Introduction
Liver cancer is the second leading cause of cancer
worldwide, with liver cancer representing ~90% of all primary liver
cancer cases in 2016 (1–3). A number of risk factors have been
identified for liver cancer, and among these factors, chronic
hepatitis B virus (HBV) infection accounts for >50% cases of
liver cancer worldwide in 2016 (4–6). Given
that HBV is one of the most common infections in the world, with
~257 million people living with chronic HBV, and that an estimated
20–30% of chronically HBV infections in adults would lead to liver
cancer in 2016, the number of HBV-associated liver cancer cases is
vast (7,8). Consequently, understanding the
pathogenesis of HBV infection-induced liver cancer is of great
importance for the development of strategies for liver cancer
diagnosis and treatment.
Numerous possible mechanisms have been proposed
underlying HBV infection-induced liver cancer so far, which can be
categorized into direct and indirect mechanisms. Directly, HBV high
level replication and genomic integration may cause chromosomal
alterations, including chromosomal instability due to viral genomic
integration occurring in random locations of any chromosome, and
increase the risk of cancer gene activation (9–12). In
addition, HBV encodes a few oncogenesis genes, such as HBx and
preS2/S, which directly induces carcinogenesis (13–16).
Indirectly, HBV infection causes long-term inflammation in the
liver, leading to increased hepatocyte proliferation, hepatic
fibrosis and cirrhosis (14,17). Furthermore, HBV can also activate a
range of antiapoptotic host proteins, such as Bcl-2 and survivin,
which promote carcinogenesis (18,19).
However, it seems that none of these mechanisms can explain all the
clinical features observed in HBV-induced liver cancer, for example
liver cancer can occur in the absence of inflammation (10,20,21), and
HBV integration is random and rarely leads to direct oncogene
activation (14). Therefore, the
mechanism underlying HBV infection-induced liver cancer is
multifaceted and more possible mechanisms are still to be
identified.
The cellular inhibitor of apoptosis protein 2
(cIAP2), a member of the IAP family, can inhibit cell apoptosis via
inhibition of caspase activity (22). cIAP2 expression is significantly
increased in a variety of human cancer types, including colorectal
cancer, lung cancer and prostate cancer, and high expression of
this protein is associated with poor outcome of these cancers
(22–24). In colorectal cancer, cIAP2 has
previously been reported to be a valuable therapeutic target, as
downregulation of this protein efficiently enhances cancer cell
apoptosis (25). In liver cancer,
however, little is known about the expression and impact of cIAP2
on HBV-induced liver cancer pathogenesis. In the present study, the
expression of cIAP2 was initially investigated in liver cancer
tissue samples and adjacent non-cancerous tissue samples with or
without HBV infection, and then the impact of HBV infection on
cIAP2 expression and underlying mechanism was explored. The
findings of the current study provide further understanding on the
pathogenesis of HBV-induced liver cancer and could also be valuable
for the development of strategies for liver cancer diagnosis and
treatment.
Materials and methods
Patient samples, cell lines and
virus
Liver and blood samples were obtained from 8
patients, including 4 male and 4 female patients (range, 46–52
years; mean, 49 years) with liver cancer who underwent resection at
the Affiliated Hospital of Jinggangshan University (Jiangxi, China)
between November 2017 and May 2018. Cancerous (CT) and
non-cancerous tissues (NCT) were then separated and frozen in −80°C
until required. The NCT samples were collected from at least 0.5 cm
away from CT samples. Blood samples were used for HSV-2 infection
determination using ELISA (HerpeSelect® 1 and 2
Immunoblot IgG; Focus Diagnostics, Inc.) and PCR (artus HSV-1/2 PCR
Kits; Qiagen China Co., Ltd.), as previously described (26). All protocols involving human samples
were reviewed and approved by the Ethical Review Board at the
Affiliated Hospital of Jinggangshan University (Jiangxi, China;
ref. JGSU-20170071), and written informed consent was provided from
all the participating patients.
The normal liver cell line, THLE-3, the non-HBV
expressing human liver cancer cell line HepG2 and persistent
HBV-expressing liver cancer cell line, HepG2.2.15 were all
purchased from the American Type Culture Collection and cultured in
DMEM (Sigma-Aldrich; Merck KGaA) supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and penicillin and streptomycin
(both at 100 U/ml; Gibco; Thermo Fisher Scientific, Inc.) at 37°C
with 5% CO2.
