Introduction
In the USA, prostate cancer (Pca) was the second
leading cause of cancer-associated mortality in men with 26,120
succumbing in 2015 (1). Specifically,
patients with metastatic castrate-resistant Pca exhibit poor
prognosis, with a median overall survival time of ~14 months
(2). The majority of available
therapies focus on controlling disease progression, including
targeted therapy. Targeted therapies are drugs that are designed to
target and inhibit cancer growth by interfering with specific
molecular signaling pathways (3).
Tyrosine kinase (TK) is an enzyme that transfers γ-phosphate groups
from ATP to the hydroxy group of tyrosine residues on signal
transduction molecules (4). Strict
regulation of TK activity controls fundamental cell processes, such
as the cell cycle, proliferation, differentiation, motility and
cell survival (5). A previous study
indicated that TKs are involved in prostate tumorigenesis and
progression through the activation of receptor and non-receptor TKs
(6). The fibroblast growth factor
(FGF) axis is required for normal prostate development; however, it
becomes aberrantly activated in Pca (7). The FGF receptor (FGFR) activates a
downstream pathway cascade, including phosphoinositide 3-kinase
(PI3K)/cell division cycle 42 (CDC42) (8). CDC42 is a member of the Rho GTPase
protein family that serves a key role in local actin organization
through a number of kinase and non-kinase effector proteins
(9). CDC42 activity is regulated by
the cell cycle and controls certain mitotic processes, including
kinetochore attachment and chromosome segregation (9). A recent study indicated that CDC42 is
involved in the cell entosis process, and knockout of CDC42
expression induces entosis in breast cancer and promotes cancer
progression (10). Known as cell
cannibalism, entosis is described as the ingestion of live cells,
resulting in the unusual appearance of whole cells contained within
large vacuoles, or so-called ‘cell-in-cell’ structures (11). Entosis is frequently identified in
human malignancies, which are associated with oncogenesis and tumor
progression, such as breast cancer (10). It has been reported previously that
entosis is particularly frequent in high-grade aggressive breast
cancers with poor prognosis (10). In
addition, enosis has also been identified in castration-resistant
Pca and is therefore associated with a poor prognosis (12). Through entosis, winner tumor cells
(engulfing cells) engulf and kill loser cells (engulfed cells), and
benefit from their death, which is a mechanism whereby cells
survive under stress and become more tumorigenic (10). Multiple molecules and pathways are
involved in the entosis process, including epithelial (E-)cadherin
and the RhoA/Rho kinase (ROCK) signaling pathway (13). RhoA is a member of the Ras superfamily
of small GTP-binding proteins, whereas ROCKs are downstream target
proteins of RhoA (13). RhoA activity
and ROCK1/2 were accumulated in internalizing cells by contributing
to myosin contraction (14).
Nintedanib, is a TK inhibitor (TKI) that can inhibit
FGFR1-3, vascular endothelial growth factor (VEGFR)1-3,
platelet-derived growth factor receptor (PDGFR)α/β, Src, Lck and
Lyn, and a Phase II clinical trial has exhibited antitumor effects
in patients with castration-resistant Pca (15). In another trial, the combination of
nintedanib, docetaxel and prednisone led to a decrease from the
baseline in prostate-specific antigen (PSA) levels and induced a
partial response (16). However, in
these two trials, certain patients did not respond to nintedanib,
and certain tumors developed resistance and then became more
aggressive, despite initially responding to nintedanib
treatment.
In the present study, we hypothesized resistance
would develop during the treatment of TKI nintedanib in Pca cells,
and entosis may contribute this process. Therefore, the present
study investigated the mechanism of resistance and the potential
involved pathways.
Materials and methods
Cell culture
The Pca cell lines 22RV1 (CRL-2505), LNCaP
(CRL-1740), DU145 (HTB-81) and PC3 (CRL-1435) and 293 cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA) and were maintained in RPMI-1640 medium (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) or Dulbecco's modified
Eagle's medium (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) that
were supplemented with 10% fetal bovine serum (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C in a humidified atmosphere with 5%
CO2. All drugs were purchased from Selleck Chemicals
(Houston, TX, USA), and Pca cells were treated at the following
concentrations: Nintedanib (3 µM for 4 weeks to develop
resistance), PI3K inhibitor buparlisib (1.5 µM for 4 weeks)
(17), protein kinase B (Akt)
inhibitor MK2206 (10 µM for 4 weeks) (18), extracellular-signal-regulated kinase
(ERK)1/2 inhibitor SCH772984 (3 µM for 4 weeks) (19), CDC42 inhibitor ML141 (2 µM for 4
weeks) (20), or ROCK1/2 inhibitor
Y-27632 (10 µM for 1 week) (21). For
drug treatment experiments, all the cells were seeded at a density
of 1×105 cells/well in 6-well culture plates in the
corresponding medium. Following 24 h of incubation at 37°C for
attachment, the cells were treated with drugs.
Cell viability assay and drug
half-maximal inhibitory concentration (IC50)
determination
Pca cells were seeded in 96-well plates (5,000
cells/well) and allowed to attach overnight at 37°C. Cells were
treated with drugs for 72 h at 37°C. Cell viability was
subsequently determined using a WST-1 assay (Roche Diagnostics,
Basel, Switzerland), according to the manufacturer's protocol.
Absorbance was determined using a Multimode Plate Reader at 440 and
690 nm (using 690 nm to remove background).
Immunofluorescence
22RV1 and DU145 cells were seeded on coverslips and
then cultured with 3 µM nintedanib until they developed resistance
(4 weeks). For the detection of RhoA, 22RV1 and DU145 cells were
fixed with 4% formaldehyde for 10 min and blocked for 20 min in PBS
containing 1% (w/v) BSA-0.1% (v/v) Triton X-100 at room
temperature. A primary antibody (mouse monoclonal anti-RhoA; cat.
no. sc-166399; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) was
diluted 1:200 and incubated at 4°C overnight. Subsequently, the
cells were incubated for 1 h with a rabbit anti-mouse
immunoglobulin G (IgG) FITC antibody (4 µg/ml; cat. no. sc-358916;
Santa Cruz Biotechnology, Inc.) at room temperature and then
mounted in antifade mounting medium with DAPI. Images were acquired
using a fluorescence microscope at ×400 magnification (Olympus
Corporation, Tokyo, Japan).
Western blotting
Pca cell pellets were lysed using
radioimmunoprecipitation buffer with a proteinase inhibitor (cat.
no. sc-24948; Santa Cruz Biotechnology, Inc.). The samples were
diluted to same quantities (20 µg) by the BCA assays and then
loaded. Protein samples were separated by SDS-PAGE on 10% gels, and
then transferred onto polyvinyl difluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
non-fat milk at room temperature for 1 h, and then incubated with
primary antibody (1:3,000) overnight at 4°C. Subsequently, the
membrane was incubated with anti-Rabbit IgG (cat. no. ab205718;
Abcam), or anti-mouse IgG (ab131368; Abcam). The proteins on the
blot were detected using the Western Bright Enhanced
Chemiluminescence western blotting detection kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Equal sample loading was
verified by the detection of GAPDH. The primary antibodies were as
follows: Mouse monoclonal anti-β-catenin (cat. no. sc-7963; Santa
Cruz Biotechnology, Inc.), mouse monoclonal anti-CDC42 (cat. no.
sc-8401; Santa Cruz Biotechnology, Inc.), rabbit monoclonal
anti-cyclin D1 (cat. no. 2978; Cell signaling Technology, Inc.,
Danvers, MA, USA), mouse monoclonal anti-Akt (cat. no. sc-377556;
Santa Cruz Biotechnology, Inc.), mouse monoclonal anti-phospho-Akt
(cat. no. sc-271966; Santa Cruz Biotechnology, Inc.), rabbit
monoclonal anti-ROCK1 (cat. no. ab45171; Abcam), rabbit monoclonal
anti-ROCK2 (cat. no. ab125025; Abcam), mouse monoclonal anti-ERK1/2
(cat. no. sc-514302; Santa Cruz Biotechnology, Inc.), mouse
monoclonal anti-phospho-ERK1/2 (cat. no. sc-81492, Santa Cruz
Biotechnology, Inc.), rabbit monoclonal anti-E-cadherin (cat. no.
EPR699; Abcam) and mouse monoclonal anti-GAPDH (cat. no. sc-47724;
Santa Cruz Biotechnology, Inc.).
