Introduction
According to the most recent global cancer data,
there were ~458,900 new cases of pancreatic cancer worldwide in
2018 and ~432,200 deaths. The mortality rate of pancreatic cancer
is the fourth highest in the USA and other Western countries
compared with all other types of cancer (1). In recent years, various therapeutic
strategies against pancreatic cancer, including surgical resection
and targeted therapy, have improved (2). However, due to early metastasis and
invasion of pancreatic cancer, only 15–20% of patients are able to
undergo surgical resection after diagnosis. Furthermore, this
treatment method has not been shown to improve the prognosis of the
remaining patients, as surgical resection does not significantly
improve the condition of patients (3). Consequently, improved understanding the
mechanisms underlying pancreatic cancer metastasis and invasion is
essential.
As previously reported, syndecan-1 (SDC1) is a cell
adhesion molecule expressed on the cell surface, which is usually
characterized by covalent bonds of heparan sulfate (HS) (4). Ibrahim et al (5) reported the involvement of SDC1 in
breast cancer, suggesting that this protein maintains cell
phenotype and inhibits cell migration, whereas when SDC1 is
isolated from the cell surface, cells may exhibit enhanced
proliferative and migratory abilities. Heparanase (HPA) is an
endoglycosidase present in mammals that is able to specifically
degrade the HS side chain of SDC1. It has been observed that HPA
may degrade this side chain in a variety of tumor cells to form the
HPA/SDC1 axis (6). Increased
expression of HPA in pancreatic cancer cells may disrupt the
extracellular matrix (ECM) and basement membrane (BM), thus
creating favorable conditions for the invasion and migration of
pancreatic cancer cells (7,8). Recently, the role of fibroblast growth
factor 2 (FGF2) in the development and progression of malignant
cancer types has attracted great interest (9). The binding of FGF2 to its receptor,
fibroblast growth factor receptor 2, may lead to the activation of
tumor-related signaling pathways, thus increasing cell
proliferation, migration and invasion (10).
Masola et al (11) previously reported that HPA may
promote the process of renal fibrosis by upregulating the
expression of FGF2. In the present study, the expression of the
HPA/SDC1 axis and FGF2 in pancreatic cancer cells was analyzed, and
the mechanisms of their interactions, and more importantly, their
effects on epithelial-mesenchymal transition (EMT) and pancreatic
cancer progression were the main areas of focus.
Materials and methods
Clinical specimens
A total of 62 primary pancreatic cancer tissues and
20 adjacent normal tissues located >2 cm away from cancer
tissues were obtained from patients (38 males and 24 females; age
range, 38–72 years; median age, 58.65 years) who underwent surgical
resection at Shanxi Dayi Hospital Affiliated to Shanxi Medical
University between January 2016 and June 2018. All tissue specimens
were examined by pathologists and diagnosed as pancreatic ductal
adenocarcinomas. No antineoplastic treatment was administered to
the patients prior to the operation. The present study was reviewed
and ethically approved by the Shanxi Dayi Hospital Ethics
Committee. Informed written consent was obtained from each patient
and his/her family.
Cell culture
Human pancreatic cancer cell lines (SW1990, PANC-1,
BxPC-3 and Aspc-1) and human pancreatic ductal epithelial cell line
(HPDE6c7) were from the American Type Culture Collection. All cell
lines were cultured in RPMI 1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 100 U/ml penicillin and 100 U/ml
streptomycin, supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) in a humidified atmosphere of 5% CO2
at 37°C.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA of pancreatic cancer cells was extracted
using TRIzol® reagent (Takara Biotechnology Co., Ltd.)
according to the instructions of the manufacturer. A PrimeScript RT
Reagent kit (Takara Biotechnology Co., Ltd.) was used for the
reverse transcription of mRNA into cDNA at 37°C for 60 min and 85°C
for 1 min. Gene amplification was then performed. qPCR was
performed using a SYBR PrimeScript RT-PCR Kit (Takara Biotechnology
Co., Ltd.) according to the instructions of the manufacturer. The
reaction started at 95°C for 5 min, followed by 35 cycles of 95°C
for 45 sec, 59°C for 30 sec, and 72°C for 45 sec. The
quantification was performed using the 2−ΔΔCq method
(12), and β-actin was used as the
internal reference gene to normalize the results. The primer
sequences are presented in Table
I.
 | Table I.Sequences of primers. |
Table I.
Sequences of primers.
| Gene | Primer sequence
(5′→3′) | Amplicon size,
bp |
|---|
| HPA | F:
GAATGGACGGACTGCTAC | 261 |
|
| R:
CCAAAGAATACTTGCCTCA |
|
| FGF2 | F:
AGCGGCTGTACTGCAAAAAC | 338 |
|
| R:
CCCAGGTCCTGTTTTGGAT |
|
| Palladin | F:
GCATCAGAGCTGACCTCAAC | 332 |
|
| R:
GGTTGATCACCGACGCTAAT |
|
| Akt | F:
GCGACAGTGGCTATTGTGA | 232 |
|
| R: AGCTGCTCAAAGATGC
CATT |
|
| SDC1 | F:
AGCTGACCTTCACACTCC | 369 |
|
| R:
TCGGCTCCTCCAAGGAGT |
|
| β-actin | F:
GTCAGTGCATCACTACTGGCAAT | 312 |
|
| R: CGATCTGGTAG
GGCCTTG |
|
Vector construction and cell
transfection
The PANC-1 cell line (2×105 cells/well)
in logarithmic growth phase was inoculated in a 6-well plate for 24
h. HPA silencing vectors were constructed using three short hairpin
(sh)RNAs (50 nM; Shanghai GeneChem Co., Ltd; Table II) specifically targeting HPA. The
HPA overexpression vector was constructed using a recombinant
plasmid encoding HPA (50 nM; cat. no. GV147; Shanghai GeneChem Co.,
Ltd.), and the empty vector was used as a negative control. The
PANC-1 cell line was then transfected to construct a silent (shHPA
group) or overexpressed (HPA group) cell line. The PANC-1 cell line
was transfected with an empty vector containing the GFP sequence as
a control (vector group). FGF2 silencing vectors were constructed
using (sh)RNA (5′-ACTACAATACTTACCGGTCAA-3′) (50 nM; Shanghai
GeneChem Co., Ltd) specifically targeting FGF2. The PANC-1 cell
line was then transfected to construct a silent (shFGF2 group) cell
line. The SDCI inhibitor synstatin (100 nM; Beyotime Institute of
Biotechnology) was added to the HPA group to form the HPA +
synstatin group. The FGF2 receptor (FGF2R) inhibitor AZD4547 and
the PI3K/Akt-specific inhibitor LY294002 were purchased from
Sigma-Aldrich; Merck KGaA. PANC-1 cells were divided into the
following groups: i) FGF2 group [PANC-1 cell line transfected with
FGF2 recombinant plasmid (100nM; cat. no. QCP5411; QCHENG BIO
Inc.)]; ii) AZD4547 group (3.0 nmol-l AZD4547 added to the FGF2
group); iii) LY294002 group (10 µmol-l LY294002 added to the FGF2
group); and iv) control group [PANC-1 cell line transfected with
empty plasmid (cat. no. QCP5400; QCHENG BIO Inc.]. Transfection was
performed using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the instructions of the
manufacturer. Subsequent experiments were performed 48 h after
transfection.
| Table II.Target sequences of shHPA. |
Table II.