HBV used in the present study was harvested from
HepG2.2.15 cells as previously described (27). In brief, cell culture media were
first filtered through a 0.45-µm filter, and then precipitated
using a PEG-it Virus Precipitation Solution (System Biosciences,
LLC) according to the manufacturer's instructions. In brief,
virus-containing medium was first mixed with cold PEG-it Virus
Precipitation Solution at the ratio of 4:1 and incubated into the
mixture overnight at 4°C. Following incubation, the virus and
PEG-it Virus Precipitation Solution mixture was centrifuged at
1,500 × g for 30 min at 4°C. Following centrifugation, all traces
of fluid were removed by aspiration and pelleted viruses were
washed with PBS twice and concentrated using Amicon 100 kD
ultracentrifugal tubes (EMD Millipore; Merck KGaA). Finally,
viruses were resuspended in PBS containing 25% FBS (Gibco; Thermo
Fisher Scientific, Inc.), aliquoted and stored at −80°C until
required. HBV titer was assessed using quantitative PCR and
quantified as genome equivalent/ml (GEq/ml), as previously
described (28).
Plasmids
pGL3-Basic (Promega Corporation), a vector with a
firefly luciferase gene under the control of a promoter of interest
to be cloned in by the user, was used to construct all plasmids
containing full length, truncated or mutated cIAP2 promoters and
NF-κB promoter. Full length cIAP2 promoter, (−2,000/+55) cIAP2-Luc,
and its truncations, (−1,000/+55) cIAP2-Luc, (−500/+55) cIAP2-Luc,
(−250/+55) cIAP2-Luc and (−100/+55) cIAP2-Luc, as well as full
length NF-κB promoter, NF-κB-Luc, sequences were synthesized by
GenScript Biotech Corporation and subcloned into pGL3-Basic using
the NEBuilder HiFi DNA Assembly master mix (New England Biolabs),
according to the manufacturer's instructions. Site-directed
mutations to the (−2,000/+55) cIAP2-Luc plasmid was introduced
using the QuikChange II Site-Directed Mutagenesis kit (Agilent
Technologies) according to the manufacturer's instructions. For
activator protein 1 (AP1) mutant (MUT), the AP1 sequence was
mutated from 5′-TTTTGGGTCATGG-3′ to 5′-TTTTGGGTCGCGG-3′. There are
3 NF-κB binding sites selected for this study in the cIAP2
promoter: NF-κB*, NF-κB** and NF-κB***. For NF-κB* MUT, NF-κB** MUT
and NF-κB*** MUT, NF-κB sequences were mutated from
5′-GGAAATCCCC-3′ to 5′-GGAAATAGCC-3′, from 5′-TGGGTTTGCC-3′ to
5-′CTCGTTTGCC-3′ and from 5′-TGGAGTTCCC-3′ to 5′-TGGAGTTAAC-3′,
respectively. For interferon regulatory factor-1 (IRF-1) MUT, the
IFR-1 sequence was mutated from 5′-TAAAAGGAAAG-3′ to
5′-TAACAGGAGAG-5′.
Transfection and luciferase reporter
gene activity assay
Transfection and luciferase reporter gene activity
assay was performed as previously described with modifications
(26). In brief, THLE-3 cells were
transfected with promoter constructs together with control plasmid
pRL-RK (Promega Corporation) using Lipofectamine 2000®
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The medium was changed, and the cells were
subsequently infected with or without HBV at 100 GEq/cell or
ascendant doses of HBV (0, 50, 100, 200 and 500 GEq/cell) for 1 h
at 37°C, 4–6 h after transfection. Fresh complete medium was added
and the cells were cultured at 37°C with 5% CO2 for
another 24 h. After which time, cells were lysed with Luciferase
Cell Culture Lysis buffer (Promega Corporation) and firefly
luciferase activity and Renilla luciferase activity were
measured using the Dual-Luciferase® Reporter Assay
System (Promega Corporation), according to the manufacturer's
protocol. The transfection efficiency was normalized to the
Renilla luciferase activity. For small interfering RNA
(siRNA)-mediated knockdown of p65 or AKT, siRNA targeting p65 (cat.
no. sc-29410), AKT (cat. no. sc-43609) or control siRNA (cat. no.
