Introduction
Leukaemia is the most common cancer among paediatric
patients with a worldwide incidence rate of ~40–50 per million
(1,2). However, even with comprehensive
treatment strategies, including chemotherapy, radiotherapy and stem
cell transplantation, the prognoses of patients with primary
resistant T-cell acute lymphoblastic leukaemia (T-ALL) who fail to
achieve complete haematological remission or who relapse following
a transient initial response remain poor (1,2).
Chemotherapeutic resistance is considered to be the primary cause
of this problem, and understanding how cancer cells acquire
resistance is the primary challenge for chemotherapy.
A number of molecular mechanisms have been suggested
to be the underlying reason for drug resistance, including enhanced
DNA damage repair ability, apoptosis inactivation, target mutation
or deletion, angiogenesis, transporter-mediated drug efflux and
autophagy (3,4). Autophagy, which plays an intricate role
in cell death and survival, has recently been considered a
potential mechanism underlying chemotherapeutic resistance in
cancer cells (5). Autophagy has dual
functions in tumour resistance: Chemotherapeutic drugs may either
induce autophagic cell death, or induce protective autophagy to
promote cell survival by recovering metabolites, saving energy and
avoiding oxidative damage (5,6). The
role of autophagy in chemotherapy is complex and depends on the
tumour and drug type as well as the basic autophagy process
(6–8).
High mobility group box 1 (HMGB1), a member of the
HMGB superfamily, has a tripartite structure composed of an A box,
a B box and a C-terminal acidic tail (9). As a chromatin-associated protein, HMGB1
is widely present in eukaryotic nuclei, and it plays an important
role in the cytoplasm and extracellular space (10,11).
HMGB1 relocation is crucial for cell survival and death (12–16). A
number of studies have demonstrated that upregulated HMGB1
expression or increased release of HMGB1 promotes drug resistance
in numerous different types of cancer, such as leukaemia, lung
cancer and osteosarcoma (10,17–19).
These findings suggest that HMGB1 is a potential target for
chemotherapy.
Unlike other secretory proteins, HMGB1 induces
atypical lysosome-mediated vesicle transport via
lysophosphatidylcholine due to the lack of signal peptides
(20,21). HMGB1 migration from the nucleus to
the cytoplasm is the most important step in this process. Among the
mechanisms regulating HMGB1 translocation, such as
post-translational modifications and the pathways for calcium
signalling, reactive oxygen species signalling, janus kinase
(JAK)-signal transducer and activator of transcription (STAT)
signalling, p53 and inflammasomes, the association between HMGB1
post-transcriptional modification and translocation is currently
the most well-known (21–25). Poly (ADP-ribose) polymerase (PARP1)
is the most important ADP-ribosomal polymerase, and it catalyses
the transfer of ADP-ribose moieties from NAD+ to itself
and other acceptor proteins (26).
PARP1 plays a critical role in the cytoplasmic translation of HMGB1
via poly (ADP-ribosylation) (16,26).
Preliminary studies have demonstrated that poly (ADP-ribosylation)
of HMGB1 promotes its acetylation and, thus, facilitates HMGB1
translocation (27,28). Furthermore, acetylation of the lysine
residues in HMGB1 is believed to be a precondition for HMGB1
translocation into the cytoplasm (29); however, the specific molecular
mechanism underlying this requires further study.
In the present study, it was demonstrated that
chemotherapeutic drugs could induce leukaemic cells to undergo
cytoprotective autophagy, thus inducing drug resistance. HMGB1
translocation represents a decisive step in chemotherapy-induced
autophagy. In addition, the association between poly
(ADP-ribosylation) and HMGB1 acetylation was investigated in the
present study, and it was revealed that poly (ADP-ribosylation) of
HMGB1 affects its acetylation and promotes HMGB1
translocation-associated chemotherapy-induced autophagy in
leukaemia cells. These results suggest that inhibiting HMGB1
translocation may increase chemotherapeutic efficacy and aid in
overcoming drug resistance.
Materials and methods
Antibodies and reagents
The antibody specific for acetylated lysine
(catalogue no. 441S) and the rabbit monoclonal antibody IgG XPTM
isotype control (catalogue no. 3900S) were obtained from Cell
Signaling Technology, Inc. The antibody specific for PAR (catalogue
no. 4336-BPC-100) was obtained from Trevigen. Antibodies specific
for HMGB1 (catalogue no. H9539), p62 (catalogue no. P0067), LC3
(catalogue no. L8918) and anti-Flag antibodies (catalogue no.
F1804) were obtained from Sigma-Aldrich; Merck KGaA.
Acetylated-lysine antibody (catalogue no. 9441S) was obtained from
Cell Signaling Technology, Inc. PAR antibody (catalogue no.
4336-BPC-100) was obtained from Trevigen. Antibodies specific for
β-actin (catalogue no. 7D2C10), laminB (catalogue no. 12987-1-AP),
and tubulin (catalogue no. 11224-1-AP) were obtained from
ProteinTech Group, Inc. All aforementioned antibodies were diluted
to 1:1,000 to detect the target protein. The secondary antibodies,
including sheep anti-mouse IgG-HRP (catalogue no. RM3001), sheep
anti-rabbit IgG-HRP (catalogue no. RM3002) were obtained from
Beijing Ray Antibody Biotech. Cy3-conjugated secondary antibody
(dilution, 1:50; catalogue no. ZF-0134) were obtained from Zsbio
Commerce Store (http://www.zsbio.com). IPKine
horseradish peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG
Light Chain (1:10,000; catalogue no. A25012; Abbkine Scientific
Co., Ltd.) was used as a secondary antibody to detect target
proteins without interference from denatured IgG in the western
blot. Daunorubicin (DNR) was purchased from MedChemExpress.
Cell culture
The Jurkat and RS4:11 human acute leukaemic cell
lines were purchased from the Type Culture Collection of the
Chinese Academy of Sciences. Jurkat and RS4:11 cells were cultured
in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.) with 10%
foetal bovine serum (HyClone; GE Healthcare Life Sciences), 2 mM
L-glutamine, and 1% penicillin/streptomycin (HyClone; GE Healthcare
Life Sciences) at 37°C in an atmosphere of 5% CO2.
Drug treatment
Jurkat cells were treated with DNR (0, 0.05, 0.1,
0.2, 0.4, 0.8, 1.6, 3.2 or 6.4 µM/ml) or DNR (0.4 µM/ml) for 24 h.
In the pre-experiment, Jurkat and RS4:11 cells were treated with
DNR for 24 h, however it was revealed that RS4:11 cells were in a
very poor state and almost all cells died, which affected the
stability of the experimental results (data not shown). Therefore,
in the subsequent experiments, RS4:11 cells were treated with DNR
(0, 0.05, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2 or 6.4 µM/ml) or DNR (0.4
µM/ml) for 12 h. Jurkat cells were transfected with lentivirus, and
HMGB1NC, HMGB1MT1, HMGB1MT2 and
HMGB1WT cells were treated with or without DNR (0.4 µM)
for 24 h.
Cell viability analysis
Cells were plated in 96-well plates at a density of
5×104/ml. Cell viability was measured using the Cell
Counting Kit-8 (Dojindo Molecular Technologies, Inc.) following
chemotherapeutic drug treatment according to the manufacturer's
protocol.
Western blot analysis
The two cell lines were subjected to the
aforementioned different DNR concentrations, collected and lysed
with RIPA buffer solution (Beyotime Institute of Biotechnology).
The protein concentration was determined by BCA method (Beyotime
Institute of Biotechnology). The samples (30 µg) were separated via
SDS-PAGE (10 or 12% gel) and transferred onto a polyvinylidene
fluoride (PVDF) membrane (EMD Millipore). After blocking with 5%
non-fat dried milk for 1 h at room temperature, the membranes were
incubated with primary antibodies, including HMGB1, p62, LC3II/I,
Acetylated-lysine antibody and PAR antibody, diluted to 1:1,000
overnight at 4°C. Secondary antibodies, including sheep anti-mouse
IgG-HRP and sheep anti-rabbit IgG-HRP were applied at a 1:5,000
dilution and IPKine horseradish peroxidase-conjugated AffiniPure
Goat Anti-Rabbit IgG Light Chain were applied at a 10,000 dilution
for 1 h at room temperature. β-actin and tubulin were used as
loading controls to detect the expression of whole protein. β-actin
was also used as an internal reference to detect the expression of
cytoplasmic protein, while laminB was used to detect the expression
of nuclear protein. The target protein expressions were detected
with an enhanced chemiluminescence reagent (EMD Millipore) using a
G:BOX XT4 system (Syngene).