Target sequences of shHPA.
| ID | Target sequence
(5′→3′) | Initial
position | GC content (%) |
|---|
| sh1 |
GCTCTGTAGATGTGCTATACA |
640 | 42.86 |
| sh2 |
GCATCACTACTATTTGAATGG | 1022 | 38.1 |
| sh3 |
GCTTGCCAGCTTTCTCATATA | 1705 | 42.86 |
Immunohistochemical staining
Tumor tissues were fixed with 10% formalin solution
for 15 min at room temperature, and then blocked in wash buffer
containing 5% normal goat serum (Shanghai GeneChem Co., Ltd.) for 2
h at room temperature. Tumor tissues were incubated with rabbit
anti-human HPA primary antibody (1:100; cat. no. PB0405) and rabbit
anti-human FGF2 primary antibody (1:100; cat. no. PB0916; both
Boster Biological Technology) at 4°C overnight, and then incubated
with goat anti-rabbit IgG secondary antibody (1:200; cat. no.
BA1003; Boster Biological Technology) for 2 h at room temperature.
The tissue was then stained with diaminobenzidine, and the nuclei
were stained with hematoxylin for 5 min at room temperature and
then observed using an optical microscope (Olympus Corporation) at
×200 magnification. Immunohistochemical staining was performed
according to the protocols of the manufacturer (Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd.; OriGene Technologies, Inc.).
The following scoring system was used to evaluate the staining
results: The staining intensity of HPA and FGF2 was rated 0–3 (0,
negative; 1, weak; 2, moderate; or 3, strong), while marker
expression was evaluated according to the percentage of the stained
area: 0, 0–10%; 1, 11–25%; 2, 26–50%; or 3, 50–100%. The final
result was calculated as the sum of the two scores. The rating
criteria were as follows: 0–3, low expression; and 4–6, high
expression. The results were evaluated by two pathologists and the
differences were re-evaluated until a consensus was reached.
Western blot analysis
Total protein was extracted by lysing the cells with
RIPA buffer (Beyotime Institute of Biotechnology) and the protein
concentration was measured using a BCA protein assay kit (Beyotime
Institute of Biotechnology). A total of 50 µg protein from each
sample was separated by SDS-PAGE (10% gel) and the proteins were
transferred to a nitrocellulose membrane by electrophoretic
transfer. After blocking with 5% skimmed milk for 2 h at room
temperature, the membranes were incubated with the following
primary antibodies: Rabbit anti-human HPA (1:1,000; cat. no.
PB0405), rabbit anti-human FGF2 (1:1,000; cat. no. PB0916), rabbit
anti-human AKT (1:5,000; cat. no. A00024-1), rabbit anti-human
E-cadherin (1:100; cat. no. BA0475), rabbit anti-human N-cadherin
(1:100; cat. no. BM3921), rabbit anti-human vimentin 1:3,000 (cat.
no. PB0378; all Boster Biological Technology) and rabbit anti-human
palladin (1:100; cat. no. PA5-65160; Thermo Fisher Scientific Inc.)
were added according to the instructions of the manufacturer and
incubated at 4°C overnight. β-actin (1:500; cat. no. BA2305; Boster
Biological Technology) was used as an internal loading control. The
goat anti-rabbit IgG secondary antibody (1:5,000; cat. no. BA1056;
Boster Biological Technology) labeled with horseradish peroxidase
was added to the membrane after washing and incubated at room
temperature. The gray value of the protein bands was analyzed by
ImageJ software (v1.8.0; National Institutes of Health) and the
ratio of the gray value of the target band to that of the internal
reference β-actin was representative of the expression level of the
protein. All assays were performed in triplicate.
Transwell chamber assay
The invasive ability of PANC-1 cells was measured
using Transwell chambers (Corning Inc.). PANC-1 cells
(2×105 cells/ml) starved for 24 h were inoculated into
the upper chamber of a 24-well chamber, and each group was analyzed
in 6 replicate wells. A total of 50 µl Matrigel solution was placed
on the polycarbonate microporous membrane between the upper and the
lower chambers, 100 µl RPMI 1640 medium was added to the upper
chamber, and a mixture of 600 µl RPMI 1640 and 20% fetal bovine
serum was added to the lower chamber. After incubation for 24 h at
room temperature, the cells were fixed with a 4% paraformaldehyde
solution for 30 min at room temperature, followed by rinsing with
PBS and staining with a 0.5% crystal violet dye solution for 10 min
at room temperature. Subsequently, the lower chamber was observed
and photographed with a light microscope at ×200 magnification, and
cell counting was performed.
Wound healing assay
PANC-1 cells were seeded in a 6-well plate at an
adjusted concentration of 1×106 cells/ml. The experiment
was performed when the cells reached 100% confluence. A wound was
created in the center of the plate with the tip of a 100-ml
pipette. The scratched cells were washed away in serum-free medium
and this process was repeated 3 times, and then cultured in regular
medium. The wound was observed at ×200 magnification with an
inverted microscope at 0 and 48 h, and images were captured.
Statistical analysis
SPSS software (version 23.0; IBM Corp.) was used for
statistical analysis throughout the study. Statistical analysis of
HPA and FGF2 expression between pancreatic cancer tissues and
paired adjacent normal tissues was evaluated using a paired
Student's t-test. One-way analysis of variance was performed on ≥3
sets of data using Tukey's post hoc test. GraphPad Prism software
(version 8.0; GraphPad Software, Inc.) was used to generate
graphical representations. Each experiment was repeated ≥3 times.
The experimental results are presented as mean ± SD values.
P<0.05 was considered to indicate a statistically significant
difference.