sc-37007) at a final concentration of 100 nM was introduced into
cells using siRNA Transfection Reagent (all from Santa Cruz
Biotechnology, Inc.) 24 h before plasmid transfection. For
signaling pathway inhibition, specific inhibitors against NF-κB
(Celastrol; 300 nM), MAPKK (PD98059; 10 µM), PI3K (LY294002; 50 µM)
or p38 (SB203580; 10 µM) was added into the medium after virus
infection and remained throughout the culture. All inhibitors were
purchased from InvivoGen and used according to the manufacturer's
instructions.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from THLE-3, HepG2 and
HepG2.215 cells using the RNeasy Mini kit (Qiagen, Inc.) and
reverse-transcribed into cDNA using the ProtoScript® II
First Strand cDNA Synthesis kit (New England Biolabs, Inc.),
according to the manufacturer's instructions. The cDNA synthesis
reaction mix was first incubated at 42°C for 1 h for cDNA synthesis
and then the enzyme was inactivated at 80°C for 5 min. Target gene
mRNA level was subsequently determined using SYBRGreen qPCR using
the SsoAdvanced Universal SYBR-Green Supermix on a Bio-Rad CFX
Connect platform (both Bio-Rad Laboratories, Inc.). The
thermocycling conditions were as follows: Polymerase activation and
DNA denaturation (95°C, 1 min); 40 cycles of denaturation (95°C, 10
sec) and annealing/extension (60°C, 30 sec), and then followed by
melt-curve analysis (65–95°C with 0.5°C increment 2–5 sec/step).
The following primers were used: cIAP2 forward,
5′-GATGTTTCAGATCTACCAGTG-3′ and reverse,
5′-GAAATGTACGAACTGTACCCT-3′; NF-κB (p65) forward,
5′-ATGGCTTCTATGAGGCTGAG-3′ and reverse, 5′-GTTGTTGTTGGTCTGGATGC-3′;
internal control β-actin forward, 5′-AAGCAGGAGTATGACGAGTCCG-3′, and
reverse, 5′-GCCTTCATACATCTCAAGTTGG-3′. Target gene mRNA level was
quantified using the 2−ΔΔCq method (29).
Isolation of cell cytoplasmic and
nuclear fractions
The cytoplasmic and nuclear fractions were isolated
from THLE-3 cells using a Cell Fractionation kit (cat. no.
ab109718; Abcam) according to the manufacturer's instructions. In
brief, cells were first harvested by trypsinization and resuspended
in buffer A, and then an equal volume of buffer B was added and
mixed for 7 min at room temperature. After centrifugation at 5,000
× g for 1 min at 4°C, the supernatant (cytoplasmic fraction) was
removed to a new tube, while the pellet was further incubated with
buffer C for 10 min at room temperature with constant mixing.
Following centrifugation at 5,000 × g for 1 min at 4°C, the
supernatant (nuclear fraction) was transferred to a new tube. All
fractionized samples were either stored at −80°C or used directly
for downstream experiments.
Western blot analysis
Western blot was performed as previously described
with modifications (30,31). Depending on the experiment, CT and
NCT tissue samples, THLE-3, HepG2 and HepG2.215 cells with or
without transfection, and cytoplasmic and nuclear fractions were
used for western blot analysis. CT and NCT tissue samples were
first homogenized in PBS supplemented with protease inhibitor
cocktail (Roche Diagnostics), and then centrifuged at 14,000 × g
for 10 min at 4°C, and then supernatants were transferred into new
tubes, mixed with 4× SDS-PAGE loading buffer. THLE-3, HepG2 and
HepG2.215 cells with or without transfection were first harvested
using non-enzymatic cell dissociation buffer (Sigma-Aldrich; Merck
KGaA), washed with PBS and then lysed using Pierce
immunoprecipitation (IP)/lysis buffer (Thermo Fisher Scientific,
Inc.) supplemented with protease inhibitor cocktail (Roche
Diagnostics). Centrifugation (14,000 × g, 10 min at 4°C) cleared
protein samples were then mixed with SDS-PAGE loading buffer. Cell
cytoplasmic and nuclear fractions were directly mixed with 4X
SDS-PAGE loading buffer and used for SDS-PAGE. All prepared samples
were separated by 12% SDS-PAGE and transferred onto PVDF membranes.
