Hepatocellular carcinoma (HCC) is one of the most
common types of cancer and the second leading cause of
cancer-associated mortality worldwide (1). Its incidence is increasing in numerous
countries (2). Progression of HCC is
characterized by abnormal cell differentiation, fast infiltrating
growth, early metastasis, high-grade malignancy and poor prognosis
(3). Liver transplantation (LT) is
considered to be one of the major treatment options for HCC
(4), as not only it eliminates the
tumor but could also cure the underlying liver disease. However,
the high relapse rate of HCC following LT, which is estimated
between 15 and 20%, remains an important clinical challenge
(5). It is therefore crucial to
determine the underlying mechanisms of HCC relapse, in order to
increase the overall survival of patients with HCC.
Transcription factors (TFs) serve crucial roles in
the regulation of tumor progression (6–8). The
study of TFs has improved our understanding of the mechanisms
underlying the dysregulation of gene expression in cancers. For
example, multitudinous compelling evidence have recently showed
that HIF-1 plays important roles in many critical aspects of HCC,
including tumorigenesis, progression, and metastasis (9–13).
Several forkhead box proteins (14)
and zinc finger proteins (ZNFs) (15–21) have
also been reported to serve crucial roles in HCC. Recently, a study
demonstrated that ZNF687 overexpression promotes HCC recurrence
(22); however, the TFs associated
with HCC relapse remain unknown (22).
Weighted correlation network analysis (WGCNA) has
emerged as a powerful technique for multi-gene analysis. This
approach is designed to uncover networks and critical genes
associated with some phenotypes of interest. WGCNA has been widely
used to detect the co-expressed modules (23,24),
driver genes (24–27) and driver TFs (28) associated with a disease. In the
present study, WGCNA was used to build TFs co-expression network
and to investigate critical TFs that may drive HCC relapse. The
results from this study may serve at understanding the role of TFs
as diagnostic markers of HCC or as therapeutic targets for HCC
treatment.
The RNA-seq data and clinical magnifestations data
of the HCC dataset SRP040998 (29)
were downloaded from recount2, which is an online RNA-seq resource
(30). The dataset consists of tumor
samples and matched adjacent normal samples from 21 patients with
HCC. Among these patients, nine presented recurrent liver tumors or
remote metastasis in the 24 months following orthotopic LT (OLT),
whereas the remaining 12 patients were tumor free following OLT
(29). Principal component analysis
clustered the analyzed samples into the tumor and adjacent groups
with only one exception per group. These two samples (sample
numbers SRR1220147 and SRR1220148), which were from the same
patient, may have been mislabeled and were therefore excluded from
this study. Genes with low counts may represent a bias of
sequencing. In order to minimize the false positive in differential
expression analysis and to speed up WGCNA analysis, only the
expressed genes in terms with total counts ≥10 in the samples were
kept.
Differential gene expression analysis was performed
using DESeq2 package (version 1.20.0) (38). A gene was defined as a differentially
expressed gene (DEG) when the false discovery rate (FDR) adjusted
P-value between the tumor and adjacent groups was ≤0.05 (FDR ≤0.05)
and the fold change (FC) is at least 2 times higher or lower
(|log2FC| ≥1). However, a transcription factor is defined as a
differentially expressed TF (DET) for the cutoff values FDR ≤0.05
and |log2FC| ≥0.6.
A signed WGCNA network between the tumor and matched
adjacent tissues was constructed based on the biweight
midcorrelation using any gene that was expressed at the total
counts value of 10 or higher in ≥90% of samples. Different from an
unsigned WGCNA network, which uses the absolute value of the
Pearson correlation as an unsigned co-expression similarity
measure, the similarity between genes in a signed WGCNA network
reflects the sign of the correlation of their expression profiles.
Therefore, highly connected hub genes in a signed networks may
upregulate adjacent genes since they are positively correlated with
them, while in unsigned networks, highly connected hub genes may
activate or repress their neighboring genes (39). The count values were normalized by
variance stabilizing transformation using DESeq2 package (38). In order to achieve a scale-free
topology, soft power parameter was selected based on the criterion
of approximate scale-free topology with mininal scale-free fit
(SFT) index R2>0.85 and used to derive a pair-wise
distance matrix for selected genes using the topological overlap
measure. The dynamic hybrid cut method was used to detect clusters
of co-expressed genes using R functions in the WGCNA package
(version 1.66) (40).
The biological pathways related to the genes in the
module of interest, including the Kyoto Encyclopedia of Genes and
Genomes (KEGG) (41), were provided
by g:Profiler (42–44).
Hub gene is defined as an abbreviation of ‘highly
connected gene’, representing a small proportion of nodes with
maximal information exchange with other nodes in a network
(45). In the present study, hub TFs
in the module of interest were defined by module connectivity that
was measured by signed eigengene-based connectivity with the
cut-off value of absolute kME >0.15. kME, representing the
module connectivity of gene k, was determined as the Pearson's
correlation coefficient between gene expression values and the
module eigengene. In addition, all genes from the module of
interest were uploaded to the Search Tool for the Retrieval of
Interacting Genes (version 11.0) database (46), by choosing a confidence >0.4 in
order to construct a protein-protein interaction (PPI) network. In
the PPI network, genes with a connectivity degree >4 (edges)
were defined as hub nodes, and TFs with a connectivity degree >4
were defined as hub TFs (47,48).
To identify which TFs were differently regulated in
the networks, the differences in connectivity (DiffK) were compared
between the relapsed vs. non-relapsed subnetworks according to the
following formula: DiffK(i)=K1(i)-K2(i), where K1(i) and K2(i)
indicate the connectivity of the gene (i) in the relapsed
subnetwork and in the non-relapsed subnetwork, respectively. The
connectivity of K1 and K2 were calculated using R functions in the
WGCNA package (version 1.66). To facilitate the comparison between
the connectivity measures of each network, standardization was
carried out in each network by dividing each TF's connectivity by
the maximum connectivity of TFs-genes co-expression sub-network
according to the following formula: K1(i)=K1(i)/max(K1).
The difference between the connectivity values of
two subnetworks was defined as DiffK. DiffK values ranged from −1
to 1. A DiffK value >0 suggested that the TFs were more highly
connected in the relapsed subnetwork compared with the non-relapsed
subnetwork, whereas a DiffK value <0 indicated that the TFs were
more highly connected in the non-relapsed subnetwork compared with
the relapsed subnetwork. TFs were defined as differentially
connected when the absolute value of DiffK was >0.4 (28).