Results
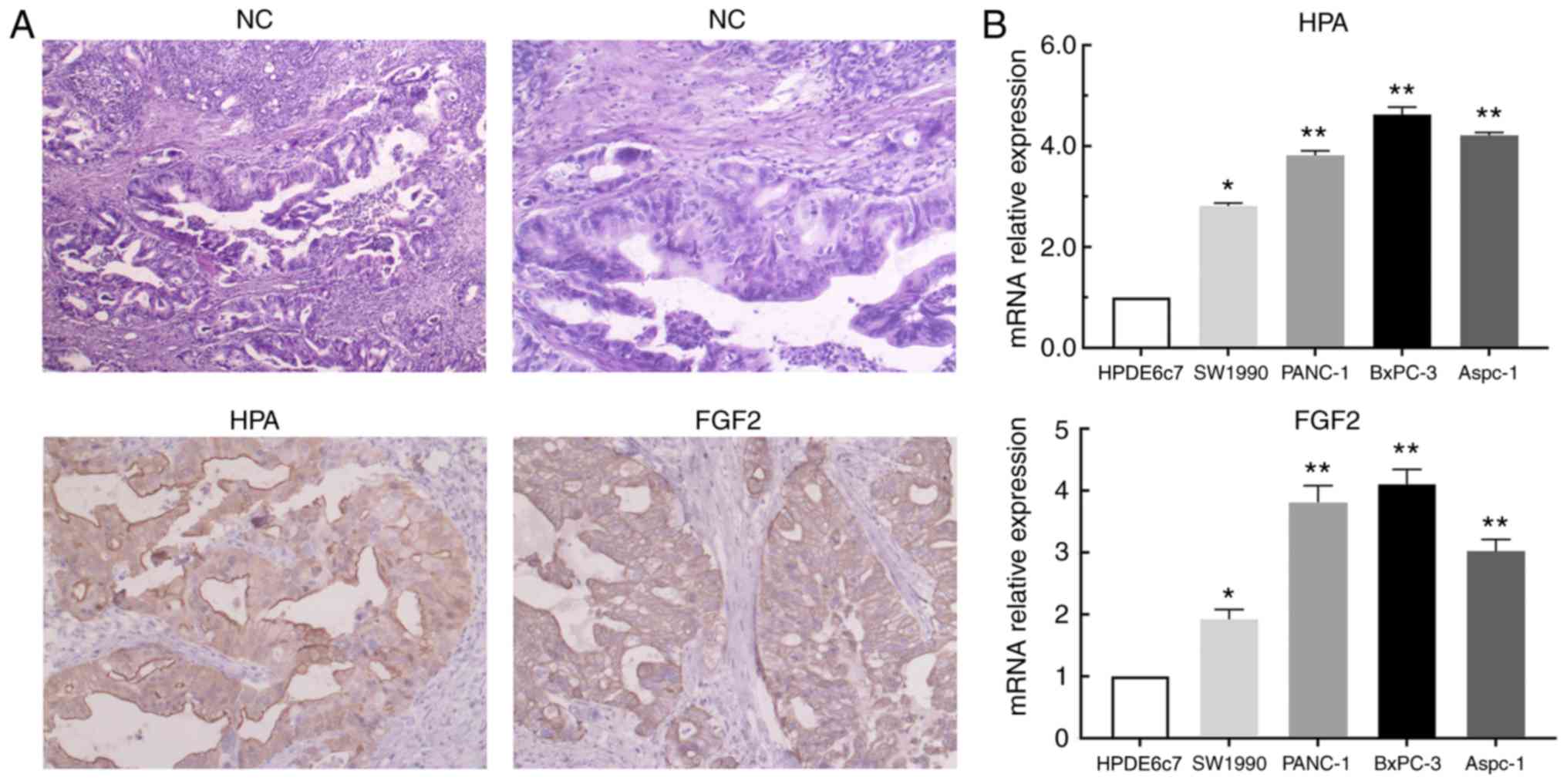
HPA and FGF2 are upregulated in
pancreatic cancer tissues and cells
In order to investigate the expression of HPA and
FGF2 in pancreatic cancer tissues and cells, immunohistochemical
staining and RT-qPCR analysis were performed. According to the
results of immunohistochemistry, significant upregulation of HPA
and FGF2 was observed in pancreatic cancer tissues compared with
adjacent non-tumor tissues (Fig.
1A). Based on the aforementioned scoring criteria, the details
of HPA and FGF2 expression are presented in Table III. The results of RT-qPCR analysis
revealed that HPA and FGF2 expression levels were significantly
higher in pancreatic cancer cell lines than in the HPDE6c7 cell
line (Fig. 1B). These results
implied that the expression of HPA and FGF2 in pancreatic cancer
was markedly upregulated.
| Table III.Expression of heparanase and
fibroblast growth factor 2 in pancreatic cancer tissues and
adjacent normal tissues. |
Table III.
Expression of heparanase and
fibroblast growth factor 2 in pancreatic cancer tissues and
adjacent normal tissues.
| Parameter | Patients, n | Positive | Negative | Positive rate
(%) |
χ2-value | P-value |
|---|
| HPA |
|
|
|
| 31.236 | <0.001 |
|
Tumor | 62 | 47 | 15 | 75.806 |
|
|
|
Non-tumor | 20 | 1 | 19 | 5.000 |
|
|
| FGF2 |
|
|
|
| 32.529 | <0.001 |
|
Tumor | 62 | 50 | 12 | 80.645 |
|
|
|
Non-tumor | 20 | 2 | 18 | 10.000 |
|
|
Association of HPA and FGF2 expression
with clinicopathological characteristics of patients with
pancreatic cancer
To further explore whether elevated HPA and FGF2
expression levels are directly associated with the
clinicopathological characteristics of pancreatic cancer, the
expression of HPA and FGF2 in different pancreatic cancer tissues
was examined via immunohistochemistry. The results revealed that
the positive rate of HPA in tissues with lymph node metastasis was
significantly higher than that in tissues without lymph node
metastasis. Tumor-node-metastasis (TNM) stage III–IV tissues were
found to have higher HPA-positive expression than TNM stage I–II
tissues. Positive expression of HPA was not associated with the
age, sex, tumor location or tumor differentiation status of
patients with pancreatic cancer. By contrast, the positive
expression rate of FGF2 was observed to increase as the degree of
tumor differentiation decreased. Moreover, similarly to HPA, the
age, sex or tumor location of the patients with pancreatic cancer
were not associated with FGF2 expression (Table IV).
| Table IV.Association between heparanase and
fibroblast growth factor 2 expression, and clinicopathological
characteristics of patients with pancreatic cancer. |
Table IV.
Association between heparanase and
fibroblast growth factor 2 expression, and clinicopathological
characteristics of patients with pancreatic cancer.
