Introduction
Osteosarcoma (OS) is a tumor characterized by the
presence of malignant mesenchymal cells produced in the bone stroma
(1). The incidence of this tumor in
the general population is 2-3 cases/million/year, and it is higher
in adolescence, when the annual incidence peaks vary from
8–11/million/year. Among adolescents age group of 10–19 years, it
represents 15% of all extracranial tumors, being 1.4 times more
frequent in men than in women (2,3). A
second peak of OS in adults over 65 years of age has been reported,
but it is likely to represent a second malignancy, often related to
Paget's disease (4). In general, OS
is most commonly characterized by an appendicular primary tumor
with a high rate of metastasis to the lungs, which usually appears
during the first or second decade of life (5,6).
Different studies point to pre-osteoblasts and
osteoblasts being the cells which give rise to tumors (7,8). As an
important component of the tumor microenvironment, mesenchymal stem
cells (MSCs) appear to play an important role in mediating and
proliferation in many cancers, including OS. The tumor
microenvironment exerts different effects on virgin MSCs, in which
some cytokines such as SDF-1, MIF, and TGF-β recruit these cells to
the tumor site, where they are stimulated by the paracrine network
and undergo a series of functional transformations. The action of
INF-γ, TNF-α and IL-1α strengthen the growth-promoting effects of
MSCs, while INF-γ, TNF-α and TGF-β enhance the ability of MSCs to
promote tumor metastases. In addition, MSCs may differentiate into
associated fibroblasts to cancer under the stimulation of TGF-β
(9).
MSCs first differentiate into chondrocytes during
the endochondral bone formation process until adolescence, which
generate new cartilage in GP and after are slowly replaced by
osteogenic progenitor cells and osteoblasts to produce bone
(10). Interestingly, the p53
cellular protein acts as a negative regulator of osteoblastogenesis
under normal conditions, repressing transcription factor such as
Osterix and Runx2 (11), which are
required in the initial osteogenesis phase in osteoprogenitor cells
(12,13). However, Runx2 may act by inhibiting
the function of p53 in activating apoptosis by inducing c-MYC
transcription by histone modifications (14). This explains the highly elevated
Runx2 expression levels in OS cells. While, Runx2 may have dual
role as a tumor suppressive and as oncoprotein, depending on its
cellular levels and context, and its regulation (15).
MSCs represent a source of osteogenic progenitor
precursor cells which give rise to osteoblasts. Thus, mutations in
the TP53 gene of these cells can lead to defects in
controlling cell growth, increasing the risk of developing OS
(16).
However, the occurrence of mutations is not the most
common event in this type of tumor. Rather, it is best
characterized by deregulation of the expression of tumor suppressor
genes such as retinoblastoma (RB1) and TP53,
aneuploidy, chromosome structure disruption and uncontrolled cell
cycles (17,18). This suggests the possibility of a
defect in surveillance or DNA repair mechanisms as one of the
possible causes of the tumor's genesis (6).
Epigenetic events are also identified as risk
factors for OS, since the DNA methylation pattern of specific genes
or gene regions and histone modifications may be involved in tumor
development (19). In addition, the
methylation levels and silencing of gene encoding tumor suppressor
micro-RNAs (miRs) have been described as specific events in human
OS cell lines (20). The
overexpression of the IGF2 growth factor and of the IRX1
gene mediated by the hypomethylation of its promoters has also been
reported as an inducer of metastasis in this tumor (21,22).
Bone tissue is highly specialized and has many
important signaling pathways to its homeostasis which require
crosstalk between bone and immune cells performed by chemical
mediators such as cytokines. This is evidenced by the fact that
osteoclast formation requires the receptor activator of nuclear
factor kappa-B (RANKL) and of macrophage colony-stimulating factor
(M-CSF). In turn, RANKL is produced by osteoblast and activated T
cells to regulate osteoclast differentiation, at the same time
M-CSF is produced by immune cells and stimulates the expression of
RANKL by osteoclast precursor cells such as monocytes and
macrophages. In addition, other factors secreted by immune cells
may promote or suppress the formation of osteoclasts. This shows
the existence of a complex network of communication between cells
triggering the immunomodulatory mechanism which may play an
important role in tumor development (23).
In this review we present some recent advances on
the biology and pathogenesis of OS, with emphasis on the probable
mechanisms involved in its initiation and progression. The
literature search was conducted using the PubMed (National
Institutes of Health; ww.ncbi.nlm.nih.gov/pubmed), Scopus (Elsevier;
www.scopus.com/scopus/home.url), and
Web of Knowledge (Thomson Reuters; wok.mimas.ac.uk) electronic databases using the
following keywords: Osteosarcoma, osteosarcoma biology,
osteosarcoma pathogenesis, osteosarcoma signaling pathways,
osteosarcoma genetics, osteosarcoma epigenetic, and cytokine
profile in osteosarcoma. Several hundred articles were found in
the surveyed databases, however only those which were considered
most relevant which had been published in impact journals and were
conducted by groups with recognized knowledge in the area were
selected.
Biology of human OS
OS is a tumor that is most frequent in the rapid
growth phase of the long bones which occurs during puberty. More
than 50% of the cases have origin in the epiphyseal GP of the
distal femur and proximal tibia where the bone growth develops,
being responsible for much of the height increase which occurs
during adolescence (4).
Among the possible mechanisms that contribute to OS
development are alterations in the differentiation pathway of MSCs
in mature osteoblasts (24).
Furthermore, abnormal expression of oncogenes and of tumor
suppressor genes triggered by genetic and epigenetic events lead to
deregulation of important cell signaling pathways, thereby creating
a favorable environment to malignant transformation (25,26).
This is because there is a greater bone turnover during the growth
phase, and so the possibility for defects to occur in the
differentiation process and in the signaling pathways is amplified
(4).
MSCs of the bone marrow stroma are undifferentiated
cells with potential for self-renewal, proliferation, and
differentiation for bone, muscle, tendon, and fat formation
(27). Many endogenous and exogenous
factors are involved in the osteogenesis process to form
osteoblasts by the osteogenic pathway which leads to the
differentiation of MSCs into osteoblasts. Deregulation of these
factors, or exposure to new non-native stimuli such as
pro-inflammatory cytokines and pro-tumor, may cause an imbalance
between cell differentiation and proliferation, contributing to a
malignant phenotype (28,29). As main component of the tumor
microenvironment, MSCs can mediate cellular proliferation and
metastasis, as well as drug resistance in OS (9).
Current knowledge indicates that OS exhibit a wide
range of genetic, epigenetic, and molecular changes, including
gains, losses, or arrangements of chromosomal regions; inactivation
of tumor suppressor genes; and deregulation of cell signaling
pathways (17). Each of the
mechanisms mentioned above will be presented with more detail in
the following sections.
Role of differentiation of mesenchymal stem
cells
The main function of MSCs is self-renewal which
requires a multi or pluripotency state, remaining undifferentiated
and proliferative to maintain homeostasis during the development
phase or even throughout life in order to maintain body homeostasis
or make repairs. Such properties are in many ways analogous to
those of cancer cells, since the unlimited potential for
proliferation, also known as immortality, is the most striking
feature of malignant tumors (30).
In addition, stemness maintenance is achieved by restricting the
differentiation, apoptosis, and cellular senescence, which are also
characteristic of transformed cells (31).
MSCs are present in many human organs and comprise a
heterogeneous population of self-renewing cells, and their
morphology, immunophenotype, and differentiation potential depend
on their tissue origin. Specific populations of the stroma maintain
the regeneration process of the tissue where they reside, but some
of them have much greater plasticity and differ across multiple
cell lineages. Thus, MSCs not only contribute to the structural
repair of tissues, but also possess strong immunomodulatory and
anti-inflammatory properties and can influence tissue repair
through modulation of the local environment (32).
In a parallel, functional and phenotypic analyses of
normal MSCs and MSCs derived from OS were performed to evaluate the
pre-malignant stages of the tumor in a murine MSC system in which
tumor development was demonstrated after grafting of transformed
MSCs. This is substantial evidence to support the hypothesis that
this tumor originates from MSCs. Analysis of different passages of
MSCs using COBRA-FISH karyotyping and CGH array revealed the
occurrence of aneuploidization, translocations, and homozygous loss
of CDKN2 region of the genome these cells, in which encoding
cyclin-2A dependent kinase inhibitor is a mediator of malignant
transformation of MSCs. Interestingly, the expression of the
CDKN2 gene product, the p16 protein, was reduced in the
samples of 88 patients with OS, confirming the results obtained by
the murine system (33).
In another study was found that that the SOX5
gene, which encodes a family of transcription factors involved in
regulating embryonic development and which determines the
destination of cells, is significantly expressed in OS tissue and
in cell line-derived tumor. In addition, the expression of
SOX5 promoted epithelial-mesenchymal transition (EMT) and
increased migration and invasion of tumor cells (34).
A recent study involving crosstalk between OS cells
and MSCs, mediated by extracellular vesicles (EVs) which play an
important role in initiating and progressing cancer, showed strong
evidence of MSCs participating in the origin of OS. MSCs and
pre-osteoblasts were treated with OS-EVs at different times, and
their epigenetic signature was evaluated through of methylation
analysis of LINE-1 (long interleaved element) and tumor suppressor
genes. This shows that OS-EVs mediate LINE-1 hypomethylation in
MSCs and LINE-1 hyper methylation in the pre-osteoblasts,
indicating that MSCs, but not pre-osteoblasts, are susceptible to
epigenetic transformation. Thus, OS-EVs modulate the fate of MSCs,
regulating epigenetic status and influencing gene expression
related to bone microenvironment remodeling. This suggests that
epigenetic regulation appears to be an early event in transforming
MSCs during OS development (35).
Role of DNA changes
The TP53 gene plays a critical role in the
regulation of both the cell cycle and apoptosis, and its product
(the p53 protein) is synthesized in response to stress situations
due to tensions such as DNA damage, hypoxia, and oncogene
activation. This gene frequently undergoes negative selection
during tumorigenic transformation. Mutations in the TP53
gene in response to DNA damage can promote uncontrolled cell
cycles, inhibit senescence and cell death by apoptosis, thereby
increasing the genomic instability. This leads to an accumulation
of mutations and cell survival, in turn increasing the risk of
malignant transformation, including OS development (36).
The occurrence of mutation in OS was investigated in
a study in which the whole-exome and RNA-sequencing of 59
tumor/normal pairs of samples revealed that only the TP53
tumor suppressor gene showed mutation with significant frequency in
all the samples. The mean non-silent somatic mutation rate was 1.2
mutations per mega base with a median of 230 somatic rearrangements
per tumor. There was great genetic intratumor heterogeneity, with
the presence of complex chains of rearrangements and hypermutation
in almost all cases (37,38).
Tumor analysis by multiregional whole-exome and
whole-genome sequencing in 86 tumor regions from 10 patients with
OS revealed an evolutionary genomic disparity between primary OS
and its pulmonary metastases, where the metastases exhibited a
higher mutational load and genomic instability compared to the
primary tumor. The mutated genes were enriched in the PI3K-Akt
pathway at both the early and late stages of tumor evolution and in
the MAPK pathway in the metastatic stage. However, metastases
showed improved immunogenicity, including increased neoantigen
loading, and also improved PD-L1 expression, an immunoglobulin
superfamily gene, and having more infiltrating lymphocytes compared
to the primary tumor. This suggests that metastases should be
treated separately from their original tumors by means of
personalized metastasis therapy, which requires real-time genetic
analysis after pulmonary metastasis (39).
Silent mutations in the TP53 and/or
RB1 genes have been reported to be the leading cause of
sporadic development of OS (11).
In vitro and in vivo study comparing MSCs with OS
malignant cells directed to the TP53 and RB1 using
transgenic mice with these silenced genes in its MSCs showed that
by only excluding TP53, the OS incidence could reach 60% of
cases (40). It has been shown that
p53 act as a guardian of the osteogenic differentiation of MSCs
into myogenic, adipogenic, hematopoietic and neural adult cells
(11).
Different studies point the pre-osteoblasts and
osteoblasts as cells which give rise to OS (8,41),
suggesting that the cellular microenvironment is critical in
determining the fate of MSCs in tumor formation (10). The osteogenic differentiation of MSCs
with defective or mutant p53 may affect signaling and its
microenvironment, and possibly contribute to tumor initiation
(11).
As MSCs represent the source of osteogenic
progenitor cells and osteoblasts, thus mutations in TP53
gene of these cells play a decisive role in proliferation,
compromising the maturation, negatively regulating their
differentiation, and interfering with cell processes such as
ontogenesis (11). This is due to
the reduced expression of key genes encoding transcription factors
involved in the early stages of osteogenic differentiation,
including Runx2 and Osterix (14,42).
Under normal conditions, Runx2 and Osterix expression
is strictly regulated during osteogenic differentiation of
progenitor cells into osteoblasts and osteocytes, ensuring balanced
bone remodeling. In vitro silencing of the TP53 gene
in embryonic mouse fibroblasts induces increased Osterix and
Runx2 expression levels in MSCs. This compromises the
differentiation of osteoblasts in mature osteocytes, causing damage
to bone remodeling, resulting in the osteosclerotic phenotype
observed in p53-deficient mice (11,42).
It has been demonstrated that p53 not only regulates
the genomic stability of MSCs, but also regulates the cell
differentiation program including osteogenesis and bone remodeling
to prevent the onset of bone tumor. The absence of the p53 function
in the regulation of the differentiation of MSCs in osteoblasts due
to mutational events or silencing can start the tumor as a result
of changes in osteogenesis, bone homeostasis, and bone remodeling
(11,40).
OS is a heterogeneous tumor containing cells at
various stages of differentiation during ontogenesis (7). Thus, it was proposed that mutation in
TP53 gene may affect osteogenic differentiation of MSCs and
significantly contribute to tumor onset by the following
mechanisms: (1) Stop acting as a
transcription factor, suppressing multipotent progenitor cell
differentiation to mediate early osteogenic differentiation
(2) promoting genomic instability
and uncontrolled proliferation of MSCs; and (3) deregulating the immune activity of MSCs,
increasing the secretion of growth factors and chemokines. This
suggests an important role for p53 function defects in OS
development. Evidence of this is that the osteosclerotic condition
imposes the OS phenotype in p53-deficient mice (11).
