Lynch syndrome: An overview
Clinical features
Lynch syndrome (LS) is the most common hereditary
form of colorectal cancer (CRC) with an incidence of 3–5% of all
CRC, followed by Familial Adenomatous Polyposis (FAP), which
accounts for less than 1% of the total CRC. LS and FAP are
autosomal dominant inheritance diseases, caused by germline
mutations in the DNA mismatch repair (MMR) genes and the tumor
suppressor gene Adenomatous Polyposis Coli (APC), respectively
(1–2).
LS is also known as hereditary non-polyposis colorectal cancer
(HNPCC) to highlight the absence of colon polyps and to distinguish
this syndrome from FAP characterized by 100–1,000 polyps (1–4) and other
hereditary syndrome of colorectal cancer, such as hamartomatous
polyposis syndrome (5–7). LS patients born with a germline mutation
in one of these MMR genes, and acquire inactivation of the second
wild-type allele in their tumoral DNA, fulfilling Knudson's two hit
hypothesis for inactivation of tumor suppressor genes. The somatic
inactivation of the corresponding wild-type allele occurs almost
exclusively by small mutations or (partial) gene loss, and
bi-allelic inactivation then leads to complete abolition of the
protein function of MMR system (3).
This results in a defective DNA MMR system. LS is characterized by
a high lifetime risk for tumor development, especially in the case
of CRC (20–70% with average age at diagnosis 44–61), endometrial
cancer (15–70% with average age at diagnosis 48–62), gastric cancer
(6–13% with average age at diagnosis 56), ovarian cancer (4–12%
with average age at diagnosis 42,5) and other extracolonic tumors
(total risk 15%) as small intestine, brain, skin hepatobiliary and
urinary tract (1). Other phenotypic
features of LS subjects are preferential tumor localization in the
right-sided colon, presence of multiple synchronous and
metachronous colorectal cancers, poorly differentiated tumors, with
a marked lymphocytic peritumoral inflammation recalling features of
so-called ‘Crohn's reaction’ and Microsatellite instability at
somatic level (8,9).
Genetic bases
LS patients present with a germline mutation in one
of the MMR genes, MLH1 on chromosome 3p21, MSH2 on
chromosome 2p16, MSH6 on chromosome 2p16, PMS2 on
chromosome 7p22, MLH3 on chromosome 2p16 and
MSH3 on chromosome 5q11. The heteroduplex MutSα that
predominantly identifies single base mispairings is formed by
MSH2 and MSH6 proteins, while MSH2 with
MSH3 form the MutSβ identifying short insertions or
deletions. imilar, the MutLα and MutLγ subunits are formed by
MLH1-PMS2 and MLH1-MLH3, respectively, they interacts
with the MutSα or MutSβ complex, stimulating excision and
resynthesis of abnormal DNA (10).
MSH2 and MLH1 are, thus essential for both complexes
to function. Therefore, the MMR genes, MLH1 and
MSH2 are defined as ‘major’ MMR genes, while the
MSH6, PMS2, MLH3 and MSH3 are known as ‘minor’
MMR genes (9). Somatic
inactivation of the corresponding wild-type allele occurs almost
exclusively through point mutations or (partial) gene loss;
bi-allelic inactivation then leads to complete abolition of the
protein function. This results in a defective DNA MMR system, since
MMR proteins are involved in the correction of single nucleotide
mismatches and small insertions or deletions that may arise during
DNA replication (11). The absence of
redundant functions for MSH2 and MLH1 proteins underlies the
importance of these two genes in MMR complex. Majority of mutations
was found in these genes (84 and 71% respectively). Carriers of
MSH2 variants show a higher incidence of extracolonic malignancy
(48–61%; endometrial, gastric, ovarian and kidney cancer) than the
carrier of MLH1 variants (11–42%) (12). Regard to minor genes, the MSH6
variants, until a few years ago, seemed to cause a form of
‘attenuated’ LS (13), PMS2 variants
were associated with combined presence of multiple colorectal
adenomas and glioblastomas (14).
Recently, also mutations in MLH3 gene have been associated with
brain tumors (15). MSH3 variants
were associated with a classic phenotype only if they were
inherited in combination with MSH2 variants (16). Recently, it was showed that biallelic
mutations in MSH3 gene are causing polyposis forms similar to FAP
phenotype (17). Sometimes
costitutional MLH1 methylation in the LS adenomas could represent
the initiation of these neoplasms and it may present as a defect
that was inherited (18).
Microsatellite instability of
related-LS tumors
The deficiency of MMR complex determines high rate
of mutations in repetitive DNA sequences known as microsatellites.
This condition is known as microsatellite instability (MSI) and is
present in approximately 95% of all LS-associated cancers (18). Many genes contain repetitive sequences
in their coding regions and some of these have an important role in
the regulation of cell growth (19).
In fact, mutations in the TGFβRII and TCF−4 genes,
that normally inhibit cell growth, and in the IGF-RII and
BAX genes involved in the apoptotic process (20) particularly predispose to colon cancer.
Moreover, the presence of polyadenine traits in the coding
sequences of the minor MMR genes, MSH6, MLH3 and
MSH3, makes the same MMR genes targets of the MSI phenotype
(21,22).
The sporadic CRC also display an MSI phenotype in
about 15%. In this case, the MSI may be result of somatic
hypermethylation of the MLH1 gene promoter. The
hypermethylation at the promoter of MLH1 allele lead to silencing
expression from that allele in all main somatic tissues. In 40–87%
of all sporadic microsatellite unstable tumors (23) with hypermethylation of the
MLH1gene is present a specific mutation in the BRAF
oncogene, usually the V600E missense mutation. This mutation is not
present in LS MSI tumors in which the MSI phenotype is due to
genetic alteration of MMR genes and it is not depend by epimutation
(24).
