to WERI-Rb1 cells.
Furthermore, the present data revealed that
exogenous c-Myc mildly reduced the viability of WERI-Rb1 and Y79
cells. However, as suggested in previous reports, c-Myc may be
highly expressed in a variety of tumour types, and may also promote
proliferation and metastasis (6–9,17). By contrast, c-Myc was revealed to
induce apoptosis in myeloid progenitor cells and fibroblasts
(45–47). Notably, c-Myc induces massive
programmed cell death (PCD) in the majority of transgenic mouse
models, with greater efficiency than other oncogenes (20). Downregulation of c-Myc protein levels
contributes to cancer cell survival under certain conditions
(21). The aforementioned studies
are consistent with the current results, which revealed that
exogenous c-Myc reduced the viability of RB cells, to a certain
extent. However, the results of c-Myc on PCD were thought to
promote cancer cells to adapt to living conditions and
environmental changes (20,21). Thus, further studies exploring the
inhibition of exogenous c-Myc on retinoblastoma may help elucidate
its role.
In conclusion, the present study indicated that
c-myc was downregulated in WERI-Rb1 and Y79 cells. Moreover,
HDAC inhibitors TSA, SAHA and MS-275 were able to transcriptionally
induce the expression of c-Myc in WERI-Rb1 cells, but not in Y79
cells. Exogenous c-myc also decreased WERI-Rb1 and Y79 cell
viability. Therefore, the current data support the hypothesis that
c-Myc regulates tumorigenesis at an epigenetic level in the
WERE-Rb1 human RB cell line.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81670848 and 81470626).
Availability of data and materials
All data generated or analyzed during the present
study are included in the published article.
Authors' contributions
KMY, JZ, JG and NY made substantial contributions to
the design of the study. NY, ML, JQ, PZ, MY, PY, YW and XH
performed the experiments. PC and QW analyzed the data. All authors
contributed to writing the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dimaras H, Kimani K, Dimba EA, Gronsdahl
P, White A, Chan HS and Gallie BL: Retinoblastoma. Lancet.
379:1436–1446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fabian ID, Onadim Z, Karaa E, Duncan C,
Chowdhury T, Scheimberg I, Ohnuma SI, Reddy MA and Sagoo MS: The
management of retinoblastoma. Oncogene. 37:1551–1560. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kivela T: The epidemiological challenge of
the most frequent eye cancer: Retinoblastoma, an issue of birth and
death. Br J Ophthalmol. 93:1129–1131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zimmerman K and Alt FW: Expression and
function of myc family genes. Crit Rev Oncog. 2:75–95.
1990.PubMed/NCBI
|
|
6
|
Nesbit CE, Tersak JM and Prochownik EV:
MYC oncogenes and human neoplastic disease. Oncogene. 18:3004–3016.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brodeur GM: Neuroblastoma: Biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bociek RG: Adult Burkitt's lymphoma. Clin
Lymphoma. 6:11–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Horiuchi D, Anderton B and Goga A: Taking
on challenging targets: Making MYC druggable. Am Soc Clin Oncol
Educ Book. e497–e502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Conacci-Sorrell M, McFerrin L and Eisenman
RN: An overview of MYC and its interactome. Cold Spring Harb
Perspect Med. 4:a0143572014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chung HJ and Levens D: c-myc expression:
Keep the noise down! Mol Cells. 20:157–166. 2005.PubMed/NCBI
|
|
12
|
Kitagawa Y, Kyo S, Takakura M, Kanaya T,
Koshida K, Namiki M and Inoue M: Demethylating reagent
5-azacytidine inhibits telomerase activity in human prostate cancer
cells through transcriptional repression of hTERT. Clin Cancer Res.
6:2868–2875. 2000.PubMed/NCBI
|
|
13
|
Grandjenette C, Schnekenburger M, Karius
T, Ghelfi J, Gaigneaux A, Henry E, Dicato M and Diederich M:
5-aza-2′- deoxycytidine-mediated c-myc Down-regulation triggers
telomere-dependent senescence by regulating human telomerase
reverse transcriptase in chronic myeloid leukemia. Neoplasia.