Short interfering RNA (siRNA)
transfection
22RV1 and DU145 cells were seeded at a density of
1×105 cells per well in 12-well culture plates, then
transfected with anti-CDC42 siRNA or a scrambled probe (cat. no.
sc-37007; Santa Cruz Biotechnology, Inc.) at a final concentration
of 40 nM using Lipofectamine™ 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. The
transfection efficiency was validated using the reverse
transcription-quantitative polymerase chain reaction (RT-qPCR). The
5′-CCUCUACUAUUGAGAAACU-3′ and 3′-GGAGAUGAUAACUCUUUGA-5′
oligoribonucleotides were used to inhibit CDC42 synthesis. At 72 h
after transfection, cells were collected and the RNA was extracted
using RNeasy Mini kit (Qiagen, Valencia, CA, USA) prior to RT-qPCR
and western blotting to detect the results of inhibition.
Immunohistochemical staining
(IHC)
Sections (5 µm) on glass slides were deparaffinized
and rehydrated (Xylene: Twice, each for 5 min, 100% ethanol for 3
min, 95% ethanol for 3 min, 70% ethanol for 3 min, 50% ethanol for
3 min, wash slides in deionized water for 3 min). The sections were
stained with the aforementioned antibodies (1:200 dilution) against
CDC42 or E-cadherin at 4°C overnight and then incubated with a
horseradish peroxidase-labeled dextran polymer coupled to an
anti-mouse antibody at room temperature for 2 h. Cytoplasmic
staining that was clearly distinguishable from the background was
considered positive. The slides were reviewed twice by two
independent investigators. Target protein expression was graded
semi-quantitatively according to the staining score results
(22), and the mean values were used
for statistical analysis.
RNA isolation and RT-qPCR
RNA from the Pca cells was extracted using a RNeasy
Mini kit (Qiagen) and cDNA was generated using a reverse
transcription kit (Invitrogen; Thermo Fisher Scientific, Inc.).
Candidate genes were measured using a RT-qPCR system according to
the manufacturer's protocol (40 cycles as 93°C, 30 sec, 95°C, 30
sec, 55°C, 1 min, 68°C, 1 min). Primers used were: CDC42,
5′-CCCTGAAACAGCGTTGGGAA-3′ (forward) and
5′-CGGATGAACGATCCCTTTAGC-3′ (reverse); ROCK1,
5′-AACATGCTGCTGGATAAATCTGG-3′ (forward) and
5′-TGTATCACATCGTACCATGCCT-3′ (reverse); ROCK2,
5′-TCAGAGGTCTACAGATGAAGGC-3′ (forward) and
5′-CCAGGGGCTATTGGCAAAGG-3′ (reverse); E-cadherin,
5′-AGAACTTACCGCTACTTCTTGC-3′ (forward) and
5′-TGCCCACATACTGATAATCGGA-3′ (reverse); RhoA,
5′-AGCCTGTGGAAAGACATGCTT-3′ (forward) and
5′-TCAAACACTGTGGGCACATAC-3′ (reverse); and GAPDH,
5′-TGTGGGCATCAATGGATTTGG-3′ (forward) and
5′-ACACCATGTATTCCGGGTCAAT-3′ (reverse). The RNA concentrations in
samples varied between 500 and 1,000 ng/µl, and the
A260/A230 ratios were more than 2. A 500 ng
amount of RNA was reverse-transcribed into cDNA. All PCRs were run
in triplicate. The relative quantification of gene expression was
calculated using the 2−ΔΔCq method and normalized to
GAPDH expression (23).
Animal xenograft study
The present study was approved by the Hebei General
Hospital Institutional Ethics Committee (Shijiazhuang, China).
DU145 cells (106 cells) were mixed with Matrigel (1:1)
and then subcutaneously injected into the right flanks of 20 male
severe combined immunodeficient mice (8 weeks old). The dimensions
of each tumor were determined directly with calipers every 3 days,
and the volume was calculated by the following formula: Tumor
volume=½ (length × width2). The care and treatment of
experimental animals were in accordance with institutional
guidelines (24). The mice were
euthanized when their tumors exceeded 1 cm, and the diameter of the
largest subcutaneous tumor was 1.3 cm. When the tumor volume
reached 200 mm3, the mice were randomized into two
groups (10 ×enografts/group) that received nintedanib (50 mg/kg/day
dissolved in 5% dimethylsulfoxide) or a vehicle control by oral
gavage (25). Tumor volumes were
determined and growth curves were generated. At the end of the
study, the animals were sacrificed via CO2 inhalation
(20% of the chamber volume/min) and the tumors were collected.
Transwell invasion assay
Cell invasion assays were performed in a 24-well
Transwell chamber (0.4 µm; Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol.
Nintedanib-resistant 22RV1 and DU145 cells or negative controls
(matched cells without treatment) were added to the upper chamber
(1×105 cells/ml). Following 24 h of incubation, the
cells that passed through the filter were fixed and stained using
0.1% crystal violet at room temperature for 20 min. The numbers of
invading cells were counted in five randomly selected fields under
an Olympus IX71 microscope under ×400 magnification.
Plasmid construction and
transfection
Full-length human CDC42 cDNAs (OriGene Technologies,
Inc., Rockville, MD, USA) were sequenced and subcloned into the
pcDNA3.1 expression vector (Thermo Fisher Scientific, Inc.). A
lentiviral vector expressing tagged CDC42 was generated using the
ViraPower™ T-Rex™ system (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Subsequently, 293
cells were transfected using the calcium phosphate precipitation
method and Pca cells were transfected using the FuGENE Transfection
Reagent (Roche Diagnostics).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean of three independent experiments. One-way analysis of
variance (ANOVA) with Fisher's least-significant difference post
hoc test, two-way repeated-measures ANOVA followed by Bonferroni
post hoc tests or Student's t-test was used for the statistical
analysis using R (version 3.4.3) developed by R Core Team.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Nintedanib inhibits Pca cell
proliferation in vitro
Different Pca cell lines were selected for
experimentation including LNCaP, PC3, 22RV1 and DU145. The data
revealed that IC50 values for nintedanib in the
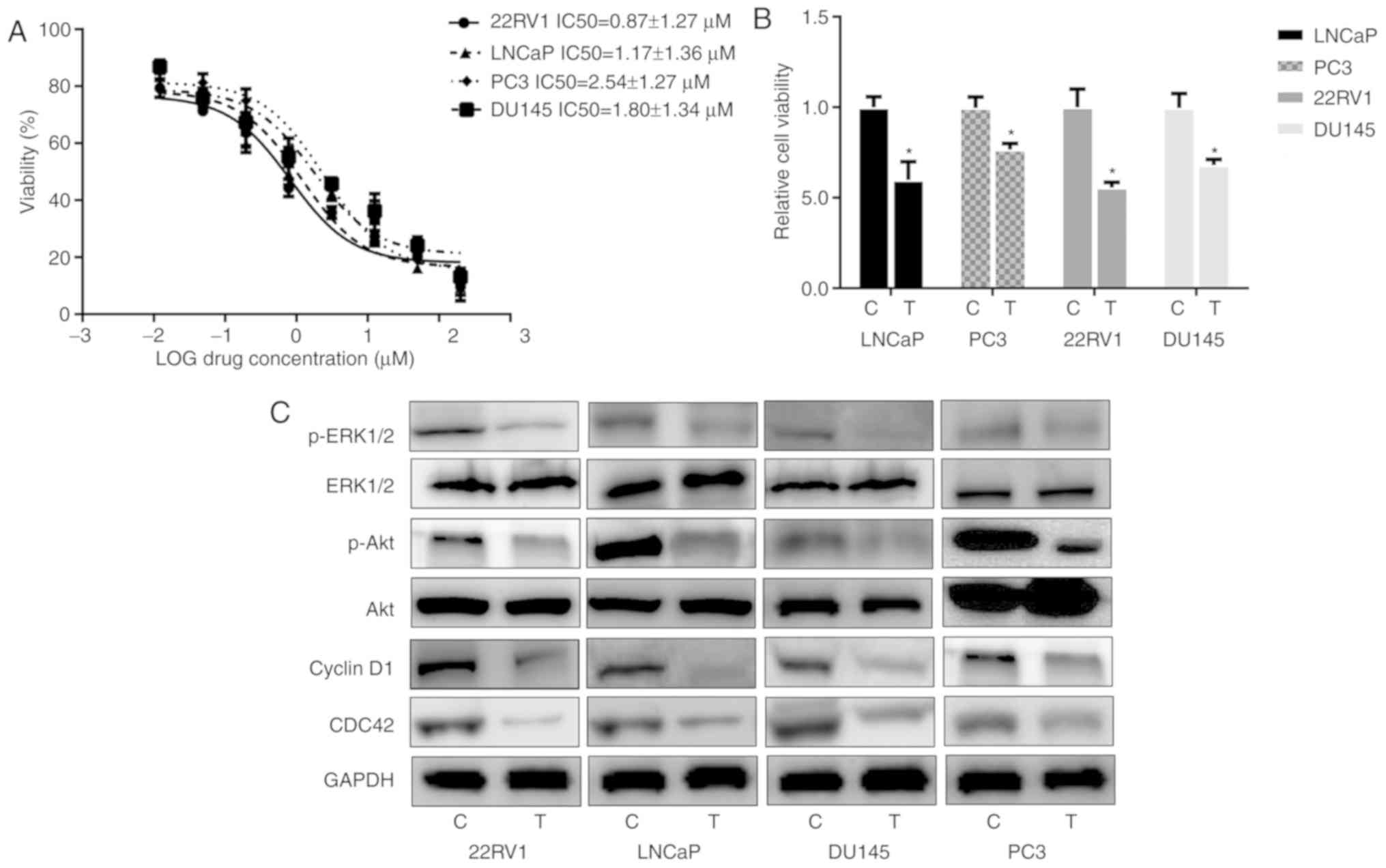
different cell lines varied between 0.87 and 2.54 µM (Fig. 1A). The Pca cells were then treated
with 3 µM nintedanib, and the viability assay revealed that the
cell proliferation was inhibited by nintedanib in vitro,
which indicated that nintedanib is capable of suppressing the
proliferation of these cells (Fig.