After blocking with 5% skimmed milk for 1 h at room temperature,
the membrane was sequentially incubated with primary antibodies and
horseradish peroxidase (HRP)-conjugated secondary antibodies,
overnight at 4°C and for 1 h at room temperature, respectively.
After extensive washes with PBS-0.1% Tween-20, the membrane was
visualized with an enhanced chemiluminescence substrate (EMD
Millipore) under a charge-coupled device camera (Bio-Rad
Laboratories, Inc.). The following primary antibodies were used:
Rabbit anti-cIAP2 (1:1,000 dilution; cat. no. ab23423; Abcam),
mouse anti-β-actin (1:1,000 dilution; cat. no. sc-47778; Santa Cruz
Biotechnology, Inc.), rabbit anti-NF-κB p65 (1:1,000 dilution; cat.
no. 8242; Cell Signaling Technology, Inc.), rabbit
anti-phosphorylated (p)-NF-κB p65 (1:1,000 dilution; cat. no. 3033;
Cell Signaling Technology, Inc.), mouse anti-PI3K (1:1,000
dilution; cat. no. 60225-1-Ig; ProteinTech Group, Inc.), rabbit
anti-AKT (1:1,000 dilution; cat. no. 9272; Cell Signaling
Technology, Inc.), rabbit anti-p-AKT (1:1,000 dilution; cat. no.
9271; Cell Signaling Technology, Inc.) and mouse anti-proliferating
cell nuclear antigen (1:1,000 dilution; cat. no. sc-56; Santa Cruz
Biotechnology, Inc.). The following secondary antibodies were used:
HRP-conjugated goat anti-mouse IgG (H+L) (1:30,000 dilution; cat.
no. SA00001-1) and HRP-conjugated goat anti-rabbit IgG (H+L)
(1:30,000 dilution; cat. no. SA00001-2; both ProteinTech Group,
Inc.). To quantify band intensity, ImageJ (version 1.51j8; National
Institutes of Health) was used.
Chromatin (Ch)IP assay
The ChIP assay was performed using a Pierce Magnetic
ChIP kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. In brief, THLE-3 cells mock-infected
with medium or infected with HBV were first washed with PBS and
then crosslinked with ChIP grade 1% formaldehyde (Thermo Fisher
Scientific, Inc.). Following crosslinking, cells were lysed and
digested with membrane extraction buffer and micrococcal nuclease,
respectively. The chromatin smear was then obtained using
sonication (3×20 sec pulses at 3W power on ice with 20 sec
incubation on ice between pulses), and subsequently incubated with
5 µg of either control Ig or ChIP grade anti-p65 antibody (both
control Ig and anti-p65 were from the same ChIP kit; cat. no.
17-10060; Sigma-Aldrich; Merck KGaA) overnight at 4°C. Following
incubation, magnetic beads were added into the mixture and
incubated for a further 2 h at 4°C. After extensive washes with
PBS-0.05% Tween-20, DNA was eluted from magnetic beads with elution
buffer. Finally, a PCR amplifying the cIAP2 promoter was performed
using Q5® High-Fidelity DNA Polymerase (New England
BioLabs. Inc) with the following primers: Forward,
5′-CCCGAGTGGGTTTGCCAG-3′ and reverse,
5′-TTTTAAATGCGTCACCCAAATCCCC-3′. The thermocycling conditions were
as follows: Initial denaturation, 98°C, 30 sec; 35 of cycles of
denaturation (98°C, 10 sec), annealing (55°C, 10 sec) and
elongation (72°C, 30 sec); and final extension (72°C, 2 min).
Statistical analysis
All experiments were repeated three times,
independently. Data are expressed as mean ± standard deviation, and
all statistical analyses were performed using GraphPad Prism v8.1.2
(GraphPad Software, Inc.). Unpaired Student's t-test and one-way
ANOVA with Student-Newman-Keuls post hoc test were used for
comparisons between two groups or more than three groups,
respectively. P<0.05 was considered to indicate a statistically
significant difference.