Finally, another type of instability, ‘elevated
microsatellite alterations at selected tetranucleotide repeats’
(EMAST), has also been identified in colon cancers. EMAST has been
associated with both MSI. One known cause of EMAST is a deficiency
or dysfunction of MSH3, which is required in the repair of
tetranucleotide repeat mismatches in complex with MSH2. The
MSH3 defect may also cause an impairment of homologous
repair and increase sensitivity to some targeted therapies, such as
poly (ADP-ribose) polymerase 1 (PARP1) inhibitors (25).
Clinical diagnosis and molecular analysis of
Lynch syndrome
Clinical criteria
Identification of families affected by LS occurs by
the Amsterdam Criteria (AC) and Bethesda guidelines. The clinical
criteria of Amsterdam were used to identify families eligible for
molecular analysis since 1990 (26).
Subsequently, these criteria were modified, the AC II in order to
include the other LS-related cancers (27). The Bethesda guidelines, which were
less restrictive than AC, were later defined (28) and take into account the MSI-status
detected at tumoral tissue. The ‘Panel of Bethesda’ recommended by
the National Cancer Institute include five microsatellites: two
mononucleotide repeats (BAT25, BAT26) and three dinucleotide
repeats (D2S123, D17S250, D5S346) (29) that are analyzed on tumoral DNA of
patients with likely LS. If at least two of these repeats (40% of
markers) are instability, tumoral DNA shows high instability
(MSI-H), while if at least 10–30% of markers are instability,
tumoral DNA shows low instability (MSI-L); when no microsatellite
is instable, tumoral DNA shows stability of microsatellite
sequences (MSS) (30). Subsequently,
other microsatellite sequences were included in the panel test:
NR21, NR22 and NR24, quasimonomorphic mononuceotide repeats in
order to improve the sensitivity rate and predictive specificity of
Bethesda guidelines (31,32); these three repeats (NR21, NR22 and
NR24) with BAT25 and BAT26 constitute the Pentaplex Panel (31).
Molecular analysis
LS is associated with mutations in MMR genes. Most
of mutations were found in the MLH1 and MSH2 genes
that account for about 50 and 40% respectively of all mutations
reported; about 15–20% of mutations were identified in the
MSH6 and in PMS2 (33,34); few
pathogenetic mutations were identified in MLH3 (15) gene and so far, only one heterozygous
variant in MSH3 gene was associated with LS phenotype
(16). The most pathogenetic variants
in MMR genes are small insertions/deletions or large genetic
rearrangements (large deletions/insertions) that, at protein level,
result in premature stop codon formation (35,36).
Moreover, several mutations identified in MMR genes are missense,
silent or intronic variants. The influence of these variants on the
development of cancer is often a controversial topic; therefore,
they are each classified as a variant of uncertain significance
(VUS) (37). According to
international recommandetions (Colon cancer Family Registry
2009, InSiGHT Variant Interpretation Committee 2011) it is
possible to use a multifactorial likelihood model in an attempt to
define a pathogenetic role of VUS (38). This approach is based on the
evaluation of both phenotipic and functional features (9,39). In
particular, the segregation analysis should be considered the ‘gold
standard’ for the validation of VUS pathogenecity (34,39).
The loss of function of one MMR protein prevents to
repair's complex to work properly and this determines a genetic
instability known as MSI at somatic level (27).
The molecular analysis to make diagnosis of LS
begins with the evaluation of the MSI status on tumoral DNA (see
above) by DNA fragment analysis using capillary electrophoresis
(38). At somatic level the MSI is
detectable by immunohistochemistry (IHC) analysis (40). Instead, the common methods for the
mutation detection analysis of MMR genes include the use of
denaturing high-performance liquid chromatography (DHPLC) and
direct sequencing for point mutations, and multiplex
ligation-dependent probe amplification (MLPA) for large
rearrangements (16,35,36). So
far, a large number of variants in Insight-group Database
(www.insightgroup.org) have been reported
in MMR genes, in particular MLH1, MSH2, MSH6 and
PMS2, Table I, while no
variants in the MLH3 and MSH3 genes have been
reported. However, literature data show cases of patients with
hereditary colorectal cancer and with mutations in these two genes
(15,16).
 | Table I.Numbers of genetic variants
identified in MMR genes. |
Table I.
Numbers of genetic variants
identified in MMR genes.
| Gene | Accession
number | Total no. of
genetic variants |
|---|
| MLH1 | NM_000249.3 | 8,023 |
| MSH2 | NM_000251.2 | 6,346 |
| MSH6 | NM_000179.2 | 2,297 |
| PMS2 | NM_000535.5 | 1,264 |
Today, high-throughput techniques, such as next
generation sequencing, have been substituted for these methods to
allow the identification of a major number of genes involved in
such hereditary cancer forms. For example, recent findings
suggested POLE and POLD1 mutations are associated with
gastrointestinal malignancies, with mutations in these genes having
been identified in subjects with a Lynch-like phenotype (41).
Recent discoveries in molecular genetics of
Lynch syndrome
New roles for MMR proteins
It is known a long time that MMR proteins have
developed various other functions in addition to the
postreplicative repair. Among these new roles (such as prevention
of reparative recombination, promotion of meiotic crossover,
expansion of repeated triplets, modulation of microRNA biogenesis)
is included the immunoglobulin (Ig) diversification based on the
‘somatic hypermutation’ (SHM) process. This process is regulated by
the MutSα -MutLα complex, in combination with two other proteins,
AID (activation-induced cytidine deaminase) and Polµ (DNA
Polymerase ‘error-prone’) (42); in
particular, MutSα deficiency is associated with neoplastic
transformation of T lymphocytes (43). Paradoxically, MMR maintains stability
throughout the genome but is responsible for up to 60% of the
mutations in V and S regions of the Ig locus that are important for
diversification antibodies (44).
Therefore, a better understanding of the intricate signaling
cascades that govern antibody diversification could help uncover
the associations between the maintenance of genomic integrity and
tumorigenesis in the adaptive immune response.