TFs regulating gene expression in the module of
interest were analyzed using the plugin iRegulon (v1.3) (49) in Cytoscape network (version 3.7.1)
(50). iRegulon is a computational
tool that can identify the upstream TFs and predict direct target
genes in a set of human, mouse and Drosophila genes. iRegulon uses
>9,000 known position weight matrices from various sources and
different species and link them to candidate binding TFs using a
‘motif2TF’ procedure. This allows to link motifs of TFs from other
species to candidate human TFs. Predicted upstream TFs are rated
and grouped according to the Normalized Enrichment Score (NES)
(28,51).
Correlation analysis for binary and continuous
variables was performed using standard screening binary trait and
standard screening numeric trait functions in the WGCNA package
(40), respectively. The output of
these functions include q-values of the correlations calculated
from the P-values using an optimised false discovery rate
approach.
Once the two mislabeled samples and lowly-expressed
genes were excluded, the remaining 26,324 genes from 58,037 genes
detected in HCC samples were grouped into 32 modules according to
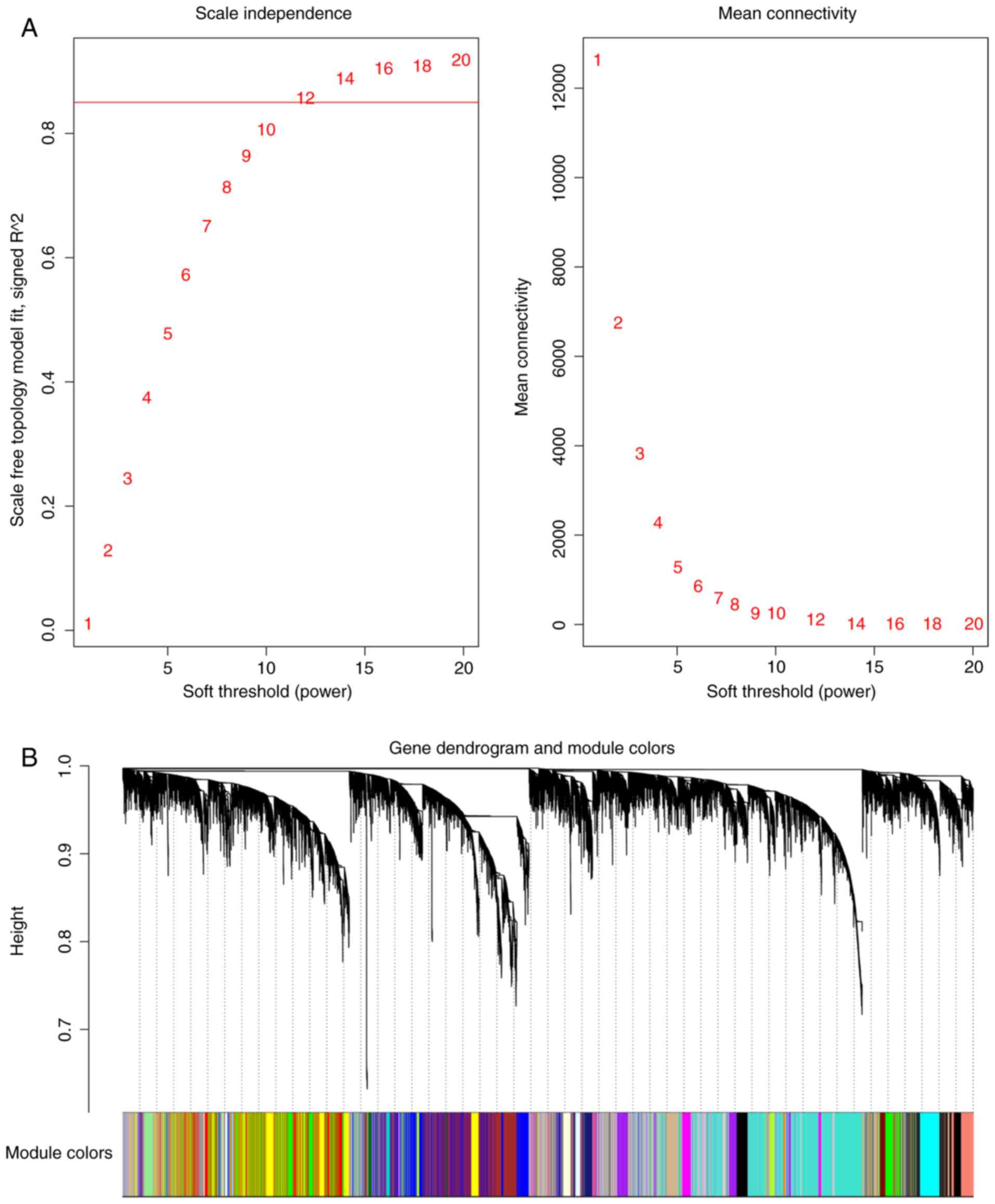
their expression profiles via hierarchical clustering (Fig. 1). To guarantee a scale-free topology
(minimal SFT R2>0.85), the soft power parameter
β-value was set at 12 (Fig. 1A). The
pairwise correlation was converted into adjacency matrix of
connection strengths through soft-thresholding approach, in order
to construct a dissimilarity matrix based on topological overlap
measure (TOM) and identify gene modules through a dynamic
tree-cutting algorithm. Each module was assigned to a corresponding
color (Fig. 1B). The module
eigengenes were calculated by the first principal component to
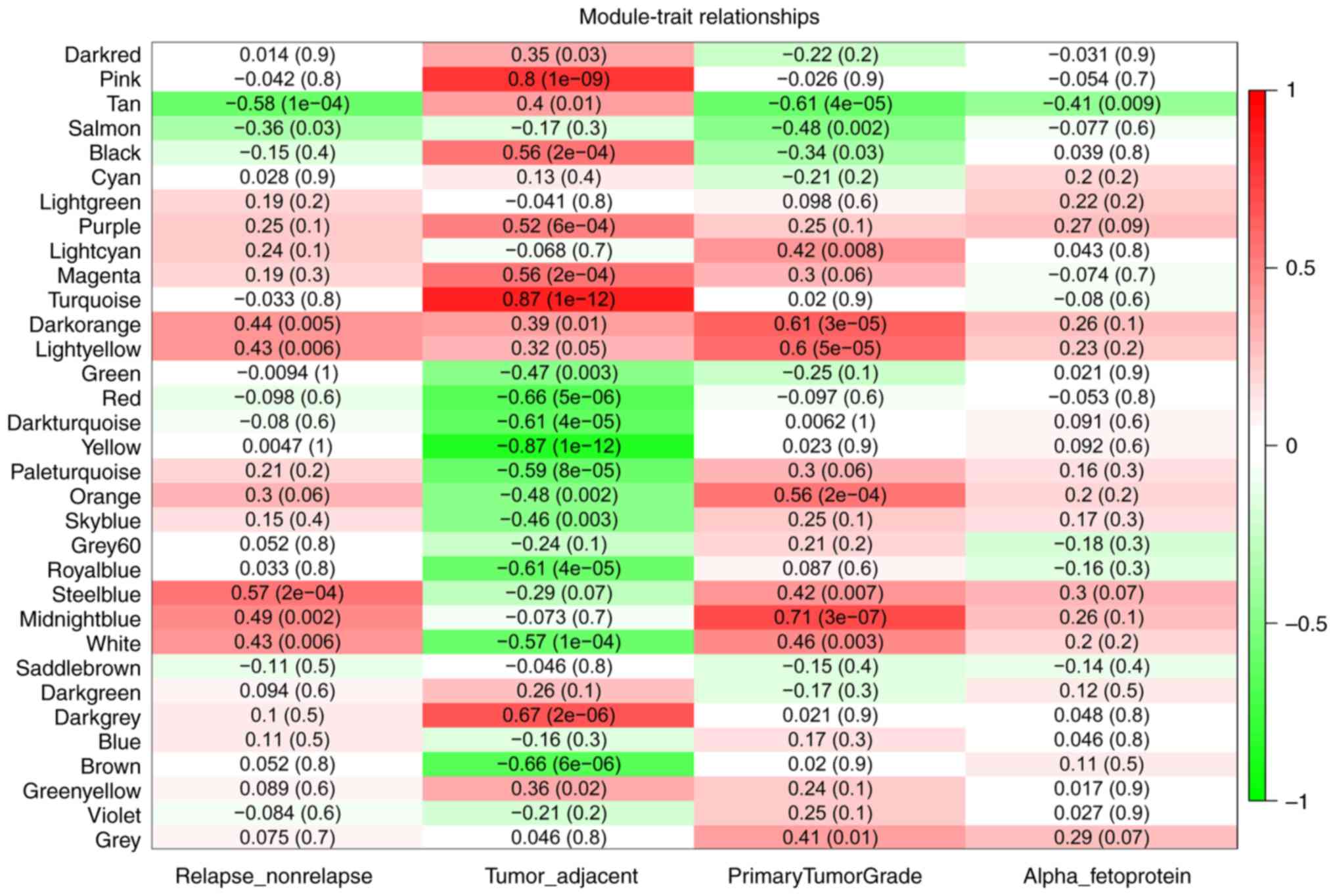
represent each module. With module eigengenes, Tan [Spearman
correlation, r=−0.58; P-value adjusted, (padj)=1×10−4]
and Steelblue (r=0.57; padj=2×10−4) modules were found
to be highly associated with HCC recurrence. The Steelblue module
contained only two TFs. Although these two TFs, poly(rC) binding
protein 2 (55) and eukaryotic
translation initiation factor 4B (56), have been reported to participate in
HCC development, the present study did not detect differences in
their expression between the tumor and adjacent normal tissues.
Subsequently, the present study focused on the Tan module, which
eigengene levels were also associated with primary tumor grade
(Spearman correlation, r=−0.61; padj=4×10−5) and
α-fetoprotein (AFP) (Pearson correlation, r=−0.41;
padj=9×10−3; Fig. 2).
The Tan module consisted of 615 genes, including 55
transcription factors. The results form KEGG pathway analysis
demonstrated that the Fanconi Anemia pathway was enriched in the
Tan module (adjusted P-values, 1.782×10−2).
Hub genes were first screened from the Tan module