Lentivirus infection
Experiments were performed in 6-well plates at a
density of 5×106 cells/well. Lentiviruses were purchased
from Obio Technology. In the present study, the lysine residues
were mutated at amino acids 28, 29, 30, 180, 182, 183, 184 and 185
in HMGB1 to alanine in order to generate mutant type-1 cells
(HMGB1MT1), and the glutamate residues were mutated at
amino acids 40, 47 and 179 to alanine to generate mutant type-2
cells (HMGB1MT2), however not all the lysine residues
and glutamate residues in HMGB1. Jurkat cells were transformed with
lentiviruses: Normal control (NC),
pLenti-EF1a-EGFP-F2A-Puro-CMV-MCS; wild type (WT),
pLenti-EF1a-EGFp-P2A-Puro-CMV-HMGB1-3Flag; mutant type 1 (MT1),
pLenti-EF1a-EGFp-P2A-Puro-CMV-HMGB1 mut1-3Flag; mutant type2 (MT2),
pLenti-EF1a-EGFp-P2A-Puro-CMV-HMGB1 mut2-3Flag. According to the
manufacturer's protocol provided by OBiO Technology Corp., Ltd.,
the transfection was performed using a lentivirus with 5 µg/ml
polybrene (OBiO Technology Corp., Ltd.) and antibiotic selection
was performed using 1 µg/ml puromycin (Beijing Solarbio Science
& Technology Co., Ltd.). Proteins were subjected to
immunoprecipitation with Protein G Magnetic Beads (Bimake;
http://biotool.cn) anti-HMGB1 antibodies (1:50; cat.
no. H9539; Sigma-Aldrich; Merck KGaA) at 4°C overnight and the
lentivirus expression was detected with anti-Flag antibodies. Cells
were cultured at 37°C in a humidified incubator containing 5%
CO2 for 3–5 generations and then used for subsequent
experiments.
Immunoprecipitation analysis
Cells subjected to the different treatments were
collected and lysed with RIPA buffer solution. The whole-cell
lysates (1,000 µg) were precleared with Protein G Magnetic Beads
for 1 h at room temperature, then incubated with normal control IgG
or anti-HMGB1 antibodies (cat. no. H9539; 1:50; Sigma-Aldrich;
Merck KGaA) at 4°C overnight to form immune complexes. The samples
were then added to a Protein G Magnetic Bead reaction to capture
the immune complexes. After washing with wash buffer, the samples
were removed under denaturing conditions in 50 µl of 2X SDS sample
buffer and boiled at 100°C for 10 min. The substrate was collected
and analysed via western blotting as described above. In this
study, the cell lysates were subjected to a pull-down assay with
Rabbit Control IgG (catalogue no. AC005; control group) or HMGB1
antibody (cat. no. H9539; 1:50; Sigma-Aldrich; Merck KGaA) at 4°C
overnight and then the cell lysates were immunoblotted with
anti-acetylated lysine (1:1,000; cat. no. 9441S; Cell Signaling
Technology, Inc.), anti-poly (ADP-ribosylation) (1:1,000; cat. no.
4336-BPC-100; Trevigen) and anti-HMGB1 antibodies (cat. no. H9539;
1:1,000; Sigma-Aldrich; Merck KGaA) 4°C overnight.
Cytoplasmic and nuclear extract
preparation
Cytoplasmic and nuclear extracts were prepared using
a Nuclear and Cytoplasmic Protein Extraction kit (Beyotime
Institute of Biotechnology). Cells subjected to the different
treatments were collected. After washing 3 times with ice-cold PBS,
the cells were resuspended in 200 µl of ice-cold cytoplasmic
extraction buffer A for 10 min. The cell lysates were incubated
with cytoplasmic extraction buffer B for 1 min in an ice bath,
vortexed for 5 sec, and centrifuged at 12,000 × g for 5 min at 4°C.
Supernatants were aliquoted and stored at −80°C. Nuclear pellets
were then resuspended in 50 µl of nuclear extraction buffer. The
lysates were vortexed four times for 15 sec at 7-min intervals, and
then centrifuged at 12,000 × g for 5 min at 4°C. Nuclear extracts
were aliquoted and stored at −80°C.
Immunofluorescence analysis
Cells were collected, fixed with 4% formaldehyde at
4°C for 15 min, permeabilised in 0.3% Triton X-100 in PBS for 10
min, and incubated in 5% BSA blocking buffer for 1 h at room
temperature. The cells were then incubated with primary antibody in
1% bovine serum albumin [Boster Biological Technology Co. Ltd.
(http://www.boster.com.cn/about/index.html)] overnight
at 4°C. The cells were washed three times with 1% Tween in PBS,
then incubated with a Cy3-conjugated secondary antibody [dilution,
1:50; catalogue no. ZF-0134; Zsbio Commerce Store (http://www.zsbio.com)] for 1 h at room temperature in
the dark, then incubated with DAPI for 5 min. The cells were
resuspended and added to a laser confocal petri dish. Images were
captured with a confocal microscope (Carl Zeiss, Inc.) at a
magnification of ×40.
The translocation of HMGB1
By examining the expression of HMGB1 in the nucleus
and cytoplasm through western blot analysis, and using
immunofluorescence technology, the translocation of HMGB1 could be
detected. When the nucleus HMGB1 translocated to the cytoplasm, the
HMGB1 protein increased in the cytoplasm, whereas it decreased in
the nucleus. Similarly, through immunofluorescence, it was observed
that when HMGB1 was transferred from nucleus to cytoplasm, the
fluorescence intensity in the cytoplasm increased, while the
fluorescence intensity decreased in the nucleus.
Transmission electron microscopy
The cells were fixed with 2% paraformaldehyde and
2.5% glutaraldehyde in 0.1 mol/l phosphate buffer (pH 7.4) at 4°C
for 2 h, followed by 1% OsO4. After dehydration and
Epon-812:100% acetone embedding at room temperature, thin sections
(50–80 nm) were stained with uranyl acetate and lead citrate
respectively at 4°C for 15 min, and viewed under a Tecnai G2 Spirit
Twin election microscope (FEI; Thermo Fisher Scientific, Inc).
Statistical analysis
All data are expressed as the mean ± standard
deviation. Paired Student's t-test was used for comparisons between
two groups and one-way analysis of variance (ANOVA) was performed
for comparisons between more than two groups. When the ANOVA was
significant, a Tukey post-hoc test was performed. P<0.05 was
considered to indicate a statistically significant result. All
statistical analyses were conducted by SPSS version 18.0 software
(SPSS, Inc.).