16:511–528. 2004. View Article : Google Scholar
|
|
14
|
Long Y, Tsai WB, Chang JT, Estecio M,
Wangpaichitr M, Savaraj N, Feun LG, Chen HH and Kuo MT:
Cisplatin-induced synthetic lethality to arginine-starvation
therapy by transcriptional suppression of ASS1 is regulated by
DEC1, HIF-1α, and c-Myc transcription network and is independent of
ASS1 promoter DNA methylation. Oncotarget. 7:82658–82670. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li H and Wu X: Histone deacetylase
inhibitor, Trichostatin A, activates p21WAF1/CIP1 expression
through downregulation of c-myc and release of the repression of
c-myc from the promoter in human cervical cancer cells. Biochem
Biophys Res Commun. 324:860–867. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dang CV, Resar LM, Emison E, Kim S, Li Q,
Prescott JE, Wonsey D and Zeller K: Function of the c-Myc oncogenic
transcription factor. Exp Cell Res. 253:63–77. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo P, Nie Q, Lan J, Ge J, Qiu Y and Mao
Q: C-Myc negatively controls the tumor suppressor PTEN by
upregulating miR-26a in glioblastoma multiforme cells. Biochem
Biophys Res Commun. 441:186–190. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Karn J, Watson JV, Lowe AD, Green SM and
Vedeckis W: Regulation of cell cycle duration by c-myc levels.
Oncogene. 4:773–787. 1989.PubMed/NCBI
|
|
19
|
Jain M, Arvanitis C, Chu K, Dewey W,
Leonhardt E, Trinh M, Sundberg CD, Bishop JM and Felsher DW:
Sustained loss of a neoplastic phenotype by brief inactivation of
MYC. Science. 297:102–104. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang C, Tai Y, Lisanti MP and Liao DJ:
c-Myc induction of programmed cell death may contribute to
carcinogenesis: A perspective inspired by several concepts of
chemical carcinogenesis. Cancer Biol Ther. 11:615–626. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okuyama H, Endo H, Akashika T, Kato K and
Inoue M: Downregulation of c-MYC protein levels contributes to
cancer cell survival under dual deficiency of oxygen and glucose.
Cancer Res. 70:10213–10223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liva KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Futamura M, Monden Y, Okabe T,
Fujita-Yoshigaki J, Yokoyama S and Nishimura S: Trichostatin A
inhibits both ras-induced neurite outgrowth of PC12 cells and
morphological transformation of NIH3T3 cells. Oncogene.
10:1119–1123. 1995.PubMed/NCBI
|
|
24
|
Kretzner L, Scuto A, Dino PM, Kowolik CM,
Wu J, Ventura P, Jove R, Forman SJ, Yen Y and Kirschbaum MH:
Combining histone deacetylase inhibitor vorinostat with aurora
kinase inhibitors enhances lymphoma cell killing with repression of
c-Myc, hTERT, and microRNA levels. Cancer Res. 71:3912–3920. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nebbioso A, Carafa V, Conte M, Tambaro FP,
Abbondanza C, Martens J, Nees M, Benedetti R, Pallavicini I,
Minucci S, et al: c-Myc modulation & acetylation is a key HDAC
inhibitor target in cancer. Clin Cancer Res. 23:2542–2555. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Napoli S, Pastori C, Magistri M, Carbone
GM and Catapano CV: Promoter-specific transcriptional interference
and c-myc gene silencing by siRNAs in human cells. EMBO J.
28:1708–1719. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Conte M, Dell'Aversana C, Sgueglia G,
Carissimo A and Altucci L: HDAC2-dependent miRNA signature in acute
myeloid leukemia. FEBS Lett. 593:2574–2584. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shan W, Jiang Y, Yu H, Huang Q, Liu L, Guo
X, Li L, Mi Q, Zhang K and Yang Z: HDAC2 overexpression correlates
with aggressive clinicopathological features and DNA-damage
response pathway of breast cancer. Am J Cancer Res. 7:1213–1226.
2017.PubMed/NCBI
|
|
29
|
Weichert W, Roske A, Gekeler V, Beckers T,
Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M
and Kristiansen G: Histone deacetylases 1, 2 and 3 are highly
expressed in prostate cancer and HDAC2 expression is associated
with shorter PSA relapse time after radical prostatectomy. Br J
Cancer. 98:604–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim JK, Noh JH, Eun JW, Jung KH, Bae HJ,
Shen Q, Kim MG, Chang YG, Kim SJ, Park WS, et al: Targeted
inactivation of HDAC2 restores p16INK4a activity and exerts
antitumor effects on human gastric cancer. Mol Cancer Res.
11:62–73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li S, Wang F, Qu Y, Chen X, Gao M, Yang J,
Zhang D, Zhang N, Li W and Liu H: HDAC2 regulates cell
proliferation, cell cycle progression and cell apoptosis in
esophageal squamous cell carcinoma EC9706 cells. Oncol Lett.