based on the eigengene-based connectivity. By using the cut-off
value of absolute kME>0.15, 17 TFs with high connectivity were
identified in the Tan module. Among them, T-cell-restricted
intracellular antigen-1 (TIA1) had the highest connectivity.
Intraflagellar transport protein 80 homolog (IFT80), mediator of
DNA damage checkpoint 1 (MDC1) and zinc finger protein 260 (ZNF260)
were differentially expressed between the tumor and adjacent
tissues (Table I).
Hub genes were also screened from the Tan module
according to the connectivity of genes in the PPI network (Table SI). By using the cutoff of
confidence >0.4 and the connectivity degree of 4 (node/edge),
nine TFs, including EP400, were identified as the hub TFs (Table SI).
These two approaches identified 23 hub TFs in total,
including TIA1, IFT80 and MDC1 that were identified by both
approaches.
Genes that are differentially co-expressed between
different sample groups are more likely to be regulators, and may
therefore explain differences between phenotypes. Differential
network analysis was performed between the relapsed and
non-relapsed subnetworks in the Tan module. The analysis identified
TIA1 and nuclear receptor subfamily 2 group C member 2 (NR2C2) as
differentially connected TFs which were more highly connected in
the relapsed subnetwork compared with the non-relapsed subnetwork.
Furuthermore, EP400 and five other TFs were differentially
connected TFs that were more highly connected in the non-relapsed
subnetwork compared with the relapsed subnetwork (Table II).
It is regarded that genes co-expressed or
participating in the same biological process may be regulated by
the same or similar TFs (51). In
order to gain insight into the upstream regulators of gene
expression in the module of interest, iRegulon was used to search
the user-defined space for motifs enriched around the transcription
start site of the genes in the Tan module. The most enriched TF
motif was ZNF143 with NES 6.39. There were 42 target genes of
ZNF143 in the Tan module. Among them, TRIM74 was negatively
correlated with ZNF143 expression level, and 15 were positively
correlated with ZNF143 expression level, including seven TFs such
as MDC1 and ZNF260 (Table
III).
iRegulon predicted 30 TFs for ZNF143. Among them, 4
were negatively correlated with ZNF143 in the gene expression
levels (Table IV), and YY1 together
with another 6 TFs were positively correlated with ZNF143
expression level.
The most enriched TF motifs were also analyzed in
the dataset consisting of 55 TFs in the Tan module, where the most
enriched TF motifs were YY1 with NES 5.54. YY1 had 36 target TFs.
Among these target TFs, 12 were positively correlated with YY1
expression level (Table V),
including MDC1 and ZNF260.
iRegulon predicted 20 TFs for YY1. Among them, one
was negatively correlated with YY expression level, and six were
positively correlated with YY1 expression level (Table VI), including YY1 itself.