It has been reported that frizzled-related secreted
protein 2 (SFRP2) has an oncogenic role in OS development
associated with TP53 gene mutation, and that the high
expression of this protein correlates with poor prognosis of OS
patients (43). Thus, induced
pluripotent stem cells (iPSCs) obtained from patients with
Li-Fraumeni syndrome that have mutations of the TP53
germline were used to analyze the role of SFRP2 in OS. It has been
found that ectopic SFRP2 overexpression in normal osteoblast
precursors containing TP53 gene mutation is enough to
suppress normal osteoblast differentiation and promote OS
phenotypes by inducing oncogenic molecules such as FOXM1 and
CYR61, independently of β-catenin. On the other hand,
inhibition of SFRP2, FOXM1 or CYR61 suppresses the
tumorigenic process. This demonstrates that the oncogenic role of
SFRP2 in OS development is due to its ability to induce
oncogenic molecules such as FOXM1 and CYR61 in the presence of
mutations in the TP53 gene (44).
A dominant subclone was identified in samples from
patients with successive recurrences after sequencing the exome and
germ cell DNA from cells collected from a patient with
chemoresistent and metastatic OS over 3 years at 3 different times
and after comparing allele frequencies of the different samples.
This clone presented two remarkable features, consisting in a novel
translocation in TP53-KPNA3 allele and the loss of the
wild-type TP53 allele. Lastly, a meta-analysis study which
included 8 publications covering 210 patients with OS evaluated the
effect of the mutations in TP53 gene and concluded that
mutations in this gene were associated with smaller 2-year overall
survival of patients. The data show that mutations in TP53
gene have one unfavorable impact on the 2-year overall survival
when compared to the wild type (45).
Role of deregulating the expression of tumor
suppressor genes
Among the tumor suppressor genes includes
WWOX, whose function is suppressed or attenuated in most
human tumors. In a Wwox-deficient mice model it was
demonstrated that these animals developed OS and a bone metabolic
disorder characterized by hypocalcemia and osteopenia. In addition,
deletion of the WWOX gene was found in 30% of OS and the
protein product of this gene was absent or reduced in about 60% of
the tumors. It has been found that the tumor suppressor function of
WWOX is exerted through its binding to RUNX2,
suppressing its transcriptional activity in osteoblasts and tumor
cells. Thus, the negative regulation of WWOX results in the
maintenance of the RUNX2 activity, creating a conducive
environment to the development of OS since low levels of
WWOX expression increase proliferation, migration, and
invasion of tumor cells (46,47).
The RB1 gene encoding the retinoblastoma
protein (pRB) plays a critical role in regulating the transition
from the G1 phase to the S phase of the cell cycle. In the absence
of mitogenic stimuli, pRB remains hypophosphorylated and bound to
E2F, a transcription factor, which prevents the pRB action on the
cell cycle progression, leading to a cycle stop in G1 (48). This function is reversed by
phosphorylation of pRB by the cyclin-dependent kinase 4 (CDK4)
during normal mitosis, which results in the release of E2F, leading
to cell cycle progression. The absence of cell cycle arrest in G1
due to mutations or RB1 silencing removes this cell cycle control
point, preventing the repair of DNA damage, and causing genomic
instability (49).
The CDKN2A gene codes two products through
alternative splicing which are functionally and structurally
distinct (p16INK4a and p14ARF). The p16INK4a is a negative
regulator of CDK4 and therefore the gift of its function
leads to an increase in CDK4 expression, which results in
inactivating the pRB function in cell cycle arrest (50). Thus, mutations or silencing of the
CDKN2A gene may lead to inactivation of the pRB function.
Curiously, control losses in the cell cycle caused by the loss of
pRB function are reported in most OS cases (18). The p14ARF protein normally acts by
removing the ubiquitin E3 MDM2 ligase from the nucleolus,
preventing its degradation action of p53 (51). Since p14ARF is expressed from the
same locus of CDKN2A encoding p16INK4a, its function in
pathway p53 is like that of p16INK4a in the pRB pathway, disrupting
the cell cycle. Thus, mutations or silencing by methylation of the
CDKN2A gene also alter p14ARF function and have
repercussions on the p53 pathway, promoting cell cycle
dysregulation, leading to genomic instability. Mutations affecting
p53 function have been described in most OS cases (Fig. 1) (18,49).
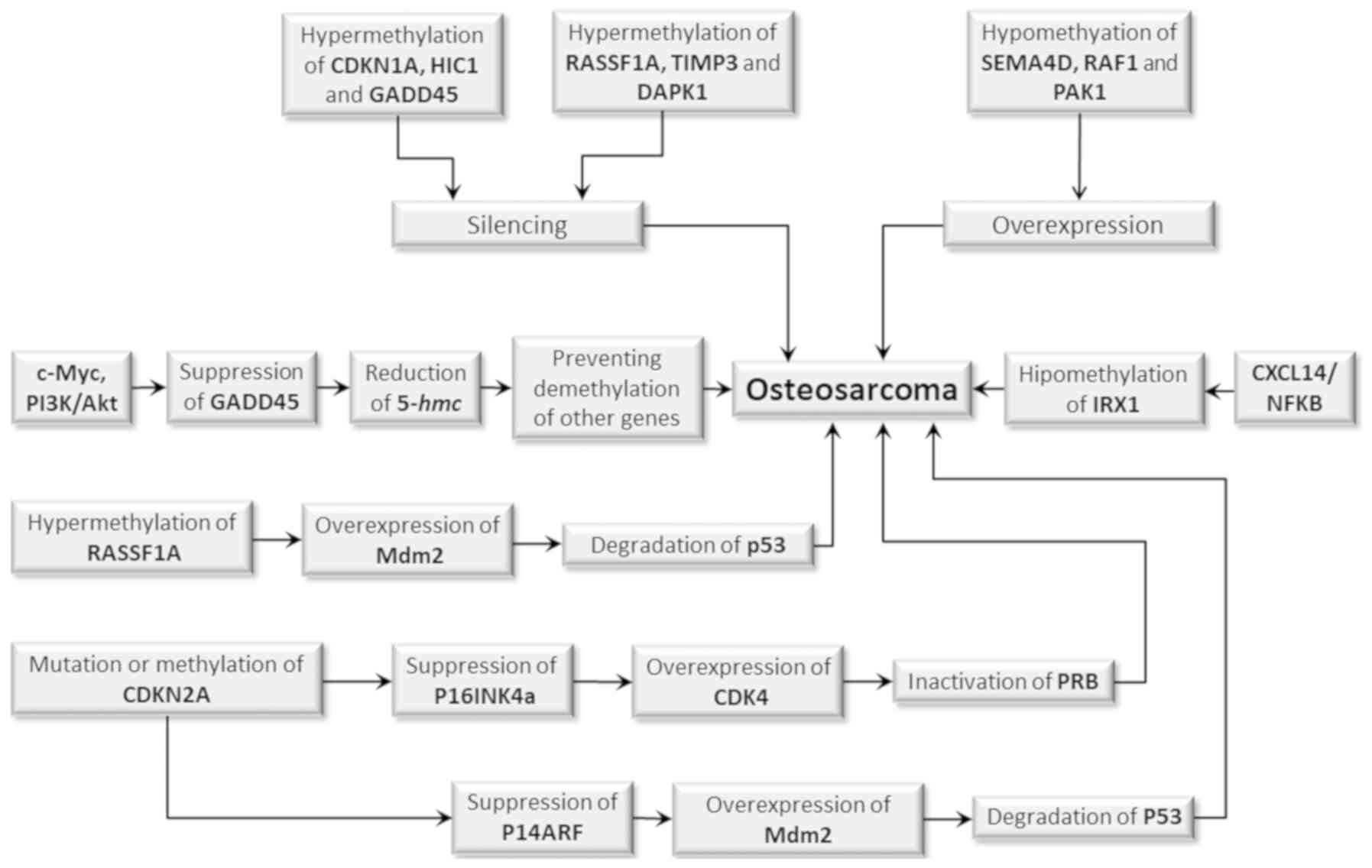 | Figure 1.Epigenetic events which may
contribute to the initiation and progression of OS. Target genes or
those which have p53-modulated activities, including GDKN1A, HIC,
GADD45, RASSF1A, TIMP3 and DAPK1, are silenced when
hypermethylated. An inverse situation occurs with SEMA4D, RAF1 and
PAK1 genes when they are hypomethylated which results in
overexpression. However, both conditions may equally favor the
development of OS. Silencing by hypermethylation of the RASSF1A
gene leads to overexpression of the MDM2 gene, whose product
promotes p53 degradation, which in turn results in uncontrolled
cell cycle and absence of DNA repair and apoptosis inhibition. The
hypomethylation of the IRX1 gene promotes activation of the
CXCL14/NFkB signaling pathway, whereas the activation of the cMyc
and PI3/Akt signaling pathways results in suppression of the GADD45
gene encoding the 5-hmC production, impeding the demethylation of
other genes, which also favors tumor development. Mutation or
methylation of the CDKN2A gene reduces the production of p16INK4a,
which leads to overexpression of CDK4 and inactivation of pRB. It
can also reduce the production of p14INK4a, which in turn
suppresses p14ARF, resulting in high Mdm2 levels which degrades
p53. Both mechanisms result in an uncontrolled cell cycle, lack of
DNA repair and apoptosis inhibition, favoring tumor initiation and
progression. OS, osteosarcoma. |
A recent study showed that functional genetic
single-nucleotide polymorphisms in the CDKN2 gene, locus A
and B (CDKN2A/B) predict susceptibility to and prognosis of OS in
Chinese individuals. The GA and AA genotypes of rs3217992 in
CDKN2A/B are related to increased risk of tumor, and the GA and AA
genotypes of rs3217992 in CDKN2A correlate with higher stage and
higher risk of pulmonary metastasis and poor prognosis (52).
Regulation of oncogene expression
Like what occurs with other tumors, the abnormal
activity of oncogenes and tumor suppressor genes is described as a
key molecular event underlying the development of OS (25). The MDM2 oncogene whose
activity may be dependent or independent of p53 presented
frequently increased expression levels in a variety of human
tumors, since it significantly impacts the p53 functions and
consequently tumorigenesis (51).
The product of this gene (the Mdm2 protein) is one of the major
negative regulators of p53 as it performs E3 ubiquitin ligase
activity which promotes inactivation of p53 function by its
degradation. Under normal conditions, Mdm2 suppresses p53 activity
to allow cell cycle continuity, but under stress conditions it
remains active and promotes cell cycle arrest for correcting
possible damage to DNA (53).
Overexpression or amplification of the MDM2 locus is detected in OS
(54).
The c-Myc oncogene stands out as one of the
most studied and whose role in OS pathogenesis is best understood.
Moreover, it is found to be overexpressed in more than 10% of cases
of the disease, being correlated with increased recurrence and
tumor invasiveness due to the activation of the MEK-ERK pathway,
leading to the reduction of apoptotic potential in tumor cells
(55). Studies show that inhibition
of c-Myc activity results in decreased proliferation, invasion, and
viability of the tumor cells (55,56).
The c-Myc oncogene is significantly overregulated in
metastatic OS samples and the high expression of this oncogene is
associated with poor survival of OS patients. Treatment of OS cells
with super enhancer inhibitors THZ1 and JQ1 effectively suppresses
OS cell proliferation, migration and invasion (57).
On the other hand, c-Fos is another oncogene which
has been found to upregulate in OS cells, being related to a higher
rate of tumor metastasis (26). The
expression of c-Fos and of Wnt2, a protein implicated in cell
oncogenesis and its Fzd9 receptor, was evaluated in OS tissue,
tumor-derived MG63 cells and in osteoblasts to investigate the role
of c-Fos in OS. MG63 cells were treated with small interference RNA
to knockdown c-Fos, aiming to confirm the relationship of c-Fos
with Wnt2/Fzd9. The expression of c-Fos, Wnt2 and Fzd9 was found to
be markedly higher in OS tissues than in adjacent non-cancerous
tissues, and their expression in MG63 was markedly increased
compared to osteoblasts. c-Fos knockdown inhibited MG63 cell
proliferation, migration and invasion, and promoted MG63 cell
apoptosis. In addition, c-Fos knockdown inhibited mRNA and protein
expression of Wnt2 and Fzd9. This indicates that the c-Fos action
in OS development is done by activating expression of Wnt2 and its
Fzd9 receptor (58).
TRIM14 is upregulated in OS samples and cell lines
derived from this tumor. Overexpression of TRIM14 increases tumor
cell proliferation, cell cycle progression, migration and invasion
in vitro and promotes tumor growth in vivo. Moreover,
TRIM14 overexpression is correlated with tumor progression and low
patient survival time. On the other hand, silencing TRIM14 has the
opposite effects. In addition, TRIM14 overexpression induced
activation of the AKT pathway, while inhibition of AKT expression
reversed TRIM14-mediated effects on cell growth and mobility, as
well as epithelial-mesenchymal transition. This indicates that
TRIM14 functions as an oncogene in OS, positively regulating the
AKT signaling pathway (59).
Role of epigenetic mechanisms
Epigenetics comprises a set of biological phenomena
triggered by environmental factors which promote gene expression
regulation at the transcription level through chemical
modifications in the DNA, such as changes in the pattern of
methylation, acetylation, phosphorylation, stable chromatin
modifications, and of histones, in addition to nucleosome
remodeling and Non-coding RNAs (60,61).
These epigenetic modifications occur in key oncogenes, tumor
suppressor genes, and transcription factors, leading to cancer
initiation and progression (61).
Such events may result in alterations in the expression or
silencing of genes and of miRs which result in the phenotypic
change of the individual without alterations in the DNA sequence
(62). Such events are crucial for
the development and normal differentiation of different cell lines
in the adult individual (63).
Unlike the genetic alterations which are irreversible, the
epigenetic changes are reversible, allowing us to intervene to
reverse the malignant characteristic of a population of cells,
returning it to its normal status (19). These chemical modifications in DNA
are constantly made and undone throughout the life of the
individual, since he or she often encounters agents which promote
these phenomena throughout their lives (6). Like what occurs with other types of
human cancers, the initiation and progression of OS can be
triggered by genetic and epigenetic events that alter the behavior
pattern of cells, altering gene expression and/or signaling
pathways, which can contribute to malignant transformation
(19). Although it exhibits a wide
range of genetic and molecular changes, including gains, losses, or
rearrangements of chromosomal regions, the more recent knowledge
suggests that OS is a disease caused by epigenetic alterations
which interrupt the osteoblastic differentiation of MSCs (17). Epigenetic studies have shown
extensive reprogramming of each component of the epigenetic
machinery of the tumor cells including DNA methylation, histone
modifications, nucleosome positioning, and miRs expression
(19).
As with other human cancers, OS appears to contain
many of these epigenome changes compared to normal osteoblasts, the
presumed target cell for transformation. Some studies have analyzed
the epigenome-like shape of this tumor, albeit with a low number of
cases (64,65). The results confirm the existence of
specific heterogeneous methylation events among the different types
of human OS and that these differences may help explain the
differences in the clinical behavior of subtypes of this tumor
(66).