Results
cIAP2 expression is increased in
HBV-infected liver tissue and liver cell lines
To determine cIAP2 expression in liver tissues in
relation to HBV infection and liver cancer, liver samples from CT
and adjacent NCT were obtained from patients with liver cancer and
who were either HBV-positive (HBV+, 2 male and 2 female)
or -negative (HBV−, 2 male and 2 female), and had
undergone surgery, and cIAP2 expression was determined using
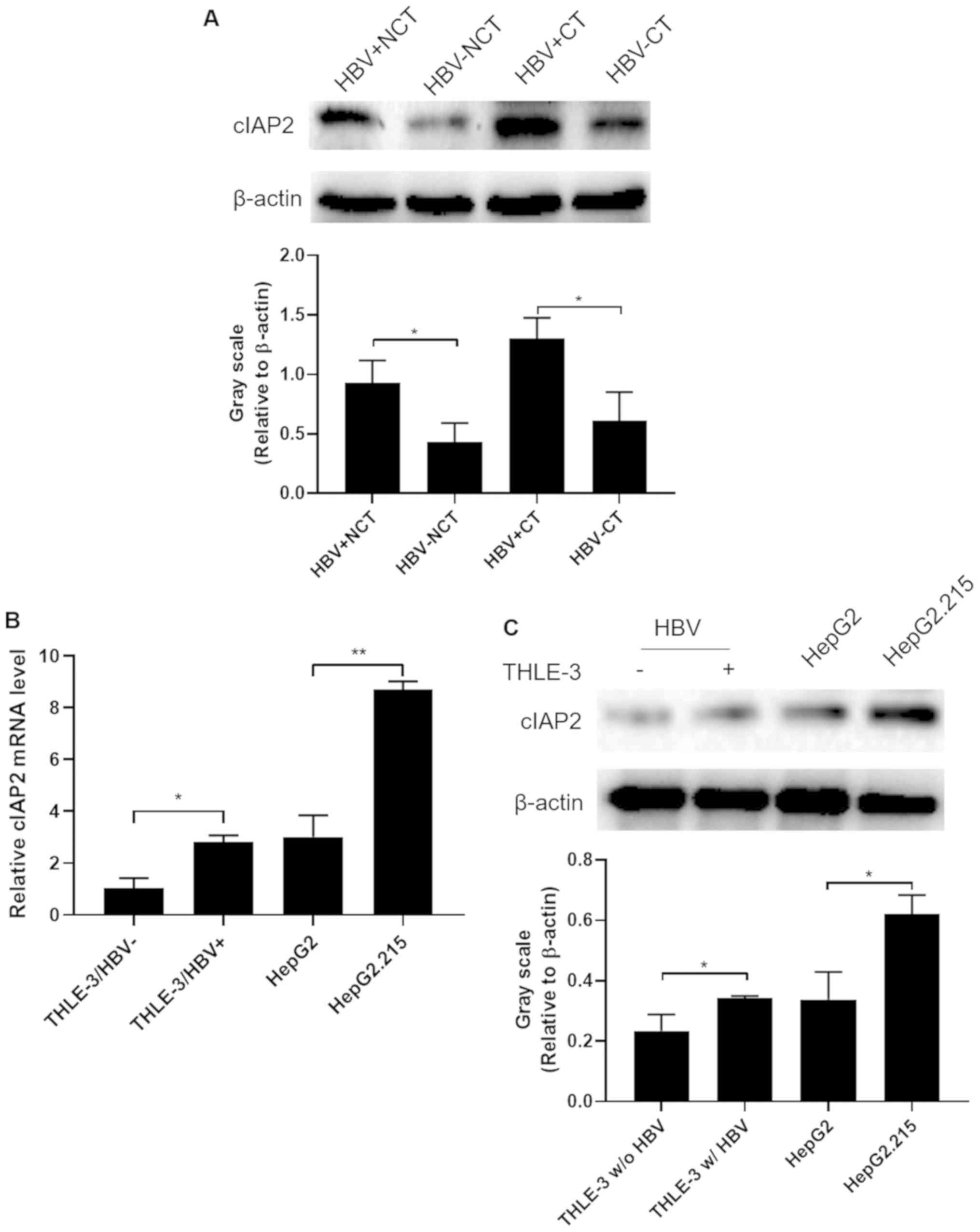
western blot analysis. As presented in Fig. 1A, cIAP2 expression in HBV+
NCT was significantly increased compared with HBV− NCT
samples, while the expression of cIAP2 in HBV+ CT was
further increased compared with HBV− CT samples
(P<0.05). These data indicate that HBV infection could increase
cIAP2 expression in liver tissue and the persistent high expression
of cIAP2 induced by HBV infection may have promoted liver cancer
carcinogenesis.