To validate the effects of the 23 hub TFs and two
master regulatory factors associated with HCC relapse, KMplot and
GEPIA were used to retrieve the RFS curves of 313 patients with HCC
and the DFS curve of 362 patients with HCC, respectively (Table SII). In order to determine the
prognostic relevance of these TFs in HCC, the OS of patients with
HCC was determined for each TF according to its expression using
UALCAN (Table SII), which contains
data from 365 patients with HCC.
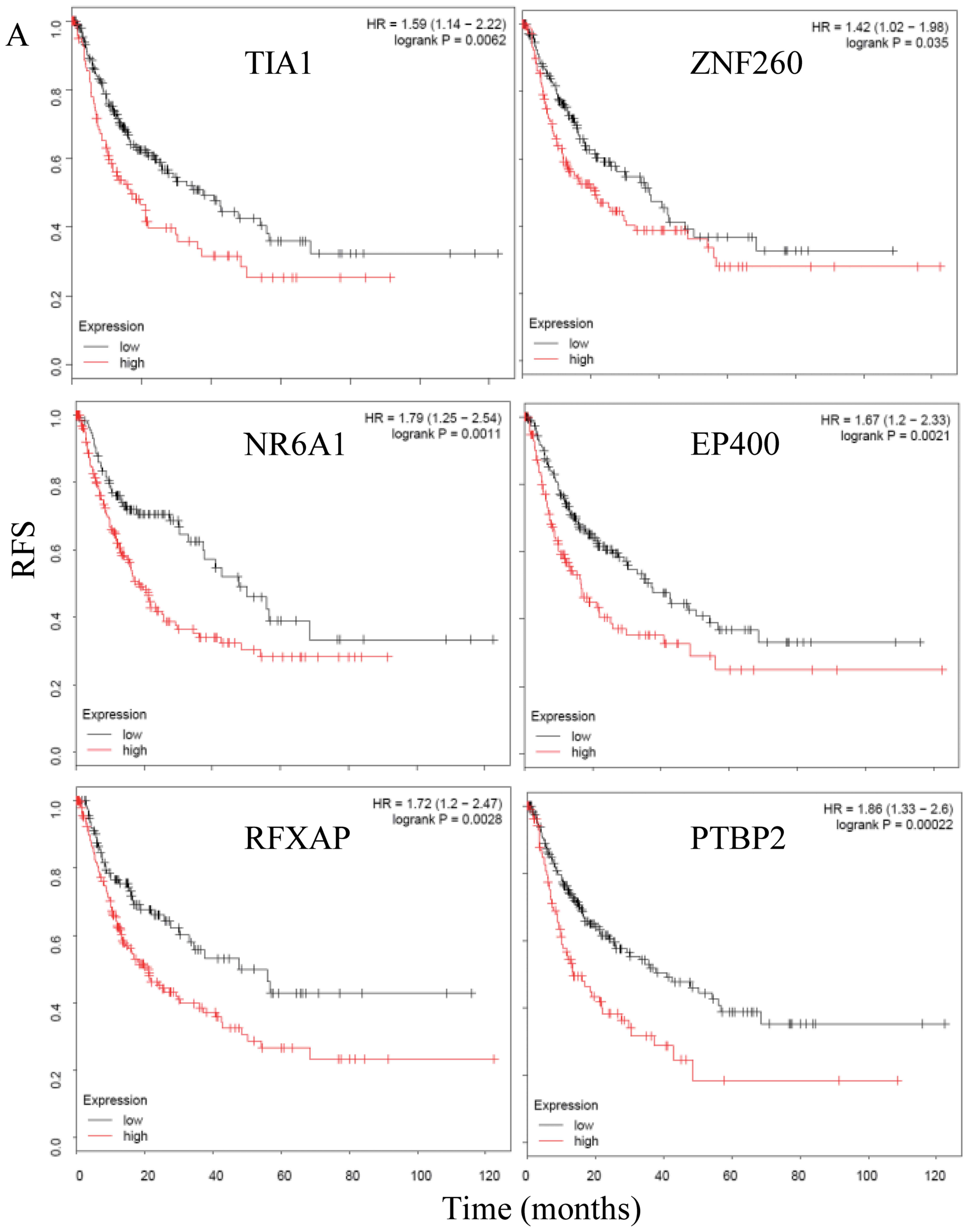
The results from KMplot and GEPIA demonstrated that
TIA1, ZNF260 and EP400 expression levels were associated with the
RFS and DFS of patients with HCC (Fig.
3A and B). Furthermore, ZNF260 and EP400 were also associated
with the OS of patients with HCC, however, TIA1 was only trend
associated with the OS of patients with HCC (P=0.09) according to
UALCAN (Table SII). In addition,
the results demonstrated that MDC1 was associated with the RFS and
DFS of patients with HCC according to KMplot and UALCAN analyses.
However, according to GEPIA, MDC1 was not associated with the DFS
of patients with HCC (P=0.07) (Table
SII).
Two master regulatory factors, ZNF143 and YY1, were
demonstrated to significantly influence the OS of patients with
HCC, but not the RFS.
Although liver resection is the most effective
curative treatment for HCC, relapse remains frequent (57,58).
Investigating the underlying mechanisms of HCC recurrence may
therefore lead to the development of novel therapeutic strategies
and prognostic biomarkers. Recent studies reported that miR-125b
(59) and miR-1246 (60) could be considered as novel biomarkers
for HCC relapse. miR-125b has been described as a tumor suppressor
that induces cellular senescence and apoptosis in hepatocellular
carcinogenesis by targeting sirtuin6 (61). Similarly, miR-1246 has been reported
to promote the cell apoptosis of HCC cells (62), and enhance migration and invasion in
HCC (63).
In addition to miRNAs, TFs may also serve important
roles in HCC relapse. In order to identify the TFs that
cooperatively drive HCC relapse, the present study constructed
signed WGCNA gene co-expression network by using the tumor and
matched adjacent normal samples of 20 patients with HCC. The Tan
module was found to be associated with HCC relapse and HCC staging,
and was enriched in the Fanconi Anemia pathway. Furthermore, 23 hub
TFs were identified, including TIA1, IFT80 and MDC1, which were
identified by both eigengene-based and PPI network approaches.
Eight TFs, including TIA1 and EP400, were found to be
differentially connected when comparing the non-relapsed and
relapsed subnetworks. ZNF143 and YY1 were detected as master
upstream regulator genes that could potentially regulate the
expression of the genes and TFs of the Tan module, respectively. KM
survival analysis demonstrated that TIA1, ZNF260 and EP400
expression levels were associated with the RFS and DFS of patients
with HCC.