1B). The antitumor effects of nintedanib could be attributed to
its selective anti-pan-receptor TKI activity. Nintedanib is already
known as an inhibitor of TKs, by binding to the ATP-binding site in
the cleft between the N- and C-terminal lobes of the kinase domain
(25). At the pharmacological level,
nintedanib can inhibit TKs, which include all three VEGFR subtypes
(IC50, 13–34 nmol/l), PDGFRα and PDGFRβ
(IC50, 59 and 65 nmol/l), and FGFR types 1, 2 and 3
(IC50, 69, 37 and 108 nmol/l), respectively (25). Marked death of Pca cells was not
observed following treatment with 3 µM nintedanib. Instead, in the
2 days following treatment, the cells maintained proliferation,
prior to suspension of cell proliferation. Consistent with the
proliferation assay results, nintedanib treatment suppressed the
expression of proteins downstream of PI3K, including CDC42,
phospho-Akt and phospho-ERK1/2 (Fig.
1C). Cyclin D1 expression was also decreased due to nintedanib
treatment.
Activation of entosis mediates
acquired resistance to nintedanib
To identify potential mechanisms of acquired
resistance to nintedanib in Pca, the present study established
different cell lines with acquired resistance by exposing the cells
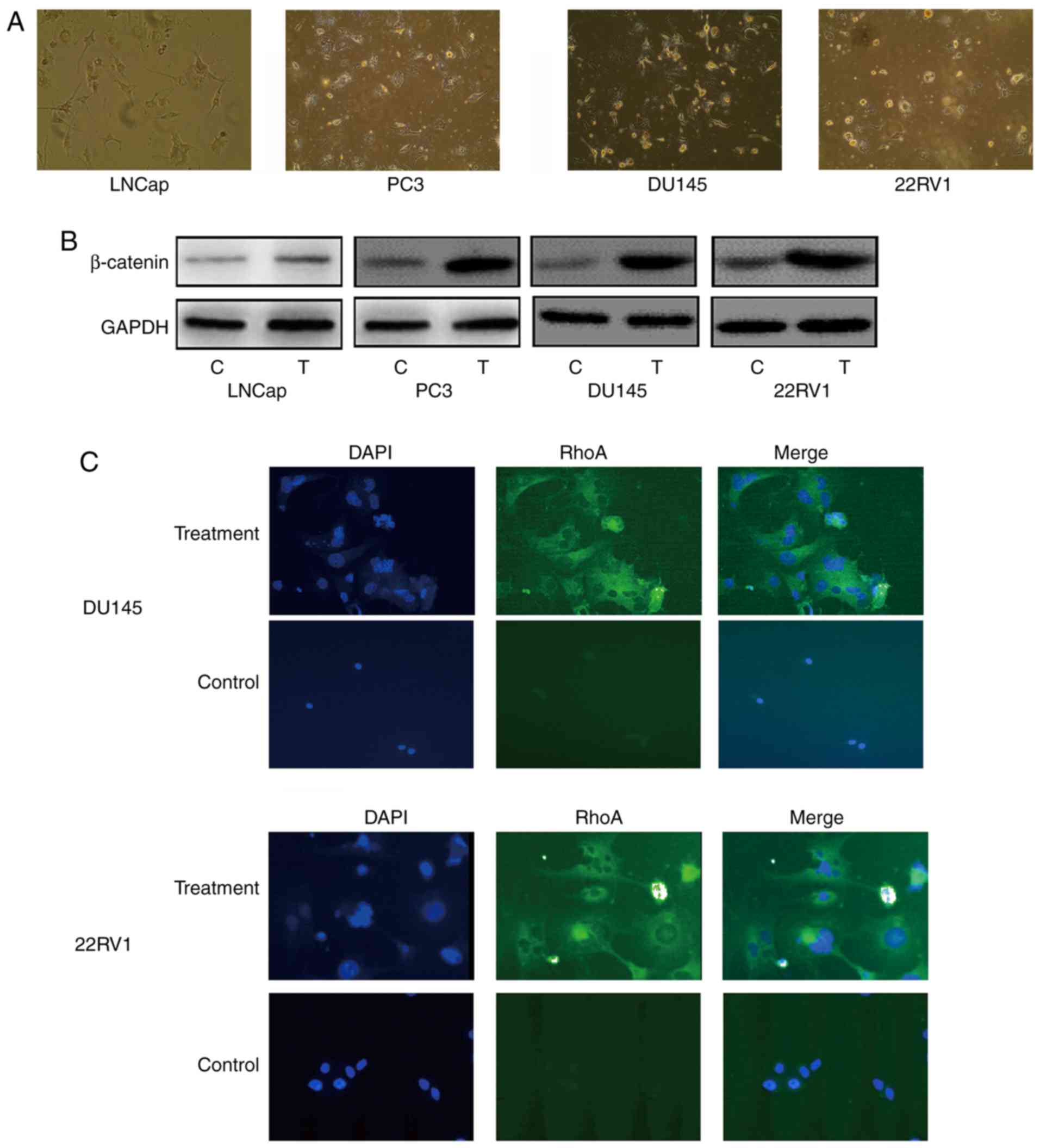
to 3 µM nintedanib. All Pca cells exhibited marked resistance to
nintedanib (Fig. 2A) at 4 weeks after
nintedanib treatment. Immunoblotting revealed that β-catenin
expression increased markedly in all cell lines examined when
compared with cells that did not undergo treatment (Fig. 2B); this is relevant as β-catenin is a
molecule that contributes to cell adherens junctions (26). The nintedanib-resistant cells were
observed under a microscope and exhibited the cell-in-cell
phenomenon morphology, which refers to one or more viable cells
present inside other cells, or more cells involved in sequential
engulfment (Fig. 2C). These were
similar to the entotic structures identified in other studies
(10,12). Notably, the engulfing cells
morphologically transformed into phagocyte-like cells, with
pseudopodia and a multipolar shape. Under a fluorescence
microscope, positive expression of RhoA in nintedanib-resistant Pca
cells was observed, which is a marker of entosis (Fig. 2B and C). In addition, the size of the
cell and the nucleus markedly increased in Pca cells treated with
nintedanib (27).
Taken together, these results demonstrated that
treatment of Pca cells with nintedanib may lead to entosis.
Therefore, in the subsequent experiments, only the 22RV1 (androgen
receptor-positive) and DU145 (androgen receptor-negative) Pca cell
lines were selected to further investigate the mechanism of entosis
induced by nintedanib.