|
|
| HPA expression | FGF2
expression |
|---|
|
|
|
|
|
|---|
| Parameter | Patients, n | Positive | Negative | χ2-value
(HPA) | P-value (HPA) | Positive | Negative | χ2
-value (FGF2) | P-value (FGF2) |
|---|
| Age, years |
|
|
| 0.067 | 0.800 |
|
|
0.223 |
0.637 |
|
≤55 | 19 | 14 | 5 |
|
| 16 | 3 |
|
|
|
>55 | 43 | 33 | 10 |
|
| 34 | 9 |
|
|
| Sex |
|
|
| 0.01 | 0.906 |
|
|
0.181 |
0.670 |
|
Male | 38 | 29 | 9 |
|
| 30 | 8 |
|
|
|
Female | 24 | 18 | 6 |
|
| 20 | 4 |
|
|
| Tumor site |
|
|
| 2.479 | 0.115 |
|
|
2.662 |
0.103 |
| Head of
Pancreas | 39 | 27 | 12 |
|
| 29 | 10 |
|
|
|
Body-Tail of Pancreas | 23 | 20 | 3 |
|
| 21 | 2 |
|
|
| Differentiation
degree |
|
|
| 1.159 | 0.560 |
|
| 21.300 | <0.001 |
|
High | 18 | 12 | 6 |
|
| 8 | 10 |
|
|
|
Moderate | 20 | 16 | 4 |
|
| 19 | 1 |
|
|
|
Poor | 24 | 19 | 5 |
|
| 23 | 1 |
|
|
| TNM stage |
|
|
| 8.961 | 0.003 |
|
|
7.436 |
0.006 |
|
I–II | 25 | 14 | 11 |
|
| 16 | 9 |
|
|
|
III–IV | 37 | 33 | 4 |
|
| 34 | 3 |
|
|
| Lymph node
metastasis |
|
|
| 4.931 | 0.026 |
|
|
7.276 |
0.007 |
|
With | 32 | 28 | 4 |
|
| 30 | 2 |
|
|
|
Without | 30 | 19 | 11 |
|
| 20 | 10 |
|
|
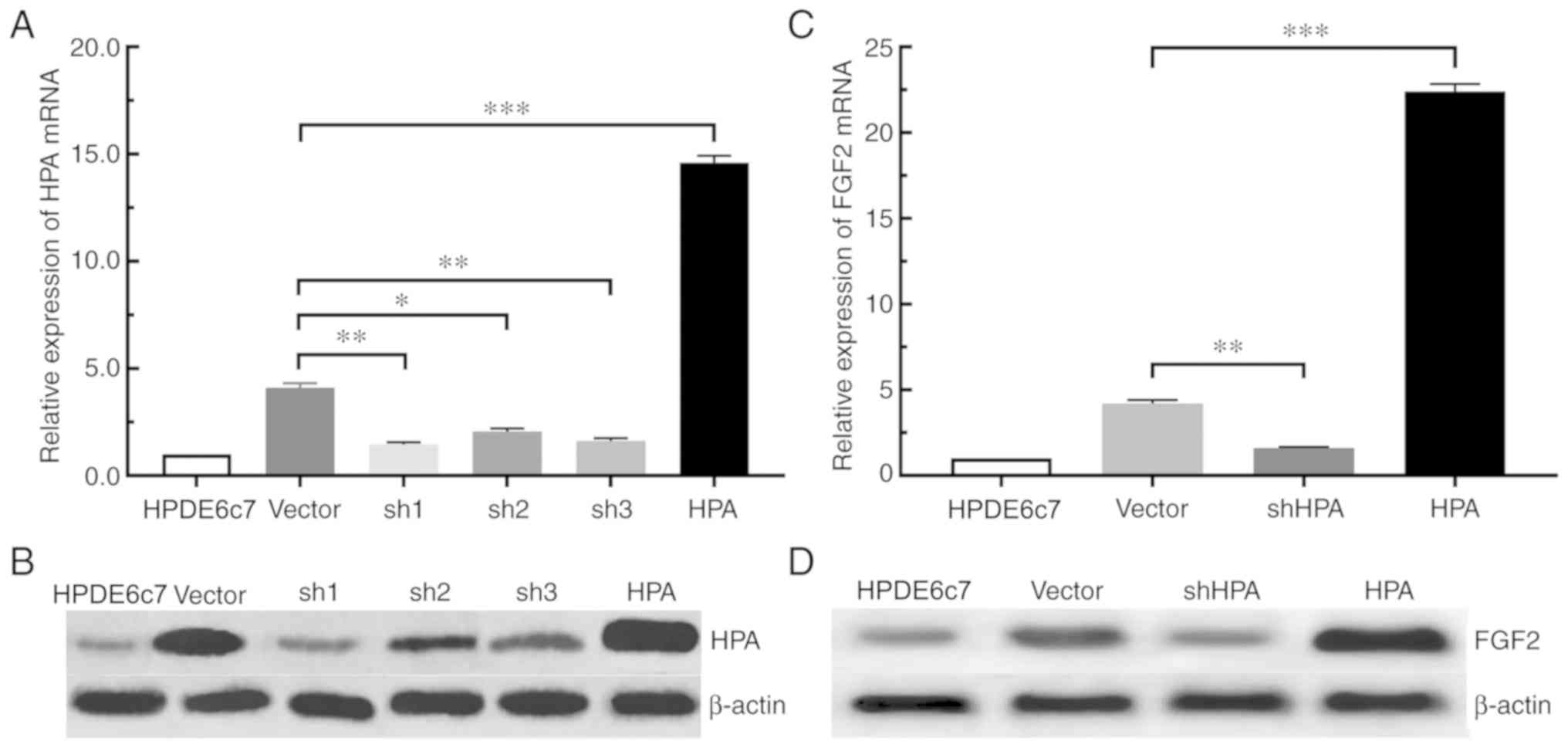
HPA is capable of upregulating the
expression of FGF2 in pancreatic cancer cells
According to the aforementioned experimental
results, the expression of HPA and FGF2 in pancreatic cancer cells
was significantly increased. For the purpose of determining whether
the expression of FGF2 was affected by HPA, the expression of FGF2
in the shHPA, HPA and vector groups was compared. Among the three
PANC-1 shHPA cell lines selected, the sh1 group had the highest
silencing efficiency, compared with the sh2 and sh3 groups
(Fig. 2A and B). The RT-qPCR and
western blot analyses demonstrated that the expression of FGF2 was
significantly increased in the HPA group compared with the vector
group. On the contrary, HPA silencing significantly inhibited the
expression of FGF2 in the shHPA group compared with the vector
group (Fig. 2C and D). Taken
together, these results suggested that HPA is able to increase the
expression of FGF2, and that HPA silencing results in the
downregulation of FGF2 in pancreatic cancer cells.