Among the 23 hub TFs, TIA1, EP400 and NR2C2 were
differentially connected when comparing the non-relapsed and
relapsed subnetworks. These TFs were less cooperative with other
genes in the Tan module of patients with HCC relapse patients
compared with patients with non-relapse, suggesting that they may
serve important role in HCC relapse.
TIA1 is a member of an RNA-binding protein family
and possesses nucleolytic activity against cytotoxic lymphocyte
target cells (68). In the present
study, TIA1 was found to be associated with the RFS and DFS of
patients with HCC. However, TIA1 was not associated with the OS of
patients with HCC. A recent study reported that high TIA-1
expression level is associated with the poor survival rate of
patients with HCC (69).
Furthermore, TIA-1 can regulate IGF binding protein-3 (IGFBP3) at
the post-transcriptional level in human HCC cells (70). IGFBP3 is the primary binding protein
of IGF-I, and IGF-I has been reported to be involved in early HCC
relapse (71). Therefore,
TIA1-mediated IGFBP3 regulation may serve an important role in HCC
relapse.
In addition to TIA1, MDC1 was found to be a hub TF
in the present study. MDC1 was also differentially expressed
between the tumor and adjacent normal tissues. MDC1 is involved in
checkpoint activation and subsequent DNA repair following DNA
damage (72,73). Previous studies reported MDC1
contributes to breast cancer (74)
and pancreatic cancer (75);
however, its role in HCC remains unknown. The result from the
present study demonstrated that MDC1 reduced the OS and RFS of
patients with HCC.
This study demonstrated that ZNF143 was a master
upstream regulator gene that could potentially regulate expression
of the genes in the Tan module. In addition, ZNF143 expression
level was correlated with 16 of its target genes, including MDC1
and ZNF260. ZNF143 is a chromatin-looping factor that contributes
to the architectural foundation of the genome by providing sequence
specificity at promoters connected with distal regulatory elements
(76). A previous study reported
that ZNF143 expression is activated following DNA damage induced by
etoposide, cisplatin and Adriamycin (77). Furthermore, ZNF143 is involved in
cellular motility via the zinc finger E-box binding homeobox
1-cadherin-linked pathway in colon cancer cells (78). ZNF143 protein level is also
correlated with clinical outcomes in patients with lung
adenocarcinoma (79). A previous
study demonstrated that ZNF143 activity inhibition by small
molecules induced tumor regression in vitro and in
vivo (80). Similarly, the
present study demonstrated that ZNF143 expression level was
associated with the OS of patients with HCC; however, ZNF143 had no
influence on the RFS and DFS of patients with HCC (Table SII).
In the present study ZNF143, MDC1 and ZNF260, and
YY1 itself, were all predicted targets of YY1. Furthermore, they
were all positively correlated with YY1 expression level (Table V). YY1 belongs to the polycomb group
of proteins; this type of protein may cause epigenetic remodeling
of the chromatin and therefore dynamically regulate expressions of
their target genes (81). YY1
overexpression observed in the majority of cancers has been
correlated with poor prognosis of patients (82). Previous studies have demonstrated
that YY1 is implicated in HCC (83–88). It
has been reported that YY1 acts predominantly as an epigenetic
modulator, influencing the activity and/or localization of
epigenetic modifiers molecules, including DNA methylation
transferases, histone deacetylases or non-coding RNAs (81). YY1 may therefore increase expressions
of ZNF143 and MDC1, and dampen DNA repair pathways in HCC
progression. Similarly, the present study demonstrated that YY1
expression level was associated with the OS of patients with HCC;
however, YY1 was not associated with the RFS of patients with
HCC.
The Fanconi anemia pathway is essential for the
repair of DNA damage and is involved in three classic DNA repair
pathways named homologous recombination, nucleotide excision repair
and mutagenic translesion synthesis (89). A recent study demonstrated that genes
from the Fanconi anemia/BReastCAncer pathway are involved in HCC
chemoresistance (90). In the
present study, the Tan module was enriched in the Fanconi Anemia
pathway, and several hub TFs involved in DNA repair, including MDC1
and EP400, were associated with the RFS of patients with HCC,
suggesting that dysfunction in DNA repair pathways may be important
mechanism involved in HCC relapse.
The DNA repair pathways may play a role in HCC
relapse. The hub TFs TIA1 and EP400 were differentially connected
between the non-relapsed and relapsed subnetworks. TIA1 and EP400
may be considered as critical drivers for HCC relapse and serve
therefore as promising targets of HCC relapse. However, further
investigation is required to confirm these in silico
results.
Not applicable.
No funding was received.
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
WH, JB and JH designed the study. WH, YH, ZF, RG and
RY performed the formal analysis. JB and JH supervised the study.
WH wrote the manuscript. WH, YH, JB and JH reviewed the manuscript.
All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jung KH, Zhang J, Zhou C, Shen H, Gagea M,
Rodriguez-Aguayo C, Lopez-Berestein G, Sood AK and Beretta L:
Differentiation therapy for hepatocellular carcinoma: Multifaceted
effects of miR-148a on tumor growth and phenotype and liver
fibrosis. Hepatology. 63:864–879. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Llovet JM, Fuster J and Bruix J;
Barcelona-Clínic Liver Cancer Group, : The Barcelona approach:
Diagnosis, staging, and treatment of hepatocellular carcinoma.
Liver Transpl. 10:S115–S120. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Welker MW, Bechstein WO, Zeuzem S and
Trojan J: Recurrent hepatocellular carcinoma after liver
transplantation - an emerging clinical challenge. Transpl Int.
26:109–118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Darnell JE Jr: Transcription factors as
targets for cancer therapy. Nat Rev Cancer. 2:740–749. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malz M, Pinna F, Schirmacher P and
Breuhahn K: Transcriptional regulators in hepatocarcinogenesis-key
integrators of malignant transformation. J Hepatol. 57:186–195.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huh HD, Kim DH, Jeong HS and Park HW:
Regulation of TEAD transcription factors in cancer biology. Cells.
8:6002019. View Article : Google Scholar
|
|
9
|
Luo D, Wang Z and Wu J, Jiang C and Wu J:
The role of hypoxia inducible factor-1 in hepatocellular carcinoma.
Biomed Res Int. 2014:4092722014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin D and Wu J: Hypoxia inducible factor
in hepatocellular carcinoma: A therapeutic target. World J
Gastroenterol. 21:12171–12178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Méndez-Blanco C, Fondevila F,
García-Palomo A, González-Gallego J and Mauriz JL: Sorafenib
resistance in hepatocarcinoma: Role of hypoxia-inducible factors.