It has recently been observed that the crosstalk
between MSCs and extracellular vesicle-mediated OS cells can
influence the epigenetic signature of cells through the methylation
of transposable elements such as LINE-1 and tumor suppressor genes,
modulating the fate of mesenchymal stem cells and the epigenetic
status of these cells by altering gene expression related to bone
turnover (35). Global changes in
epigenetic patterns are a hallmark of cancer, as disturbances in
epigenetic processes can lead to altered genetic function and
malignant transformation of the cell. The initiation and
progression of cancer, traditionally considered as a genetic
disease, is now understood as a complex process involving
epigenetic abnormalities along with genetic alterations. A better
understanding of epigenetic mechanisms in the development of cancer
and the role of some of these components in relation to OS is
discussed below.
DNA methylation
DNA methylation is a chemical change involving the
addition of a methyl group to the cytosine DNA nucleotides which
typically occurs in CpG dinucleotides not randomly distributed in
the genome that represents an important epigenetic mechanism used
for the prolonged silencing of gene expression (67). The human genome contains long
stretches of CpG islands, with unusually elevated levels of CpG
dinucleotides, concentrated in the promoters of the genes. In
general, the CpG islands of normal gene promoters are not
methylated and they are normally expressed (68).
The methylation pattern of a given mammal is
established during its development and is normally maintained
throughout life, being regulated by the enzymes DNA
methyltransferase (DNMT) and demethylase. Changes in the expected
pattern of methylation by either hypomethylation or
hypermethylation can lead to genomic instability and trigger
tumorigenesis (69).
There are three classes of DNMTs in eukaryotes
(Dnmt1, Dnmt2, Dnmt3a/Dnmt3b) (70).
DNMT1 is the most important and responsible for maintaining DNA
methylation levels, while DNMT3a/DNMT3b is involved in methylation
again, being responsible for establishing DNA methylation patterns
during embryogenesis and setting up genomic imprints during germ
cell development (71). Although
DNMT2 is not currently considered to be a DNA methylase, this
enzyme methylates small transfer RNAs (tRNAs) (72).
Changes in the expected pattern of methylation are
it for hypomethylation or hypermethylation can lead to genomic
instability and trigger tumorigenesis or maintain the malignant
state of cancer cells. Hypermethylation of the promoter of a gene
is responsible for its transcriptional inactivation, a common event
in cancers. The silencing or activation of genes mediated by
aberrant DNA methylation, can affect almost all cell signaling
pathways, including those of DNA repair, cell cycle regulation,
promotion of apoptosis or control of signaling networks relevant to
tumor development (73).
Methylation as the consequent silencing of tumor
suppressor genes has been reported in OS. Although RB and
TP53 genes are not frequent targets of silencing by
methylation, changes in pRB and p53 pathways have been pointed as
pathogenic methylation targets, specifically the CDKN2A locus,
which encodes the cyclin-dependent kinase inhibitor, p16INK4a, and
the inhibitor of Mdm2, p14ARF (7).
Several gene targets of p53, or which have its p53-modulated
activity, have been found in the methylated form and silenced on OS
or xenograft cell lines, including CDKN1A, HIC1 and
GADD45 (74). In addition,
many other tumor suppressor genes are silenced by hypermethylation
of their promoter in OS-derived cell lines, including RASSF1A,
TIMP3, DAPK1, and others (Fig.
1) (6).
It was found that GADD45 gene encoding
proteins of the 5-hydroxymethylcytosine (5 hmC) family, which
mediates the methylation in osteogenic differentiation, is
co-operatively repressed by the c-Myc and PI3K/Akt pathways in OS
cells (75). The repression of
GADD45 may be due to the aberrant methylation pattern of its
promoters. Interestingly, p53 reduce methylation of promoters of
tumor suppressor genes, among them RASSF1A (6). However, there are cancer cells that
have wild-type TP53, which is down-regulated by means of a
p53-RASSF1A-Mdm2 feedback loop which results in hypermethylation of
RASSF1A, leading to MDM2 expression which remains
bound to p53, promoting its degradation (76). On the other hand, RASSF1A promotes
Mdm2 degradation in a p53-dependent manner, preventing degradation
of p53 by Mdm2. The silencing of RASSF1A due to DNA
methylation is the explanation for the fact that there are cancer
cells which have wild TP53 (77).
A study of gene expression associated with
metastasis in OS identified the IRX1 gene as a candidate to
be gene pro-metastatic when it undergoes little methylation.
IRX1 encodes a member of the iroquois homeobox protein 1
family, a transcription factor which plays a crucial role in
embryonic development and was previously pointed out as a potential
tumor suppressor in gastric cancer (78). It was hypothesized that the
hypomethylation of IRX1 gene promotes pulmonary metastasis
of the OS, since overexpression of this gene was strongly
associated with the hypomethylation of its promoter in both
OS-derived cell lines and in clinical samples obtained from the
tumor. These pro-metastatic effects of IRX1 are due to its role as
positive regulator of the CXCL14/NF-kB cell signaling pathway. In
addition, it has been shown that IRX1 can increase tumor cell
metastatic activity both in vitro and in vivo,
favoring migration and invasion, as well as promoting resistance to
anoikis in the murine model (Fig. 1)
(22).
The degree of methylation of more than 1.1 million
loci was tested on biopsy samples obtained from patients with OS
and analyzed in function of relapse or not of the disease. It was
found that patients who had tumor recurrence were more methylated
in more than 17% of the samples, whereas less than 1% of patients
who did not have relapse had high methylation. Moreover,
hypermethylation was found in genetic bodies, intragenic regions,
and promoters in patients with recurrent disease. It was
demonstrated that in 6.6% of the patients who had relapsed, the
promoters of the candidate gene were hypermethylated and 2% were
hypomethylated. A locus at the TLR4 gene demonstrates one of
strongest positive associations between DNA methylation and 5 y
event-free survival (66). Several
candidate oncogenes including SEMA4D, RAF1 and PAK1
are also hypomethylated and overexpressed in human OS compared to
normal osteoblasts (79).
Furthermore, some of these epigenetic changes, including repression
or aberrant activation, are associated with loss of expression
control at specific loci in OS cells (Fig. 1) (66,79).
A comparative study of the DNA methylation degree of
normal samples with those obtained from OS revealed that the
promoters of some genes are differentially methylated in the tumor.
The pathways and functions affected by these genes were identified
through protein-protein interaction (PPI), followed by the
identification of genes associated with cancer which had their
promoters differentially methylated, wherein 1379 hypermethylated
regions and 169 hypomethylated regions were identified.
Differential hypomethylation was significantly greater in the toll
receptor signaling pathway. In the PPI network, the MAXI interactor
signals transducer 1 (MXI1), the transcription activator
STAT3 and the T-cell acute lymphocytic leukemia 1
(TAL1) had the highest degree of hypermethylation. These
genes were identified as being associated with cancer and were
hypermethylated in OS cells (80).
The HOTAIR gene has been shown to be highly
expressed in OS cells, while knockdown of this gene results in down
regulation of DNMT1 with a reduction in overall DNA methylation
level. It was further seen that the HOTAIR product represses CDKN2A
expression by inhibiting CDKN2A promoter activity by DNA
hypermethylation. Mechanistically, HOTAIR acts in OS by suppressing
miR-126 expression, which is the negative regulator of DNMT1. Thus,
DNMT1 occurs in the absence of miR-126 overexpression, leading to
silencing of CDKN2A due to hypermethylation of DNA its promoter,
thus favoring tumor development (81).
A methylation status analysis of the whole genome of
19 different OS-derived cell lines and of 6 normal controls was
performed and the comparison between the two cell types was
established. The differentially methylated sites in tumor cells
were analyzed with the CpG assoc package and a total of 75 sites
were methylated in transcription factor binding regions to which 83
transcription factors can bind, which may lead to alteration in the
expression of 75 genes being differentially expressed in tumor
cells. In addition, several differentially methylated sites have
been associated with up-regulation of genes such as SEZ6L2, KIRREL,
CEP72 and CDK4, which may play an important role in OS pathogenesis
(82).
The hypermethylation of DNA from two CpG islands
adjacent to miR-449c genomic locus results in inhibition of its
expression, and consequently abolishes the function of miR-449c as
a negative regulator of c-Myc oncogene expression. In this
condition, c-Myc passes to be overexpressed, leading to activation
of downstream targets, contributing to OS tumorigenesis (83). Analysis of over 11,000 genes for
differential methylation level and over 3,000 genes for
differential expression in the OS revealed that the functions of
genes related to this tumor were mainly enriched in biological
processes related to inflammatory/immune response, Pertussis
pathways and hematopoietic cell lineage pathways. UBS and NRF were
found to be regulated by multiple genes in the OS. Kaplan Meier
analysis of genes to OS-associated identified BHMT2, DOCK2, DNALI1
and RIPK3 as significant survival-related genes. SEMA3A and PRAME
are included in the 40 genes and within the top 10 of the most
differentially expressed genes in OS (84).
Histone modification
Covalent modifications of the amino termini of the
histones in nucleosomes play a critical role in the regulation of
gene expression (85). Such
modifications are even more complex than DNA methylation because
they include acetylation, methylation, phosphorylation,
ubiquitination, and sumoylation (86). The amino-terminal modifications of
these proteins affect the affinities of the chromatin-associated
proteins and influence regulation of the dynamic transitions
between transcriptionally active or silent chromatin states. Thus,
the normal state of acetylation of histones and other transcription
factors bound to the promoter determines the dynamic equilibrium
that is regulated by acetyltransferase and histone deacetylase
(HDAC) enzymes. Aberrant acetylation with histone modifications are
implicated in anomalous expression of oncogenes and tumor
suppressor genes, which ultimately leads to tumorigenesis (87).
Unlike the dynamic equilibrium of acetylation
observed in normal cells, histones are typically hypoacetylated in
tumor cells (88). Histone
methylation may activate or inactivate gene transcription,
depending on where methylation occurs. Generally, H3K4, H3K36 and
H3K79 methylations are related to active gene transcription, while
methylations of H3K9, H3K27 and H4K20 are associated to gene
silencing. Thus, modifications of histones interact with DNA
methylation and the combined action of the two mechanisms plays a
key role in gene expression (89).
WNT5A is a family of genes which encode
signaling glycoprotein and its altered expression is associated
with various types of cancer. Expression of promoters A and B of
the WNT5A gene was studied in normal human osteoblasts, in
two SaOS-2 and U2OS OS cell lines, and in tumor tissue. It has been
found that both promoters A and B are active in normal osteoblasts,
being that promoter B was nearly 11 times more active than promoter
A. Three regions enriched with CpG islands of exon 1β of promoter B
are highly methylated in both SaOS-2 and U2OS cells. Histone
modifications were examined for their involvement in the activity
of promoters A and B. It was found that H3K4me3, a marker of
histone activation, showed a high level of histone modifications in
promoter A and a reduced level of promoter B modifications in cell
U20S, suggesting that H3K4me3 plays a repressive role, reducing the
activity of the promoter B. It has also been found that promoter B
is less enriched with the active H3K4me3 compared to promoter A in
U2OS and SaOS-2 cells. In addition, there is increased enrichment
of the repressive H3K27me3 in the promoter B in SaOS-2 cells.
Inhibition of promoter B of the WNT5A gene appears to be an
OS characteristic and involves both DNA methylation and histone
modifications. These results indicate that histone modifications in
the WNT5A gene promoter B reduce the transcription activity
of this gene in OS cells (90).
Histone demethylases KDM6A and KDM6B, associated
with the demethylation of histone H3 lysine trimethylation
(H3K27me3) were found to be upregulated in OS cells after treatment
with cisplatin. Cisplatin-resistant tumors had lower levels of
H3K27me3 than sensitive OS specimens. In vitro inhibition of
histone methyltransferase EZH2 in OS cells decreased H3K27me3
levels and led to cisplatin resistance. On the other hand,
inhibition KDM6A and KDM6B demethylases increased H3K27me3 levels
and reversed cisplatin resistance in vitro and in
vivo. This indicates that H3K27me3 acts in reducing KDM6A and
KDM6B expression by increasing tumor cell sensitivity to cisplatin
(91).
Nucleosome remodeling
The conformational changes and changes in position
of the nucleosomes along the DNA strand alter the interactions
between DNA and histones and interfere in the affinities of
transcription factors to DNA (92).
Thus, aberrant nucleosome remodeling can cause great damage to the
correct functioning of the cell. Remodeling of the nucleosomes has
a critical role in the process of normal differentiation and is
controlled by ATP-dependent chromatin-remodeling complexes (CRCs).
Such complexes act by regulating a wide range of cellular
processes, including transcription regulation in response to DNA
damage, DNA replication, and determination of cellular identity. In
this way, the deregulation of any of these processes can contribute
to the cellular transformation and tumorigenesis (90).
It has been shown that both DNA methylation and
histone modification as nucleosome remodeling may contribute to
transcriptional suppression and gene silencing in human OS. When
the CpG island of the promoter is methylated, the methyl-CpG
binding domain proteins (MBDs) will bind to this site instead of
the transcriptional activator complex. MBDs will recruit histone
deacetylase (HDAC), and consequently the histones are deacetylated.
Histone deacetylation increases the overall positive charge of
histone tails, which is associated with a more compact
heterochromatin structure, causing condensed chromatin (69).
Alterations in the RB-E2F signaling pathway are
known to be found in virtually all cases of OS, showing its
importance in this tumor development. It is also known that
lymphoid-specific helicase (HELLS) participates as a critical
effector of chromatin remodeling downstream of the RB-E2F signaling
pathway in various cancers, and has its expression regulated by the
RB-E2F pathway. A study using an OS model in genetically modified
mice revealed that the loss of the E2F1 and E2F3 transcription
factors significantly delays tumor progression and increases the
overall survival of mice with p53/Rb1 deficient OS. On the other
hand, it has been seen that HELLS mRNA is upregulated and its
protein is overexpressed in OS, but has no effect on tumor
proliferation and migration. In addition, loss of HELLS in OS has
no effect on tumor onset and overall survival of mice. The authors
concluded that while HELLS may serve as a biomarker for
tumorigenesis and for RB-E2F pathway status, it is unlikely to
serve as a target for therapeutics in OS (93).
Role of non-coding RNAs
Current knowledge reveals that most of the genes
that make up the human genome are transcribed into non-coding RNAs
(ncRNAs) which play important roles in the normal functioning of
the cell, but are also associated with pathological processes,
including cancer and infectious diseases (94,95).
Although ncRNAs are not translated into protein, they perform
important regulatory functions within the cell, and today are
recognized as causing huge changes in all fields of biology and
medicine due to its role in gene expression regulation (96). Increasing evidence has shown that
ncRNAs, including miRNAs, non-coding long RNAs (lncRNAs) and
circular RNAs (circRNAs) play important roles in regulating a wide
range of biological processes involved in human disease etiology,
including tumors (94). Some aspects
related to these non-coding RNAs in OS development are subsequently
presented.