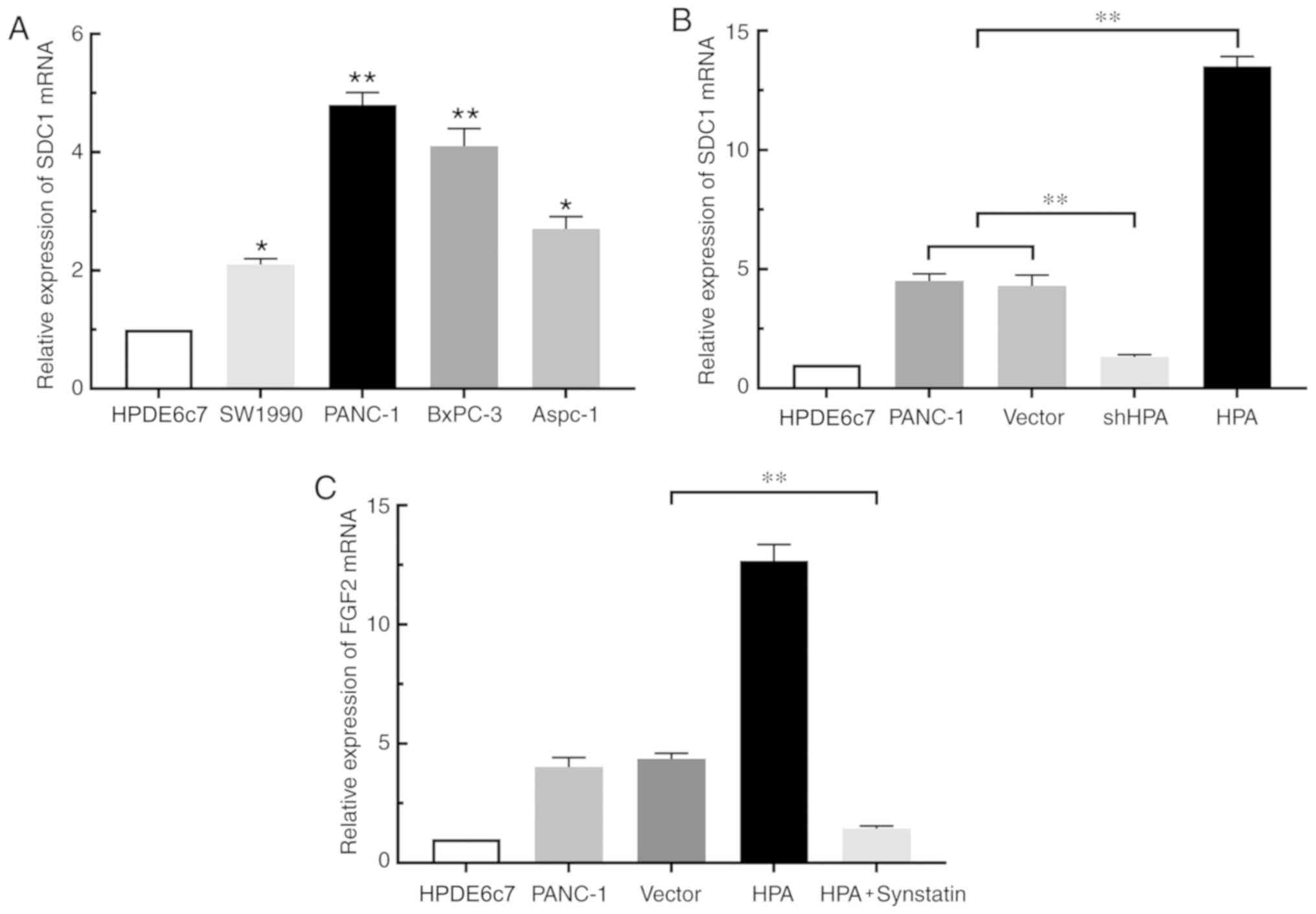
HPA regulates FGF2 expression via the
HPA/SDC1 axis in pancreatic cancer cells
By binding to SDC1, HPA removes this proteoglycan
from the cell surface and promotes its expression (13). To confirm the study hypothesis that
HPA affects the expression of FGF2 in pancreatic cancer cells by
binding to SDC1, RT-qPCR was performed. The results revealed that
SDC1 expression was increased in pancreatic cancer cell lines
compared with the HPDE6c7 cell line. (Fig. 3A). Overexpression of HPA resulted in
a significant increase in SDC1 expression in the HPA group compared
with the vector group, whereas HPA silencing considerably
suppressed the expression of SDC1 (Fig.
3B). Moreover, the expression of FGF2 was markedly decreased in
the HPA + synstatin group compared with that in the vector group
(Fig. 3C). Taken together, these
findings indicated that HPA may promote the expression of FGF2 by
upregulating SDC1 in pancreatic cancer cells. Notably, neither
overexpression nor silencing of FGF2 inflenced HPA expression
(Fig. S3)
HPA promotes pancreatic cancer cell
migration and invasion by upregulating FGF2 to activate the
PI3K/Akt pathway and EMT process
Increased expression of Palladin in
pancreatic cancer tissues and cells
A previous study demonstrated that Palladin is
highly expressed in pancreatic cancer cells (14). Moreover, the PI3K/Akt pathway has
been observed to maintain the stability of Palladin and promote its
expression (15). It was
hypothesized that FGF2 promotes the expression of Palladin via the
PI3K/Akt pathway, thereby promoting the invasion and migration of
pancreatic cancer cells. According to the western blot results, the
expression of Palladin in pancreatic cancer cells was significantly
higher than in the HPDE6c7 cell line (Fig. 4A). This result was further confirmed
via RT-qPCR analysis (Fig. 4B).
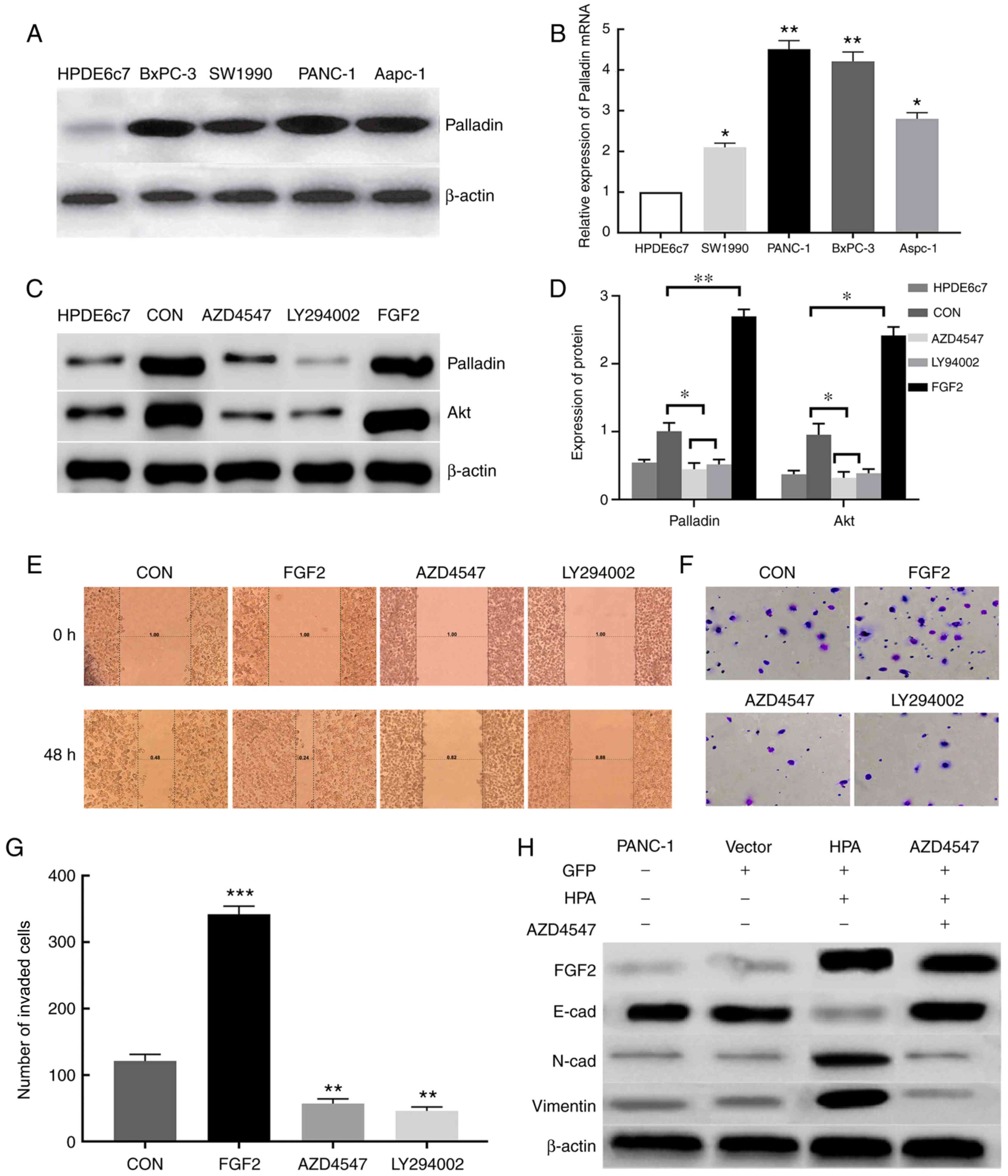 | Figure 4.HPA promotes cell migration and
invasion by activating the PI3K/Akt signaling pathway and EMT
processes via FGF2 upregulation. (A) Expression of Palladin protein
in various pancreatic cancer cell lines was analyzed by western
blot assay. β-actin was used as an internal control. (B) Analysis
of the relative expression of Palladin mRNA in pancreatic cancer
cell lines by reverse transcription-quantitative PCR. (C) Western
blot analysis of the effect of FGF2 on Palladin and Akt. (D)
Protein expression of Palladin and Akt. β-actin was used as an
internal control. (E) Effect of FGF2, AZD4547 and LY294002 on the
migratory ability of PANC-1 cells assessed by wound healing assay.
Magnification, ×200. (F) Transwell assays were performed to
determine the invasive ability of PANC-1 cells treated with FGF2,
AZD4547 and LY294002. (G) Number of invaded PANC-1 cells treated
with FGF2, AZD4547 and LY294002. CON, PANC-1 cell line transfected
with empty plasmid. (H) Western blot analysis of the effect of HPA
and FGF2 on EMT. Vector, PANC-1 cell line transfected with empty
plasmid. β-actin was used as an internal control. *P<0.05,
**P<0.01, ***P<0.001. HPA, heparanase; FGF2, fibroblast
growth factor 2; EMT, epithelial-mesenchymal transition; CON,
PANC-1 cell line transfected with empty plasmid; Vector, PANC-1
cell line transfected with empty plasmid containing the GFP
sequence. |
FGF2 promotes pancreatic cancer cell
invasion and migration via the PI3K/Akt pathway
Successful overexpression of FGF2 in FGF2 group
cells was determined via RT-qPCR and western blot analyses
(Fig. S1). According to the western
blot analysis, the expression of Palladin and Akt proteins in the
FGF2 group was significantly increased compared with the other
groups (Fig. 4C and D). This result
was also confirmed by RT-qPCR (Fig.
S2). The invasive and migratory abilities of pancreatic cancer