Results
Chemotherapeutic drugs induce
autophagy in leukaemia cells
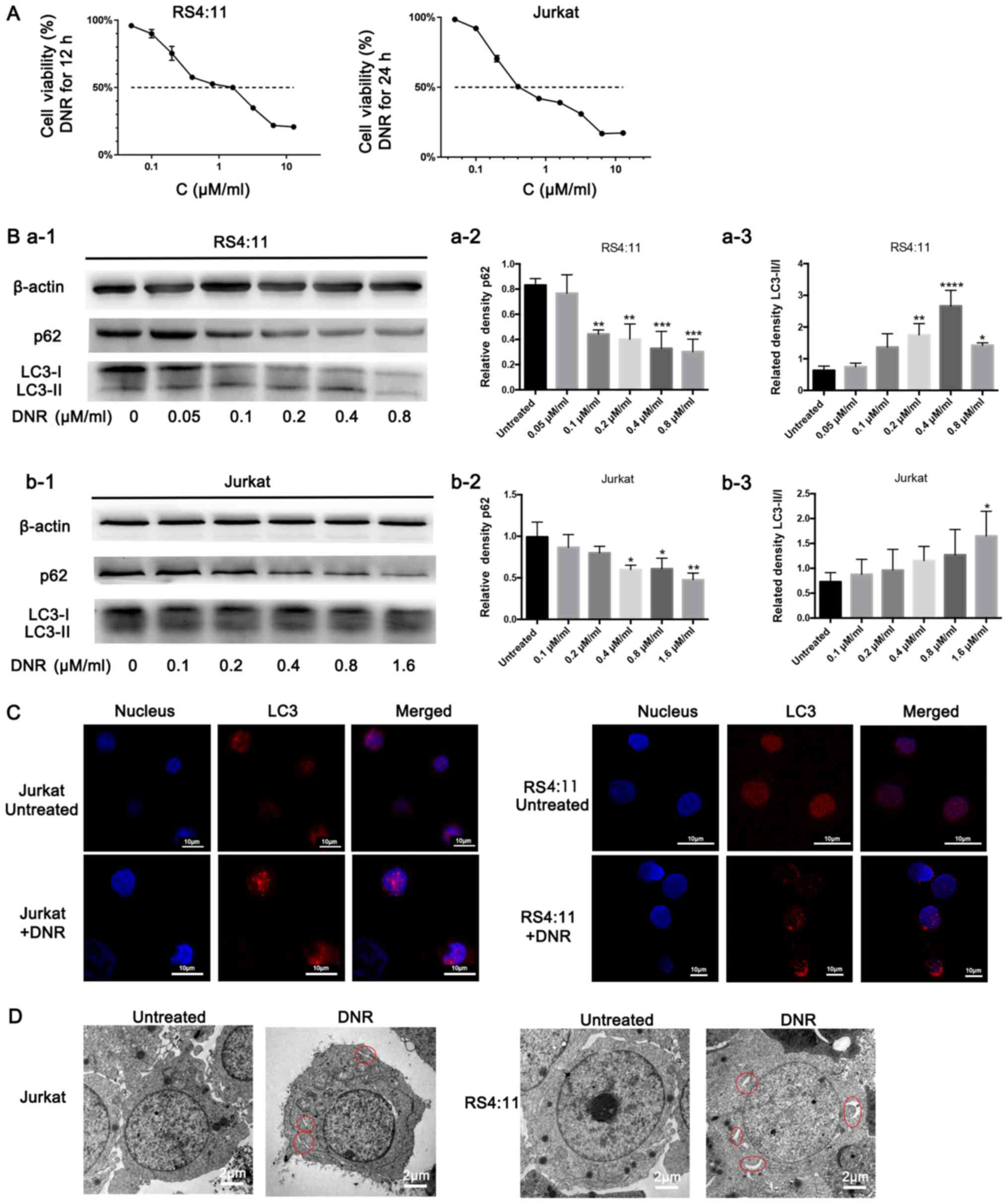
DNR is a major anti-tumour agent that is widely used
to treat leukaemia. As presented in Fig.
1A, DNR significantly damaged the Jurkat and RS4:11 cells in a
dose-dependent manner, particularly the RS4:11 cells, as RS4:11
cells were treated with DNR for 12 h only, while Jurkat cells were
treated with DNR for 24 h. In order to investigate whether
autophagy occurred when the leukaemia cells were treated with
chemotherapeutic drugs, the present study conducted relevant
experiments, and the results are presented below. Based on the
western blot analysis, the chemotherapy-induced LC3-II/I ratio
increased as the DNR concentration increased. The level of p62, an
adaptor between the autophagy machinery and its substrates,
gradually decreased as the drug concentrations increased (Fig. 1B). The immunofluorescence analysis
revealed that the changes in endogenous LC3 puncta were consistent
with the western blot analysis results in the two cell lines
(Fig. 1C). Furthermore, the
ultrastructural analysis revealed that chemotherapy-treated cells
had more autophagosomes and autophagolysosomes during chemotherapy
compared with the untreated cells in both cell lines (Fig. 1D). Fig.
1 demonstrates that the chemotherapeutic drugs induced
autophagy in the leukaemia cells.
Translocation of HMGB1 is associated
with chemotherapy-induced autophagy
In order to investigate the potential role of HMGB1
in regulating chemotherapeutic drug anticancer activity, the
present study focused on HMGB1 localisation. HMGB1 expression was
detected in both the nucleus and cytoplasm separately via western
blotting. In Jurkat cells, HMGB1 expression decreased in the
nucleus but increased in the cytoplasm following DNR treatment. In
the RS4:11 cells, HMGB1 expression decreased in the nucleus
following DNR treatment. In addition, the expression of HMGB1 in
the cytoplasm decreased at 0.05 and 0.2 µM/ml DNR concentration,
but not significantly, and there was no significant change in HMGB1
expression in the cytoplasm at other DNR concentrations (Fig. 2A b-1 and b-2). This subcellular
localisation of HMGB1 was detected indirectly using
immunofluorescence techniques, through which red fluorescence was
observed in the nucleus and cytoplasm following DNR treatment in
the two cell lines, but the red fluorescence remained primarily in
the nucleus without DNR treatment (Fig.
2B). After repeated experiments and statistical analyses, the
results presented in Fig. 2A and B
suggested that HMGB1 may be transferred from the nucleus to the
cytoplasm in Jurkat and RS4:11 cells during chemotherapeutic
treatment. In the present study, HMGB1 expression did not increase
in Jurkat or RS4:11 cells following DNR stimulation. By contrast,
Fig. 2C demonstrates that the HMGB1
protein levels in the Jurkat and RS4:11 cells decreased gradually
as the DNR concentration increased (P<0.05). Therefore, it was
speculated that the HMGB1 that had been released early was not a
newly synthesised protein in the cytoplasm, but it was transferred
from the nucleus to the cytoplasm and eventually released from the
cell (10,30). These results suggest that HMGB1
translocation is necessary for chemotherapy-induced autophagy.
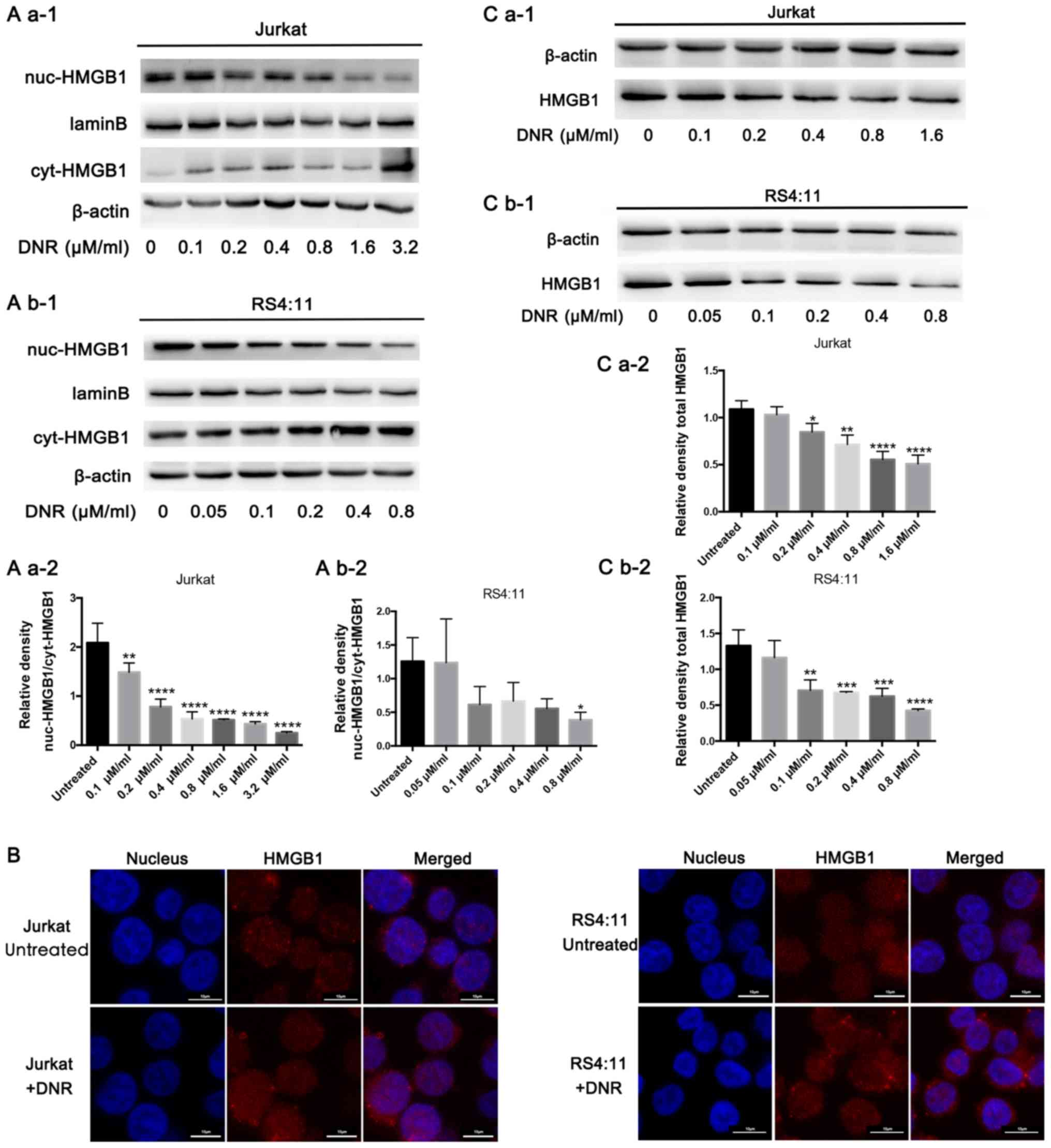 | Figure 2.HMGB1 translocation is associated
with chemotherapeutic drug-induced autophagy. (A) Cell lysates were
separated into cytosolic and nuclear fractions. Cytosolic and
nuclear HMGB1 levels were assayed via western blotting. β-actin was
used as a loading control to detect the expression of cytoplasmic
protein, while laminB was used to detect the expression of nuclear
protein. Quantified data are presented
[(nuc-HMGB1/laminB)/(cyt-HMGB1/β-actin)]. (a-1) Western blot
diagram of Jurkat cells. (a-2) nuc-HMGB1/cyt-HMGB1 quantitative
data of Jurkat cells. (b-1) Western blot diagram of RS4:11 cells.