Immune-response in LS colorectal
cancer
LS cancers are usually referred to as MSI-H or MSI
and conceptually display a very interesting biology and clinical
behavior that is governed by the underlying mutational mechanism of
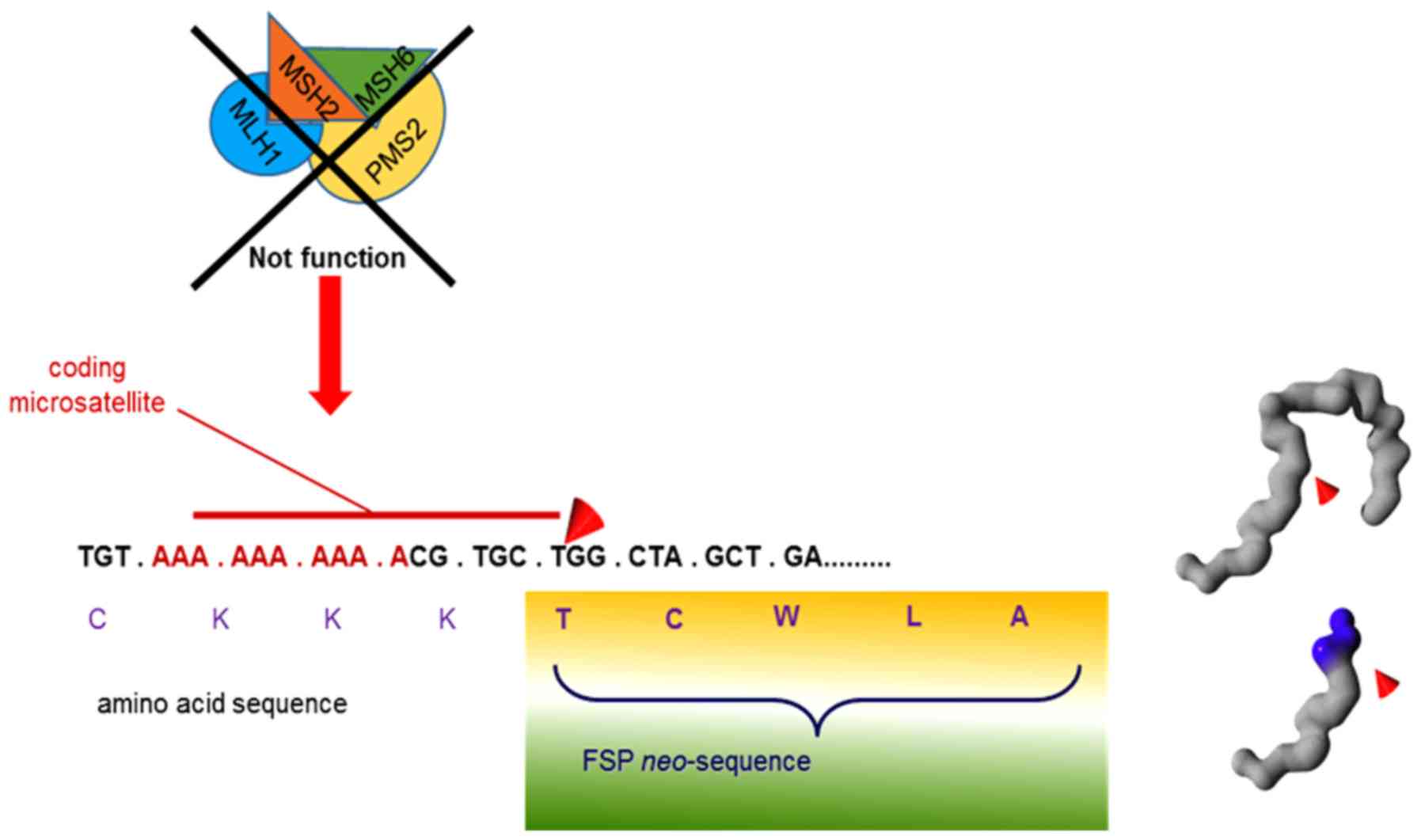
these tumors. MMR-deficient cells accumulate an abundance of
mutations at coding microsatellites, also found in tumor-relevant
genes. These mutations may give rise to a loss of function of the
respective proteins but may also trigger the translation of highly
immunogenic frameshift neo-peptides or -antigens (FSPs) (45). Such FSP antigens may be shared if they
occur in genes for which the mutational inactivation has a
growth-promoting impact that is supportive for the development of a
neoplasm. Such shared antigens may thus occur in multiple and
independently arising MMR-deficient tumors. FSP neo-antigens are
highly immunogenic due to long mutational antigens that encompass
multiple potential epitopes (46). As
these FSPs are derived from real shifts of the reading frame of the
respective gene, they usually have a completely novel, and for the
affected organism, foreign amino acid sequence, that results from
insertions or deletions of single, individual nucleotides that
alter the reading frame of the affected genes (Fig. 1). Such frame-shift mutations therefore
generated substantially more immunogenic antigens in comparison,
for example, to single missense mutations as they, for example,
frequently occur in mutant P53 or KRAS genes. If one
considers MMR deficiency as a unique mechanism of carcinogenesis,
one can imagine that at the beginning of the carcinogenic process
cell clones are generated that acquire mutations in coding
microsatellites on a more random basis. Only cells in which the
mutational spectrum favours neoplastic growth features will survive
and further expand, whereas other cells with less favourable
mutational spectra will be lost. Over time, this mechanism drives
and shapes the mutational spectrum of the surviving cell clones
into a better and better adapted status for local growth
requirements. Thus, loss of the MMR system could represent an
efficient mutational mechanism allows for a Darwinian selection