Increased Rho/ROCK and E-cadherin in
Pca cells treated with nintedanib
It is well established that entosis is regulated by
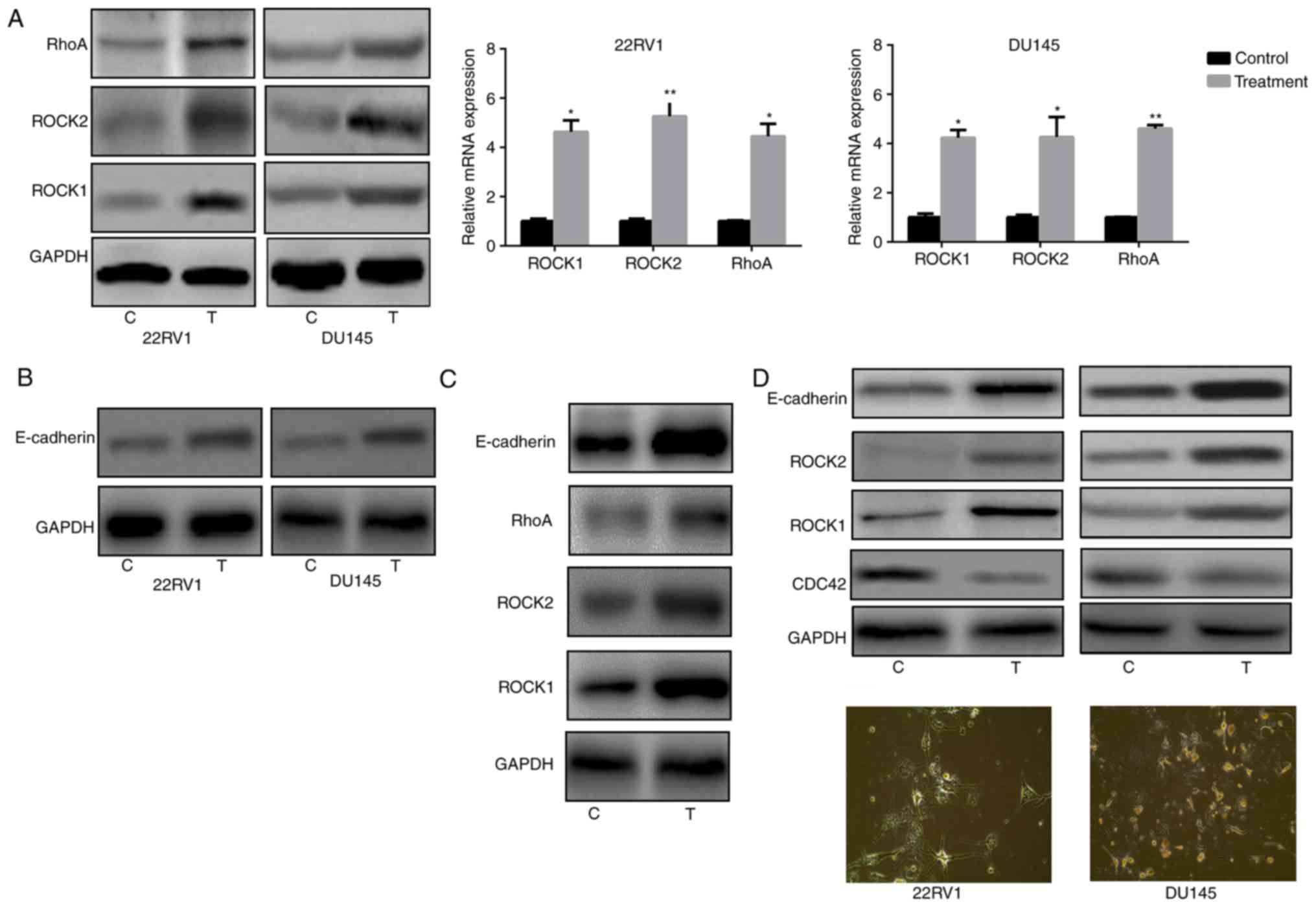
the RhoA/ROCK1/2 signaling pathway and E-cadherin (28). Therefore, RhoA and ROCK1/2 expression
was analyzed in Pca cells and it was observed that nintedanib
treatment of 22RV1 and DU145 cells led to an increase in the
expression of RhoA, ROCK1 and ROCK2 (Fig.
3A). The present study also evaluated E-cadherin expression in
Pca cells following treatment with nintedanib. The results
indicated that E-cadherin expression was also significantly
increased (Fig. 3B). Since E-cadherin
is another biomarker of the entosis phenomenon, the experiment
presented in Fig. 3B was attempted to
verify that entosis developed following treatment with nintedanib,
accompanied by an increased E-cadherin level. Although PC3 is a Pca
cell line with a deep PTEN deletion and Akt activation, treatment
of PC3 cells with nintedanib still increased the expression of
ROCK1/2 and E-cadherin (Fig. 3C).
Nintedanib regulates entosis via the
PI3K/CDC42 signaling pathway
Nintedanib inhibits FGFR in Pca cells, and inhibits
the activation of its downstream pathways, including PI3K. The data
indicated that nintedanib inhibited CDC42, Akt and ERK1/2
expression (downstream of PI3K) in 22RV1 and DU145 cells (Fig. 1C). In addition, CDC42 was inhibited
when the cells were treated with the PI3K inhibitor buparlisib
(Fig. 3D). However, cells in which
either Akt or ERK1/2 were inhibited neither developed entosis nor
exhibited an increase in ROCK1/2 or E-cadherin expression (Fig. 4A). Therefore, we hypothesized that
nintedanib could induce entosis via the CDC42 pathway.
Consequently, Pca cells were treated with a CDC42 inhibitor, and
observed that entosis morphology developed following 2 weeks of
treatment along with increased ROCK1/2 and E-cadherin expression
(Fig. 4B and C). Furthermore,
increased ROCK1/2 and E-cadherin expression were also observed in
CDC42-knockdown cells (Fig. 4D and
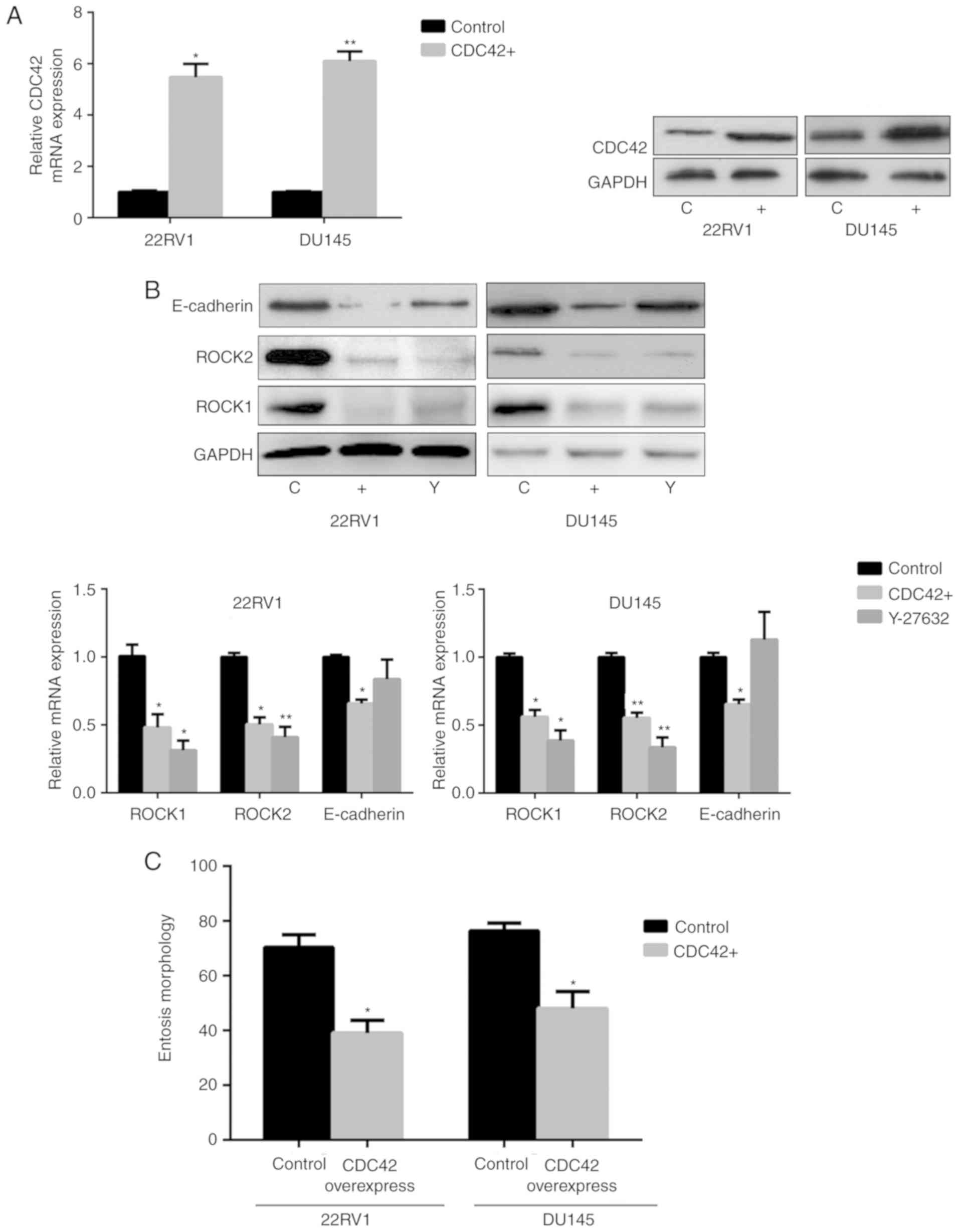
E). Subsequently CDC42 was overexpressed in Pca cells via
transfection (Fig. 5A). The results
demonstrated that ROCK1/2 expression decreased under CDC42
overexpression, which was also observed in cells treated with the
ROCK1/2 inhibitor Y-27632. E-cadherin expression also decreased in
cells overexpressing CDC42 (Fig. 5B).