MicroRNAs
MicroRNAs (miRNAs) are a class of small non-coding
RNA endogenous containing 20–30 nucleotides which play important
regulatory roles in various biological processes including
differentiation, cell proliferation, cell cycle control, apoptosis,
embryonic development and innate immunity (97,98).
miRNAs most often interact with the 3′untranslated regions (3′UTR)
of target mRNAs to induce their mRNA degradation or translational
repression. The interaction of miRNAs with other regions, including
the 5′UTR gene promoter sequence, has also been reported. In
addition, miRNAs may also activate translation or regulate
transcription under certain conditions (99).
Some miRs are implicated in OS and may act as a
factor protection or contribute to tumor initiation and
progression. Evidence of this was obtained in an in vitro
and in vivo functional validation study in tumor cell lines
obtained in which the tumor suppressor role of miR-16 and the
pro-metastatic role of miR-27a were confirmed (20).
It has been demonstrated that low levels of
miR-200b expression have been associated with advanced clinical
stage and metastasis in OS, and that its expression is
down-regulated in tumor-derived U2OS, Saos2, HOS, and MG63 cell
lines compared to normal osteoblasts. Restoring miR-200b expression
led to a significant decrease in proliferation, migration, and
invasion of tumor cells. In addition, ZEB1 gene encoding is a
transcription factor which suppresses the interleukin 2 (IL-2) gene
in specific T lymphocytes. It was identified as miR-200b target and
its expression were down-regulated by miR-200b in OS. ZEB1
expression has also been shown to be significantly increased in
tumor cells, while inhibition of ZEB1 expression has reduced
proliferation, migration, and invasion of tumor cells. The results
show that miR-200b inhibits proliferation of migration and invasion
of tumor cells by inhibiting ZEB1 expression (100).
A recent study in MG-63 cells lines derived of OS
showed that overexpression of miR-101 significantly suppressed the
expression of ROCK1, a gene encoding a serine/threonine kinase
signaling protein compared to knockdown of miR-101 in MG-63 cells.
Overexpression of miR-101 reduced the viability, migration, and
invasion of MG-63 cells and promoted apoptosis. Independent
inhibition of ROCK1 and reduction of miR-101 expression
levels increased proliferation, migration, and invasion of MG-63
cells and inhibited apoptosis. In addition, the miR-101 inhibitory
effect upon proliferation, migration, and invasion of MG-63 cells,
and the activation of apoptosis were reversed in knockdown of
ROCK1 in MG-63 cells. These results show that miR-101 plays
a tumor suppressor role in OS by targeting the ROCK1 gene,
and that overexpression of miR-101 inhibits tumor growth and tumor
cell movement by inactivating the PI3K/AKT and JAK/STAT signaling
pathways by down-regulation of ROCK1 gene expression
(Fig. 2) (101).
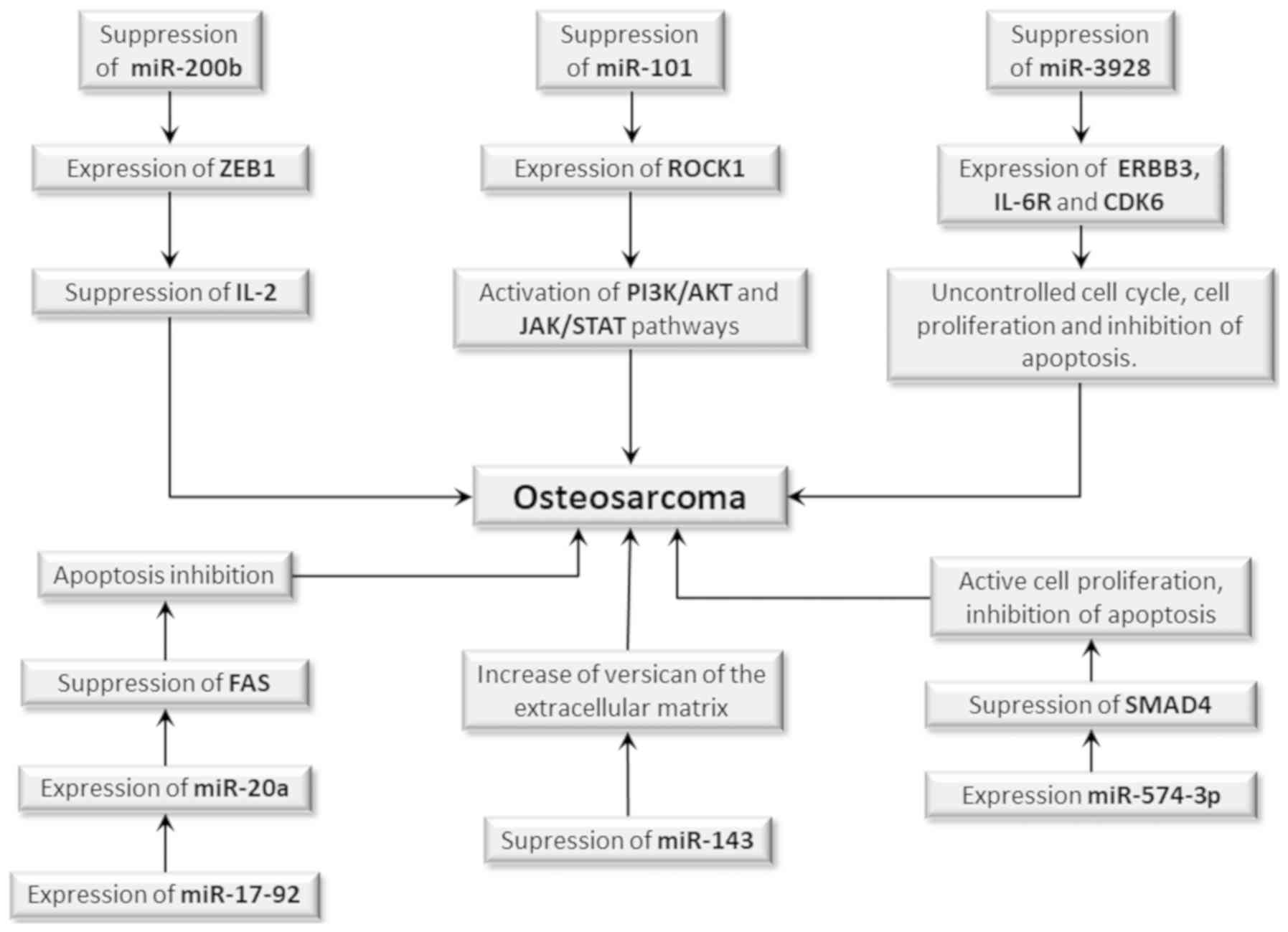 | Figure 2.Participation of miRs in OS
development. The suppression or activation of miR expression is an
epigenetic event which may be involved in OS development.
Suppression of miR-200b results in overexpression of the ZEB1 gene,
whose product suppresses IL-2 gene expression, whereas the
suppression of miR-101 results in overexpression of the ROCK1 gene
which actuates in the PI3K/AKT and JAK/STAT signaling pathways.
Both cases may increase the risk of initiation and progression of
OS, since they cause impairment of the immune response. Suppression
of miR-3928 increases the expression of the ERBB3, IL-6R and CDK6
genes, which may favor tumor development, promoting cell
proliferation, uncontrolled cell cycle and apoptosis inhibition.
The suppression of miR-143 results in the expression of versican
and proteoglycan of extracellular matrix, favoring tumor
progression. However, the expression of miR-17-92 increases the
expression of miR-20a, which in turn reduces Fas expression, a cell
death receptor, and inhibits apoptosis, whereas miR-574-3p
expression suppresses expression of the SMAD4 tumor suppressor
gene, which leads to increased cell proliferation and apoptosis
inhibition. Therefore, both mechanisms may increase the risk of OS.
miR, microRNA; OS, osteosarcoma. |
In another essay with cell lines derived from human
OS and normal osteoblasts, transfection for up-regulation or
down-regulation was used to measure the expression miR-3928. It was
found that miR-3928 inhibited tumor growth, induced cell apoptosis,
increased the percentage of cells in the G1 phase, and decreased
the percentage of cells in the S phase in the up-regulation
condition, whereas it promoted cellular proliferation and tumor
growth in the down-regulation condition. This suggests that
miR-3928 acts as a tumor suppressor, having the ERBB3,
IL-6R, and CDK6, gene encoders of the tyrosine-protein
kinase receptor, IL-6 receptor, and cyclin-dependent kinase 6 as
targets, respectively (Fig. 2)
(102).
It has been previously reported that pulmonary
metastasis formation in OS is inversely correlated with Fas (a type
II transmembrane protein of the TNF family) expression on the cell
surface. Interestingly, expression levels of miR-17-92 group
members, including miR-20a and miR-19a, were observed to be higher
in LM7 lineage metastatic cells expressing low Fas when compared to
non-metastatic lines which present high Fas expression levels. An
inverse correlation between Fas expression and miR-20a was observed
in all analyzed tumor-derived cells. Overexpression of miR-20a
resulted in a consistent and sustained negative regulation of Fas
expression in SAOS-2 cells. Inhibition of miR-20a in LM7 cells
increased Fas expression levels and reduced metastasis in mice
injected with LM7 stably transfected with anti-miR-20a. This
suggests that miR-20a encoded by the miR-17-92 gene
negatively regulates Fas expression in OS, increasing its
metastatic potential (Fig. 2)
(103).
Another miR strongly associated with OS development
is miR-574-3p, whose expression levels are increased very much in
the tissue obtained from tumors, as well as in U2OS, SAOS, and MG63
OS-derived cell lines compared to normal osteoblasts. Negative
regulation of miR-574-3p by antisense mi-574-3p resulted in cell
growth inhibition and induced cellular apoptosis. Furthermore,
overexpression of miR-574-3p by transfection with miR-574-3p mimics
promoted a proliferation of U2OS cells. Functional analysis
identified the decapentaplegic homologue 4 (SMAD4), which
encodes a family of signaling proteins, is a target of miR-574-3p,
since this gene function was suppressed in miR-574-3p transfected
cells. It has also been shown that overexpression of SMAD4
was able to neutralize the promoter effects of miR-574-3p on the
growth of cancer cells. Thus, it has been established that
miR-574-3p exerts a tumor-promoting function in OS by
down-regulating the expression of the SMAD4 tumor
suppression gene (Fig. 2) (104).
In 40 OS tissue samples it was shown that
miR-140 expression is reduced and that restoration of its
expression in OS-derived cells has a marked effect on inhibiting
cell proliferation and invasion, inducing apoptosis in
vitro, and suppressing tumor growth in vivo. A
bioinformatics study revealed that miR-140 has the gene encoding
histone deacetylase 4 (HDAC4) as target, and in this case
miR-140 acts as a tumor suppressor gene (105). Another study involving 85 patients
with resectable OS and 56 patients with un-resectable OS showed a
shorter disease-free survival in patients with low levels of
expression of miR-125b. The low miR-125b expression was
associated with advanced tumors in patients with un-resectable OS.
The results suggest that low expression of circulating
miR-125b may be a potential marker of poor prognosis in
patients with OS (106).
In OS metastatic cell models obtained by exogenous
transfection of F5M2 cells, a low level of miR-150
expression and significantly increased Ezrin (a gene encoding the
protein-tyrosine kinase) expression were found. The exogenous
transfection of miR-150 mimics in F5M2 cells resulted in
reduced Ezrin gene expression. In addition, overexpression of this
gene has been shown to promote a significant suppression of the
invasion and metastasis capability of F5M2 cells. The upregulation
of miR-150 results in down-regulation of Ezrin, which leads
to a reduction in the invasion and metastasis capacity of tumor
cells, indicating that miR-150 acts as a tumor suppressor in OS
(107).
It was found that that miR-449c is significantly
down-regulated in OS cells and presented hypermethylation of the
DNA in two CpG islands adjacent to the miR-449c genomic locus in OS
cells. Ectopic expression of miR-449c significantly inhibited OS
cell proliferation, colony formation and caused cell cycle arrest
in the G1 phase. miR-449c was able to negatively regulate c-Myc
oncogene expression. On the other hand, overexpression c-Myc
partially reversed cell proliferation and colony formation
inhibited by miR-449c. This shows that miR-449c acts as a tumor
suppressor, inhibiting c-Myc expression and that, in the OS
miR-449c is down-regulated due to DNA methylation (83).
Long non-coding RNAs (lncRNAs)
LncRNAs are transcribed with more than 200
nucleotides that play critical roles in different biological
processes such as cell growth, transcription, and translation,
epigenetic regulation of gene expression, splicing, nuclear
cytoplasmic traffic, and cell cycle control (108). Recent studies show that lncRNAs can
epigenetically regulate oncogenesis, which can prevent (109) favoring initiation and progression
of OS (110,111). Thus, lncRNAs can contribute to the
development and progression of OS by acting as tumor suppressors or
as oncogenes inducing tumor formation (109,111).
Therefore, they can modulate cancer pathogenesis in many aspects
including proliferation migration, metastasis, invasion and
cellular apoptosis (108).
It has been seen that lncRNAs can regulate OS by at
least two mechanisms that target mRNA: By activating signaling
pathways or by acting as a miRNA sponge. Positive regulation of the
Hedgehog (Hh) signaling pathway, which is implicated in the
regulation of differentiation, proliferation, cell polarity and
carcinogenesis, has been shown to promote the expression of
YAP1, a candidate human oncogene in multiple tumors, which
in turn is responsible for aberrant expression of lncRNA H19 in
malignant OS. In addition, lncRNAs may also regulate gene
expression at post-transcriptional levels, acting as an endogenous
‘sponge’ and under regulation of a microRNA chain (108).
Several lncRNAs have been reported as important
regulators in initiating and progressing OS, among them
MALAT1 which has been found to be upregulated in tumor
tissues compared to adjacent non-tumor soft tissues. Overexpression
of this lncRNA results in tumor cell proliferation, migration and
invasion in vitro and enhances tumor growth in a mouse
xenograft model, and has also been correlated with poor prognosis.
In OS, MALAT1 modulates RET proto-oncogene expression
by sponging miR-129-5p, increasing protein expression downstream of
the RET-Akt pathway, and its expression was positively correlated
with RET and negatively correlated with miR-129-5p in
original OS and in xenografted tumor cells. This shows that
MALAT1 act as oncogenic lncRNA in OS by regulating
RET via miR-129-5p suppression, thereby activating the
PI3K-Akt signaling pathway (112).