process of carcinogenesis. The extensive generation of these
neo-antigens in MSI cancers explains the pathological finding that
MSI cancers are characterized by massive infiltration of
lymphocytes and other immune-related cells that point to the strong
immunogenicity of such cancers. MSI cancer cells can grow out to
clinically manifest cancers if local T cells in their environment
become exhausted. Alternatively, MSI cancer cells that have
undergone immune evasion due to a loss of HLA-mediated antigen
presentation may grow out irrespective of local T cell surveillance
(47). Indeed, direct and indirect
molecular mechanisms do not structurally interfere with the tumor
cells' capacity to present FSP neoantigens, but influence the T
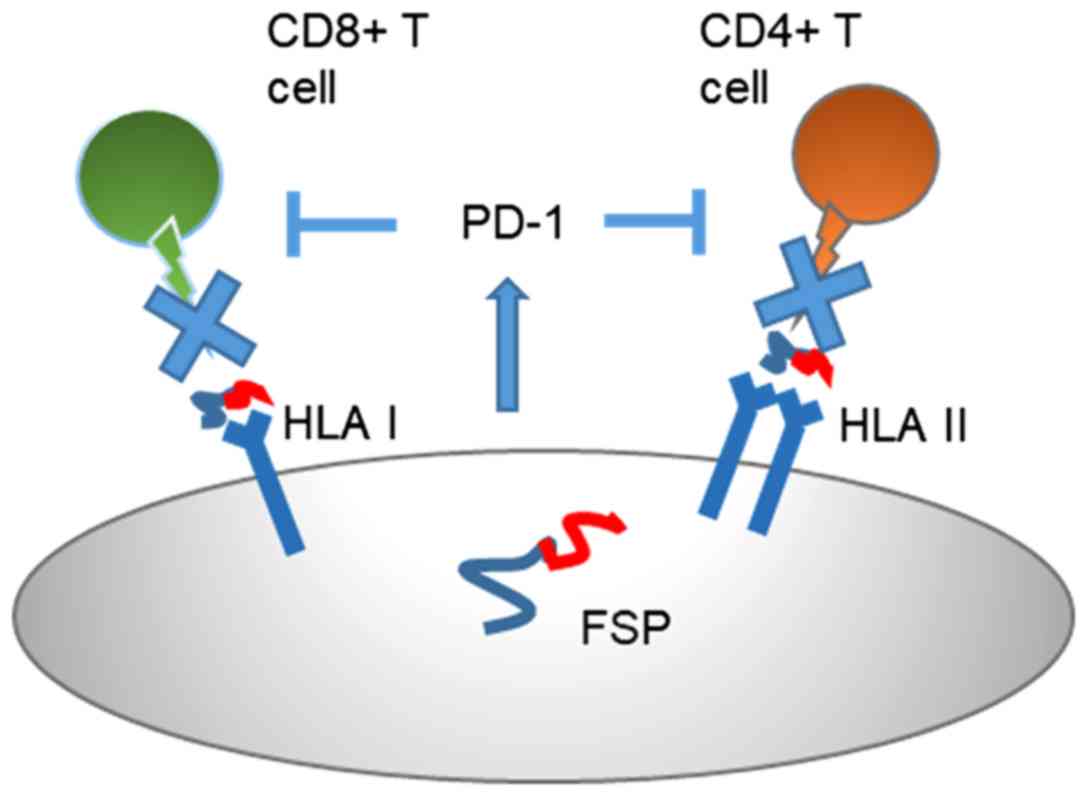
cell activation status. In approximately 30% LS tumoral tissue
mutation-induced loss of Beta 2 Microglobulin (B2M), the essential
light chain of HLA class I antigens, induces a complete lack of
assembled HLA class I antigens on the tumor cell surface. As a
consequence, CD8-positive T cells cannot attack B2M-mutant LS
cancer cells. Even mutations of the genes CIITA and RFX5, which are
required for functional HLA class II antigen expression on the
tumor cell surface, are found in up to 20% of MSI colorectal
cancers and associated with a complete loss of HLA class II
antigens on the tumor cell surface, consequently the inactivation
of CD4-positive T cell (48). This
may justify an endogenous immune antitumor response,
counterbalanced by the expression of inhibitory immune signals,
such as PD-1 binding to the PD-L1 receptor present on the
lymphocyte membrane, inhibiting its production (Fig. 2). Immune checkpoints play a key role
in limiting antitumor immunologic responses, such as those directed
against cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed
cell death-1 (PD-1) receptor and its ligand, PD-L. The ligation of
T-cell PD-1 by the tumor results in the downregulation of T-cell
effector functions that can destroy tumor tissue. Therefore, the
blockade of this pathway by anti-PD-1 antibodies prevents this
downregulation, and allows T cells to maintain their antitumor
functionality and ability to mediate tumor cell death (48,49).