In addition, nintedanib-associated entosis was significantly
inhibited in cells overexpressing CDC42 compared with controls
(P<0.05, Fig. 5C). Therefore,
PI3K/CDC42 negatively regulated ROCK1/2 and E-cadherin
expression.
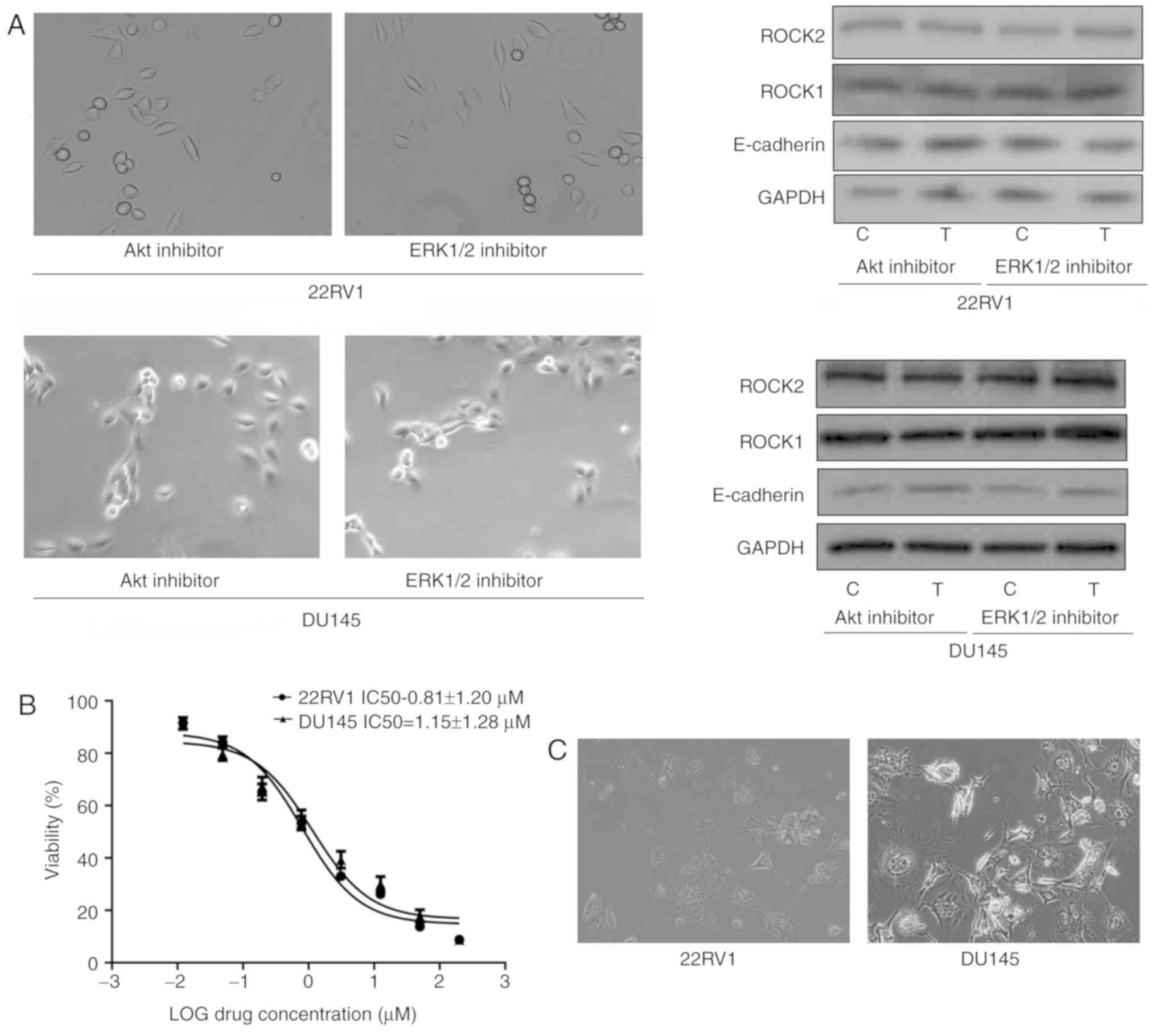 | Figure 4.Decreased CDC42 induces entosis by
promoting the ROCK1/2 signaling pathway and E-cadherin expression
in Pca cells (×400 magnification). (A) No entosis morphology or
increased ROCK1/2 or E-cadherin expression was observed in Pca
cells treated with Akt inhibitor (MK2206; 10 µM) or ERK1/2
inhibitor (SCH772984; 3 µM) for 4 weeks, respectively. (B)
IC50 values of the CDC42 inhibitor ML141 in the Pca cell
lines. (C) Entosis-like morphology of Pca cells under ML141
pressure (2 µM for 4 weeks). (D) siRNA knockdown of CDC42
expression at the mRNA and protein level. *P<0.05 vs. control
(Student's t-test) (E) CDC42 siRNA and ML141 (2 µM) promote ROCK1/2
expression and E-cadherin expression (following treatment for 1
week). *P<0.05 and **P<0.01 vs. control (one-way analysis of
variance followed by Fisher's least-significant difference test).