cells were assessed by Transwell chamber and wound healing assays.
When FGF2 was overexpressed in the FGF2 group, the invasive and
migratory abilities of pancreatic cancer cells were significantly
enhanced compared with the control group. However, these abilities
of pancreatic cancer cells were markedly reduced in the AZD4547 and
LY294002 groups compared with in the FGF2 and control groups.
(Fig. 4E-G). These results indicated
that FGF2 may regulate the invasion and migration of pancreatic
cancer cells via the PI3K/Akt signaling pathway, thus promoting the
progression of pancreatic cancer.
HPA is able to activate the EMT
process by upregulating FGF2
EMT is closely associated with cell migration and
invasion. To examine the effects of HPA and FGF2 on EMT, western
blot analysis was conducted in order to detect the markers of EMT.
The expression of E-cadherin was found to be inhibited, whereas the
expression of N-cadherin and vimentin was considerably increased
after HPA overexpression; these effects were reversed by adding
AZD4547 (Fig. 4H). These results
demonstrated that HPA may activate the EMT process by upregulating
the expression of FGF2.
Discussion
ECM and BM degradation is a prerequisite for tumor
metastasis (16). The secretion of
HPA is important for tumors to break down ECM and BM (7). HPA is the only endoglycosidase in the
human body that is able to degrade the HS chain in
glycosaminoglycans (8). It
specifically recognizes sites on the HS side chain and cleaves HS,
resulting in the formation of short fragments with a length of
10–20 sugar units, which release certain growth factor (including
fibroblast growth factor and hepatocyte growth factor) bound to HS,
thereby promoting tumor invasion and metastasis (16,17).
HPA-mediated tumor metastasis may be influenced by a number of
factors, including the following: i) Dysregulation of HPA may
mediate the release of angiogenic factors including vascular
endothelial growth factor and cyclooxygenase-2, thus facilitating
the regeneration of tumor microenvironment vessels (18,19); ii)
HPA promotes fibrosis, and regulates the expression of transforming
growth factor-β and other factors, creating a hypoxic
microenvironment that is conducive to tumor growth and resulting in
degradation of the normal tissue structure (while preventing drug
uptake), further promoting the chemoresistance of tumors due to the
fibrosis surrounding them (20); and
iii) HPA cleaves the HS proteoglycan, consequently destroying the
normal structure of ECM and BM, which are protective structures
that hinder tumor cell migration (21). In addition, HPA may enhance tumor
invasion and metastasis via various mechanisms, which include
promoting the progression of inflammation and the formation of
exosomes and increasing the chemical resistance of tumors (22,23).
In recent years, the role of growth factors in the
progression of tumors is an area that has gained great interest
(24,25). After binding to their specific
receptors, growth factors may affect a series of biological
activities by activating downstream signaling pathways (26). As a member of the growth factor
family, FGF2 has been detected in several types of cancer,
including breast and gastric cancer, and FGF2 is highly expressed
in these types of cancers (27,28). A
limited number of studies have also reported the detection of FGF2
in pancreatic cancer (29,30). The present study demonstrated that
HPA and FGF2 were highly expressed in pancreatic cancer tissues and
cell lines. HPA overexpression resulted in increased FGF2
expression. A study by Li et al (31) also confirmed the present study's
hypothesis; it revealed that in rheumatoid arthritis, upregulation
of HPA can significantly promote the expression of FGF2.