13:403–409. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Mei DT and Zeng Y: HDAC2 promotes
the migration and invasion of non-small cell lung cancer cells via
upregulation of fibronectin. Biomed Pharmacother. 84:284–290. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao H, Yu Z, Zhao L, He M, Ren J, Wu H,
Chen Q, Yao W and Wei M: HDAC2 overexpression is a poor prognostic
factor of breast cancer patients with increased multidrug
resistance-associated protein expression who received
anthracyclines therapy. Jpn J Clin Oncol. 46:893–902. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krämer OH, Zhu P, Ostendorff HP,
Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I,
Heinzel T and Göttlicher M: The histone deacetylase inhibitor
valproic acid selectively induces proteasomal degradation of HDAC2.
EMBO J. 22:3411–3420. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kwiecińska P, Wróbel A, Taubøll E and
Gregoraszczuk EŁ: Valproic acid, but not levetiracetam, selectively
decreases HDAC7 and HDAC2 expression in human ovarian cancer cells.
Toxicol Lett. 224:225–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jones J, Juengel E, Mickuckyte A, Hudak L,
Wedel S, Jonas D and Blaheta RA: The histone deacetylase inhibitor
valproic acid alters growth properties of renal cell carcinoma in
vitro and in vivo. J Cell Mol Med. 13:2376–2385. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Travaglini L, Vian L, Billi M, Grignani F
and Nervi C: Epigenetic reprogramming of breast cancer cells by
valproic acid occurs regardless of estrogen receptor status. Int J
Biochem Cell Biol. 41:225–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nebbioso A, Clarke N, Voltz E, Germain E,
Ambrosino C, Bontempo P, Alvarez R, Schiavone EM, Ferrara F,
Bresciani F, et al: Tumor-selective action of HDAC inhibitors
involves TRAIL induction in acute myeloid leukemia cells. Nat Med.
11:77–84. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Noh JH, Jung KH, Kim JK, Eun JW, Bae HJ,
Xie HJ, Chang YG, Kim MG, Park WS, Lee JY and Nam SW: Aberrant
regulation of HDAC2 mediates proliferation of hepatocellular
carcinoma cells by deregulating expression of G1/S cell cycle
proteins. PLoS One. 6:e281032011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ecker J, Oehme I, Mazitschek R, Korshunov
A, Kool M, Hielscher T, Kiss J, Selt F, Konrad C, Lodrini M, et al:
Targeting class I histone deacetylase 2 in MYC amplified group 3
medulloblastoma. Acta Neuropathol Commun. 3:222015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Glaser KB, Staver MJ, Waring JF, Stender
J, Ulrich RG and Davidsen SK: Gene expression profiling of multiple
histone deacetylase (HDAC) inhibitors: Defining a common gene set
produced by HDAC inhibition in T24 and MDA carcinoma cell lines.
Mol Cancer Ther. 2:151–163. 2003.PubMed/NCBI
|
|
42
|
Bookstein R, Lee EY, To H, Young LJ, Sery
TW, Hayes RC, Friedmann T and Lee WH: Human retinoblastoma
susceptibility gene: Genomic organization and analysis of
heterozygous intragenic deletion mutants. Proc Natl Acad Sci USA.
85:2210–2214. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Busch M, Philippeit C, Weise A and Dünker
N: Re-characterization of established human retinoblastoma cell
lines. Histochem Cell Biol. 143:325–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ross DT, Scherf U, Eisen MB, Perou CM,
Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M,
et al: Systematic variation in gene expression patterns in human
cancer cell lines. Nat Genet. 24:227–235. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Askew DS, Ashmun RA, Simmons BC and
Cleveland JL: Constitutive c-Myc expression in an IL-3 dependent
myeloid cell line suppresses cell cycle arrest and accelerates
apoptosis. Oncogene. 6:1915–1922. 1991.PubMed/NCBI
|
|
46
|
Evan GI, Wyllie AH, Gilbert CS, Littlewood
TD, Land H, Brooks M, Waters CM, Penn LZ and Hancock DC: Induction
of apoptosis in fibroblasts by c-Myc protein. Cell. 69:119–128.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Harrington EA, Bennett MR, Fanidi A and
Evan GI: c-Myc-induced apoptosis in fibroblasts is inhibited by
specific cytokines. EMBO J. 13:3286–3295. 1994. View Article : Google Scholar : PubMed/NCBI
|