CDC42, cell division cycle 42; ROCK, Rho kinase; Pca, prostate
cancer; Akt, protein kinase B; ERK, extracellular-signal-regulated
kinase; IC50, half-maximal inhibitory concentration;
siRNA, short interfering RNA; E-cadherin, epithelial cadherin; C,
control; T, treatment; M, ML141 treatment; S/Si, siRNA treatment;
Sc, scrambled. |
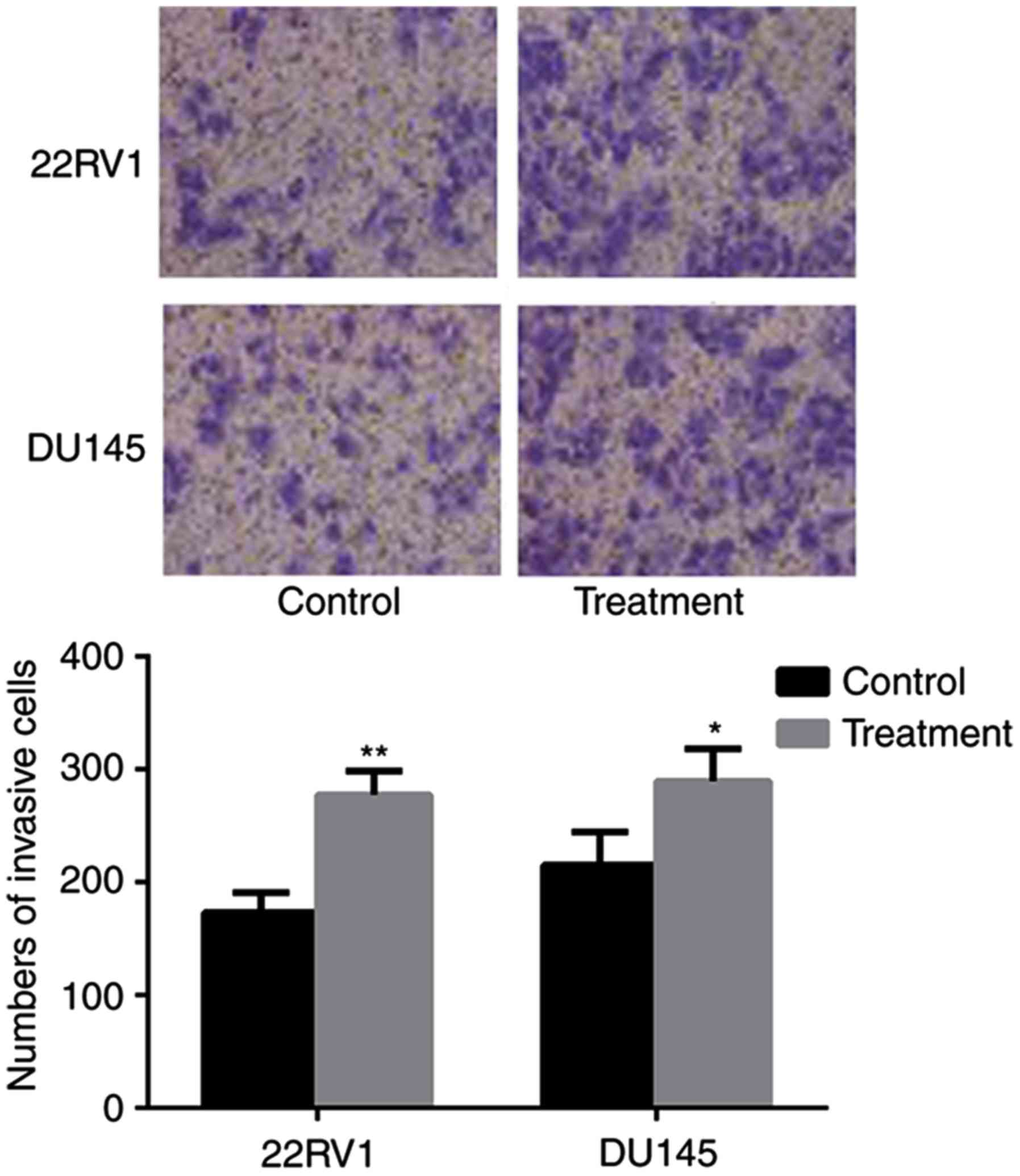
Entosis promotes invasion in under
nintedanib stress
The consequences of nintedanib-induced entosis on
cell invasion ability were investigated. Over the extended period
(8 weeks) of treatment, the cell population was continuously
decreased by the frequent occurrence of entosis, apoptosis and
necrosis, until the cells developed nintedanib resistance and
avoided cell death. Pca cells with passage-matched resistant cells
as controls were cultured, and the Transwell invasion assay
indicated that the invasive ability of nintedanib-resistant Pca
cells had significantly increased (P<0.05; Fig. 6).
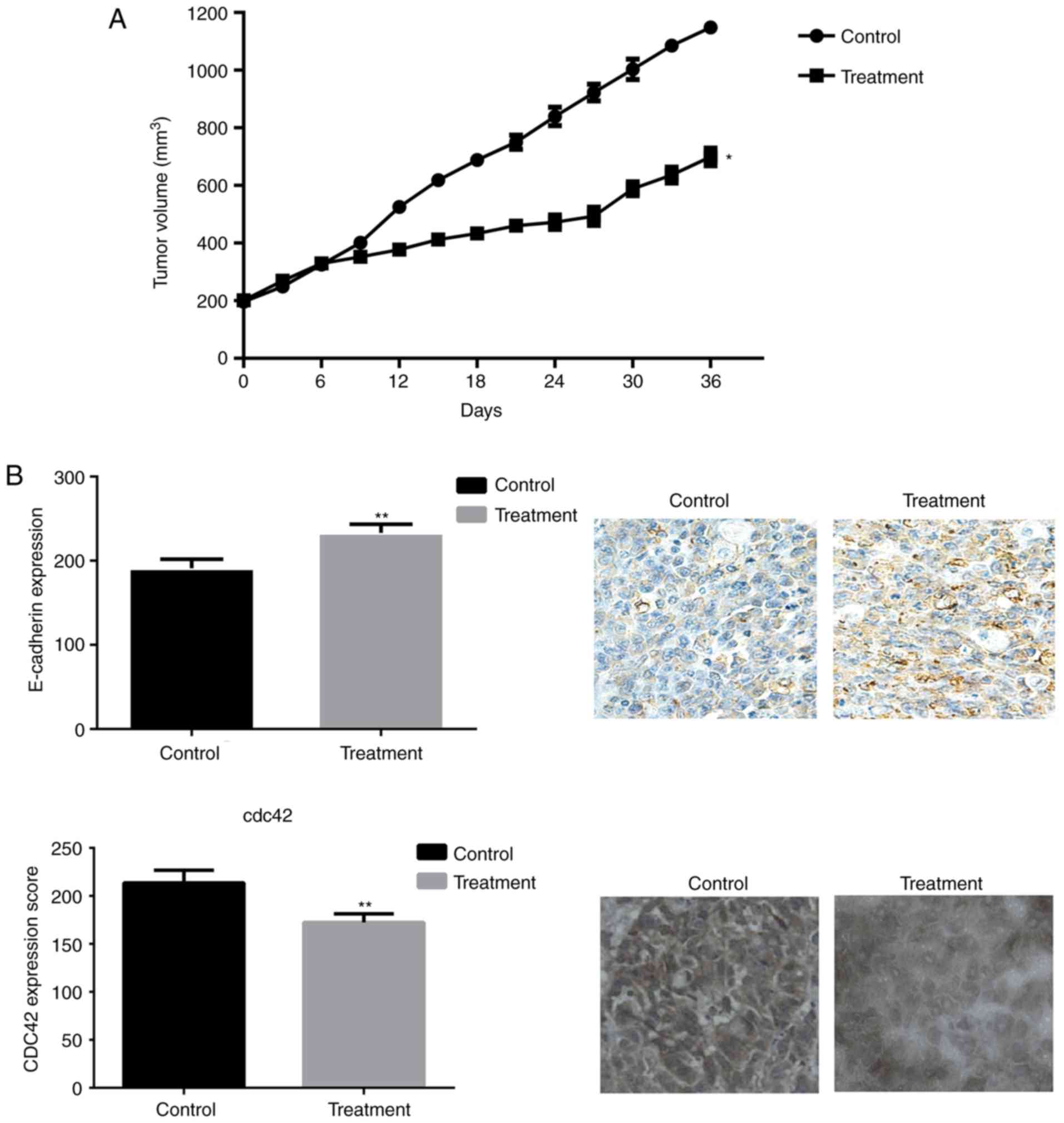
Entosis in a mouse Pca xenograft
To further investigate the role of nintedanib in Pca
cell entosis, mouse xenografts by were created by subcutaneously
injecting DU145 cells. Mice were treated with nintedanib, and it
was observed that nintedanib can attenuate the growth of tumors
compared with that using the placebo. IHC indicated that the
expression of E-cadherin was increased in the nintedanib-treated
tumors compared with in the controls, whereas CDC42 expression was
markedly decreased in nintedanib-treated tumors (Fig. 7). These results were consistent with
the data obtained from the in vitro cell lines, which
revealed that nintedanib could induce entosis via the upregulation
of E-cadherin expression and the ROCK1/2 signaling pathway.
Discussion
Nintedanib, a pan-inhibitor of TKs including FGFR,
has been evaluated in clinical trials for several types of cancer,
including prostate, lung and colorectal cancer (15,29,30). In a
randomized Phase II trial, nintedanib combined with afatinib
decreased PSA levels in ~50% of patients with castration-resistant
Pca (15). In another study,
nintedanib attenuated Pca progression in transgenic adenocarcinoma
of the mouse prostate mice (31).
However, it is unknown how Pca cells survive and develop resistance
under nintedanib pressure. The results of the present study
indicated that: i) Nintedanib is able to inhibit Pca cell
proliferation and decrease the growth of xenografts; ii) resistance
to nintedanib will develop during in vitro and vivo
treatment; and iii) nintedanib induces Pca cell entosis via the
upregulation of E-cadherin and ROCK1/2 through the PI3K/CDC42
signaling pathway.
It was observed multiple cancer cells were treated
with nintedanib at concentrations ranging between 1 and 5 µM
(32), the results revealed that
nintedanib inhibited cell proliferation without a toxic response.