Management of Lynch syndrome patient with
CRC
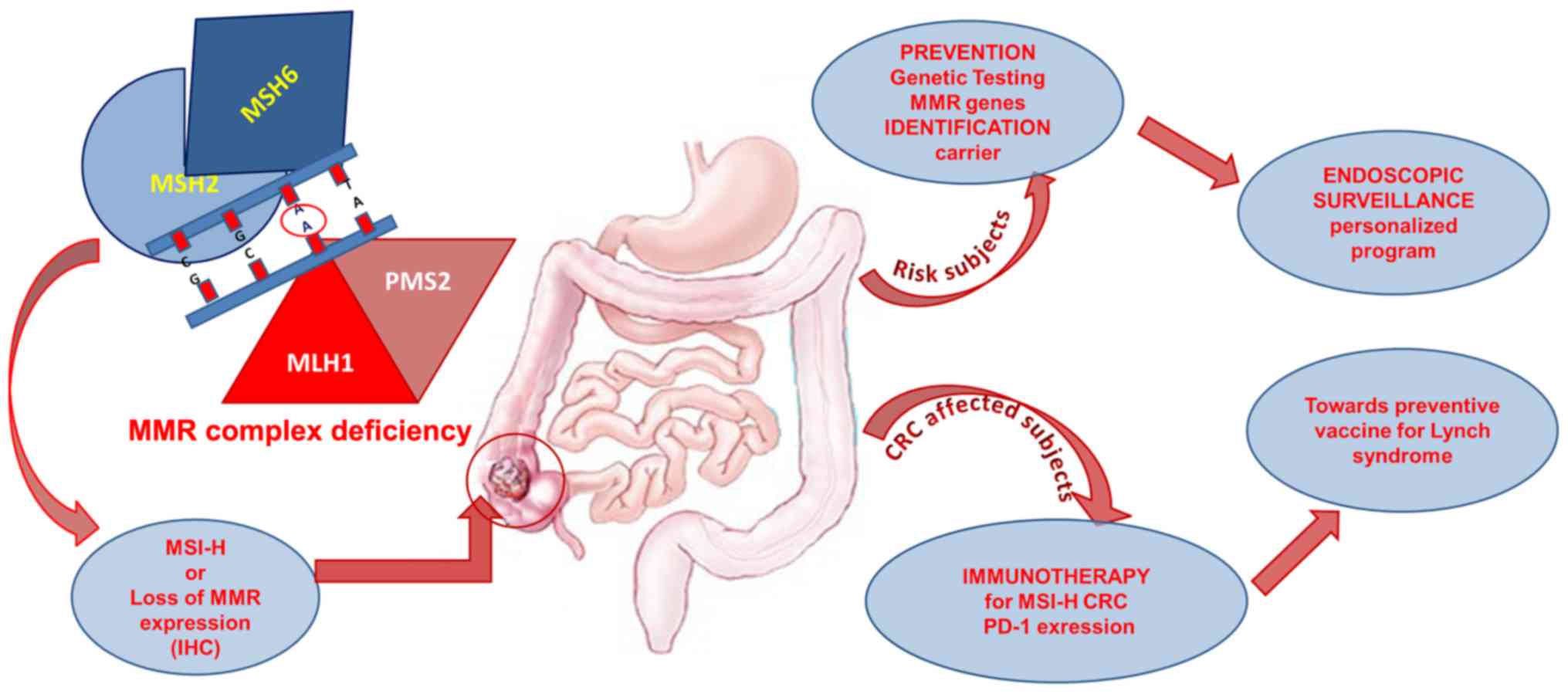
The early detection of LS-mediated CRC
progression
To improve the quality of care of patients and
families with any hereditary condition resulting in
gastrointestinal tumours as Lynch syndrome is the identification of
carriers of relevant predisposition alleles (50). The purpose of this is reducing MMR
associated hereditary colorectal cancer mortality. It is known a
long time that carrier subjects of pathogenetic mutation in a MMR
gene undergone to recommends annual surveillance colonoscopy from
age 25 years (51). In order to
include the mutation carriers in the endoscopy surveillance
programs more suited to them, a crucial point is represented by
correct definition of the pathogenecity of MMR genetic variants
identified in the mutation detection analysis (52,53). Thus,
this knowledge may helpful to improve the related-LS cancer
prevention programs. Recently, MSH6 and PMS2 mutation carriers have
been reported to have a lower risk of CRC with a later age of
presentation (54). Indeed,
literature data support a move to commence colonoscopy surveillance
in MSH6 and PMS2 mutation carriers at the older age of 30 years,
providing no young index CRC, and extend the interval to 2 years
(55). Therefore, the classification
of MMR genetic variants is many important to choose the most
appropriate endoscopic surveillance program and to precede towards
a personalized medicine (56)
(Fig. 3).
Therapeutic approaches of LS-related
colon cancer
The choose of the optimal treatment approach for
patients with metastatic colorectal cancer is based on evaluation
of clinical and genomic features of tumors. It is important to take
into account of the side of the colon in which the primary tumor
originates, the sites and burden of metastatic disease, by
mutational status of some genes, as KRAS, BRAF (57) and the MSI status on tumoral DNA
(58). The most applied protocol of
adjuvant chemotherapy for colorectal cancer not metastatic (stage
II) involves the administration of 5-fluorouracil (5FU). Instead in
some metastatic CRC cases (stage III), systemic therapy with a
FOLFOX- or CAPOX (capecitabine and oxaliplatin) regimen is the
standard of care in these patients. Patients with left-sided and
RAS wild-type tumors receive anti-epidermal growth factor receptor
(EGFR)-directed therapy, while patients with right-sided tumors or
those with RAS mutations receiving bevacizumab (59). In patients with tumors that manifest
microsatellite instability or deficient mismatch repair, adjuvant
chemotherapy with 5-fluorouracil did not result in a survival
benefit in subgroup analyses of patients with colon cancer without
metastasis. While, among patients with metastatic colon cancer who
received the treatment with capecitabine and oxaliplatin, survival
was significantly longer among those who had deficient mismatch
repair than among those who had proficient mismatch repair.
These different features are probably related to the
lymphocytic infiltrate characteristic of MMR-deficient tumors that
determines an antitumor immune response that may be abrogated by
the immunosuppressive effects of chemotherapy (60). Despite this enhanced immunogenicity, T
cells are unable to eradicate these tumors, likely due to
overexpression of immune checkpoint proteins that can be
antagonized by checkpoint inhibitors (Fig. 2). Recently, immune
checkpoint-inhibiting agents have been developed as antitumor drugs
and appear promising, especially in sporadic CRC patients with MSI.
Pembrolizumab (P) is an anti-PD-1 antibody that blocks the
interaction between PD-1 on T-cells, and PD-L1 and PD-L2 on tumor
cells. The antibody pembrolizumab has been evaluated in patients
with metastatic colorectal cancer and MSI in whom previous
treatment with cytotoxic agents had failed. The response to
treatment was similar in patients with LS-related CRC and those
sporadic CRC. Moreover, the combination of nivolumab, another
anti-PD-1 antibody, plus ipilimumab, an anti-cytotoxic T-lymphocyte
antigen 4 (CTLA-4) antibody, resulted in response rates and
disease-control rates that were higher than those previously
reported with nivolumab alone (61).