One study shows that lncRNA HOXD-AS1 encoded by
HOXD genes was significantly over-regulated in OS tissues and cells
derived from this tumor, and that its overexpression was associated
with poor prognosis of OS patients. HOXD-AS1 silencing resulted in
inhibition of tumor cell proliferation, induced cycle arrest in the
G1/G0 phase in vitro, and suppressed tumor cell growth in
vivo. It was confirmed that HOXD-AS1 could interact with
homologous zest enhancer (EZH2) of the p57 gene promoter,
inhibiting its tumor suppressive action, favoring OS oncogenesis
(113). It has also been shown that
expression of the TUG1 gene encoding lncRNA taurine 1 (TUG1)
was significantly higher in tumor tissues than in adjacent normal
bone tissues. Overexpression of TUG1 results in down-regulation of
miR-212-3p expression causing increased tumor size, advanced lymph
node metastasis, and decreased overall survival time of OS patients
(114).
Another non-coding long RNA involved in initiating
and progressing OS is lncRNA SNHG1, having miRNA-101-3p as targets
since it is up-regulated, while miRNA 101-3p is down-regulated in
tumoral tissue and tumor-derived cell lines. In addition, lncRNA
SNHG1 knockdown resulted in cell apoptosis and kept the cell cycle
in the G0/G1 phase, with reduced overall cell viability. Under
normal conditions, miRNA-101-3p acts by suppressing proliferation,
migration and cell invasion. Thus, down-regulated expression of
miRNA-101-3p enhances the expression of Rho-associated
coiled-coil-containing protein kinase 1 (ROCK1) and promotes cell
proliferation, migration and invasion. Overexpression of lncRNA
SNHG1 results in inactivation of the phosphoinositide 3-kinase/ATK
pathway and activation of the epithelial-mesenchymal transition of
the OS-derived cell lines. Thus, lncRNA SNHG1 behaves as an
oncogene, while miRNA-101-3p acts as a tumor suppressor (110).
On the other hand, RNA-steroid receptor RNA
activator 1 (SRA1) plays a protective role against OS in targeting
miRNA-208a, since its expression is down-regulated in tumor tissues
compared to normal bone tissue, while expression of microRNA-208a
was up-regulated in the tumor tissues. In addition, restoration of
expression of this lncRNA inhibited proliferation, migration and
invasion of tumor cells by increasing the apoptosis rate of these
cells. Up-regulation of microRNA-208a played a similar role in
silencing RNA-steroid receptor RNA activator 1, leading to
inhibition of apoptosis and increasing tumor cell proliferation,
migration and invasion (109).
Circular RNAs (circRNAs)
Circular RNAs (circRNAs) are a class of endogenous
non-coding RNAs generated from back-splicing, which are covalently
closed in forming a circular loop structure, with high stability
that can act in gene regulation (115,116).
Recent studies show that such molecules can regulate
transcriptional or post-transcriptional gene expression by acting
as miRNA sponges and are involved in the regulation of many
important biological processes (117). CircRNAs have been shown to play a
critical role in regulating gene expression in eukaryotes and
therefore may play central roles in initiating and progressing
cancer in humans (118).
In a microarray-based circRNA expression study
performed on OS-derived cell lines and compared to normal cells, 12
differentially expressed circRNAs were found; among them,
up-regulated hsa_circRNA_103801 and down-regulated
hsa_circRNA_104980. The potential targets of hsa_circRNA_103801
include hsa-miR-370-3p, hsa-miR-338-3p and hsa-miR-877-3p, while
the potential targets of hsa_circRNA_104980 were hsa-miR-1298-3p
and hsa-miR-660-3p. Functional analysis showed that
hsa_circRNA_103801 was involved in cancer signaling pathways such
as HIF-1, VEGF and angiogenesis pathway, the Rap1 signaling pathway
and the PI3K-Akt signaling pathway, while hsa_circRNA_104980 was
related to some pathways such as the tight junction pathway
(Fig. 3) (119).
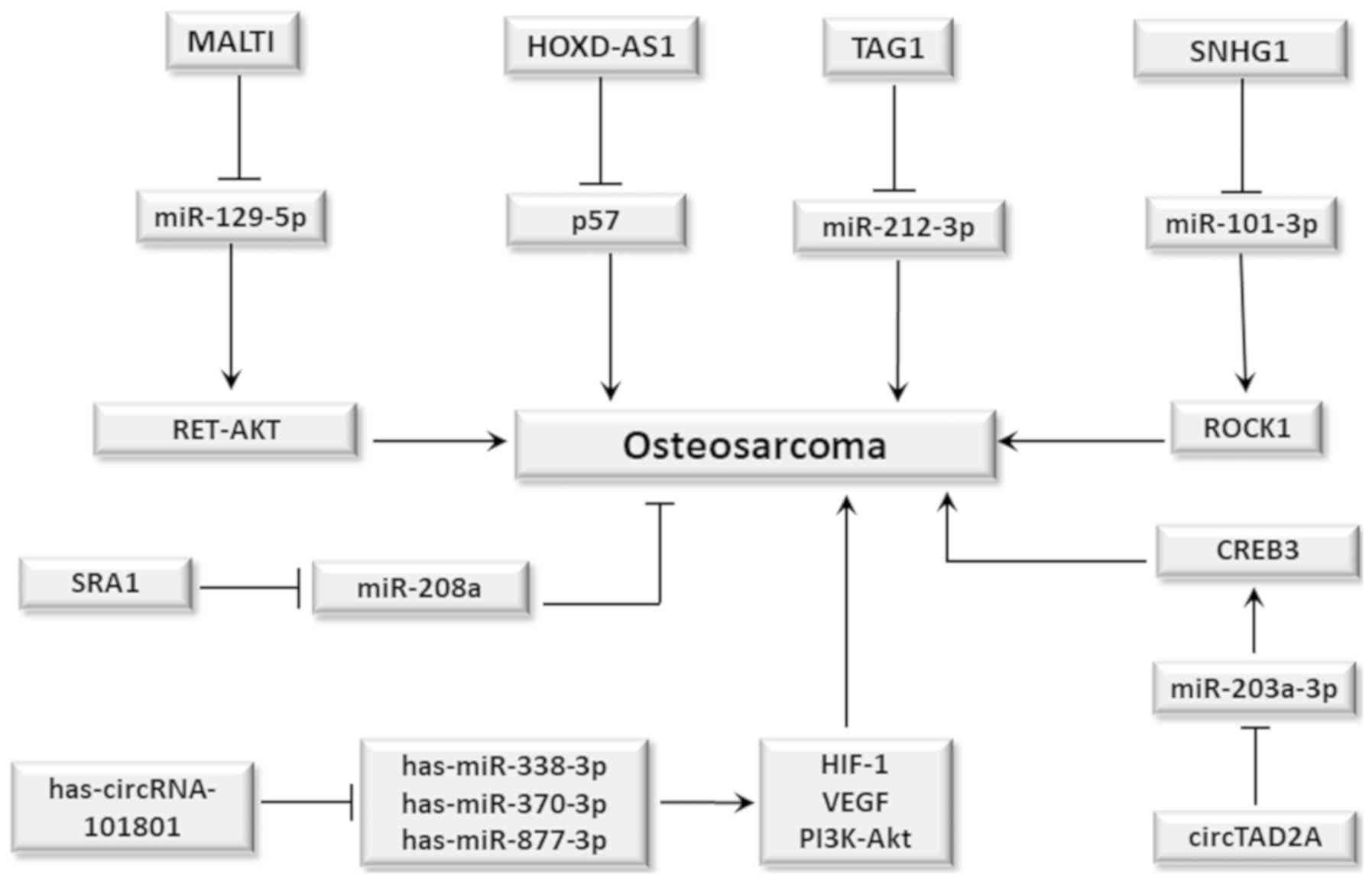 | Figure 3.Role of lncRNAs and circRNAs in
osteosarcoma. In general, lncRNAs and circRNAs act as sponges for
miRNAs, causing its inactivation and favoring tumor development.
Therefore, MAULTI acts by suppressing miR-129-5p which leads to
activation of the RET-AKT signaling pathway; HOXD-AS1 suppresses
the tumor suppressing action of p57 protein; TAG1 suppresses
miR-212-3p, whereas SNHG1 suppresses miR-101-3p, which results in
activation of ROCK1 gene expression. All these events favor the
development of osteosarcoma. However, lncRNA SRA1 acts by
suppressing miR-208a, inhibiting its tumorigenic action.
Additionally, has-circRNA-101801 circRNA acts by suppressing
miRNAs: has-miR-338-3p, has-miR-370-3p and has-miR-877-3p resulting
in increased expression of HIF-1 and VEGF, and PI3K-Akt signaling
pathway activation, thus favoring angiogenesis. Furthermore,
circRNA TAD2A suppresses miR-203a-3p, which leads to increased CREB
expression. This favors tumor development in both cases. circRNA,
circular RNA; lncRNA, long non-coding RNA. |
The high expression of CircTADA2A was found in both
OS tissue and tumor-derived cell lines. The inhibition of
circTADA2A expression attenuated tumor cell proliferation,
migration and invasion in vitro, as well as tumorigenesis
and metastasis in vivo. It has been shown that circTADA2A
acts as a sponge, absorbing miR-203a-3p to regulate CREB3
expression, which has been identified as an OS-conducting gene. In
addition, the inhibition miR-203a-3p, or CREB3
overexpression could reverse the circTADA2A silencing-induced
impairment in malignant tumor behavior (Fig. 3) (118).
Role of cytokines
Cytokine is a generic term used to denote a large
group of signaling proteins secreted by specific cells in response
to stressful conditions which mediate and regulate immunity,
inflammation, and hematopoiesis. Such molecules are also designated
as the basis in their presumed function, secretion cell, or target
of their action. For example, cytokines produced by lymphocytes may
be referred to as lymphokines, which are also known as interleukins
(ILs), since they are not only secreted by leukocytes, but are also
capable of affecting leukocyte cellular responses (120).) Although the main function of
cytokines is to seek homeostasis in conditions of stress and tissue
damage when there are failures in this process and the stressful
condition remains for a long time, the persistence of cytokines
will increase the risk of malignant transformation. Thus, host
responses to stress can affect various stages of cancer initiation
and tumor progression (121).
Therefore, it is very important to understand the deep and complex
interaction between the different cytokines in the oncogenesis
process, including those which occur in OS development (23). Next, we present some cytokines whose
actions are cited as possible mechanisms involved in OS
development.
Interleukin 6 (IL-6)
IL-6 is among the possible cytokines involved in OS
development, and is a pro-inflammatory cytokine which activates
Janus kinase (JAK), promoting the phosphorylation of transcription
activator 3 (STAT3), which in turn signals for increased cell
proliferation and inhibits apoptosis of the MSCs and of OS-derived
cells (122). High expression
levels of SOX18, IL-6 and p-STAT3 are found in OS, compared with
normal bone tissue (123). It has
been shown that neutralization of IL-6 with antibody or by the
STAT3 inactivation reduces tumor progression, besides inhibiting
JAK2 preventing lung metastases and increasing survival in animals
(122). In addition, IL-6
contributes to bone degradation by promoting the osteoclast
differentiation and expression of proteins which act on bone
resorption and induces the expression of the vascular endothelial
growth factor (VEGF) in OS cells (124,125).
Additionally, IL-6 can function as a mediator of pulmonary tropism
of cell of the OS favoring the installation of metastases in these
organs (126).
Transforming growth factor beta
(TGFβ)
TGFβ is linked to the dedifferentiation of MSCs in
OS, a dynamic population of cells associated with tumor invasion
and radio-and chemoresistance with poor prognosis (127). TGFβ is produced by autocrine
signaling of cancer cells which enhance the migration potential of
OS cells through the activation of the MAPK pathway (128). Activation of TGFβ signal
transduction activates pleiotropic functions involved in regulating
cell proliferation and differentiation, apoptosis, cell migration
and invasion, extracellular matrix production, angiogenesis and
immune response (129,130).
Due to its complex activity, TGFβ plays an
ambiguous role in tumors in humans. It acts as a tumor suppressor
in the early stages of tumorigenesis, inhibiting cell proliferation
and immortalization, and promoting apoptosis. In later stages it
promotes metastasis, migration, invasion and chemotaxis, and its
functions are associated with aggressive and invasive tumors
(131,132). Regarding OS, it was demonstrated
in vitro that tumor cells secrete TGFβ by activating the
TGFβ/SMAD-2/-3 signaling pathway, keeping MSCs in an
undifferentiated state and producing higher levels of pro-tumor
cytokines such as IL- 6 and VEGF (28). High TGFβ mRNA levels were also
reported in OS-derived cells and are associated with aggressive
behavior and lung metastases (133). In addition, an association was
observed between a significant increase in the activation of SMAD3
signaling pathway and high TGFβ1 levels in serum with a higher risk
of developing lung metastasis in OS patients (28).
An in vitro assay showed that TGF-β1
promoted OS cell migration and invasion by up-regulating the
expression of versican, an extracellular matrix proteoglycan, whose
expression is down-regulated by miR-143. TGF-β mechanistically
activates the MAPK pathway, which in turn activates the
TGF-β/SMAD-2/-3 pathway, leading to miR-143 suppression which leads
to an increase of versican in the extracellular matrix, thus
contributing to tumor progression since this favors tumor cell
migration and invasion (Fig. 4)
(129).