Conversely, HPA silencing was observed to mediate a reduction of
FGF2. The present study also analyzed the impact of FGF2
dysregulation on HPA. Notably, neither overexpression nor silencing
of FGF2 had an effect on HPA expression, a result that differed
from the findings reported by Masola et al (11). Masola et al believe that HPA
can promote the expression of FGF2 in renal fibrosis, and the high
expression of FGF2 can further promote the expression of HPA. This
difference may be due to the pancreatic cancer cell lines selected
for the present study. Kato et al (32) revealed that HPA is capable of
converting SDC1 into an activator of FGF2 in certain cases. It was
hypothesized that HPA regulated FGF2 via SDC1 in pancreatic cancer.
In the PANC-1 cell line, the expression level of SDC1 increased
when HPA was overexpressed, whereas HPA silencing had the opposite
effect. SDC1 inhibitors were added to the HPA overexpression group
to detect the effect of SDC1 on FGF2, and it was observed that the
expression of FGF2 significantly decreased, a result which was
consistent with the study hypothesis.
It has been reported that intracellular FGF2
signaling is activated by mitogen-activated protein kinases,
PI3K/Akt and phosphatase Cγ signaling pathways. In these pathways,
PI3K/Akt signaling influences tumor cell invasion and migration,
and is closely associated with FGF2-induced EMT (33,34).
Treatment of PANC-1 cells with FGF2 was found to significantly
enhance cell migration and invasion. By contrast, the invasion and
migration of PANC-1 cells were markedly suppressed after treatment
with the FGF2R inhibitor AZD4547 and the PI3K/Akt signaling
pathway-specific inhibitor LY294002, respectively. The present
results indicated that HPA is able to induce cell invasion and
migration by upregulating FGF2, which in turn activates the
PI3K/Akt signaling pathway in pancreatic cancer cells.
The cell scaffold protein Palladin serves an
important role in wound healing, development of chronic
inflammation and progression of malignant tumors (35,36). A
previous study showed that Palladin protein is highly expressed in
breast cancer tissues, and its expression level is linked to the
degree of invasion and metastasis of breast cancer (37). PI3K/Akt signaling induces the
expression of Palladin, thereby enhancing cell motility, and
promoting the migration and invasion of cancer cells (15). In the present study, PANC-1 cells
were divided into groups, subjected to different interventions and
compared. FGF2 was able to promote the expression of Palladin by
activating the PI3K/Akt signaling pathway, a result which is
consistent with the study hypothesis.
Previous reports have demonstrated that, during EMT
activation, the expression of E-cadherin and certain cytokeratins
(vimentin, fbronectin, twist and slug) is diminished, whereas the
expression of mesenchymal markers is activated (38,39). The
most prominent mesenchymal markers are N-cadherin, vimentin,
fibronectin and integrins (40,41).
Western blot analysis was conducted to detect the expression of the
epithelial phenotype marker E-cadherin, and the mesenchymal
phenotype markers N-cadherin and vimentin. According to the current
results, using FGF2R inhibitor AZD4547 to prevent the binding of
FGF2 to FGF2R, was capable of reversing the downregulation of
E-cadherin and the upregulation of N-cadherin and vimentin induced
by HPA overexpression, thus inhibiting the activation of EMT.
According to Thompson et al (42), EMT serves a major role in tumor
development. In conclusion, it was hypothesized that HPA activates
EMT by upregulating the expression of FGF2, thereby promoting the
migration and invasion of pancreatic cancer cells.
However, the present study had certain limitations,
including the small sample of matched paracancerous tissues, lack
of correlation analysis between target gene/protein and prognosis
and an absence of in vivo experiments. Future studies should
further verify the relevant mechanisms, use animal models to
confirm the relevant pathways and conduct prognostic correlation
analysis.
In conclusion, the present study demonstrated that
HPA and FGF2 are highly expressed in pancreatic cancer tissues and
cells, and are closely associated with the clinicopathological
characteristics of pancreatic cancer. Moreover, the HPA/SDC1 axis
was observed to mediate the upregulation of FGF2, which in turn
activated the EMT process, and promoted the invasion and migration
of pancreatic cancer cells. The results of the present study
indicate that the HPA/SDC1/FGF2 axis may be a valuable therapeutic
target for pancreatic cancer.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr. Yumeng Jing (The
First Affiliated Hospital of Shanxi Medical University, Taiyuan
China) for critically reviewing the pathology results. The authors
would also like to thank Mr. Yuan Jia of the general surgery
laboratory teacher of Shanxi University Hospital (Taiyuan China),
for his guidance and technical support.
Funding
The present study was supported by the Shanxi
Province ‘136’ Xingyi Medical Engineering Leading Specialist and
Shanxi Basic Research Project (grant no. 2015011131).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HLZ and XC designed the study. XC, CC, JL and HCZ
performed the experiments. XC wrote the paper. HLZ, JH and XF
reviewed and edited the manuscript. JH and XF conducted statistical
analysis of the data. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The present study was reviewed and ethically
approved by the Shanxi Dayi Hospital Ethics Committee. Informed
written consent was obtained from each patient and his/her
family.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McGuigan A, Kelly P, Turkington RC, Jones
C, Coleman HG and McCain RS: Pancreatic cancer: A review of
clinical diagnosis, epidemiology, treatment and outcomes. World J
Gastroenterol. 24:4846–4861. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Puleo F, Maréchal R, Demetter P, Bali MA,
Calomme A, Closset J, Bachet JB, Deviere J and Van Laethem JL: New
challenges in perioperative management of pancreatic cancer. World
J Gastroenterol. 21:2281–2293. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Szatmári T, Mundt F, Kumar-Singh A, Möbus
L, Ötvös R, Hjerpe A and Dobra K: Molecular targets and signaling
pathways regulated by nuclear translocation of syndecan-1. BMC Cell
Biol. 18:342017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ibrahim SA, Gadalla R, El-Ghonaimy EA,
Samir O, Mohamed HT, Hassan H, Greve B, El-Shinawi M, Mohamed MM
and Götte M: Syndecan-1 is a novel molecular marker for triple
negative inflammatory breast cancer and modulates the cancer stem
cell phenotype via the IL-6/STAT3, Notch and EGFR signaling
pathways. Mol Cancer. 16:572017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jung O, Trapp-Stamborski V, Purushothaman
A, Jin H, Wang H, Sanderson RD and Rapraeger AC: Heparanase-induced
shedding of syndecan-1/CD138 in myeloma and endothelial cells
activates VEGFR2 and an invasive phenotype: Prevention by novel
synstatins. Oncogenesis. 5:e2022016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meirovitz A, Hermano E, Lerner I, Zcharia
E, Pisano C, Peretz T and Elkin M: Role of heparanase in
radiation-enhanced invasiveness of pancreatic carcinoma. Cancer
Res. 71:2772–2780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Quiros RM, Rao G, Plate J, Harris JE,
Brunn GJ, Platt JL, Gattuso P, Prinz RA and Xu X: Elevated serum
heparanase-1 levels in patients with pancreatic carcinoma are
associated with poor survival. Cancer. 106:532–540. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hegab AE, Ozaki M, Kameyama N, Gao J,
Kagawa S, Yasuda H, Soejima K, Yin Y, Guzy RD, Nakamura Y, et al:
Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its
cancer-associated fibroblasts. J Pathol. 249:193–205. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
La Venuta G, Zeitler M, Steringer JP,
Müller HM and Nickel W: The startling properties of fibroblast
growth factor 2: How to exit mammalian cells without a signal
peptide at hand. J Biol Chem. 290:27015–27020. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Masola V, Zaza G, Secchi MF, Gambaro G,
Lupo A and Onisto M: Heparanase is a key player in renal fibrosis
by regulating TGF-β expression and activity. Biochim Biophys Acta.