In the present study that cells that have developed nintedanib
resistance display entosis. Nintedanib could block FGFR and then
inhibit the downstream PI3K/CDC42 signaling pathway to promote
entosis. A previous study identified that the activated PI3K
signaling pathway promotes Pca cell proliferation and facilitates
cell survival (33). In addition,
activated PI3K was observed to promote aerobic glycolysis in cancer
cells to tolerate nutrient starvation (34). In the present study, treatment with
nintedanib and blocking FGFR downregulated PI3K, and also blocked
its downstream pathways. CDC42 is an important molecule in the PI3K
downstream signaling pathway, and the results of the present study
have demonstrated that treatment with nintedanib decreased the
expression of CDC42, and this effect was also observed in Pca cells
treated with the PI3K inhibitor buparlisib. There are two isoforms
produced by alternative splicing from CDC42 gene: CDC42a and CDC42b
and to date, the functional differences between the two isoforms
remains unclear; however, it has been established that the two
isoforms can stimulate filopodia formation (35). In the present study, the primers used
reflect the total expression level of the two isoforms of CDC42
under nintedanib pressure. However, as the focus of the present
study was on the resistance of Pca cells to the nintedanib and the
entosis phenomenon, differences in the expression of the CDC42
isoforms were not investigated. The PI3K/CDC42 is a classic
signaling pathway in multiple cell events, including cell
proliferation and movement, and serves an important role in cancer
by promoting cancer progression and metastasis (36). Studies have demonstrated that blocking
CDC42 can inhibit tumor growth and prolong patient survival. A
study by Humphries-Bickley et al (37) suggested that blocking CDC42 can
inhibit breast cancer cell migration, viability and
epithelial-mesenchymal transition. A study of Guo et al
(38) demonstrated that R-ketorolac
blocked CDC42 and Rac1 and then inhibited ovarian cancer growth,
adhesion, migration and invasion.
PI3K/CDC42 can regulate phagosomes and promote
entotic vacuole maturation by regulating the ROCK signaling pathway
and E-cadherin expression (10,27,39–41).
Durgan et al (10) revealed
that knockout of CDC42 expression induced entosis in breast cancer,
and significant mitotic deadhesion and rounding was also observed
following the depletion of CDC42, and these phenotypes were
associated with a prominent increase in RhoA/ROCK1/2 activity. As a
downregulator of the RhoA/ROCK1/2 signaling pathway, blocking CDC42
may induce RhoA/ROCK1/2 activation and promote entosis. The
activated ROCK pathway would induce the accumulation of actomyosin,
increase actomyosin contractility, and be indispensable for
cell-in-cell formation (39). Instead
of engulfing cells, the increased RhoA/ROCK1/2 activity is located
in internalizing cells. In addition, actin and myosin contractility
are the driving force for entosis within ‘loser’ cells (40), whereas a ROCK1/2 inhibitor can abolish
the entosis process between cells in mixed culture experiments
(27), which was also observed in the
present study. Additionally, E-cadherin expression was also
increased in Pca cells when CDC42 was blocked. The study of Izumi
et al (41) identified that
CDC42 downregulation increased E-cadherin expression and in turn
promoted cancer progression. E-cadherin is one of the principal
components of adherens junctions in epithelial cells that connects
the actin cytoskeleton of neighboring cells (42). It has previously been established that
E-cadherin is involved in cancer progression, metastasis,
epithelial-mesenchymal transition and the cancer engulfment process
(43). In breast cancer, researchers
identified that entosis is induced by the establishment of
epithelial adhesion through the expression of E-/placental
cadherins (28). In addition, another
study focused on the cell entosis association in natural killer
cells and tumor cells, and observed that E-cadherin-mediated cell
junctions were required for the engulfment process (44).
CDC42 serves a role in cell division, and when CDC42
is blocked, cell division is disrupted (45). However, blocking cell mitosis would
allow for cell entosis. Division in engulfing cells can be
disrupted by entosis by blocking CDC42; thus, entosis would be
completed during engulfing cells mitosis (10,46). The
engulfed nucleus and mitosis-disrupted cells would create
multi-nucleated cells that can contribute to the generation of
aneuploidy, particularly when multipolar spindles are involved
(47). Aneuploidy cells have more
genetic instability, randomly distributed chromosomes and
chromosome breakage (48). Aneuploidy
also facilities cancer cells to gain extra copies to resist drug
pressures (49). Gene copy number
alterations are a major mechanism for signaling pathway
modifications, as increasing the copy number can amplify gene
expression, which would promote cell survival under the pressure of
specific inhibitors and allow the cells to tolerate the inhibition
(49). For example, in a previous
study, lung cancer was treated with an epidermal growth factor
receptor TKI, and the results demonstrated that MET amplification,
accompanied by KRAS amplification, can result in TKI therapy
failure (50). Owing to the induced
aneuploidy, entosis could contribute to cancer progression. The
study of Schwegler et al (51)
identified that entosis occurs most frequently in high-grade breast
cancers with increased recurrence and decreased overall patient
survival rates. In the present study, the results of the Transwell
assay indicated that entosis-like cells have increased invasive
abilities.
However, there are some limitations to the present
study. Namely, the anti-TK activity of nintedanib on Pca cells was
not investigated, which requires further research in order to
explain the effect of anti-TK on the entosis phenomenon.
Nintedanib is effective for inhibiting Pca cell
proliferation; however, resistance develops during the course of
treatment through the activation of entosis. In addition, the
nintedanib-induced entosis status is determined by ROCK activity
and E-cadherin protein expression levels.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Key Technology
Research and Development Program of Hebei Province (grant no.
14277774D).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
The present study was approved by the Hebei General
Hospital Institutional Ethics Committee (Shijiazhuang, China).
Patient consent for publication
Not applicable.
Authors' contributions
JL performed certain experiments and wrote the
paper. LW and YZ completed the majority of the experiments. SL, FS,
GW, TY, DW, LG and HX contributed to the idea and conception of
this study. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Madan RA, Gulley JL, Schlom J, Steinberg
SM, Liewehr DJ, Dahut WL and Arlen PM: Analysis of overall survival
in patients with nonmetastatic castration-resistant prostate cancer
treated with vaccine, nilutamide, and combination therapy. Clin
Cancer Res. 14:4526–4531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hait WN and Hambley TW: Targeted cancer
therapeutics. Cancer Res. 69:1263–1267. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schlessinger J: Cell signaling by receptor
tyrosine kinases. Cell. 103:211–225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Prenzel N, Fischer OM, Streit S, Hart S
and Ullrich A: The epidermal growth factor receptor family as a
central element for cellular signal transduction and
diversification. Endocr Relat Cancer. 8:11–31. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai B, Chen H, Guo S, Yang X, Linn DE, Sun
F, Li W, Guo Z, Xu K, Kim O, et al: Compensatory upregulation of
tyrosine kinase Etk/BMX in response to androgen deprivation
promotes castration-resistant growth of prostate cancer cells.
Cancer Res. 70:5587–5596. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Daniele G, Corral J, Molife LR and de Bono
JS: FGF receptor inhibitors: Role in cancer therapy. Curr Oncol
Rep. 14:111–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Muha V and Müller HA: Functions and
mechanisms of fibroblast growth factor (FGF) signalling in
drosophila melanogaster. Int J Mol Sci. 14:5920–5937. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qadir MI, Parveen A and Ali M: Cdc42: Role
in cancer management. Chem Biol Drug Des. 86:432–439. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Durgan J, Tseng YY, Hamann JC, Domart MC,
Collinson L, Hall A, Overholtzer M and Florey O: Mitosis can drive
cell cannibalism through entosis. Elife. 6:e271342017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun Q, Huang H and Overholtzer M:
Cell-in-cell structures are involved in the competition between
cells in human tumors. Mol Cell Oncol. 2:e10027072015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wen S, Shang Z, Zhu S, Chang C and Niu Y:
Androgen receptor enhances entosis, a non-apoptotic cell death,
through modulation of Rho/ROCK pathway in prostate cancer cells.
Prostate. 73:1306–1315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sahai E, Olson MF and Marshall CJ:
Cross-talk between Ras and Rho signalling pathways in
transformation favours proliferation and increased motility. EMBO
J. 20:755–766. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krishna S and Overholtzer M: Mechanisms
and consequences of entosis. Cell Mol Life Sci. 73:2379–2386. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Molife LR, Omlin A, Jones RJ, Karavasilis
V, Bloomfield D, Lumsden G, Fong PC, Olmos D, O'Sullivan JM, Pedley
I, et al: Randomized phase II trial of nintedanib, afatinib and
sequential combination in castration-resistant prostate cancer.