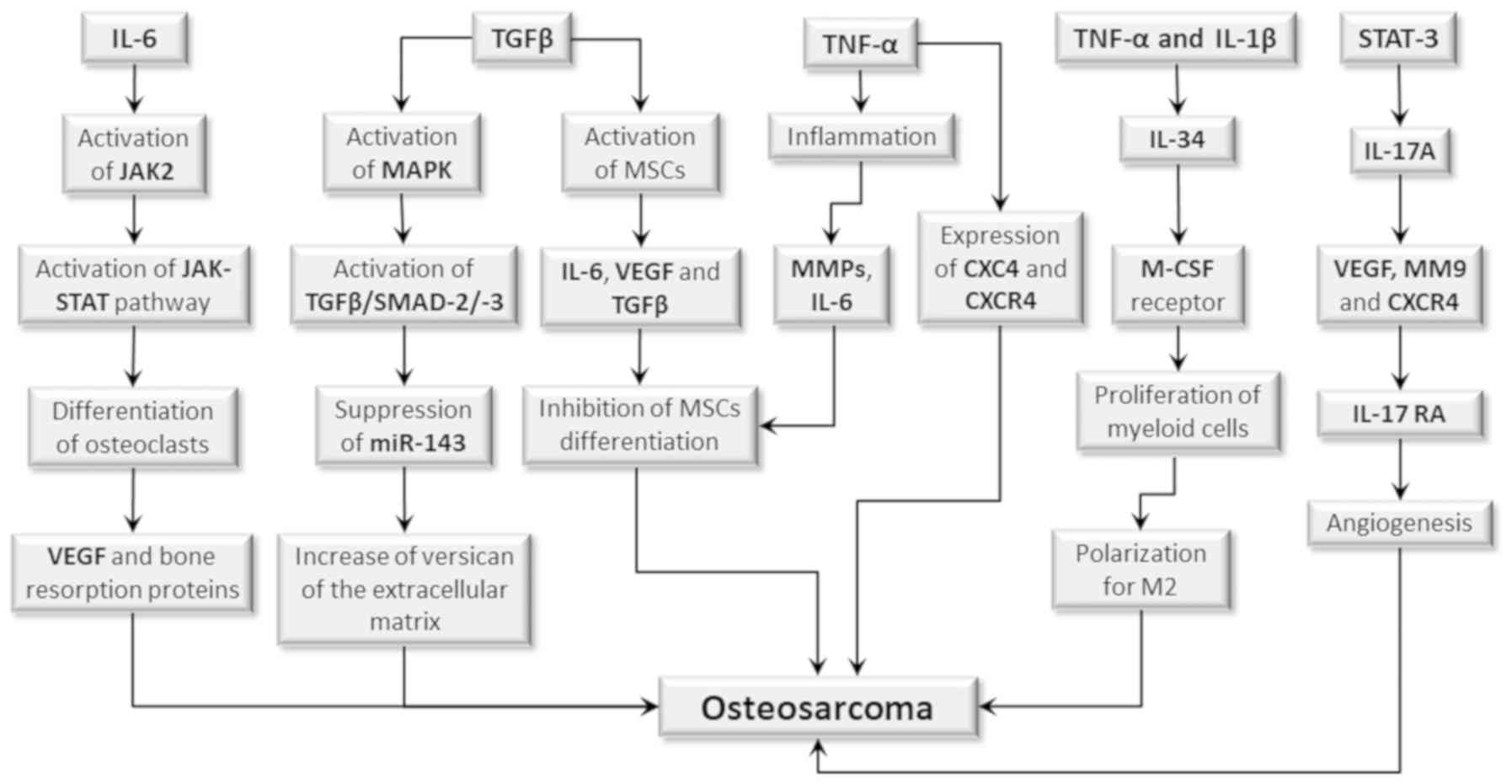 | Figure 4.Role of cytokines in human OS.
Certain cytokines may contribute to OS development by activating
cell signaling pathways, or by interfering in the differentiation
process of MSCs. IL-6 activates the expression of JAK2 signal
transducer, which in turn activates JAK-STAT signaling pathway that
induces the differentiation of MSCs into osteoclasts. This leads to
the production of VEGF and of proteins which act on bone resorption
and induces angiogenesis, contributing to tumor development. TGFβ
activates the MAPK pathway, which in turn activates the
TGF-β/SMAD-2/-3 pathway, resulting in miR-143 suppression, with a
consequent increase in versican expression of the extracellular
matrix, thus contributing to tumor progression. TGF-β also acts on
MSCs, inducing the production of IL-6, VEGF and additional TGF-β.
IL-6 together with TNFα induces inflammation and the production of
MMPs which (in synergistic action) inhibits MSC differentiation,
increasing the risk of initiating OS. Furthermore, TNFα also
increases the expression of CXC4 chemokine and its CXCR4 receptor,
a condition which also favors tumor development. TNFα together with
IL1β activates the production of IL-34 which binds to the M-CSF
receptor, promoting growth and survival of myeloid cells and
macrophage polarization for the M2 profile with recruitment of
these cells into the tumor environment, therefore contributing to
its progression. The STST3 transcription factor, upon being
phosphorylated, activates cytokine IL-17 production, which (if
connected to its IL-17RA receptor) stimulates VEGF, MM9 and CXCR4
production and promotes angiogenesis, thus contributing to tumor
progression and formation of metastases. MSCs, mesenchymal stem
cells; OS, osteosarcoma. |
TGF-β signaling under hypoxia conditions
dramatically increases the self-renewal capacity of MSCs in OS,
resulting in an increased potential for tumorigenesis,
neovasculogenesis, and metastasis. The blockade of TGF-β1 signaling
inhibited the differentiation and clonogenicity of tumor cells and
reduced hypoxia-mediated self-renewal of MSCs. These findings
suggest that a dynamic balance exists between stem cells and
non-stem cells within the cell population of the OS, and that MSCs
can be generated from differentiated cancer cells (127).
It has been shown that the Saos-2 and U2-OS cell
lines derived from OS produce high TGFβ levels as they activate
MSCs to produce IL-6 and VEGF, inhibiting the osteogenic
differentiation of MSCs. In addition, treatment with the anti-TGF-β
antibody significantly reduced the IL-6 and VEGF production by MSCs
and induced their osteogenic differentiation, showing that TGFβ
plays an important role in tumor initiation (28). In an orthotopic xenograft mouse model
of OS, tumor cells have been shown to incorporate TGFβ in the form
associated with membrane, which induces IL-6 production by tumor
mesenchymal stem cells promoting tumor growth, accompanied by the
intratumor activation of STAT3 and formation of pulmonary
metastases (Fig. 4) (132). In addition, TGFβ induces
mesenchymal epithelial transition by inhibiting of miR-499a
expression, interacting with the Snail1/Zeb1 of miR-499a promoter.
This result in phenotypic conversion of primary tumor cells that
acquire the ability to migrate and generate pulmonary metastases
(134).
Tumor necrosis factor (TNF-α)
TNF-α is a pro-inflammatory cytokine produced by
lymphocytes and macrophages which, although it can induce apoptosis
of tumor cells, is associated with progression of several types of
tumors, including OS (29). TNF-α
increases pulmonary metastasis in OS by increasing CXC 4 (CXCR4)
chemokine receptor expression. In a mouse model, infliximab
treatment, a TNF-α inhibitor decreased CXCR4 expression and
significantly reduced cellular mobility and lung metastases
(135). In an OS murine model
induced by the transfer of AX MSCs of INK4a-deficient to wild type
mice, the production of NF-α resulted in tumor growth and
maintaining cells in the undifferentiated state by means of
extracellular signal-regulated protein kinases (136). The treatment with TNF-α inhibitor
resulted in reduced tumor growth, increased osteoblast
differentiation, and the survival of the animals, highlighting the
pro-tumorigenic effect of TNF-α on OS (29).
Interleukin 34 (IL-34)
IL-34 has recently been identified and
characterized by its ability to form macrophage colonies in human
bone marrow cell cultures, constituting a similar function to that
of the macrophage colony stimulating factor (M-CSF) including its
synergistic action on inflammation (137). IL-34 signaling occurs by its
binding to the M-CSF receptor, which is expressed in human
mononuclear phagocytes (138).
Similarly, to M-CSF, IL-34 stimulates growth and survival of
myeloid cells and induces macrophage polarization to the profile of
M2 tumor-associated macrophages (139,140).
High levels of IL-34 expression are reported in
several types of cancers and are associated with poor prognosis. In
OS, IL-34 increases the blood supply, recruits and polarizes
macrophages to the M2 profile functioning as an angiogenic,
pro-metastatic factor and stimulator of tumor progression. The
paratibial inoculation of human OS cells which resulted in
overexpressing IL-34 in a murine model revealed that this cytokine
is correlated with tumor progression, promoting angiogenesis, tumor
growth, and pulmonary metastasis formation. In addition, IL-34
promoted M2 recruitment to inside the tumor. It has also been shown
that IL-34 is expressed in OS cells regulated by TNF-α, IL-1β, and
contributes to tumor growth by promoting M2 macrophage recruitment
(Fig. 4) (140).
Interleukin 17 (IL-17)
IL-17 is a cytokine produced by Th17 cells and
other cell types, including neutrophils, NK, TCD8+, and Tγδ cells,
whose role in cancer development is still controversial, although
it is correlated with poor prognosis in many types of human
cancers. In stromal cells, it has been shown that IL-17 induces
angiogenesis stimulators including VEGF, and that IL-17 receptor
expression is associated with VEGF production by OS-derived cell
lines (141,142). In the murine model, IL-17A
interaction with its IL-17RA receptor has been shown to promote
metastasis in nude mice (non-T-cell athymic) inoculated with
OS-derived cell lines expressing high levels of IL-17RA and then
transfected with IL-17 gene encoding (142). In addition, clinical trials have
shown that patients with OS had higher IL-17 serum levels when
compared to healthy subjects, and that IL-17 levels were even
higher in patients with metastases (141).
IL-17RA expression was also higher in the tumor
tissue of patients with metastatic OS and in the U-2 cell line
derived from the tumor, but not in MG63 cells. Interestingly,
negative IL-17RA regulation in U-2 cells was able to nullify the
increase in IL-17A-induced metastasis, while upregulation of
IL-17RA in MG63 cells increased the ability of these cells to
produce metastasis in response to IL-17A. The increased metastasis
may be due to the interaction of IL-17A with its IL-17RA receptor,
resulting in increased VEGF, MMP9, and CXCR4 expression in tumor
cells. In addition, STAT3 activity was shown to be crucial in the
interaction of IL-17A/IL-17RA to promote metastasis in OS (Fig. 4) (142).
Conclusions
OS, is a disease of multifactorial origin which
involves a complex interaction between a wide variety of factors
and mechanisms that when acting together promotes the deregulation
of cellular signaling pathways, causing disturbances in bone tissue
homeostasis. Bone tissue renewal requires an intensification of the
differentiation process of precursor cells into the bone formation.
As most cases of OS begin on the bone growth plate, disturbances in
differentiation of precursor cells play a role in tumor initiation.
It is believed that MSCs, osteogenic precursor cells which give
rise to osteoblasts for bone formation, are a key component in OS
initiation. This hypothesis is reinforced by experimental evidence
involving the premalignant stages of the disease through the
functional and phenotypic parallel analysis of normal MSCs,
transformed MSCs, and MSCs derived from the tumor. The karyotyping
of different MSCs revealed the occurrence of aneuploidization,
translocations, and homozygous loss of the Cdkn2 region of the
genome of those cells which controls the CDKN2A/p16 singling
pathway, and plays a crucial role in malignant transformation of
MSCs.
Another important aspect is the changes in the
functions of tumor suppressor genes and/or oncogenes, either due to
the occurrence of mutations, interference of epigenetic mechanisms
or even by the action of cytokines. Such events can alter the
crosstalk between cells and promote changes in their behavior,
leading to activation or deactivation of signaling pathways which
regulate cellular processes such as differentiation, proliferation,
migration, and apoptosis, increasing the risk of suffering
malignant transformation. Experimental evidence obtained through an
in vivo study in transgenic animals in which the TP53
and RB1 genes of MSCs cells were silenced reinforced the
importance of the dysfunction of these genes in the development of
OS. In addition, expression deregulation of tumor suppressor genes
and of other genes, including oncogenes, may also occur due to
epigenetic mechanisms triggered by environmental factors. These
events influence gene activation and silencing, and are frequently
found in cells obtained from tumors. Epigenetic mechanisms such as
DNA methylation, histone modifications, nucleosome remodeling, and
the action non-coding RNAs are often implicated in OS progression.
It is believed that epigenetic mechanisms act individually or
together to change the expression of tumor suppressor genes and/or
oncogenes, activating or deactivating the transcription of these
genes, resulting in the deregulation of cell signaling pathways
which, in some way, triggers the tumor initiation and progression
process.
Major scientific and conceptual advances have been
made in recent years, especially in the field of biology, focusing
on the mechanisms of tumor initiation, progression, metastasis and
heterogeneity. This is due to technological advances which have
enabled developing experimental models of tumors that mimic the
disease in humans. Along with the growing knowledge about the
mechanisms involved in tumor pathogenesis, several researchers are
devoting themselves to try to find ways to infer these mechanisms
in order to discover new options for treating the disease. Thus, we
believe that the prospects are very promising to achieve more
advances soon, as well as in the clinical area.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
JWVDA was involved in drafting the manuscript and
was a major contributor in writing the manuscript. TAADMF was
involved in literature review and revising the manuscript
critically for important intellectual content, and was a major
contributor in writing the manuscript. JVFJr was involved in
drafting the manuscript and literature review. JCVDA was involved
in literature review and revising the manuscript critically for
important intellectual content. DCFL was involved in literature
review and revising the manuscript critically for important
intellectual content. CMB was involved in drafting the manuscript
and literature review. VSA was involved in the conception of the
study and revising the manuscript critically for important
intellectual content. JMGDA was involved in literature review and
drafting the manuscript. JVF was involved in the conception and
design of the study, drafting the manuscript and was the major
contributor in writing the manuscript. All authors have read and
approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Klein MJ and Siegal GP: Osteosarcoma:
Anatomic and histologic variants. Am J Clin Pathol. 125:555–581.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stiller CA, Desandes E, Danon SE,
Izarzugaza I, Ratiu A, Vassileva-Valerianova Z and
Steliarova-Foucher E: Cancer incidence and survival in European
adolescents (1978–1997)-report from the automated childhood cancer
information system project. Eur J Cancer. 42:2006–2018. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nie Z and Peng H: Osteosarcoma in patients
below 25 years of age-An observational study of incidence,
metastasis, treatment and outcomes. Oncol Lett. 16:6502–6514.
2018.PubMed/NCBI
|
|
4
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Geller DS and Gorlick R: Osteosarcoma: A
review of diagnosis, management, and treatment strategies. Clin Adv
Hematol Oncol. 8:705–718. 2010.PubMed/NCBI
|
|
6
|
Morrow JJ and Khanna C: Osteosarcoma
genetics and epigenetics: Emerging biology and candidate therapies.
Crit Rev Oncog. 20:173–197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rao-Bindal K and Kleinerman ES: Epigenetic
regulation of apoptosis and cell cycle in osteosarcoma. Sarcoma.