(b-2) nuc-HMGB1/cyt-HMGB1 quantitative data of RS4:11 cells. (B)
Intracellular HMGB1 was stained via indirect immunofluorescence and
analysed under a confocal microscope to detect the location of
HMGB1 (HMGB1, Cy3 staining; nucleus, DAPI staining). (C) Cell
lysates were subjected to western blotting to detect HMGB1
expression. β-actin was used as a loading control. Quantified data
are presented (HGMB1/β-actin). Data are the mean ± standard
deviation of three independent experiments. (a-1) Western blot
diagram of Jurkat cells. (a-2) Total HMGB1 quantitative data of
Jurkat cells. (b-1) Western blot diagram of RS4:11 cells. (b-2)
Total HMGB1 quantitative data of RS4:11 cells. *P<0.05,
**P<0.01, ***P<0.001 and ****P<0.0001, compared with the
untreated group. HMGB1, high mobility group box protein 1; DNR,
daunorubicin; Cyt, cytoplasm; Nuc, nucleus. |
Poly (ADP-ribosylation) of HMGB1
facilitates its acetylation
The present study then sought to further define the
mechanisms by which HMGB1 translocation occurs. Previous studies
have demonstrated that post-translational modifications, such as
acetylation and ribosylation, are critical for HMGB1 translocation
from the nucleus to the cytoplasm (31–33).
Therefore, the present study immunoprecipitated the cell lysates
with an HMGB1-specific antibody, and these immunoprecipitated
proteins were immunoblotted with specific anti-poly
(ADP-ribosylation) and anti-acetylation antibodies. Fig. 3A demonstrates that the total HMGB1
was decreased and the poly (ADP-ribosylation) and acetylation of
HMGB1 increased significantly in both cell lines following DNR
treatment. The aforementioned lysine and glutamate residues are
primarily located near nuclear localisation sequences (NLS), which
are frequently modified by acetylation and poly (ADP-ribosylation),
respectively, and previous studies have demonstrated that they may
be associated with the re-localization of HMGB1 (26,29,34).
HMGB1 was enriched by immunoprecipitation, and specific anti-Flag
antibodies were used to detect lentiviral expression via western
blotting (Fig. 3B).
HMGB1MT1, HMGB1MT2 and wild-type Jurkat cells
(HMGB1WT) were successfully constructed. Theoretically,
the expression level of HMGB1 protein in HMGB1MT1,
HMGB1MT2 and HMGB1WT cells should be higher
than that in normal control Jurkat cells (HMGB1NC)
group, but in fact, this was not the case. Considering that HMGB1
is highly expressed in a number of different types of tumour cell,
including leukaemia, these results indicated insignificant HMGB1
overexpression (10). Fig. 3C demonstrates that the total HMGB1 in
different cells presented a decreasing trend following DNR
treatment. However, HMGB1 acetylation and poly (ADP-ribosylation)
were significantly increased following DNR treatment in
HMGB1WT cells and normal control Jurkat cells
(HMGB1NC). By contrast, following DNR treatment, the
HMGB1 acetylation level remained low, while the HMGB1 poly
(ADP-ribosylation) expression was increased in HMGB1MT1
cells, indicating that blocking HMGB1 acetylation did not affect
its poly (ADP-ribosylation). In HMGB1MT2 cells, HMGB1
poly (ADP-ribosylation) did not increase following DNR treatment,
and the degree of HMGB1 acetylation increased slightly, but there
was no statistical significance. In addition, considering the data
presented in Fig. 3A and C, the
present study concluded that the poly (ADP-ribosylation) of HMGB1
may facilitate its acetylation.
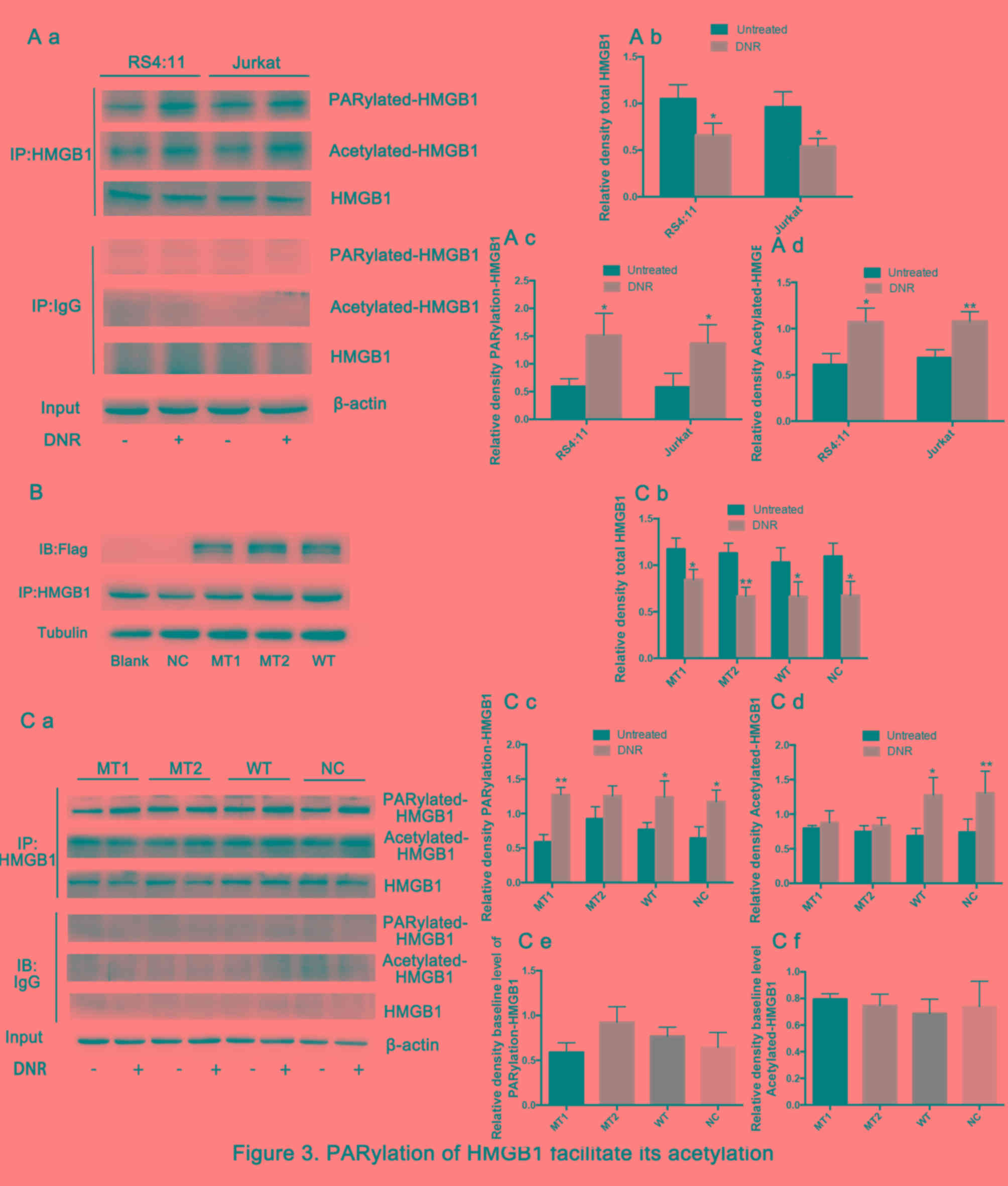 | Figure 3.Poly (ADP-ribosylation) of HMGB1
facilitates its acetylation. (A) Cell lysates were
immunoprecipitated with an HMGB1 antibody, followed by western blot
analysis. The acetylation or poly (ADP-ribosylation) levels were
measured using antibodies specific for acetylated lysine or poly
(ADP-ribosylation). β-actin was used as a loading control.