Exp Mol Med. 50:1342018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Matteis S, Scarpi E, Granato AM,
Vespasiani-Gentilucci U, La Barba G, Foschi FG, Bandini E, Ghetti
M, Marisi G, Cravero P, et al: Role of SIRT-3, p-mTOR and HIF-1α in
hepatocellular carcinoma patients affected by metabolic
dysfunctions and in chronic treatment with metformin. Int J Mol
Sci. 20:2019. View Article : Google Scholar
|
|
13
|
Wen Y, Zhou X, Lu M, He M, Tian Y, Liu L,
Wang M, Tan W, Deng Y, Yang X, et al: Bclaf1 promotes angiogenesis
by regulating HIF-1α transcription in hepatocellular carcinoma.
Oncogene. 38:1845–1859. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang G and Zhang G: Upregulation of FoxP4
in HCC promotes migration and invasion through regulation of EMT.
Oncol Lett. 17:3944–3951. 2019.PubMed/NCBI
|
|
15
|
Wang Q, Tan YX, Ren YB, Dong LW, Xie ZF,
Tang L, Cao D, Zhang WP, Hu HP and Wang HY: Zinc finger protein
ZBTB20 expression is increased in hepatocellular carcinoma and
associated with poor prognosis. BMC Cancer. 11:2712011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kan H, Huang Y, Li X, Liu D, Chen J and
Shu M: Zinc finger protein ZBTB20 is an independent prognostic
marker and promotes tumor growth of human hepatocellular carcinoma
by repressing FoxO1. Oncotarget. 7:14336–14349. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Z, Sun B, Li Y, Zhao X, Zhao X, Gu Q,
An J, Dong X, Liu F and Wang Y: ZEB2 promotes vasculogenic mimicry
by TGF-β1 induced epithelial-to-mesenchymal transition in
hepatocellular carcinoma. Exp Mol Pathol. 98:352–359. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu D, Liu G, Liu Y, Saiyin H, Wang C, Wei
Z, Zen W, Liu D, Chen Q, Zhao Z, et al: Zinc finger protein 191
inhibits hepatocellular carcinoma metastasis through discs large
1-mediated yes-associated protein inactivation. Hepatology.
64:1148–1162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yi PS, Wu B, Deng DW, Zhang GN and Li JS:
Positive expression of ZNF689 indicates poor prognosis of
hepatocellular carcinoma. Oncol Lett. 16:5122–5130. 2018.PubMed/NCBI
|
|
20
|
Xiang Q, Zhou D, He X, Fan J, Tang J, Qiu
Z, Zhang Y, Qiu J, Xu Y and Lai G: The zinc finger protein GATA4
induces MET and cellular senescence through the NF-kappaB pathway
in hepatocellular carcinoma. J Gastroenterol Hepatol. 2019.
View Article : Google Scholar
|
|
21
|
Wang N, Wang S, Yang SL, Liu LP, Li MY,
Lai PBS and Chen GG: Targeting ZBP-89 for the treatment of
hepatocellular carcinoma. Expert Opin Ther Targets. 22:817–822.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang T, Huang Y, Liu W, Meng W, Zhao H,
Yang Q, Gu SJ, Xiao CC, Jia CC, Zhang B, et al: Overexpression of
zinc finger protein 687 enhances tumorigenic capability and
promotes recurrence of hepatocellular carcinoma. Oncogenesis.
6:e3632017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu X, Zhou Y, Miao R, Chen W, Qu K, Pang Q
and Liu C: Transcriptional modules related to hepatocellular
carcinoma survival: Coexpression network analysis. Front Med.
10:183–190. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yin L, Cai Z, Zhu B and Xu C:
Identification of key pathways and genes in the dynamic progression
of HCC based on WGCNA. Genes (Basel). 9:922018. View Article : Google Scholar
|
|
25
|
Xu W, Rao Q, An Y, Li M and Zhang Z:
Identification of biomarkers for Barcelona Clinic Liver Cancer
staging and overall survival of patients with hepatocellular
carcinoma. PLoS One. 13:e02027632018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li B, Pu K and Wu X: Identifying novel
biomarkers in hepatocellular carcinoma by weighted gene
co-expression network analysis. J Cell Biochem. Feb 11–2019.(Epub
ahead of print). doi: 10.1002/jcb.28420.
|
|
27
|
Zhang C, Peng L, Zhang Y, Liu Z, Li W,
Chen S and Li G: The identification of key genes and pathways in
hepatocellular carcinoma by bioinformatics analysis of
high-throughput data. Med Oncol. 34:1012017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Skinkyte-Juskiene R, Kogelman LJA and
Kadarmideen HN: Transcription factor co-expression networks of
adipose RNA-Seq data reveal regulatory mechanisms of obesity. Curr
Genomics. 19:289–299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xue F, Higgs BW, Huang J, Morehouse C, Zhu
W, Yao X, Brohawn P, Xiao Z, Sebastian Y, Liu Z, et al: HERC5 is a
prognostic biomarker for post-liver transplant recurrent human
hepatocellular carcinoma. J Transl Med. 13:3792015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Collado-Torres L, Nellore A, Kammers K,
Ellis SE, Taub MA, Hansen KD, Jaffe AE, Langmead B and Leek JT:
Reproducible RNA-seq analysis using recount2. Nat Biotechnol.
35:319–321. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harrow J, Frankish A, Gonzalez JM,
Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa
A, Searle S, et al: GENCODE: The reference human genome annotation
for The ENCODE Project. Genome Res. 22:1760–1774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng G, Tu K, Yang Q, Xiong Y, Wei C, Xie
L, Zhu Y and Li Y: ITFP: An integrated platform of mammalian
transcription factors. Bioinformatics. 24:2416–2417. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marbach D, Lamparter D, Quon G, Kellis M,
Kutalik Z and Bergmann S: Tissue-specific regulatory circuits
reveal variable modular perturbations across complex diseases. Nat
Methods. 13:366–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Neph S, Stergachis AB, Reynolds A,
Sandstrom R, Borenstein E and Stamatoyannopoulos JA: Circuitry and
dynamics of human transcription factor regulatory networks. Cell.