1843:2122–2128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang Y, MacLeod V, Miao HQ, Theus A, Zhan
F, Shaughnessy JD Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I and
Sanderson RD: Heparanase enhances syndecan-1 shedding: A novel
mechanism for stimulation of tumor growth and metastasis. J Biol
Chem. 282:13326–13333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goicoechea SM, García-Mata R, Staub J,
Valdivia A, Sharek L, McCulloch CG, Hwang RF, Urrutia R, Yeh JJ,
Kim HJ and Otey CA: Palladin promotes invasion of pancreatic cancer
cells by enhancing invadopodia formation in cancer-associated
fibroblasts. Oncogene. 33:1265–1273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chin YR and Toker A: Akt2 regulates
expression of the actin-bundling protein Palladin. FEBS Lett.
584:4769–4774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bame KJ: Heparanases: Endoglycosidases
that degrade heparan sulfate proteoglycans. Glycobiology.
11:91R–98R. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nasser NJ: Heparanase involvement in
physiology and disease. Cell Mol Life Sci. 65:1706–1715. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Purushothaman A, Uyama T, Kobayashi F,
Yamada S, Sugahara K, Rapraeger AC and Sanderson RD:
Heparanase-enhanced shedding of syndecan-1 by myeloma cells
promotes endothelial invasion and angiogenesis. Blood.
115:2449–2457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Okawa T, Naomoto Y, Nobuhisa T, Takaoka M,
Motoki T, Shirakawa Y, Yamatsuji T, Inoue H, Ouchida M, Gunduz M,
et al: Heparanase is involved in angiogenesis in esophageal cancer
through induction of cyclooxygenase-2. Clin Cancer Res.
11:7995–8005. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masola V, Zaza G, Onisto M, Lupo A and
Gambaro G: Impact of heparanase on renal fibrosis. J Transl Med.
13:1812015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gingis-Velitski S, Zetser A, Kaplan V,
Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY,
Vlodavsky I and Ilan N: Heparanase uptake is mediated by cell
membrane heparan sulfate proteoglycans. J Biol Chem.
279:44084–44092. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meirovitz A, Goldberg R, Binder A,
Rubinstein AM, Hermano E and Elkin M: Heparanase in inflammation
and inflammation-associated cancer. FEBS J. 280:2307–2319. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sanderson RD, Elkin M, Rapraeger AC, Ilan
N and Vlodavsky I: Heparanase regulation of cancer, autophagy and
inflammation: New mechanisms and targets for therapy. FEBS J.
284:42–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rugo HS, Delord JP, Im SA, Ott PA,
Piha-Paul SA, Bedard PL, Sachdev J, Tourneau CL, van Brummelen EMJ,
Varga A, et al: Safety and antitumor activity of pembrolizumab in
patients with estrogen receptor-positive/human epidermal growth
factor receptor 2-negative advanced breast cancer. Clin Cancer Res.
24:2804–2811. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qu Y, Zhang H, Sun W, Han Y, Li S, Qu Y,
Ying G and Ba Y: MicroRNA-155 promotes gastric cancer growth and
invasion by negatively regulating transforming growth factor-β
receptor 2. Cancer Sci. 109:618–628. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nandy D and Mukhopadhyay D: Growth factor
mediated signaling in pancreatic pathogenesis. Cancers (Basel).
3:841–871. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Khurana A, Liu P, Mellone P, Lorenzon L,
Vincenzi B, Datta K, Yang B, Linhardt RJ, Lingle W, Chien J, et al:
HSulf-1 modulates FGF2- and hypoxia-mediated migration and invasion
of breast cancer cells. Cancer Res. 71:2152–2161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li T, Meng XL and Yang WQ: Long noncoding
RNA PVT1 acts as a ‘Sponge’ to inhibit microRNA-152 in gastric
cancer cells. Dig Dis Sci. 62:3021–3028. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Coleman SJ, Chioni AM, Ghallab M, Anderson
RK, Lemoine NR, Kocher HM and Grose RP: Nuclear translocation of
FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic
cancer cell invasion. EMBO Mol Med. 6:467–481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kleeff J, Friess H, Buechler M, Kothari N,
Datta S, Fan H and Korc M: Adenovirus mediated transfer of a
truncated fibroblast growth factor (FGF) typ I receptor blocks FGF2
signaling in pancreatic cancer cells. Gastroenterology.
124:A1042003. View Article : Google Scholar
|
|
31
|
Li RW, Freeman C, Yu D, Hindmarsh EJ,
Tymms KE, Parish CR and Smith PN: Dramatic regulation of heparanase
activity and angiogenesis gene expression in synovium from patients
with rheumatoid arthritis. Arthritis Rheum. 58:1590–1600. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kato M, Wang H, Kainulainen V, Fitzgerald
ML, Ledbetter S, Ornitz DM and Bernfield M: Physiological
degradation converts the soluble syndecan-1 ectodomain from an
inhibitor to a potent activator of FGF-2. Nat Med. 4:691–697. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dailey L, Ambrosetti D, Mansukhani A and
Basilico C: Mechanisms underlying differential responses to FGF
signaling. Cytokine Growth Factor Rev. 16:233–247. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Katoh M and Katoh M: Cross-talk of WNT and
FGF signaling pathways at GSK3beta to regulate beta-catenin and
SNAIL signaling cascades. Cancer Biol Ther. 5:1059–1064. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Boukhelifa M, Hwang SJ, Valtschanoff JG,
Meeker RB, Rustioni A and Otey CA: A critical role for Palladin in
astrocyte morphology and response to injury. Mol Cell Neurosci.
23:661–668. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Najm P and El-Sibai M: Palladin regulation
of the actin structures needed for cancer invasion. Cell Adh Migr.
8:29–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Goicoechea SM, Bednarski B, García-Mata R,
Prentice-Dunn H, Kim HJ and Otey CA: Palladin contributes to
invasive motility in human breast cancer cells. Oncogene.
28:587–598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: A role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995. 2005.
View Article : Google Scholar : PubMed/NCBI
|