Quantified data are presented (HMGB1/β-actin, PARylation-HMGB1 or
Acetylated-HMGB1/HMGB1/β-actin). (a Western blot diagram of RS4:11
and Jurkat cells. (b) Total HMGB1 quantitative data of RS4:11 and
Jurkat cells. (c) PARylation-HMGB1 quantitative data of RS4:11 and
Jurkat cells. (d) Aceytlation-HMGB1 quantitative data of RS4:11 and
Jurkat cells. (B) HMGB1MT1, HMGB1MT2 and
HMGB1WT cells were successfully constructed. The lysine
residues at amino acids 28, 29, 30, 180, 182, 183, 184 and 185 of
HMGB1 in HMGB1MT1 cells were mutated to alanine, and the
glutamate residues at 40, 47 and 179 of HMGB1 in
HMGB1MT2 cells were mutated to alanine. The whole
protein of HMGB1MT1, HMGB1MT2,
HMGB1WT, HMGB1NC and Jurkat cells were
subjected to immunoprecipitation with anti-HMGB1 antibodies and
then subjected to western blotting to detect the expression of
lentivirus with anti-Flag antibodies. Tubulin was used as a loading
control. (C) Cell lysates were subjected to a pull-down assay with
an HMGB1 antibody and immunoblotted with anti-acetylated lysine,
anti-poly (ADP-ribosylation) and anti-HMGB1 antibodies. β-actin was
used as a loading control. Quantified data are presented
(HMGB1/β-actin, PARylation-HMGB1 or
Acetylated-HMGB1/HMGB1/β-actin). Data are the mean ± standard
deviation of three independent experiments. (a) Western blot
diagram of MT1, MT2, WT and NC cells. (b) Total HMGB1 quantitative
data of MT1, MT2, WT and NC cells. (c) PARylation-HMGB1
quantitative data of MT1, MT2, WT and NC cells. (d)
Aceytlation-HMGB1 quantitative data of MT1, MT2, WT and NC cells.
*P<0.05, **P<0.01, Fig. 3Cb-d
compared with the untreated group and Fig. 3Ce and f compared with the NC group.
HMGB1, high mobility group box protein 1; DNR, daunorubicin; MT,
mutant type; WT, wild type; NC, normal control; UT, untreated
group. |
Poly (ADP-ribosylation) and
acetylation play important roles in HMGB1 translocation, which is
required for chemotherapeutic drug-induced autophagy
HMGB1 expression in the nucleus and cytoplasm was
detected via western blotting. The results revealed that HMGB1
expression was significantly increased in the cytoplasm of
HMGB1WT and HMGB1NC cells following DNR
treatment; however, the HMGB1 expression in the nucleus and
cytoplasm of HMGB1MT1 and HMGB1MT2 cells did
not change significantly (Fig. 4A).
In addition, following immunofluorescence labelling of HMGB1, red
fluorescence was observed in the nucleus and cytoplasm of the
HMGB1WT and HMGB1NC cells following DNR
treatment. However, in the HMGB1MT1 and
HMGB1MT2 cells, the red fluorescence in the cytoplasm
was weaker than that in the HMGB1WT and
HMGB1NC cells following DNR treatment (Fig. 4B). To further demonstrate that HMGB1
translocation is associated with chemotherapy-induced autophagy,
the present study conducted relevant experiments. The western blot
analysis revealed that the LC3-II/I ratio increased (but not
significantly) and that p62 levels decreased following DNR
treatment in the HMGB1WT and HMGB1NC cells,
while the LC3-II/I ratio and p62 level did not change significantly
in the HMGB1MT1 or HMGB1MT2 cells (Fig. 4C). Immunofluorescence revealed that
the changes in endogenous LC3 puncta indicated increased autophagy
levels in the HMGB1WT and HMGB1NC cells,
while the autophagy levels of the HMGB1MT1 and
HMGB1MT2 cells remained low (Fig. 4D). Compared with the
HMGB1WT and HMGB1NC cells, fewer
autophagosomes and autophagolysosomes were observed in the
HMGB1MT1 and HMGB1MT2 cells following
chemotherapy, which was the same as the trend observed in the
immunofluorescence analysis (Fig.
4E). Blocking the acetylation and poly (ADP-ribosylation) of
HMGB1 significantly decreased the translocation of HMGB1 from the
nucleus to the cytoplasm and inhibited the induction of autophagy
during chemotherapy in Jurkat cells (Fig. 4). Combined with the data in Figs. 3 and 4, these results demonstrate that poly
(ADP-ribosylation) of HMGB1 facilitated its acetylation, thereby
inducing HMGB1 translocation and ultimately promoting
chemotherapy-induced autophagy in leukaemia cells.
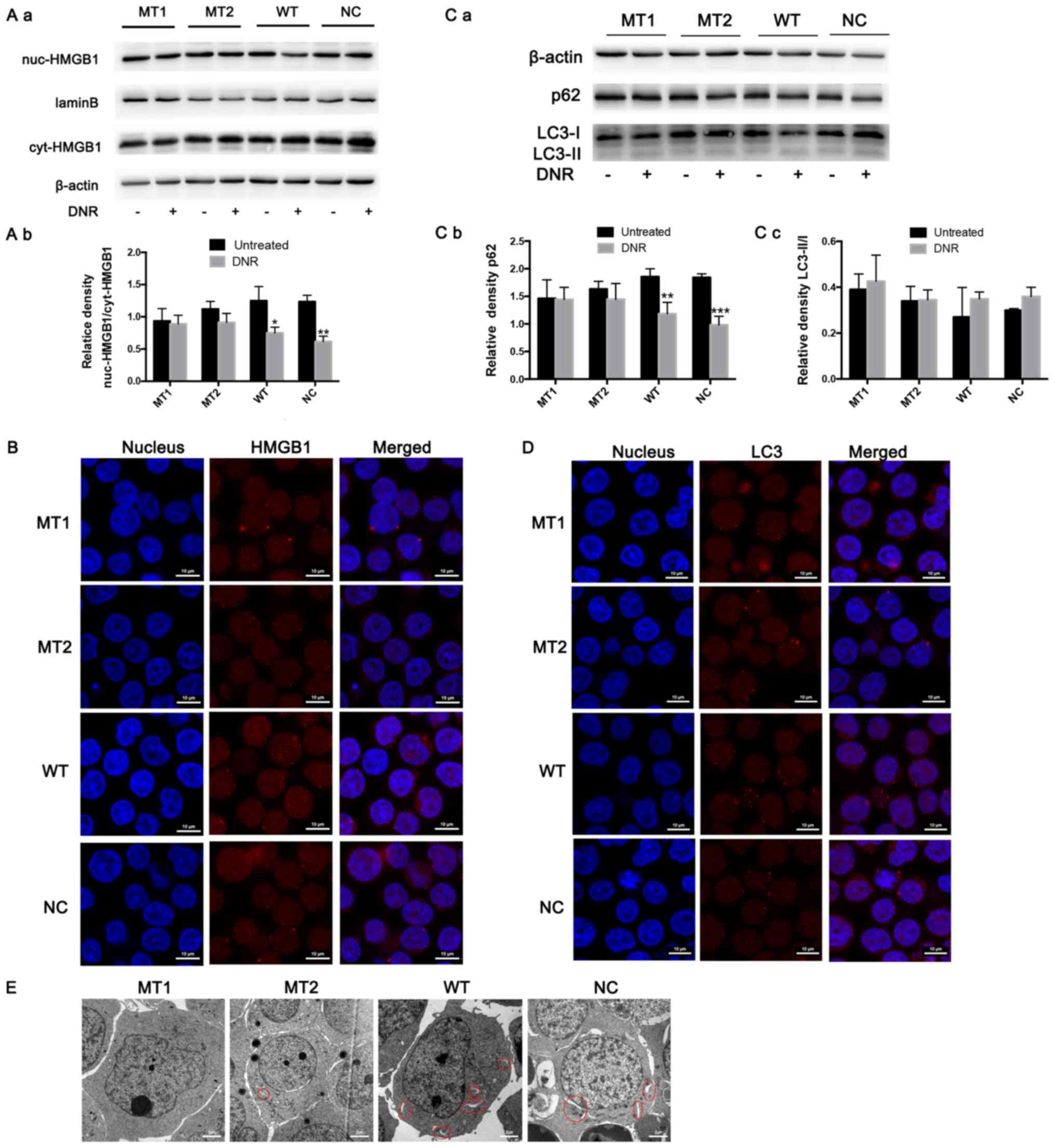 | Figure 4.HMGB1 translocation is associated
with chemotherapeutic drug-induced autophagy. (A) Cell lysates were
separated into cytosolic and nuclear fractions. Cytosolic and
nuclear HMGB1 were assayed via western blotting. β-actin was used
as a loading control to detect the expression of cytoplasmic
protein, while laminB was used to detect the expression of nuclear
protein. Quantified data are presented
[(nuc-HMGB1/laminB)/(cyt-HMGB1/β-actin)]. (a) Western blot diagram
of MT1, MT2, WT and NC cells. (b) nuc-HMGB1/cyt-HMGB1 quantitative
data of J MT1, MT2, WT and NC cells. (B) Intracellular HMGB1 was
stained via indirect immunofluorescence and analysed under a
confocal microscope to detect the location of HMGB1 (HMGB1, Cy3
staining; nucleus, DAPI staining). (C) The cell lysates were
subjected to western blotting to detect LC3-II/I and p62
expression. β-actin was used as a loading control. Quantified data
are presented (p62 or LC3-II/I/β-actin). (a) Western blot diagram
of MT1, MT2, WT and NC cells. (b) p62 quantitative data of MT1,
MT2, WT and NC cells. (c) LC3II/I quantitative data of MT1, MT2, WT
and NC cells. (D) LC3 was stained via indirect immunofluorescence
and analysed under a confocal microscope to measure the LC3 puncta
(LC3, staining with Cy3; nucleus, staining with DAPI). (E) Cells
were subjected to transmission electron microscopy to observe
autophagosome-like structures (indicated by red circles). Data are
the mean ± standard deviation of three independent experiments.