In this context, it is interesting to note that this drugs show
good results in the treatment of tumors with MSI (Fig. 3).
Conclusions
Identifying the mutation that causes clinical
manifestations of Lynch syndrome is crucial given the relatively
early onset of the disease, the high penetrance of mutations, as
well as the proven efficiency of surveillance strategies.
Furthermore, the studies carried out over the years on the
molecular mechanisms underlying the onset of LS-related colorectal
cancer have allowed us to make significant advances also in the
therapeutic treatments of these tumors. Recently, immune
checkpoint-inhibiting agents have been developed as antitumor drugs
and appear promising, especially in sporadic CRC patients with MSI.
Precisely because the subjects with Lynch syndrome show in 95% of
cases MSI-H on the tumor tissue, we could say that they are ideal
candidates for immunotherapeutic treatment. Finally, we hope in the
near future in the context of this research will be possible to
establish a preventive cancer vaccine for Lynch syndrome as
recently reported by studies on the preclinical mouse model
(62) (Fig.
3).
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
FD and PI designed the review. RL and MDR preformed
the literature search. FD and PI interpreted the scientific
articles, and FD wrote the first draft of the manuscript. FD
developed the structure of the paper and the discussion. RL, MDR
and PI critically revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kohlmann W and Gruber SB: Lynch Syndrome.
Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K
and Amemiya A: GeneReviews® [Internet]. Seattle (WA).
University of Washington; Seattle: 1993-2018
|
|
2
|
Dodaro C, Grifasi C, Florio J, Santangelo
ML, Duraturo F, De Rosa M, Izzo P and Renda A: The role of mutation
analysis of the APC gene in the management of FAP patients. A
controversial issue. Ann Ital Chir. 87:321–325. 2016.PubMed/NCBI
|
|
3
|
Lynch HT, Snyder CL, Shaw TG, Heinen CD
and Hitchins MP: Milestones of Lynch syndrome: 1895–2015. Nat Rev
Cancer. 15:181–194. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Rosa M, Galatola M, Borriello S,
Duraturo F, Masone S and Izzo P: Implication of adenomatous
polyposis coli and MUTYH mutations in familial colorectal
polyposis. Dis Colon Rectum. 52:268–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galatola M, Paparo L, Duraturo F, Turano
M, Rossi GB, Izzo P and De Rosa M: Beta catenin and cytokine
pathway dysregulation in patients with manifestations of the ‘PTEN
hamartoma tumor syndrome’. BMC Med Genet. 13:282012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paparo L, Rossi GB, Delrio P, Rega D,
Duraturo F, Liccardo R, Debellis M, Izzo P and De Rosa M:
Differential expression of PTEN gene correlates with phenotypic
heterogeneity in three cases of patients showing clinical
manifestations of PTEN hamartoma tumour syndrome. Hered Cancer Clin
Pract. 11:82013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carlomagno N, Duraturo F, Candida M, De
Rosa M, Varone V, Ciancia G, Calogero A and Santangelo ML: Multiple
splenic hamartomas and familial adenomatous polyposis: A case
report and review of the literature. J Med Case Rep. 9:1542015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cudia B, Liccardo R, Di Carlo G, Damiano
G, Lo Monte AI, Izzo P and Duraturo F: Clinical and anamnestic
evaluation rôle for the diagnosis and treatment of families
affected by Lynch syndrome. Case report and review of the
literature. Eur J Oncol. 19:265–271. 2014.
|
|
9
|
Liccardo R, De Rosa M, Izzo P and Duraturo
F: Novel implications in molecular diagnosis of Lynch syndrome.
Gastroenterol Res Pract. 2017:25950982017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peltomäki P: Deficient DNA mismatch
repair: A common etiologic factor for colon cancer. Hum Mol Genet.
10:735–740. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gupta R, Sinha S and Paul RN: The impact
of microsatellite stability status in colorectal cancer. Curr Probl
Cancer. 42:548–559. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yurgelun MB, Kulke MH, Fuchs CS, Allen BA,
Uno H, Hornick JL, Ukaegbu CI, Brais LK, McNamara PG, Mayer RJ, et
al: Cancer susceptibility gene mutations in individuals with
colorectal cancer. J Clin Oncol. 35:1086–1095. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lucci-Cordisco E, Rovella V, Carrara S,
Percesepe A, Pedroni M, Bellacosa A, Caluseriu O, Forasarig M, Anti
M, Neri G, et al: Mutations of the ‘minor’ mismatch repair gene
MSH6 in typical and atypical hereditary nonpolyposis colorectal
cancer. Fam Cancer. 1:93–99. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Agostini M, Tibiletti MG, Lucci-Cordisco
E, Chiaravalli A, Morreau H, Furlan D, Boccuto L, Pucciarelli S,
Capella C, Boiocchi M and Viel A: Two PMS2 mutations in a Turcot
syndrome family with small bowel cancers. Am J Gastroenterol.
100:1886–1891. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duraturo F, Liccardo R and Izzo P:
Coexistence of MLH3 germline variants in colon cancer patients
belonging to families with Lynch syndrome-associated brain tumors.
J Neurooncol. 129:577–578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Grosso M and Izzo P: Association of low-risk MSH3 and MSH2
variant alleles with Lynch syndrome: Probability of synergistic
effects. Int J Cancer. 129:1643–1650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Adam R, Spier I, Zhao B, Kloth M, Marquez
J, Hinrichsen I, Kirfel J, Tafazzoli A, Horpaopan S, Uhlhaas S, et
al: Exome sequencing identifies biallelic MSH3 germline mutations
as a recessive subtype of colorectal adenomatous polyposis. Am J
Hum Genet. 99:337–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boland CR: Recent discoveries in the
molecular genetics of Lynch syndrome. Fam Cancer. 15:395–403. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Cheong N, Miura M and Iliakis G:
Overexpression of insulin-like growth factor (IGF)-I receptor
enhances inhibition of DNA replication in mouse cells exposed to
x-rays. Radiat Environ Biophys. 36:117–123. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Plaschke J, Krüger S, Jeske B, Theissig F,
Kreuz FR, Pistorius S, Saeger HD, Iaccarino I, Marra G and
Schackert HK: Loss of MSH3 protein expression is frequent in
MLH1-deficient colorectal cancer and is associated with disease
progression. Cancer Res. 64:864–870. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loukola A, Vilkki S, Singh J, Launonen V
and Aaltonen LA: Germline and somatic mutation analysis of MLH3 in
MSI-positive colorectal cancer. Am J Pathol. 157:347–352. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alhopuro P, Sammalkorpi H, Niittymäki I,
Biström M, Raitila A, Saharinen J, Nousiainen K, Lehtonen HJ,
Heliövaara E, Puhakka J, et al: Candidate driver genes in
microsatellite-unstable colorectal cancer. Int J Cancer.