To determine if this HBV infection-induced cIAP2
expression could also be observed in liver cell lines, the normal
liver cell line, THLE-3 with or without HBV infection, the non-HBV
expressing human liver cancer cell line, HepG2 and the persistent
HBV-expressing liver cancer cell line, HepG2.2.15 were analyzed to
determine the cIAP2 expression at both the mRNA and protein level.
The data demonstrated that cIAP2 expression was increased in
HBV+ THLE-3 cells and HepG2.215 at both the mRNA
(Fig. 1B) and protein levels
(Fig. 1C), compared with that in
HBV- THLE-3 and HepG2 cells, respectively.
HBV induces cIAP2 expression through
transactivation of the cIAP2 promoter
The THLE-3 cell line was used for subsequent
experiments to determine whether HBV-induced cIAP2 expression was
due to the transactivation of the cIAP2 promoter. Therefore, a
firefly luciferase reporter gene plasmid under the control of the
cIAP2 promoter, designated as (−2,000/+55) cIAP2-Luc was
constructed, and the luciferase expression in response to HBV
infection was determined. As shown in Fig. 2A, luciferase activity was markedly
increased in a HBV infection dose-dependent manner. Serial 5′
flanking region deletions were then created on the
(−2,000/+55)cIAP2-Luc and their responses to HBV infection were
also investigated. The results showed that (−100/+55) cIAP2-Luc
variant had significantly lower luciferase activity, indicating
that the deletion of the −250 to −100 nucleotide sequence abolished
luciferase expression, and that this region is essential for cIAP2
promoter activation by HBV infection (Fig. 2B).
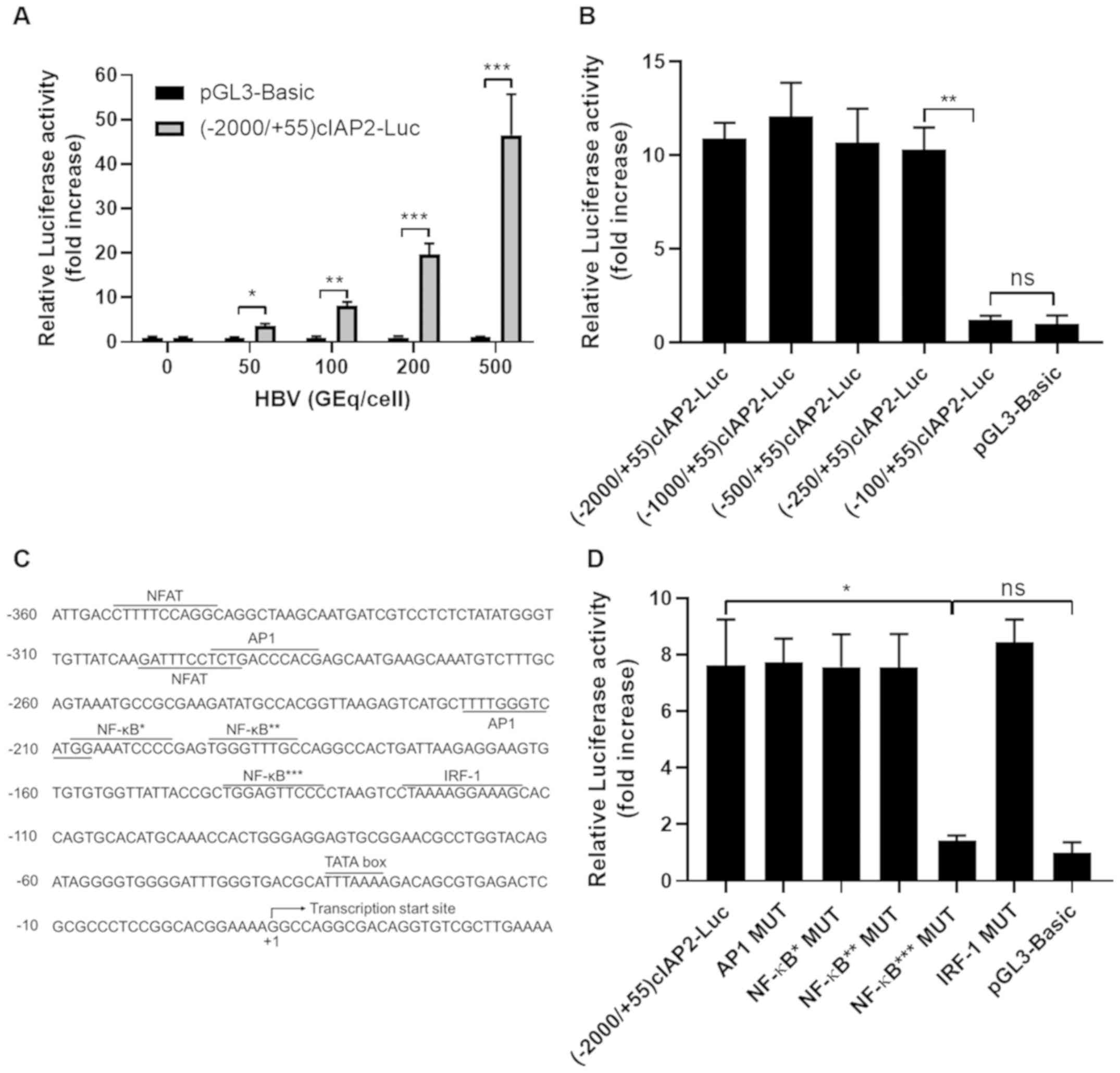 | Figure 2.A NF-κB binding site in cIAP2
promoter is essential for HBV-induced cIAP2 promoter
transactivation. (A) THLE-3 cells transfected with (−2,000/+55)
cIAP2-Luc or pGL3-Basic and then infected with increasing doses of
HBV. (B) THLE-3 cells transfected with serially truncated cIAP
promoter constructs and then infected with 100 GEq/cell of HBV.
Cells were lysed, and luciferase activity was measured 24 h later.
(C) Predicted transcription factor binding sites within cIAP2
promoter sequence. The following transcription factor binding sites