*P<0.05, **P<0.01, ***P<0.001 compared with the untreated
group. HMGB1, high mobility group box protein 1; MT, mutant type;
WT, wild type; NC, normal control; UT, untreated group; DNR,
daunorubicin. |
Discussion
Drug resistance in leukaemia cells is a primary
factor leading to the development of refractory and recurrent
leukaemia. A number of studies have demonstrated that drug
resistance in leukaemia is partly attributable to autophagy induced
by multiple chemotherapeutic drugs. For example, inhibiting
autophagy by pharmacological inhibitors or genetic knockdown of
critical autophagy-associated genes, such as Atg5 and Atg7, could
enhance the anticancer effects of chemotherapeutic drugs (35,36). DNR
is one of the most widely used chemotherapy drugs for leukaemia,
and the cytotoxicity mediated by DNR is thought to result from
drug-induced DNA damage (37). Chen
et al (38) and Kudoh et
al (39) suggested that DNR also
triggers a pathway that negatively regulates apoptosis, and the
phospholipase C-dependent diacylglycerol
(DAG)/raf-1/mitogen-activated protein kinase (MEK) cascade and the
DAG independent phosphoinositide 3-kinase (PI3K)/protein kinase C ζ
type cascade play significant roles in this process. raf-1/MEK and
PI3K are believed to be involved in autophagy signalling (40,41). Han
et al (35) demonstrated for
the first time that DNR can induce cytoprotective autophagy in K562
cells by activating the MEK/extracellular signal-regulated kinase-1
signalling pathway.
In the present study, it was difficult to detect
changes in LC3-II protein levels in leukaemia cells via western
blotting (Figs. 1B and 4C). Cytoplasmic LC3 forms LC3-I by
enzymatic hydrolysis of a small segment of polypeptide, which then
binds to Phosphatidylethanolamine (PE) and converts to membrane
LC3-II (36). Therefore, it was
speculated that the LC3-II protein was difficult to detect for the
following reasons: i) The cytoplasm of leukaemia cells is small and
the membrane protein is difficult to dissolve in the conventional
RIPA list; ii) LC3-II, as a part of autophagy, fuses with lysosome
to form autophagic lysosome and degrades due to the autophagy
(42). Autophagy is a highly
dynamic, multi-step process. Although it is difficult to obtain a
satisfactory and convincing result regarding the increase in the
chemotherapy-induced LC3-II/I using western blotting alone as
indicated in Fig. 1B, by combining
the results of the p62 level via western blotting (Fig. 1B), immunofluorescence (Fig. 1C) and transmission electron
microscopy (Fig. 1D), conclusions
could be drawn that indicated that the level of
chemotherapeutic-induced autophagy was increased in leukaemia
cells. Consistent with these results, the present study revealed
that DNR triggered both apoptosis and autophagy in leukaemia cells
(Fig. 1).
HMGB1 acts as both a tumour suppressor and an
oncogenic factor in tumourigenesis and cancer therapy (43). Bell et al (44) demonstrated that HMGB1 appears in the
medium of Jurkat and U937 cells time-dependently following
chemotherapeutic drug treatment. In addition, high HMGB1 expression
is suggested to be closely associated with tumour occurrence, and
plays an important role in regulating tumour cell autophagy and
apoptosis (10,45). Tang et al (46) demonstrated that in human pancreatic
and colon cancer cells, anticancer drugs such as melphalan and
paclitaxel could enhance the autophagy production of tumour cells
by increasing the release of HMGB1 and its binding to the receptor
for advance glycation endproducts. Zhan et al (47) demonstrated that the chemotherapeutic
drug vincristine can promote the release of HMGB1 in gastric cancer
cells and upregulate the expression of Mcl-1 protein in the Bcl-2
protein family, thereby producing anti-apoptotic effects. HMGB1 in
breast cancer cells can promote cell tolerance in chemotherapy and
chemoradiotherapy. Luo et al (48) demonstrated that miR-129-5p can
enhance the efficacy of radiotherapy by targeting HMGB1 to decrease
autophagy caused by breast cancer radiotherapy. Liu et al
(13) and Hu et al (49) demonstrated that treatment with an
HMGB1-neutralising antibody improved the sensitivity of leukaemia
cells to chemotherapy, while exogenous HMGB1 made the cells more
resistant to drug-induced cytotoxicity. In the present study, there
was no significant upregulation of HMGB1 observed in the whole-cell
protein samples following DNR treatment, but the HMGB1 protein may
have transferred from the nucleus to the cytoplasm (Figs. 2, 3A and
C, and 4A and B).
Previous studies have demonstrated that HMGB1 is
highly expressed in a number of different types of tumour,
including leukaemia, and it is more abundant on the surface of
metastatic tumour cell membranes (10) and is closely associated with
chemotherapy-induced drug resistance (12). HMGB1 is believed to regulate
autophagy at multiple levels via different subcellular
localisations (17,46,50).
Although the function of HMGB1 in the cytoplasm remains unclear,
evidence suggests that the primary function of HMGB1 in the
cytoplasm is to provide positive regulatory factors for autophagy,
as was first reported in 2010 (45).
A previous study demonstrated that HMGB1 is translocated from the
nucleus to the cytoplasm following chemotherapeutic treatment in
leukaemia cells, and cytoplasmic HMGB1 then promotes the
dissociation of Beclin1-Bcl-2 complexes and modifies Beclin1
binding to PI3k catalytic subunit 3, thus initiating autophagosome
formation and upregulating autophagy (17,25).
Figs. 1, 2 and 4
suggest that HMGB1 is an important regulator of
chemotherapy-induced autophagy and that HMGB1 translocation is a
key step in this mechanism.
HMGB1 contains two NLS and two nuclear export
sequences (NES), and post-translational modification of amino acids
on NLS and NES can result in the relocation of HMGB1 (20,32). In
the present study, acetylation and poly (ADP-ribosylation) of HMGB1
were increased during chemotherapy-induced autophagy (Fig. 3A). This drew focus towards the
post-transcriptional modifications of HMGB1 in the present study,
including methylation, acetylation, phosphorylation and poly
(ADP-ribosylation), which are key steps in HMGB1 translocation from
the nucleus to the cytoplasm and its eventual release into the
extracellular space (31). Different
post-transcriptional modifications play important but varied roles
in localising HMGB1 in the cytoplasm, but it is currently unclear
which modification is dominant. Acetylation is regarded as a
prerequisite for HMGB1 migration to the cytoplasm. Sterner et
al (51) and Bonaldi et
al (29) observed that the
acetylation of certain lysine residues in NLS promotes HMGB1
migration from the nucleus to the cytoplasm and active secretion of
HMGB1 to the extracellular space. Lu et al (24) demonstrated that the JAK/STAT1
signalling pathway played a key role in HMGB1 hyperacetylation and
cytoplasmic accumulation. Pan et al (52) and Dhupar et al (53) revealed that interferon regulatory
factor 1 promoted HMGB1 acetylation through histone
acetyltransferases, which was required for lipopolysaccharide
(LPS)-induced HMGB1 cytoplasmic accumulation and release.