2011:6794572011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mutsaers AJ and Walkley CR: Cells of
origin in osteosarcoma: Mesenchymal stem cells or osteoblast
committed cells? Bone. 62:56–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng Y, Wang G, Chen R, Hua Y and Cai Z:
Mesenchymal stem cells in the osteosarcoma microenvironment-their
biological properties, influence on tumor growth, and therapeutic
implications. Stem Cell Res Ther. 9:222018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Alfranca A, Martinez-Cruzado L, Tornin J,
Abarrategi A, Amaral T, de Alava E, Menendez P, Garcia-Castro J and
Rodriguez R: Bone microenvironment signals in osteosarcoma
development. Cell Mol Life Sci. 72:3097–3113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Velletri T, Xie N, Wang Y, Huang Y, Yang
Q, Chen X, Chen Q, Shou P, Gan Y, Cao G, et al: P53 functional
abnormality in mesenchymal stem cells promotes osteosarcoma
development. Cell Death Dis. 7:e20152016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han Y, Kim YM, Kim HS and Lee KY:
Melatonin promotes osteoblast differentiation by regulating Osterix
protein stability and expression. Sci Rep. 7:57162017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Komori T: Runx2, an inducer of osteoblast
and chondrocyte differentiation. Histochem Cell Biol. 149:313–323.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shin MH, He Y, Marrogi E, Piperdi S, Ren
L, Khanna C, Gorlick R, Liu C and Huang J: A RUNX2-mediated
epigenetic regulation of the survival of p53 defective cancer
cells. PLoS Genet. 12:e10058842016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martin JW, Zielenska M, Stein GS, van
Wijnen AJ and Squire JA: The role of RUNX2 in osteosarcoma
oncogenesis. Sarcoma. 2011:2827452011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Molchadsky A, Shats I, Goldfinger N,
Pevsner-Fischer M, Olson M, Rinon A, Tzahor E, Lozano Gina G,
Zipori D, Sarlig R and Rotter V: p53 plays a role in mesenchymal
differentiation programs, in a cell fate dependent manner. PLoS
One. 3:e37072008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang N, Song WX, Luo J, Haydon RC and He
TC: Osteosarcoma development and stem cell differentiation. Clin
Orthop Relat Res. 466:2114–2130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martin JW, Squire JA and Zielenska M: The
genetics of osteosarcoma. Sarcoma. 2012:6272542012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jones KB, Salah Z, Del Mare S, Galasso M,
Gaudio E, Nuovo GJ, Lovat F, LeBlanc K, Palatini J, Randall RL, et
al: MicroRNA signatures associate with pathogenesis and progression
of osteosarcoma. Cancer Res. 72:1865–1877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Meng G, Huang L and Guo QN:
Hypomethylation of the P3 promoter is associated with up-regulation
of IGF2 expression in human osteosarcoma. Hum Pathol. 40:1441–1447.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu J and Wang J: IRX1 hypomethylation in
osteosarcoma metastasis. Oncotarget. 6:16802–16803. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lopes-Júnior LC, Silveira DSC, Vulczak A,
Santos JC, Veronez LC, Fisch A, Flória-Santos M, Lima RAG and
Pereira-da-Silva G: Emerging cytokine networks in osteosarcoma.
Oncol Commun. 2:e11672016.
|
|
24
|
Yang Y, Yang R, Roth M, Piperdi S, Zhang
W, Dorfman H, Rao P, Park A, Tripathi S, Freeman C, et al:
Genetically transforming human osteoblasts to sarcoma: Development
of an osteosarcoma model. Genes Cancer. 8:484–494. 2017.PubMed/NCBI
|
|
25
|
Broadhead ML, Clark JCM, Myers DE, Dass CR
and Choong PFM: The molecular pathogenesis of osteosarcoma: A
review. Sarcoma. 2011:9592482011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Denduluri SK, Wang Z, Yan Z, Wang J, Wei
Q, Mohammed MK, Haydon RC, Luu HH and He TC: Molecular pathogenesis
and therapeutic strategies of human osteosarcoma. J Biomed Res.
30:5–18. 2016.
|
|
27
|
Deng ZL, Sharff KA, Tang N, Song WX, Luo
J, Luo X, Chen J, Bennett E, Reid R, Manning D, et al: Regulation
of osteogenic differentiation during skeletal development. Front
Biosci. 13:2001–2021. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tu B, Peng ZX, Fan QM, Du L, Yan W and
Tang TT: Osteosarcoma cells promote the production of pro-tumor
cytokines in mesenchymal stem cells by inhibiting their osteogenic
differentiation through the TGF-β/Smad2/3 pathway. Exp Cell Res.
320:164–173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mori T, Sato Y, Miyamoto K, Kobayashi T,
Shimizu T, Kanagawa H, Katsuyama E, Fujie A, Hao W, Tando T, et al:
TNFα promotes osteosarcoma progression by maintaining tumor cells
in an undifferentiated state. Oncogene. 33:4236–4246. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tung PY and Knoepfler PS: Epigenetic
mechanisms of tumorigenicity manifesting in stem cells. Oncogene.
34:2288–2296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Klimczak A and Kozlowska U: Mesenchymal
stromal cells and tissue-specific progenitor cells: their role in
tissue homeostasis. Stem Cells Int. 2016:42852152016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mohseny AB, Szuhai K, Romeo S, Buddingh
EP, Briaire-de Bruijn I, de Jong D, van Pel M, Cleton-Jansen AM and
Hogendoorn PC: Osteosarcoma originates from mesenchymal stem cells
in consequence of aneuploidization and genomic loss of Cdkn2. J
Pathol. 219:294–305. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang D and Liu S: SOX5 promotes
epithelial-mesenchymal transition in osteosarcoma via regulation of
Snail. J BUON. 22:258–264. 2017.PubMed/NCBI
|
|
35
|
Mannerström B, Kornilov R, Abu-Shahba AG,
Chowdhury IM, Sinha S, Seppänen-Kaijansinkko R and Kaur S:
Epigenetic alterations in mesenchymal stem cells by
osteosarcoma-derived extracellular vesicles. Epigenetics.
14:352–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Z, Guo J, Zhang K and Guo Y: TP53
mutations and survival in osteosarcoma patients: A meta-analysis of
published data. Dis Markers. 2016:46395752016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Perry JA, Kiezun A, Tonzi P, Van Allen EM,
Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS,
et al: Complementary genomic approaches highlight the PI3K/mTOR
pathway as a common vulnerability in osteosarcoma. Proc Natl Acad
Sci USA. 111:E5564–E5573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kovac M, Blattmann C, Ribi S, Smida J,
Mueller NS, Engert F, Castro-Giner F, Weischenfeldt J, Kovacova M,
Krieg A, et al: Exome sequencing of osteosarcoma reveals mutation
signatures reminiscent of BRCA deficiency. Nat Commun. 6:89402015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang D, Niu X, Wang Z, Song CL, Huang Z,
Chen KN, Duan J, Bai H, Xu J, Zhao J, et al: Multiregion sequencing
reveals the genetic heterogeneity and evolutionary history of
osteosarcoma and matched pulmonary metastases. Cancer Res. 79:7–20.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin PP, Pandey MK, Jin F, Raymond AK,
Akiyama H and Lozano G: Targeted mutation of p53 and Rb in
mesenchymal cells of the limb bud produces sarcomas in mice.
Carcinogenesis. 30:1789–1795. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rutkovskiy A, Stensløkken Ko and Vaage IJ:
Osteoblast differentiation at a glance. Med Sci Monit Basic Res.
22:95–106. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Long F: Building strong bones: Molecular
regulation of the osteoblast lineage. Nat Rev Mol Cell Biol.
13:27–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Techavichit P, Gao Y, Kurenbekova L, Shuck
R, Donehower LA and Yustein JT: Secreted Frizzled-Related Protein 2
(sFRP2) promotes osteosarcoma invasion and metastatic potential.
BMC Cancer. 16:8692016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim H, Yoo S, Zhou R, Xu A, Bernitz JM,
Yuan Y, Gomes AM, Daniel MG, Su J, Demicco EG, et al: Oncogenic
role of SFRP2 in p53-mutant osteosarcoma development via autocrine
and paracrine mechanism. Proc Natl Acad Sci USA. 115:E11128–E11137.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen KS, Kwon WS, Kim J, Heo SJ, Kim HS,
Kim HK, Kim SH, Lee WS, Chung HC, Rha SY and Hwang TH: A novel
TP53-KPNA3 translocation defines a de novo treatment-resistant
clone in osteosarcoma. Cold Spring Harb Mol Case Stud.
2:a0009922016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Del Mare S, Kurek KC, Stein GS, Lian JB
and Aqeilan RI: Role of the WWOX tumor suppressor gene in bone
homeostasis and the pathogenesis of osteosarcoma. Am J Cancer Res.
1:585–594. 2011.PubMed/NCBI
|
|
47
|
Del Mare S and Aqeilan RI: Tumor
Suppressor WWOX inhibits osteosarcoma metastasis by modulating
RUNX2 function. Sci Rep. 5:129592015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Henley SA and Dick FA: The retinoblastoma
family of proteins and their regulatory functions in the mammalian
cell division cycle. Cell Div. 7:102012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Abreu Velez AM and Howard MS:
Tumor-suppressor genes, cell cycle regulatory checkpoints, and the
skin. N Am J Med Sci. 7:176–188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Min EY, Kim IH, Lee J, Kim EY, Choi YH and
Nam TJ: The effects of fucodian on senescence are controlled by the
p16INK4a-pRb and p14Arf-p53 pathways in hepatocellular carcinoma
and hepatic cell lines. Int J Oncol. 45:47–56. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shi D and Gu W: Dual roles of MDM2 in the
regulation of p53: Ubiquitination dependent and ubiquitination
independent mechanisms of MDM2 repression of p53 activity. Genes
Cancer. 3:240–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang H, Mao JS and Hu WF: Functional
genetic single-nucleotide polymorphisms (SNPs) in cyclin-dependent
kinase inhibitor 2A/B (CDKN2A/B) locus are associated with risk and
prognosis of osteosarcoma in chinese populations. Med Sci Monit.
25:1307–1313. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhao Y, Yu H and Hu W: The regulation of
MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys
Sin (Shanghai). 46:180–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yu Q, Li Y, Mu K, Li Z, Meng Q, Wu X, Wang
Y and Li L: Amplification of Mdmx and overexpression of MDM2
contribute to mammary carcinogenesis by substituting for p53
mutations. Diagn Pathol. 9:712014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Han G, Wang Y and Bi W: C-Myc
overexpression promotes osteosarcoma cell invasion via activation
of MEK-ERK pathway. Oncol Res. 20:149–156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wu X, Cai ZD, Lou LM and Zhu YB:
Expressions of p53, c-Myc, Bcl-2 and apoptotic index in human
osteosarcoma and their correlations with prognosis of patients.
Cancer Epidemiol. 36:212–216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen D, Zhao Z, Huang Z, Chen DC, Zhu XX,
Wang YZ, Yan YW, Tang S, Madhavan S, Ni W, et al: Super enhancer
inhibitors suppress MYC driven transcriptional amplification and
tumor progression in osteosarcoma. Bone Res. 6:112018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang Q, Liu H, Wang Q, Zhou F, Liu Y,
Zhang Y, Ding H, Yuan M, Li F and Chen Y: Involvement of c-Fos in
cell proliferation, migration, and invasion in osteosarcoma cells
accompanied by altered expression of Wnt2 and Fzd9. PLoS One.
12:e01805582017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xu G, Guo Y, Xu D, Wang Y, Shen Y, Wang F,
Lv Y, Song F, Jiang D, Zhang Y, et al: TRIM14 regulates cell
proliferation and invasion in osteosarcoma via promotion of the AKT
signaling pathway. Sci Rep. 7:424112017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nowacka-Zawisza M and Wiśnik E: DNA
methylation and histone modifications as epigenetic regulation in
prostate cancer. Oncol Rep. 38:2587–2596. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shanmugam MK, Arfuso F, Arumugam S,
Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS,
Sethi G, et al: Role of novel histone modifications in cancer.
Oncotarget. 9:11414–11426. 2017.PubMed/NCBI
|
|
62
|
Sachdeva M, Dodd RD, Huang Z, Grenier C,
Ma Y, Lev DC, Cardona DM, Murphy SK and Kirsch DG: Epigenetic
silencing of Kruppel like factor-3 increases expression of
pro-metastatic miR-182. Cancer Lett. 369:202–211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kanherkar RR, Bhatia-Dey N and Csoka AB:
Epigenetics across the human lifespan. Front Cell Dev Biol.
2:492014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sadikovic B, Yoshimoto M, Al-Romaih K,
Maire G, Zielenska M and Squire JA: In vitro analysis of integrated
global high-resolution DNA methylation profiling with genomic
imbalance and gene expression in osteosarcoma. PLoS One.
3:e28342008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kresse SH, Rydbeck H, Skårn M, Namløs HM,
Barragan-Polania AH, Cleton-Jansen AM, Serra M, Liestøl K,
Hogendoorn PC, Hovig E, et al: Integrative analysis reveals
relationships of genetic and epigenetic alterations in
osteosarcoma. PLoS ONE. 7:e482622012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Rosenblum JM, Wijetunga NA, Fazzari MJ,
Krailo M, Barkauskas DA, Gorlick R and Greally JM: Predictive
properties of DNA methylation patterns in primary tumor samples for
osteosarcoma relapse status. Epigenetics. 10:31–39. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Varshney J, Scott MC, Largaespada DA and
Subramanian S: Understanding the osteosarcoma pathobiology-a
comparative oncology approach. Vet Sci. 3(pii): E32016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Jeziorska DM, Murray RJS, De Gobbi M,
Gaentzsch R, Garrick D, Ayyub H, Chen T, Li E, Telenius J, Lynch M,
et al: DNA methylation of intragenic CpG islands depends on their
transcriptional activity during differentiation and disease. Proc
Natl Acad Sci USA. 114:E7526–E7535. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li B and Ye Z: Epigenetic alterations in
osteosarcoma: Promising targets. Mol Biol Rep. 41:3303–3315. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hamidi T, Singh AK and Chen T: Genetic
alterations of DNA methylation machinery in human diseases.
Epigenomics. 7:247–265. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li E and Zhang Y: DNA methylation in
mammals. Cold Spring Harb Perspect Biol. 6:a0191332014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Goll MG, Kirpekar F, Maggert KA, Yoder JA,
Hsieh CL, Zhang X, Golic KG, Jacobsen SE and Bestor TH: Methylation
of tRNA Asp by the DNA methyltransferase homolog Dnmt2. Science.
311:395–398. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pfeifer GP: Defining driver DNA
methylation changes in human cancer. Int J Mol Sci. 19(pii):
E11662018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Al-Romaih K, Sadikovic B, Yoshimoto M,
Wang Y, Zielenska M and Squire JA: Decitabine-induced demethylation
of 5′ CpG island in GADD45A leads to apoptosis in osteosarcoma
cells. Neoplasia. 10:471–480. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Amente S, Zhang J, Lavadera ML, Lania L,
Avvedimento EV and Majello B: Myc and PI3K/AKT signaling
cooperatively repress FOXO3a-dependent PUMA and GADD45a gene
expression. Nucleic Acids Res. 39:9498–9507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang H, He J, Li J, Tian D, Gu L and Zhou
M: Methylation of RASSF1A gene promoter is regulated by p53 and
DAXX. FASEB J. 27:232–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Song MS, Song SJ, Kim SY, Oh HJ and Lim
DS: The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination
by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 27:1863–1874.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Guo X, Liu W, Pan Y, Ni P, Ji J, Guo L,
Zhang J, Wu J, Jiang J, Chen X, et al: Homeobox gene IRX1 is a
tumor suppressor gene in gastric carcinoma. Oncogene. 29:3908–3920.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Moriarity BS, Otto GM, Rahrmann EP, Rathe
SK, Wolf NK, Weg MT, Manlove LA, LaRue RS, Temiz NA, Molyneux SD,
et al: A sleeping beauty forward genetic screen identifies new
genes and pathways driving osteosarcoma development and metastasis.