150:1274–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stergachis AB, Neph S, Sandstrom R, Haugen
E, Reynolds AP, Zhang M, Byron R, Canfield T, Stelhing-Sun S, Lee
K, et al: Conservation of trans-acting circuitry during mammalian
regulatory evolution. Nature. 515:365–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang C, Xuan Z, Zhao F and Zhang MQ:
TRED: A transcriptional regulatory element database, new entries
and other development. Nucleic Acids Res. 35:D137–D140. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han H, Shim H, Shin D, Shim JE, Ko Y, Shin
J, Kim H, Cho A, Kim E, Lee T, et al: TRRUST: A reference database
of human transcriptional regulatory interactions. Sci Rep.
5:114322015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mason MJ, Fan G, Plath K, Zhou Q and
Horvath S: Signed weighted gene co-expression network analysis of
transcriptional regulation in murine embryonic stem cells. BMC
Genomics. 10:3272009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Reimand J, Arak T, Adler P, Kolberg L,
Reisberg S, Peterson H and Vilo J: g:Profiler-a web server for
functional interpretation of gene lists (2016 update). Nucleic
Acids Res. 44:W83–W89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Reimand J, Arak T and Vilo J:
g:Profiler--a web server for functional interpretation of gene
lists (2011 update). Nucleic Acids Res. 39:W307–W315. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Raudvere U, Kolberg L, Kuzmin I, Arak T,
Adler P, Peterson H and Vilo J: g:Profiler: A web server for
functional enrichment analysis and conversions of gene lists (2019
update). Nucleic Acids Res. 47:W191–W198. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang F, Yuan JH, Wang SB, Yang F, Yuan SX,
Ye C, Yang N, Zhou WP, Li WL, Li W and Sun SH: Oncofetal long
noncoding RNA PVT1 promotes proliferation and stem cell-like
property of hepatocellular carcinoma cells by stabilizing NOP2.
Hepatology. 60:1278–1290. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
von Mering C, Jensen LJ, Snel B, Hooper
SD, Krupp M, Foglierini M, Jouffre N, Huynen MA and Bork P: STRING:
Known and predicted protein-protein associations, integrated and
transferred across organisms. Nucleic Acids Res. 33:D433–D437.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yuan L, Qian G, Chen L, Wu CL, Dan HC,
Xiao Y and Wang X: Co-expression network analysis of biomarkers for
adrenocortical carcinoma. Front Genet. 9:3282018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen PF, Wang F, Nie JY, Feng JR, Liu J,
Zhou R, Wang HL and Zhao Q: Co-expression network analysis
identified CDH11 in association with progression and prognosis in
gastric cancer. Onco Targets Ther. 11:6425–6436. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Janky R, Verfaillie A, Imrichová H, Van de
Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval
Sanchez M, Potier D, et al: iRegulon: From a gene list to a gene
regulatory network using large motif and track collections. PLoS
Comput Biol. 10:e10037312014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yin L, Guo X, Zhang C, Cai Z and Xu C: In
silico analysis of expression data during the early priming stage
of liver regeneration after partial hepatectomy in rat. Oncotarget.
9:11794–11804. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Menyhárt O, Nagy Á and Győrffy B:
Determining consistent prognostic biomarkers of overall survival
and vascular invasion in hepatocellular carcinoma. R Soc Open Sci.
5:1810062018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang X, Hua L, Yan D, Zhao F, Liu J, Zhou
H, Liu J, Wu M, Zhang C, Chen Y, et al: Overexpression of PCBP2
contributes to poor prognosis and enhanced cell growth in human
hepatocellular carcinoma. Oncol Rep. 36:3456–3464. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Golob-Schwarzl N, Krassnig S, Toeglhofer
AM, Park YN, Gogg-Kamerer M, Vierlinger K, Schröder F, Rhee H,
Schicho R, Fickert P and Haybaeck J: New liver cancer biomarkers:
PI3K/AKT/mTOR pathway members and eukaryotic translation initiation
factors. Eur J Cancer. 83:56–70. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Poon RT: Prevention of recurrence after
resection of hepatocellular carcinoma: A daunting challenge.
Hepatology. 54:757–759. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vilarinho S and Calvisi DF: New advances
in precision medicine for hepatocellular carcinoma recurrence
prediction and treatment. Hepatology. 60:1812–1814. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shimagaki T, Yoshizumi T, Harimoto N,
Yoshio S, Naito Y, Yamamoto Y, Ochiya T, Yoshida Y, Kanto T and
Maehara Y: MicroRNA-125b expression and intrahepatic metastasis are
predictors for early recurrence after hepatocellular carcinoma
resection. Hepatol Res. 48:313–321. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chuma M, Toyoda H, Matsuzaki J, Saito Y,
Kumada T, Tada T, Kaneoka Y, Maeda A, Yokoo H, Ogawa K, et al:
Circulating microRNA-1246 as a possible biomarker for early tumor
recurrence of hepatocellular carcinoma. Hepatol Res. 49:810–822.
2019.PubMed/NCBI
|
|
61
|
Song S, Yang Y, Liu M, Liu B, Yang X, Yu
M, Qi H, Ren M, Wang Z, Zou J, et al: MiR-125b attenuates human
hepatocellular carcinoma malignancy through targeting SIRT6. Am J
Cancer Res. 8:993–1007. 2018.PubMed/NCBI
|
|
62
|
Zhang Q, Cao LY, Cheng SJ, Zhang AM, Jin
XS and Li Y: p53-induced microRNA-1246 inhibits the cell growth of
human hepatocellular carcinoma cells by targeting NFIB. Oncol Rep.
33:1335–1341. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sun Z, Meng C, Wang S, Zhou N, Guan M, Bai
C, Lu S, Han Q and Zhao RC: MicroRNA-1246 enhances migration and
invasion through CADM1 in hepatocellular carcinoma. BMC Cancer.
14:6162014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chan HM, Narita M, Lowe SW and Livingston
DM: The p400 E1A-associated protein is a novel component of the p53
-->p21 senescence pathway. Genes Dev. 19:196–201. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Shi M, Chen MS, Sekar K, Tan CK, Ooi LL
and Hui KM: A blood-based three-gene signature for the non-invasive
detection of early human hepatocellular carcinoma. Eur J Cancer.