Consistent with these results, the present study revealed that in
HMGB1MT1 cells, HMGB1 translocation was inhibited
(Fig. 4A and B).
Poly (ADP-ribosylation) is one of the most important
methods of protein posttranslational modification (54). Ditsworth et al (34) suggested that in a DNA-alkylating
damage model, PARP1 activity played a role in the
nuclear-to-cytosolic translocation of HMGB1. Davis et al
(26) reported that LPS and
alkylating agents could induce PARP1 activation and
ADP-ribosylation to ultimately release HMGB1. Consistent with these
results, the present study revealed that the level of HMGB1 poly
(ADP-ribosylation) was not upregulated in HMGB1MT2
cells, and HMGB1 expression in the cytoplasm did not change
significantly compared with the untreated group. Furthermore,
immunofluorescence in the HMGB1 sublocalisation did not change.
Therefore, it can be concluded that HMGB1 translocation from the
nucleus to the cytoplasm depends on the poly (ADP-ribosylation) of
HMGB1 (Figs. 3C and 4A and B).
An increasing number of studies have demonstrated
that acetylation or poly (ADP-ribosylation) can promote HMGB1
translocation, but few studies have addressed the interaction
between these modifications. Recently, Yang et al (27) reported for the first time that PARP1
can promote HMGB1 acetylation through poly (ADP-ribosylation).
However, the association between poly (ADP-ribosylation) and
acetylation and the role of these modifications in HMGB1
translocation in leukaemia remain unclear. Figs. 3B and 4A
and B demonstrate that in HMGB1MT1 cells, although
the level of poly (ADP ribosylation) modification increased, HMGB1
expression in the cytoplasm did not increase compared with that in
the untreated group, and no changes occurred in the sublocalisation
of HMGB1 via immunofluorescence. This suggests that poly (ADP
ribosylation) modification of HMGB1 facilitates its acetylation but
is insufficient to induce HMGB1 translocation. The present study
hypothesised that the possible mechanism for this poly
(ADP-ribosylation)/acetylation-controlled distribution was that the
poly ADP-ribosylation of the glutamic acid residues altered the
conformation of the A box, thereby exposing the acetylation site
and promoting acetylation of the lysine residues in HMGB1. These
HMGB1 modifications decrease its ability to bind to DNA and promote
its translocation.
Combining the results of previous studies with those
of the present, the process of chemotherapy-induced autophagy in
leukaemic cells can be elucidated; revealing that poly
(ADP-ribosylation) of HMGB1 enhanced HMGB1 acetylation and
ultimately promoted chemotherapy-induced autophagy of leukaemic
cells mediated by the HMGB1 translocation. Chemotherapeutic drugs
are thought to induce apoptosis that is dependent on nuclear HMGB1
induction (50).
The association between HMGB1 and chemotherapeutic
drug-induced autophagy has been extensively studied and understood,
but the role of HMGB1 in the cytoplasm remains to be poorly
understood (17,45). In addition, studies focussing on the
underlying molecular mechanism of influencing HMGB1 translocation
primarily concentrated on inflammation, sepsis and other diseases,
and rarely focussed on leukaemia (16,26). As
a specific type of tumour, leukaemia is different from solid
tumours. Particularly in T-ALL, despite targeted gene therapy and
chimeric antigen receptor T-cell therapy, the prognosis of children
with drug-resistant or relapsed leukaemia remains poor (55,56). In
the present study, Jurkat cells, an acute T-lymphoblastic leukaemia
cell line, were mutated to verify that poly (ADP-ribosylation) of
HMGB1 facilitated its acetylation and promoted HMGB1
translocation-associated chemotherapy-induced autophagy, which will
ultimately provide a theoretical basis and new therapeutic targets
for drug resistance in T-ALL. Therefore, the present study is of
great significance in the clinical treatment of T-ALL and the
therapeutic strategies that inhibit HMGB1 transfer to decrease
HMGB1 expression in the cytoplasm and extracellular space are
promising and may improve the effectiveness of chemotherapy.
The present study demonstrated that poly
(ADP-ribosylation) of HMGB1 facilitates its acetylation, thereby
inducing HMGB1 translocation and ultimately promoting
chemotherapy-induced autophagy in leukaemic cells.
Acknowledgements
The authors would like to thank Dr Sarah Conte, for
her assistance in revising an earlier version of this
manuscript.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81570140) and the
Doctoral Start-up Fund of Natural Science Foundation of Guangdong
Province (grant no. 2016A030310161).
Availability of data and materials
The datasets used and/or analysed during the present
are available from the corresponding author on reasonable
request.
Authors' contributions
YL and JF designed the study. YL and JX collated and
analysed the data. YL wrote the manuscript. XL and JF participated
in revising manuscript. XL also performed the study and collected
background information. All authors read and approved the final
submitted manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Belver L and Ferrando A: The genetics and
mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer.
16:494–507. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Richter-Pechańska P, Kunz JB, Hof J,
Zimmermann M, Rausch T, Bandapalli OR, Orlova E, Scapinello G, Sagi
JC, Stanulla M, et al: Identification of a genetically defined
ultra-high-risk group in relapsed pediatric T-lymphoblastic
leukemia. Blood Cancer J. 7:e5232017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khamisipour G, Jadidi-Niaragh F, Jahromi
AS, Zandi K and Hojjat-Farsangi M: Mechanisms of tumor cell
resistance to the current targeted-therapy agents. Tumour Biol.
37:10021–10039. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nencioni A, Cea M, Montecucco F, Longo VD,
Patrone F, Carella AM, Holyoake TL and Helgason GV: Autophagy in
blood cancers: Biological role and therapeutic implications.
Haematologica. 98:1335–1343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Evangelisti C, Evangelisti C, Chiarini F,
Lonetti A, Buontempo F, Neri LM, McCubrey JA and Martelli AM:
Autophagy in acute leukemias: A double-edged sword with important
therapeutic implications. Biochim Biophys Acta. 1853:14–26. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gump JM, Staskiewicz L, Morgan MJ, Bamberg
A, Riches DW and Thorburn A: Autophagy variation within a cell
population determines cell fate through selective degradation of
Fap-1. Nat Cell Biol. 16:47–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lange SS and Vasquez KM: HMGB1: The
jack-of-all-trades protein is a master DNA repair mechanic. Mol
Carcinog. 48:571–580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao
L, Huang J, Yu Y, Fan XG, Yan Z, et al: HMGB1 in health and
disease. Mol Aspects Med. 40:1–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bianchi ME and Agresti A: HMG proteins:
Dynamic players in gene regulation and differentiation. Curr Opin
Genet Dev. 15:496–506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang D, Kang R, Zeh HJ III and Lotze MT:
High-mobility group box 1 and cancer. Biochim Biophys Acta.
1799:131–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stevens NE, Chapman MJ, Fraser CK, Kuchel
TR, Hayball JD and Diener KR: Therapeutic targeting of HMGB1 during
experimental sepsis modulates the inflammatory cytokine profile to
one associated with improved clinical outcomes. Sci Rep.
7:58502017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee W, Kwon OK, Han MS, Lee YM, Kim SW,
Kim KM, Lee T, Lee S and Bae JS: Role of moesin in HMGB1-stimulated
severe inflammatory responses. Thromb Haemost. 114:350–363. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu B, Wang H, Andersson U and Tracey KJ:
Regulation of HMGB1 release by inflammasomes. Protein Cell.
4:163–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kong Q, Xu LH, Xu W, Fang JP and Xu HG:
HMGB1 translocation is involved in the transformation of autophagy
complexes and promotes chemoresistance in leukaemia. Int J Oncol.
47:161–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu L and Yang L: The function and
mechanism of HMGB1 in lung cancer and its potential therapeutic
implications. Oncol Lett. 15:6799–6805. 2018.PubMed/NCBI
|
|
19
|
Livingston JA, Wang WL, Tsai JW, Lazar AJ,
Leung CH, Lin H, Advani S, Daw N, Santiago-O'Farrill J, Hollomon M,
et al: Analysis of HSP27 and the autophagy marker LC3B+
puncta following preoperative chemotherapy identifies high-risk
osteosarcoma patients. Mol Cancer Ther. 17:1315–1323. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gardella S, Andrei C, Ferrera D, Lotti LV,
Torrisi MR, Bianchi ME and Rubartelli A: The nuclear protein HMGB1
is secreted by monocytes via a non-classical, vesicle-mediated
secretory pathway. EMBO Rep. 3:995–1001. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ito I, Fukazawa J and Yoshida M:
Post-translational methylation of high mobility group box 1 (HMGB1)
causes its cytoplasmic localization in neutrophils. J Biol Chem.