Nat Genet. 47:615–624. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Xu J, Li D, Cai Z, Zhang Y, Huang Y, Su B
and Ma R: An integrative analysis of DNA methylation in
osteosarcoma. J Bone Oncol. 9:34–40. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li X, Lu H, Fan G, He M, Sun Y, Xu K and
Shi F: A novel interplay between HOTAIR and DNA methylation in
osteosarcoma cells indicates a new therapeutic strategy. J Cancer
Res Clin Oncol. 143:2189–2200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang Q: CpG methylation patterns are
associated with gene expression variation in osteosarcoma. Mol Med
Rep. 16:901–907. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li Q, Li H, Zhao X, Wang B, Zhang L, Zhang
C and Zhang F: DNA methylation mediated downregulation of miR-449c
controls osteosarcoma cell cycle progression by directly targeting
oncogene c-Myc. Int J Biol Sci. 13:1038–1050. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tian W, Li Y, Zhang J, Li J and Gao J:
Combined analysis of DNA methylation and gene expression profiles
of osteosarcoma identified several prognosis signatures. Gene.
650:7–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Bannister AJ and Kouzarides T: Regulation
of chromatin by histone modifications. Cell Res. 21:381–395. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Li S: Implication of posttranslational
histone modifications in nucleotide excision repair. Int J Mol Sci.
13:12461–12486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kelly TK, De Carvalho DD and Jones PA:
Epigenetic modifications as therapeutic targets. Nat Biotechnol.
28:1069–1078. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ropero S and Esteller M: The role of
histone deacetylases (HDACs) in human cancer. Mol Oncol. 1:19–25.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Black JC, Van Rechem C and Whetstine JR:
Histone lysine methylation dynamics: Establishment, regulation, and
biological impact. Mol Cell. 48:491–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Vaidya H, Rumph C and Katula KS:
Inactivation of the WNT5A alternative promoter B is associated with
DNA methylation and histone modification in osteosarcoma cell lines
U2OS and SaOS-2. PLoS One. 11:e01513922016. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
He C, Sun J, Liu C, Jiang Y and Hao Y:
Elevated H3K27me3 levels sensitize osteosarcoma to cisplatin. Clin
Epigenetics. 11:82019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lawlor ER and Thiele CJ: pigenetic changes
in pediatric solid tumors: Promising new targets. Clin Cancer Res.
18:2768–2779. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wu SC and Benavente CA: Chromatin
remodeling protein HELLS is upregulated by inactivation of the
RB-E2F pathway and is nonessential for osteosarcoma tumorigenesis.
Oncotarget. 9:32580–32592. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Nie JH, Li TX, Zhang XQ and Liu J: Roles
of non-coding RNAs in normal human brain development, brain tumor,
and neuropsychiatric disorders. Noncoding RNA. 5(pii):
E362019.PubMed/NCBI
|
|
95
|
Fernandes JCR, Acuña SM, Aoki JI,
Floeter-Winter LM and Muxel SM: Long non-coding RNAs in the
regulation of gene expression: Physiology and disease. Noncoding
RNA. 5(pii): E172019.PubMed/NCBI
|
|
96
|
Calin GA: The noncoding RNA
revolution-three decades and still going strong! Mol Oncol.
13(3)2019.
|
|
97
|
Patil VS, Zhou R and Rana TM: Gene
regulation by non-coding RNAs. Crit Rev Biochem Mol Biol. 49:16–32.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mens MMJ and Ghanbari M: Cell cycle
regulation of stem cells by microRNAs. Stem Cell Rev Rep.
14:309–322. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
O'Brien J, Hayder H, Zayed Y and Peng C:
Overview of microRNA biogenesis, mechanisms of actions, and
circulation. Front Endocrinol (Lausanne). 9:4022018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li Y, Zeng C, Tu M, Jiang W, Dai Z, Hu Y,
Deng Z and Xiao W: MicroRNA-200b acts as a tumor suppressor in
osteosarcoma via targeting ZEB1. Onco Targets Ther. 9:3101–3111.
2016.PubMed/NCBI
|
|
101
|
Jiang R, Zhang C, Liu G, Gu R and Wu H:
MicroRNA-101 inhibits proliferation, migration and invasion in
osteosarcoma cells by targeting ROCK1. Am J Cancer Res. 7:88–97.
2017.PubMed/NCBI
|
|
102
|
Xu H, Liu X and Zhao J: Down-regulation of
miR-3928 promoted osteosarcoma growth. Cell Physiol Biochem.
33:1547–1556. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Huang G, Nishimoto K, Zhou Z, Hughes D and
Kleinerman ES: miR-20a encoded by the miR-17-92 cluster increases
the metastatic potential of osteosarcoma cells by regulating Fas
expression. Cancer Res. 72:908–916. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Xu H, Liu X, Zhou J, Chen X and Zhao J:
miR-574-3p acts as a tumor promoter in osteosarcoma by targeting
SMAD4 signaling pathway. Oncol Lett. 12:5247–5253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Xiao Q, Huang L, Zhang Z, Chen X, Luo J,
Zhang Z, Chen S, Shu Y, Han Z and Cao K: Overexpression of miR-140
inhibits proliferation of osteosarcoma cells via suppression of
histone deacetylase 4. Oncol Res. 25:267–275. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Luo Z, Liu M, Zhang H and Xia Y:
Association of circulating miR-125b and survival in patients with
osteosarcoma-A single center experience. J Bone Oncol. 5:167–172.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Qu Y, Pan S, Kang M, Dong R and Zhao J:
MicroRNA-150 functions as a tumor suppressor in osteosarcoma by
targeting IGF2BP1. Tumour Biol. 37:5275–5284. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Ali A, Hu L, Qian A, Chen C and Yang T:
Long noncoding RNAs and human osteosarcoma. J Stem Cell Res Ther.
8:32018. View Article : Google Scholar
|
|
109
|
Guo W, Jiang H, Li H, Li F, Yu Q, Liu Y,
Jiang W and Zhang M: LncRNA-SRA1 suppresses osteosarcoma cell
proliferation while promoting cell apoptosis. Technol Cancer Res
Treat. 18:1–11. 2019. View Article : Google Scholar
|
|
110
|
Deng R, Zhang J and Chen J: lncRNA SNHG1
negatively regulates miRNA-101-3p to enhance the expression of
ROCK1 and promote cell proliferation, migration and invasion in
osteosarcoma. Int J Mol Med. 43:1157–1166. 2019.PubMed/NCBI
|
|
111
|
Zhou Y, Yin L, Li H, Liu LH and Xiao T:
The LncRNA LINC00963 facilitates osteosarcoma proliferation and
invasion by suppressing miR-204-3p/FN1 axis. Cancer Biol Ther.
20:1141–1148. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chen Y, Huang W, Sun W, Zheng B, Wang C,
Luo Z, Wang J and Yan W: LncRNA MALAT1 promotes cancer metastasis
in osteosarcoma via activation of the PI3K-Akt signaling pathway.
Cell Physiol Biochem. 51:1313–1326. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gu W, Zhang E, Song L, Tu L, Wang Z, Tian
F, Aikenmu K, Chu G and Zhao J: Long noncoding RNA HOXD-AS1
aggravates osteosarcoma carcinogenesis through epigenetically
inhibiting p57 via EZH2. Biomed Pharmacother. 106:890–895. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Yu X, Hu L, Li S, Shen J, Wang D, Xu R and
Yang H: Long non-coding RNA Taurine upregulated gene 1 promotes
osteosarcoma cell metastasis by mediating HIF-1α via miR-143-5p.
Cell Death Dis. 10:2802019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kun-Peng Z, Xiao-Long M and Chun-Lin Z:
Overexpressed circPVT1, a potential new circular RNA biomarker,
contributes to doxorubicin and cisplatin resistance of osteosarcoma
cells by regulating ABCB1. Int J Biol Sci. 14:321–330. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao
L, Zhang Y, Wu YM, Dhanasekaran SM, Engelke CG, Cao X, et al: The
landscape of circular RNA in cancer. Cell. 176:869–881.e13. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wang C, Ren M, Zhao X, Wang A and Wang J:
Emerging roles of circular RNAs in osteosarcoma. Med Sci Monit.
24:7043–7050. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wu Y, Xie Z, Chen J, Chen J, Ni W, Ma Y,
Huang K, Wang G, Wang J, Ma J, et al: Circular RNA circTADA2A
promotes osteosarcoma progression and metastasis by sponging
miR-203a-3p and regulating CREB3 expression. Mol Cancer. 18:732019.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Liu W, Zhang J, Zou C, Xie X, Wang Y, Wang
B, Zhao Z, Tu J, Wang X, Li H, et al: Microarray expression profile
and functional analysis of circular RNAs in osteosarcoma. Cell
Physiol Biochem. 43:969–985. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Goldszmid RS and Trinchieri G: The price
of immunity. Nat Immunol. 13:932–938. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Turtle CJ, Hudecek M, Jensen MC and
Riddell SR: Engineered T cells for anti-cancer therapy. Curr Opin
Immunol. 24:633–639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Tu B, Du L, Fan QM, Tang Z and Tang TT:
STAT3 activation by IL-6 from mesenchymal stem cells promotes the
proliferation and metastasis of osteosarcoma. Cancer Lett.
325:80–88. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wu Z, Yang W, Liu J and Zhang F:
Interleukin-6 upregulates SOX18 expression in osteosarcoma. Onco
Targets Ther. 10:5329–5336. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Tzeng HE, Tsai CH, Chang ZL, Su CM, Wang
SW, Hwang WL and Tang CH: Interleukin-6 induces vascular
endothelial growth factor expression and promotes angiogenesis
through apoptosis signal-regulating kinase 1 in human osteosarcoma.
Biochem Pharmacol. 85:531–540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wu Q, Zhou X, Huang D, Ji Y and Kang F:
IL-6 enhances osteocyte-mediated osteoclastogenesis by promoting
JAK2 and RANKL activity in vitro. Cell Physiol Biochem.
41:1360–1369. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Gross AC, Cam H, Phelps DA, Saraf AJ, Bid
HK, Cam M, London CA, Winget SA, Arnold MA, Brandolini L, et al:
IL-6 and CXCL8 mediate osteosarcoma-lung interactions critical to
metastasis. JCI Insight. 3(pii): 997912018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang H, Wu H, Zheng J, Yu P, Xu L, Jiang
P, Gao J, Wang H and Zhang Y: Transforming growth factor β1 signal
is crucial for dedifferentiation of cancer cells to cancer stem
cells in osteosarcoma. Stem Cells. 31:433–446. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Lamora A, Talbot J, Mullard M, Brounais-Le
Royer B, Redini F and Verrecchia F: TGF-β signaling in bone
remodeling and osteosarcoma progression. J Clin Med. 5(pii):
E962016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Li F, Li S and Cheng T: TGF-β1 promotes
osteosarcoma cell migration and invasion through the
miR-143-versican pathway. Cell Physiol Biochem. 34:2169–2179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Verrecchia F and Rédini F: Transforming
growth factor-β signaling plays a pivotal role in the interplay
between osteosarcoma cells and their microenvironment. Front Oncol.
8:1332018. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lebrun JJ: The dual role of TGFβ in human
cancer: From tumor suppression to cancer metastasis. ISRN Mol Biol.
2012:3814282012.PubMed/NCBI
|
|
132
|
Cantelli G, Crosas-Molist E, Georgouli M
and Sanz-Moreno V: TGFΒ-induced transcription in cancer. Semin
Cancer Biol. 42:60–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Baglio SR, Lagerweij T, Pérez-Lanzón M, Ho
XD, Léveillé N, Melo SA, Cleton-Jansen AM, Jordanova ES, Roncuzzi
L, Greco M, et al: Blocking tumor-educated MSC paracrine activity
halts osteosarcoma progression. Clin Cancer Res. 23:3721–3733.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang T, Wang D, Zhang L, Yang P, Wang J,
Liu Q, Yan F and Lin F: The TGFβ-miR-499a-SHKBP1 pathway induces
resistance to EGFR inhibitors in osteosarcoma cancer stem cell-like
cells. J Exp Clin Cancer Res. 38:2262019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Kato H, Wakabayashi H, Naito Y, Kato S,
Nakagawa T, Matsumine A and Sudo A: Anti-tumor necrosis factor
therapy inhibits lung metastasis in an osteosarcoma cell line.
Oncology. 88:139–146. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Ishikawa T, Shimizu T, Ueki A, Yamaguchi
SI, Onishi N, Sugihara E, Kuninaka S, Miyamoto T, Morioka H,
Nakayama R, et al: Twist2 functions as a tumor suppressor in murine
osteosarcoma cells. Cancer Sci. 104:880–888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Lin H, Lee E, Hestir K, Leo C, Huang M,
Bosch E, Halenbeck R, Wu G, Zhou A, Behrens D, et al: Discovery of
a cytokine and its receptor by functional screening of the
extracellular proteome. Science. 320:807–811. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Wang Y, Szretter KJ, Vermi W, Gilfillan S,
Rossini C, Cella M, Barrow AD, Diamond MS and Colonna M: IL-34 is a
tissue-restricted ligand of CSF1R required for the development of
Langerhans cells and microglia. Nat Immunol. 13:753–760. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Foucher ED, Blanchard S, Preisser L, Garo
E, Ifrah N, Guardiola P, Delneste Y and Jeannin P: IL-34 induces
the differentiation of human monocytes into immunosuppressive
macrophages-Antagonistic effects of GM-CSF and IFNγ. PLoS One.
8:e560452013. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ségaliny AI, Mohamadi A, Dizier B,
Lokajczyk A, Brion R, Lanel R, Amiaud J, Charrier C, Boisson-Vidal
C and Heymann D: Interleukin-34 promotes tumor progression and
metastatic process in osteosarcoma through induction of
angiogenesis and macrophage recruitment. Int J Cancer. 137:73–85.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Honorati MC, Cattini L and Facchini A:
Possible prognostic role of IL-17R in osteosarcoma. J Cancer Res
Clin Oncol. 133:1017–1021. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Wang M, Wang L, Ren T, Xu L and Wen Z:
IL-17A/IL-17RA interaction promoted metastasis of osteosarcoma
cells. Cancer Biol Ther. 14:155–163. 2013. View Article : Google Scholar : PubMed/NCBI
|