50:928–936. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Montalvá EM, Cantos M, Boscà A, Rubín A,
Vinaixa C, Granero P, Maupoey J and López-Andújar R: Prognostic
value of pre-transplantation serum alpha-fetoprotein levels in
hepatocellular carcinoma recurrence. Transplant Proc. 48:2966–2968.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tsukamoto M, Nitta H, Imai K, Higashi T,
Nakagawa S, Okabe H, Arima K, Kaida T, Taki K, Hashimoto D, et al:
Clinical significance of half-lives of tumor markers α-fetoprotein
and des-γ-carboxy prothrombin after hepatectomy for hepatocellular
carcinoma. Hepatol Res. 48:E183–E193. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Carrascoso I, Alcalde J, Tabas-Madrid D,
Oliveros JC and Izquierdo JM: Transcriptome-wide analysis links the
short-term expression of the b isoforms of TIA proteins to
protective proteostasis-mediated cell quiescence response. PLoS
One. 13:e02085262018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tak H, Kang H, Ji E, Hong Y, Kim W and Lee
EK: Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27
as a novel marker for hepatocellular carcinoma. Biochem Biophys Res
Commun. 497:1117–1122. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Subramaniam K, Ooi LL and Hui KM:
Transcriptional down-regulation of IGFBP-3 in human hepatocellular
carcinoma cells is mediated by the binding of TIA-1 to its AT-rich
element in the 3′-untranslated region. Cancer Lett. 297:259–268.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yao Y, Mao W, Dong M, Yang D, Li W and
Chen Y: Serum insulin-like growth factor-1 (IGF-1): A novel
prognostic factor for early recurrence of hepatocellular carcinoma
(HCC). Clin Lab. 63:261–270. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li Z, Shao C, Kong Y, Carlock C, Ahmad N
and Liu X: DNA damage response-independent role for MDC1 in
maintaining genomic stability. Mol Cell Biol. 37:2017. View Article : Google Scholar
|
|
73
|
Lee JH, Park SJ, Hariharasudhan G, Kim MJ,
Jung SM, Jeong SY, Chang IY, Kim C, Kim E, Yu J, et al: ID3
regulates the MDC1-mediated DNA damage response in order to
maintain genome stability. Nat Commun. 8:9032017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang S, Zou Z, Luo X, Mi Y, Chang H and
Xing D: LRH1 enhances cell resistance to chemotherapy by
transcriptionally activating MDC1 expression and attenuating DNA
damage in human breast cancer. Oncogene. 37:3243–3259. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhou H, Qin Y, Ji S, Ling J, Fu J, Zhuang
Z, Fan X, Song L, Yu X and Chiao PJ: SOX9 activity is induced by
oncogenic Kras to affect MDC1 and MCMs expression in pancreatic
cancer. Oncogene. 37:912–923. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bailey SD, Zhang X, Desai K, Aid M,
Corradin O, Cowper-Sal Lari R, Akhtar-Zaidi B, Scacheri PC,
Haibe-Kains B and Lupien M: ZNF143 provides sequence specificity to
secure chromatin interactions at gene promoters. Nat Commun.
2:61862015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ishiguchi H, Izumi H, Torigoe T, Yoshida
Y, Kubota H, Tsuji S and Kohno K: ZNF143 activates gene expression
in response to DNA damage and binds to cisplatin-modified DNA. Int
J Cancer. 111:900–909. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Paek AR, Lee CH and You HJ: A role of
zinc-finger protein 143 for cancer cell migration and invasion
through ZEB1 and E-cadherin in colon cancer cells. Mol Carcinog. 53
(Suppl 1):E161–E168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kawatsu Y, Kitada S, Uramoto H, Zhi L,
Takeda T, Kimura T, Horie S, Tanaka F, Sasaguri Y, Izumi H, et al:
The combination of strong expression of ZNF143 and high MIB-1
labelling index independently predicts shorter disease-specific
survival in lung adenocarcinoma. Br J Cancer. 110:2583–2592. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Haibara H, Yamazaki R, Nishiyama Y, Ono M,
Kobayashi T, Hokkyo-Itagaki A, Nishisaka F, Nishiyama H, Kurita A,
Matsuzaki T, et al: YPC-21661 and YPC-22026, novel small molecules,
inhibit ZNF143 activity in vitro and in vivo. Cancer Sci.
108:1042–1048. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ruosi C, Colella G, Fazioli F, Miceli R,
Gallo M, Di Salvatore MG, Cimmino A and de Nigris F: Yin Yang I as
an epimodulator of miRNAs in the metastatic cascade. Crit Rev
Oncog. 22:99–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cho AA and Bonavida B: Targeting the
overexpressed YY1 in cancer inhibits EMT and metastasis. Crit Rev
Oncog. 22:49–61. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Han J, Meng J, Chen S, Wang X, Yin S,
Zhang Q, Liu H, Qin R, Li Z, Zhong W, et al: YY1 complex promotes
quaking expression via super-enhancer binding during EMT of
hepatocellular carcinoma. Cancer Res. 79:1451–1464. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Dong S, Ma X, Wang Z, Han B, Zou H, Wu Z,
Zang Y and Zhuang L: YY1 promotes HDAC1 expression and decreases
sensitivity of hepatocellular carcinoma cells to HDAC inhibitor.
Oncotarget. 8:40583–40593. 2017.PubMed/NCBI
|
|
85
|
Kim JS, Son SH, Kim MY, Choi D, Jang IS,
Paik SS, Chae JH, Uversky VN and Kim CG: Diagnostic and prognostic
relevance of CP2c and YY1 expression in hepatocellular carcinoma.
Oncotarget. 8:24389–24400. 2017.PubMed/NCBI
|
|
86
|
Tsang DP, Wu WK, Kang W, Lee YY, Wu F, Yu
Z, Xiong L, Chan AW, Tong JH, Yang W, et al: Yin Yang 1-mediated
epigenetic silencing of tumour-suppressive microRNAs activates
nuclear factor-κB in hepatocellular carcinoma. J Pathol.
238:651–664. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang S, Jiang T, Feng L, Sun J, Lu H,
Wang Q, Pan M, Huang D, Wang X, Wang L and Jin H: Yin Yang-1
suppresses differentiation of hepatocellular carcinoma cells
through the downregulation of CCAAT/enhancer-binding protein alpha.
J Mol Med (Berl). 90:1069–1077. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Teng CF, Wu HC, Tsai HW, Shiah HS, Huang W
and Su IJ: Novel feedback inhibition of surface antigen synthesis
by mammalian target of rapamycin (mTOR) signal and its implication
for hepatitis B virus tumorigenesis and therapy. Hepatology.
54:1199–1207. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Moldovan GL and D'Andrea AD: How the
fanconi anemia pathway guards the genome. Annu Rev Genet.
43:223–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ferroudj S, Yildiz G, Bouras M, Iscan E,
Ekin U and Ozturk M: Role of Fanconi anemia/BRCA pathway genes in
hepatocellular carcinoma chemoresistance. Hepatol Res.
46:1264–1274. 2016. View Article : Google Scholar : PubMed/NCBI
|