130:1558–1566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ryan E, Sheahan K, Creavin B, Mohan HM and
Winter DC: The current value of determining the mismatch repair
status of colorectal cancer: A rationale for routine testing. Crit
Rev Oncol Hematol. 116:38–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Cunningham JM, Winters JL,
Guenther JC, French AJ, Boardman LA, Burgart LJ, McDonnell SK,
Schaid DJ and Thibodeau SN: BRAF mutations in colon cancer are not
likely attributable to defective DNA mismatch repair. Cancer Res.
63:5209–5212. 2003.PubMed/NCBI
|
|
25
|
Carethers JM, Koi M and Tseng-Rogenski SS:
EMAST is a form of microsatellite instability that is initiated by
inflammation and modulates colorectal cancer progression. Genes
(Basel). 6:185–205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vasen HF, Mecklin JP, Watson P, Utsunomiya
J, Bertario L, Lynch P, Svendsen LB, Cristofaro G, Müller H, Khan
PM, et al: Surveillance in hereditary nonpolyposis colorectal
cancer: An international cooperative study of 165 families. The
International Collaborative Group on HNPCC. Dis Colon Rectum.
36:1–4. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vasen HF, Watson P, Mecklin JP and Lynch
HT: New clinical criteria for hereditary nonpolyposis colorectal
cancer (HNPCC, Lynch syndrome) proposed by the International
Collaborative group on HNPCC. Gastroenterology. 116:1453–1456.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boland CR, Thibodeau SN, Hamilton SR,
Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA,
Fodde R, Ranzani GN and Srivastava S: A National Cancer Institute
Workshop on Microsatellite Instability for cancer detection and
familial predisposition: Development of international criteria for
the determination of microsatellite instability in colorectal
cancer. Cancer Res. 58:5248–5257. 1998.PubMed/NCBI
|
|
29
|
Umar A, Boland CR, Terdiman JP, Syngal S,
de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ,
Hamelin R, et al: Revised bethesda guidelines for hereditary
nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite
instability. J Natl Cancer Inst. 96:261–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vilar E, Mork ME, Cuddy A, Borras E,
Bannon SA, Taggart MW, Ying J, Broaddus RR, Luthra R,
Rodriguez-Bigas MA, et al: Role of microsatellite instability-low
as a diagnostic biomarker of Lynch syndrome in colorectal cancer.
Cancer Genet. 207:495–502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Suraweera N, Duval A, Reperant M, Vaury C,
Furlan D, Leroy K, Seruca R, Iacopetta B and Hamelin R: Evaluation
of tumor microsatellite instability using five quasimonomorphic
mononucleotide repeats and pentaplex PCR. Gastroenterology.
123:1804–1811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Buhard O, Suraweera N, Lectard A, Duval A
and Hamelin R: Quasimonomorphic mononucleotide repeats for
high-level microsatellite instability analysis. Dis Markers.
20:251–257. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lynch PM: The hMSH2 and hMLH1 genes in
hereditary nonpolyposis colorectal cancer. Surg Oncol Clin N Am.
18:611–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liccardo R, De Rosa M, Rossi GB,
Carlomagno N, Izzo P and Duraturo F: Incomplete segregation of MSH6
frameshift variants with phenotype of lynch syndrome. Int J Mol
Sci. 18(pii): E9992017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Duraturo F, Cavallo A, Liccardo R, Cudia
B, De Rosa M, Diana G and Izzo P: Contribution of large genomic
rearrangements in Italian Lynch syndrome patients: Characterization
of a novel alu-mediated deletion. Biomed Res Int. 2013:2198972013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liccardo R, De Rosa M, Rossi GB, Rigler G,
Izzo P and Duraturo F: Characterization of novel, large
duplications in the MSH2 gene of three unrelated Lynch syndrome
patients. Cancer Genet. 221:19–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rasmussen LJ, Heinen CD, Royer-Pokora B,
Drost M, Tavtigian S, Hofstra RM and de Wind N: Pathological
assessment of mismatch repair gene variants in Lynch syndrome:
Past, present, and future. Hum Mutat. 33:1617–1625. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Rossi GB and Izzo P: Multivariate analysis as a method for
evaluating the pathogenicity of novel genetic MLH1 variants in
patients with colorectal cancer and microsatellite instability. Int
J Mol Med. 36:511–517. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Goldgar DE, Easton DF, Byrnes GB, Spurdle
AB, Iversen ES and Greenblatt MS; IARC Unclassified Genetic
Variants Working Group, : Genetic evidence and integration of
various data sources for classifying uncertain variants into a
single model. Hum Mutat. 29:1265–1272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kheirelseid EA, Miller N, Chang KH, Curran
C, Hennessey E, Sheehan M and Kerin MJ: Mismatch repair protein
expression in colorectal cancer. J Gastrointest Oncol. 4:397–408.