Future Oncol. 10:219–231. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bousquet G, Alexandre J, Le Tourneau C,
Goldwasser F, Faivre S, de Mont-Serrat H, Kaiser R, Misset JL and
Raymond E: Phase I study of BIBF 1120 with docetaxel and prednisone
in metastatic chemo-naive hormone-refractory prostate cancer
patients. Br J Cancer. 105:1640–1645. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu L and Dong X: Complex impacts of
PI3K/AKT inhibitors to androgen receptor gene expression in
prostate cancer cells. PLoS One. 9:e1087802014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Somnay Y, Simon K, Harrison AD,
Kunnimalaiyaan S, Chen H and Kunnimalaiyaan M: Neuroendocrine
phenotype alteration and growth suppression through apoptosis by
MK-2206, an allosteric inhibitor of AKT, in carcinoid cell lines in
vitro. Anticancer Drugs. 24:66–72. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morris EJ, Jha S, Restaino CR, Dayananth
P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al:
Discovery of a novel ERK inhibitor with activity in models of
acquired resistance to BRAF and MEK inhibitors. Cancer Discov.
3:742–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maes H, Van Eygen S, Krysko DV,
Vandenabeele P, Nys K, Rillaerts K, Garg AD, Verfaillie T and
Agostinis P: BNIP3 supports melanoma cell migration and
vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell
Death Dis. 5:e11272014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chatterjee S, Schmidt S, Pouli S, Honisch
S, Alkahtani S, Stournaras C and Lang F: Membrane androgen receptor
sensitive Na+/H+ exchanger activity in
prostate cancer cells. FEBS Lett. 588:1571–1579. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Detre S, Saclani Jotti G and Dowsett M: A
‘quickscore’ method for immunohistochemical semiquantitation:
Validation for oestrogen receptor in breast carcinomas. J Clin
Pathol. 48:876–878. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hathcock KS, Farrington L, Ivanova I,
Livak F, Selimyan R, Sen R, Williams J, Tai X and Hodes RJ: The
requirement for pre-TCR during thymic differentiation enforces a
developmental pause that is essential for V-DJbeta rearrangement.
PLoS One. 6:e206392011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wallace J: Humane endpoints and cancer
research. ILAR J. 41:87–93. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hilberg F, Roth GJ, Krssak M, Kautschitsch
S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel
A, Quant J, et al: BIBF 1120: Triple angiokinase inhibitor with
sustained receptor blockade and good antitumor efficacy. Cancer
Res. 68:4774–4782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamann JC, Surcel A, Chen R, Teragawa C,
Albeck JG, Robinson DN and Overholtzer M: Entosis is induced by
glucose starvation. Cell Rep. 20:201–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun Q, Luo T, Ren Y, Florey O, Shirasawa
S, Sasazuki T, Robinson DN and Overholtzer M: Competition between
human cells by entosis. Cell Res. 24:1299–1310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun Q, Cibas ES, Huang H, Hodgson L and
Overholtzer M: Induction of entosis by epithelial cadherin
expression. Cell Res. 24:1288–1298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Riesco-Martinez MC, Sanchez-Torre A and
Garcia-Carbonero R: Safety and efficacy of nintedanib for the
treatment of metastatic colorectal cancer. Expert Opin Investig
Drugs. 26:1295–1305. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gabasa M, Ikemori R, Hilberg F, Reguart N
and Alcaraz J: Nintedanib selectively inhibits the activation and
tumour- promoting effects of fibroblasts from lung adenocarcinoma
patients. Br J Cancer. 117:1128–1138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
da Silva RF, Nogueira-Pangrazi E, Kido LA,
Montico F, Arana S, Kumar D, Raina K, Agarwal R and Cagnon VHA:
Nintedanib antiangiogenic inhibitor effectiveness in delaying
adenocarcinoma progression in Transgenic Adenocarcinoma of the
Mouse Prostate (TRAMP). J Biomed Sci. 24:312017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kutluk Cenik B, Ostapoff KT, Gerber DE and
Brekken RA: BIBF 1120 (nintedanib), a triple angiokinase inhibitor,
induces hypoxia but not EMT and blocks progression of preclinical
models of lung and pancreatic cancer. Mol Cancer Ther. 12:992–1001.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Raja Singh P, Sugantha Priya E,
Balakrishnan S, Arunkumar R, Sharmila G, Rajalakshmi M and
Arunakaran J: Inhibition of cell survival and proliferation by
nimbolide in human androgen-independent prostate cancer (PC-3)
cells: Involvement of the PI3K/Akt pathway. Mol Cell Biochem.
427:69–79. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Elstrom RL, Bauer DE, Buzzai M, Karnauskas
R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM and
Thompson CB: Akt stimulates aerobic glycolysis in cancer cells.
Cancer Res. 64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
He X, Yuan C and Yang J: Regulation and
functional significance of CDC42 alternative splicing in ovarian
cancer. Oncotarget. 6:29651–29663. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Murga C, Zohar M, Teramoto H and Gutkind
JS: Rac1 and RhoG promote cell survival by the activation of PI3K
and Akt, independently of their ability to stimulate JNK and
NF-kappaB. Oncogene. 21:207–216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Humphries-Bickley T, Castillo-Pichardo L,
Hernandez-O'Farrill E, Borrero-Garcia LD, Forestier-Roman I, Gerena
Y, Blanco M, Rivera-Robles MJ, Rodriguez-Medina JR, Cubano LA, et
al: Characterization of a Dual Rac/Cdc42 inhibitor MBQ-167 in
metastatic cancer. Mol Cancer Ther. 16:805–818. 2017.PubMed/NCBI
|
|
38
|
Guo Y, Kenney SR, Muller CY, Adams S,
Rutledge T, Romero E, Murray-Krezan C, Prekeris R, Sklar LA, Hudson
LG and Wandinger-Ness A: R-ketorolac targets Cdc42 and Rac1 and
alters ovarian cancer cell behaviors critical for invasion and
metastasis. Mol Cancer Ther. 14:2215–2227. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yamada S and Nelson WJ: Localized zones of
Rho and Rac activities drive initiation and expansion of epithelial
cell-cell adhesion. J Cell Biol. 178:517–527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Purvanov V, Holst M, Khan J, Baarlink C
and Grosse R: G-protein-coupled receptor signaling and polarized
actin dynamics drive cell-in-cell invasion. Elife. 32014
|
|
41
|
Izumi G, Sakisaka T, Baba T, Tanaka S,
Morimoto K and Takai Y: Endocytosis of E-cadherin regulated by Rac
and Cdc42 small G proteins through IQGAP1 and actin filaments. J
Cell Biol. 166:237–248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Baum B and Georgiou M: Dynamics of
adherens junctions in epithelial establishment, maintenance, and
remodeling. J Cell Biol. 192:907–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Petrova YI, Schecterson L and Gumbiner BM:
Roles for E-cadherin cell surface regulation in cancer. Mol Biol
Cell. 27:3233–3244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang S, Guo Z, Xia P, Liu T, Wang J, Li S,
Sun L, Lu J, Wen Q, Zhou M, et al: Internalization of NK cells into
tumor cells requires ezrin and leads to programmed cell-in-cell
death. Cell Res. 19:1350–1362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Melendez J, Grogg M and Zheng Y: Signaling
role of Cdc42 in regulating mammalian physiology. J Biol Chem.
286:2375–2381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Krajcovic M and Overholtzer M: Mechanisms
of ploidy increase in human cancers: A new role for cell
cannibalism. Cancer Res. 72:1596–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Krajcovic M, Johnson NB, Sun Q, Normand G,
Hoover N, Yao E, Richardson AL, King RW, Cibas ES, Schnitt SJ, et
al: A non-genetic route to aneuploidy in human cancers. Nat Cell
Biol. 13:324–330. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Guerrero AA, Gamero MC, Trachana V,
Fütterer A, Pacios-Bras C, Díaz-Concha NP, Cigudosa JC, Martínez-AC
and van Wely KH: Centromere-localized breaks indicate the
generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci
USA. 107:4159–4164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tang YC and Amon A: Gene copy-number
alterations: A cost-benefit analysis. Cell. 152:394–405. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schwegler M, Wirsing AM, Schenker HM, Ott
L, Ries JM, Büttner-Herold M, Fietkau R, Putz F and Distel LV:
Prognostic value of homotypic cell internalization by
nonprofessional phagocytic cancer cells. Biomed Res Int.
2015:3593922015. View Article : Google Scholar : PubMed/NCBI
|