282:16336–16344. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Wheeler D, Tang Y, Guo L, Shapiro
RA, Ribar TJ, Means AR, Billiar TR, Angus DC and Rosengart MR:
Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates
nucleocytoplasmic shuttling and release of HMGB1 during
lipopolysaccharide stimulation of macrophages. J Immunol.
181:5015–5023. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsung A, Klune JR, Zhang X, Jeyabalan G,
Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR and Billiar TR:
HMGB1 release induced by liver ischemia involves Toll-like receptor
4 dependent reactive oxygen species production and calcium-mediated
signaling. J Exp Med. 204:2913–2923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu B, Antoine DJ, Kwan K, Lundbäck P,
Wähämaa H, Schierbeck H, Robinson M, Van Zoelen MA, Yang H, Li J,
et al: JAK/STAT1 signaling promotes HMGB1 hyperacetylation and
nuclear translocation. Proc Natl Acad Sci USA. 111:3068–3073. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livesey KM, Kang R, Vernon P, Buchser W,
Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ III, Li L, et
al: p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer
Res. 72:1996–2005. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davis K, Banerjee S, Friggeri A, Bell C,
Abraham E and Zerfaoui M: Poly(ADP-ribosyl)ation of high mobility
group box 1 (HMGB1) protein enhances inhibition of efferocytosis.
Mol Med. 18:359–369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Z, Li L, Chen L, Yuan W, Dong L,
Zhang Y, Wu H and Wang C: PARP-1 mediates LPS-induced HMGB1 release
by macrophages through regulation of HMGB1 acetylation. J Immunol.
193:6114–6123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang M, Liu L, Xie M, Sun X, Yu Y, Kang R,
Yang L, Zhu S, Cao L and Tang D: Poly-ADP-ribosylation of HMGB1
regulates TNFSF10/TRAIL resistance through autophagy. Autophagy.
11:214–224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bonaldi T, Talamo F, Scaffidi P, Ferrera
D, Porto A, Bachi A, Rubartelli A, Agresti A and Bianchi ME:
Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect
it towards secretion. EMBO J. 22:5551–5560. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ugrinova I, Pasheva EA, Armengaud J and
Pashev IG: In vivo acetylation of HMG1 protein enhances its binding
affinity to distorted DNA structures. Biochemistry. 40:14655–14660.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Q and Wang Y: HMG modifications and
nuclear function. Biochim Biophys Acta. 1799:28–36. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Richard SA, Jiang Y, Xiang LH, Zhou S,
Wang J, Su Z and Xu H: Post-translational modifications of high
mobility group box 1 and cancer. Am J Transl Res. 9:5181–5196.
2017.PubMed/NCBI
|
|
33
|
VanPatten S and Al-Abed Y: High mobility
group box-1 (HMGb1): Current wisdom and advancement as a potential
drug target. J Med Chem. 61:5093–5107. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ditsworth D, Zong WX and Thompson CB:
Activation of poly(ADP)-ribose polymerase (PARP-1) induces release
of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol
Chem. 282:17845–17854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han W, Sun J, Feng L, Wang K, Li D, Pan Q,
Chen Y, Jin W, Wang X, Pan H and Jin H: Autophagy inhibition
enhances daunorubicin-induced apoptosis in K562 cells. PLoS One.
6:e284912011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livesey KM, Tang D, Zeh HJ and Lotze MT:
Autophagy inhibition in combination cancer treatment. Curr Opin
Investig Drugs. 10:1269–1279. 2009.PubMed/NCBI
|
|
37
|
Laurent G and Jaffrézou JP: Signaling
pathways activated by daunorubicin. Blood. 98:913–924. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen JJ, Wu R, Yang PC, Huang JY, Sher YP,
Han MH, Kao WC, Lee PJ, Chiu TF, Chang F, et al: Profiling
expression patterns and isolating differentially expressed genes by
cDNA microarray system with colorimetry detection. Genomics.
51:313–324. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kudoh K, Ramanna M, Ravatn R, Elkahloun
AG, Bittner ML, Meltzer PS, Trent JM, Dalton WS and Chin KV:
Monitoring the expression profiles of doxorubicin-induced and
doxorubicin-resistant cancer cells by cDNA microarray. Cancer Res.
60:4161–4166. 2000.PubMed/NCBI
|
|
40
|
Klionsky DJ: Autophagy: From phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang Z and Klionsky DJ: An overview of the
molecular mechanism of autophagy. Curr Top Microbiol Immunol.
335:1–32. 2009.PubMed/NCBI
|
|
42
|
Jacquin E, Leclerc-Mercier S, Judon C,
Blanchard E, Fraitag S and Florey O: Pharmacological modulators of
autophagy activate a parallel noncanonical pathway driving
unconventional LC3 lipidation. Autophagy. 13:854–867. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kang R, Zhang Q, Zeh HJ III, Lotze MT and
Tang D: HMGB1 in cancer: Good, bad, or both? Clin Cancer Res.
19:4046–4057. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bell CW, Jiang W, Reich CF III and
Pisetsky DS: The extracellular release of HMGB1 during apoptotic
cell death. Am J Physiol Cell Physiol. 291:C1318–C1325. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III
and Lotze MT: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tang D, Kang R, Cheh CW, Livesey KM, Liang
X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et
al: HMGB1 release and redox regulates autophagy and apoptosis in
cancer cells. Oncogene. 29:5299–5310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhan Z, Li Q, Wu P, Ye Y, Tseng HY, Zhang
L and Zhang XD: Autophagy-mediated HMGB1 release antagonizes
apoptosis of gastric cancer cells induced by vincristine via
transcriptional regulation of Mcl-1. Autophagy. 8:109–121. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Luo J, Chen J and He L: mir-129-5p
attenuates irradiation-induced autophagy and decreases
radioresistance of breast cancer cells by targeting HMGB1. Med Sci
Monit. 21:4122–4129. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hu YH, Yang L and Zhang CG: HMGB1-a as
potential target for therapy of hematological malignancies.
Zhongguo Shi Yan Xue Ye Xue Za Zhi. 22:560–564. 2014.(In Chinese).
PubMed/NCBI
|
|
50
|
Tang D, Kang R, Livesey KM, Kroemer G,
Billiar TR, Van Houten B, Zeh HJ III and Lotze MT: High-mobility
group box 1 is essential for mitochondrial quality control. Cell
Metab. 13:701–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sterner R, Vidali G and Allfrey VG:
Studies of acetylation and deacetylation in high mobility group
proteins. Identification of the sites of acetylation in HMG-1. J
Biol Chem. 254:11577–11583. 1979.PubMed/NCBI
|
|
52
|
Pan PH, Cardinal J, Li ML, Hu CP and Tsung
A: Interferon regulatory factor-1 mediates the release of high
mobility group box-1 in endotoxemia in mice. Chin Med J (Engl).
126:918–924. 2013.PubMed/NCBI
|
|
53
|
Dhupar R, Klune JR, Evankovich J, Cardinal
J, Zhang M, Ross M, Murase N, Geller DA, Billiar TR and Tsung A:
Interferon regulatory factor 1 mediates acetylation and release of
high mobility group box 1 from hepatocytes during murine liver
ischemia-reperfusion injury. Shock. 35:293–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hassa PO, Haenni SS, Elser M and Hottiger
MO: Nuclear ADP-ribosylation reactions in mammalian cells: Where
are we today and where are we going? Microbiol Mol Biol Rev.
70:789–829. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vadillo E, Dorantes-Acosta E, Pelayo R and
Schnoor M: T cell acute lymphoblastic leukemia (T-ALL): New
insights into the cellular origins and infiltration mechanisms
common and unique among hematologic malignancies. Blood Rev.
32:36–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yuan T, Yang Y, Chen J, Li W, Li W, Zhang
Q, Mi Y, Goswami RS, You JQ, Lin D, et al: Regulation of PI3K
signaling in T cell acute lymphoblastic leukemia: A novel
PTEN/Ikaros/miR-26b mechanism reveals a critical targetable role
for PIK3CD. Leukemia. 31:2355–2364. 2017. View Article : Google Scholar : PubMed/NCBI
|