2013.PubMed/NCBI
|
|
41
|
Jansen AM, van Wezel T, van den Akker BE,
Ventayol Garcia M, Ruano D, Tops CM, Wagner A, Letteboer TG,
Gómez-García EB, Devilee P, et al: Combined mismatch repair and
POLE/POLD1 defects explain unresolved suspected Lynch syndrome
cancers. Eur J Hum Genet. 24:1089–1092. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zanotti KJ and Gearhart PJ: Antibody
diversification caused by disrupted mismatch repair and promiscuous
DNA polymerases. DNA Repair (Amst). 38:110–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Davari K, Frankenberger S, Schmidt A, Tomi
NS and Jungnickel B: Checkpoint kinase 2 is required for efficient
immunoglobulin diversification. Cell Cycle. 13:3659–3669. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bak ST, Sakellariou D and Pena-Diaz J: The
dual nature of mismatch repair as antimutator and mutator: For
better or for worse. Front Genet. 5:2872014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schwitalle Y, Linnebacher M, Ripberger E,
Gebert J and von Knebel Doeberitz M: Immunogenic peptides generated
by frameshift mutations in DNA mismatch repair-deficient cancer
cells. Cancer Immun. 4:142004.PubMed/NCBI
|
|
46
|
Schwitalle Y, Kloor M, Eiermann S,
Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A and
von Knebel Doeberitz M: Immune response against frameshift-induced
neopeptides in HNPCC patients and healthy HNPCC mutation carriers.
Gastroenterology. 134:988–997. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Koi M, Tseng-Rogenski SS and Carethers JM:
Inflammation-associated microsatellite alterations: Mechanisms and
significance in the prognosis of patients with colorectal cancer.
World J Gastrointest Oncol. 10:1–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bilgin B, Sendur MA, Bülent Akıncı M,
Şener Dede D and Yalçın B: Targeting the PD-1 pathway: A new hope
for gastrointestinal cancers. Curr Med Res Opin. 33:749–759. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Link JT and Overman MJ: Immunotherapy
progress in mismatch repair-deficient colorectal cancer and future
therapeutic challenges. Cancer J. 22:190–195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dominguez-Valentin M, Nakken S, Tubeuf H,
Vodak D, Ekstrøm PO, Nissen AM, Morak M, Holinski-Feder E, Martins
A, Møller P and Hovig E: Identification of genetic variants for
clinical management of familial colorectal tumors. BMC Med Genet.
19:262018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Giardiello FM, Allen JI, Axilbund JE,
Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA,
Kaltenbach T, et al: Guidelines on genetic evaluation and
management of Lynch syndrome: A consensus statement by the US
Multi-society task force on colorectal cancer. Am J Gastroenterol.
109:1159–1179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kanga-Parabia A, Gaff C, Flander L,
Jenkins M and Keogh LA: Discussions about predictive genetic
testing for Lynch syndrome: The role of health professionals and
families in decisions to decline. Fam Cancer. 17:547–555. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lindberg LJ, Ladelund S, Frederiksen BL,
Smith-Hansen L and Bernstein I: Outcome of 24 years national
surveillance in different hereditary colorectal cancer subgroups
leading to more individualised surveillance. J Med Genet.
54:297–304. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aissaoui S, Cartellier C, Seytier T,
Giraud S and Calender A: Genetic mutation risk calculation in Lynch
syndrome inheritance: Evaluating the utility of the
PREMM1,2,6 model in Lyon: The first French study. Bull
Cancer. 104:288–294. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dillon JL, Gonzalez JL, DeMars L, Bloch KJ
and Tafe LJ: Universal screening for Lynch syndrome in endometrial
cancers: Frequency of germline mutations and identification of
patients with Lynch-like syndrome. Hum Pathol. 70:121–128. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
De Rosa M, Rega D, Costabile V, Duraturo
F, Niglio A, Izzo P, Pace U and Delrio P: The biological complexity
of colorectal cancer: Insights into biomarkers for early detection
and personalized care. Therap Adv Gastroenterol. 9:861–886. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wojas-Krawczyk K, Kalinka-Warzocha E,
Reszka K, Nicoś M, Szumiło J, Mańdziuk S, Szczepaniak K, Kupnicka
D, Lewandowski R, Milanowski J and Krawczyk P: Analysis of KRAS,
NRAS, BRAF, and PIK3CA mutations could predict metastases in
colorectal cancer: A preliminary study. Adv Clin Exp Med.
7:Aug;2018.[Epub ahead of print].
|
|
58
|
Murcia O, Juárez M, Rodríguez-Soler M,
Hernández-Illán E, Giner-Calabuig M, Alustiza M, Egoavil C,
Castillejo A, Alenda C, Barberá V, et al: Colorectal cancer
molecular classification using BRAF, KRAS, microsatellite
instability and CIMP status: Prognostic implications and response
to chemotherapy. PLoS One. 13:e02030512018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Romera A, Peredpaya S, Shparyk Y,
Bondarenko I, Mendonça Bariani G, Abdalla KC, Roca E, Franke F,
Melo Cruz F, Ramesh A, et al: Bevacizumab biosimilar BEVZ92 versus
reference bevacizumab in combination with FOLFOX or FOLFIRI as
first-line treatment for metastatic colorectal cancer: A
multicentre, open-label, randomised controlled trial. Lancet
Gastroenterol Hepatol. 3:845–855. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Li LS, Morales JC, Veigl M, Sedwick D,
Greer S, Meyers M, Wagner M, Fishel R and Boothman DA: DNA mismatch
repair (MMR)-dependent 5-fluorouracil cytotoxicity and the
potential for new therapeutic targets. Br J Pharmacol. 158:679–692.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
O'Neil BH, Wallmark JM, Lorente D, Elez E,
Raimbourg J, Gomez-Roca C, Ejadi S, Piha-Paul SA, Stein MN, Abdul
Razak AR, et al: Safety and antitumor activity of the anti-PD-1
antibody pembrolizumab in patients with advanced colorectal
carcinoma. PLoS One. 12:e01898482017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
von Knebel Doeberitz M and Kloor M:
Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam
Cancer. 12:307–312. 2013. View Article : Google Scholar : PubMed/NCBI
|