were detected in this region: 2 NFAT, 2 AP1, 3 NF-κB, 1 IRF-1 and 1
TATA box. Transcription start site is also indicated. (D) THLE-3
cells transfected with wild type or mutated (−2,000/+55)cIAP2-Luc
(namely, AP1 MUT, NF-κB* MUT, NF-κB** MUT, NF-κB*** MUT and IRF-1
MUT or pGL3-Basic were infected with 100 GEq/cell of HBV. Cells
were lysed, and luciferase activity was measured 24 h later. Data
are presented as the mean ± standard deviation of three independent
experiments. *P<0.05. **P<0.01. ***P<0.001. ns, not
statistically significant; cIAP2, cellular inhibitor of apoptosis
protein 2; HBV, hepatitis B virus; NFAT, nuclear factor of
activated T-cells; AP1, activator protein 1; IRF-1, interferon
regulatory factor-1; MUT, mutant; GEq, genome equivalent. |
Previous bioinformatics analysis showed several
transcription factor binding sites, including one AP1, three NF-κB
sites and one IRF-1 site, in the region of −250 nt to −100 nt in
the cIAP2 promoter sequence (32)
(Fig. 2C). To further determine if
one or more transcription factor binding sites may be involved in
HBV-induced cIAP2 promoter transactivation, mutations to these
transcription binding sites were created and their activation by
HBV infection was investigated. The data showed that only the
mutation to the NF-κB binding site closest to the transcription
start site (designated as NF-κB***) completely abolished luciferase
expression, while the other mutations did not show any reduction in
signal (Fig. 2D). These data herein
indicate that NF-κB*** is involved in the cIAP2 transactivation by
HBV infection.
HBV infection induces NF-κB binding
onto cIAP2 promoter
Since NF-κB*** is essential for HBV to transactivate
cIAP2 promoter, it was investigated whether NF-κB could bind to
cIAP2 promoter and activate cIAP2 transcription. At the luciferase
reporter gene level, p65 siRNA knockdown significantly decreased
luciferase expression, which was induced by HBV infection,
comparing to control siRNA transfected group (Fig. 3A). Similar results were observed when
cIAP2 protein level was determined in cells infected with HBV in
the presence or absence of p65 siRNA (Fig. 3B). These data indicate that NF-κB is
involved in HBV-induced cIAP2 transcription and expression.
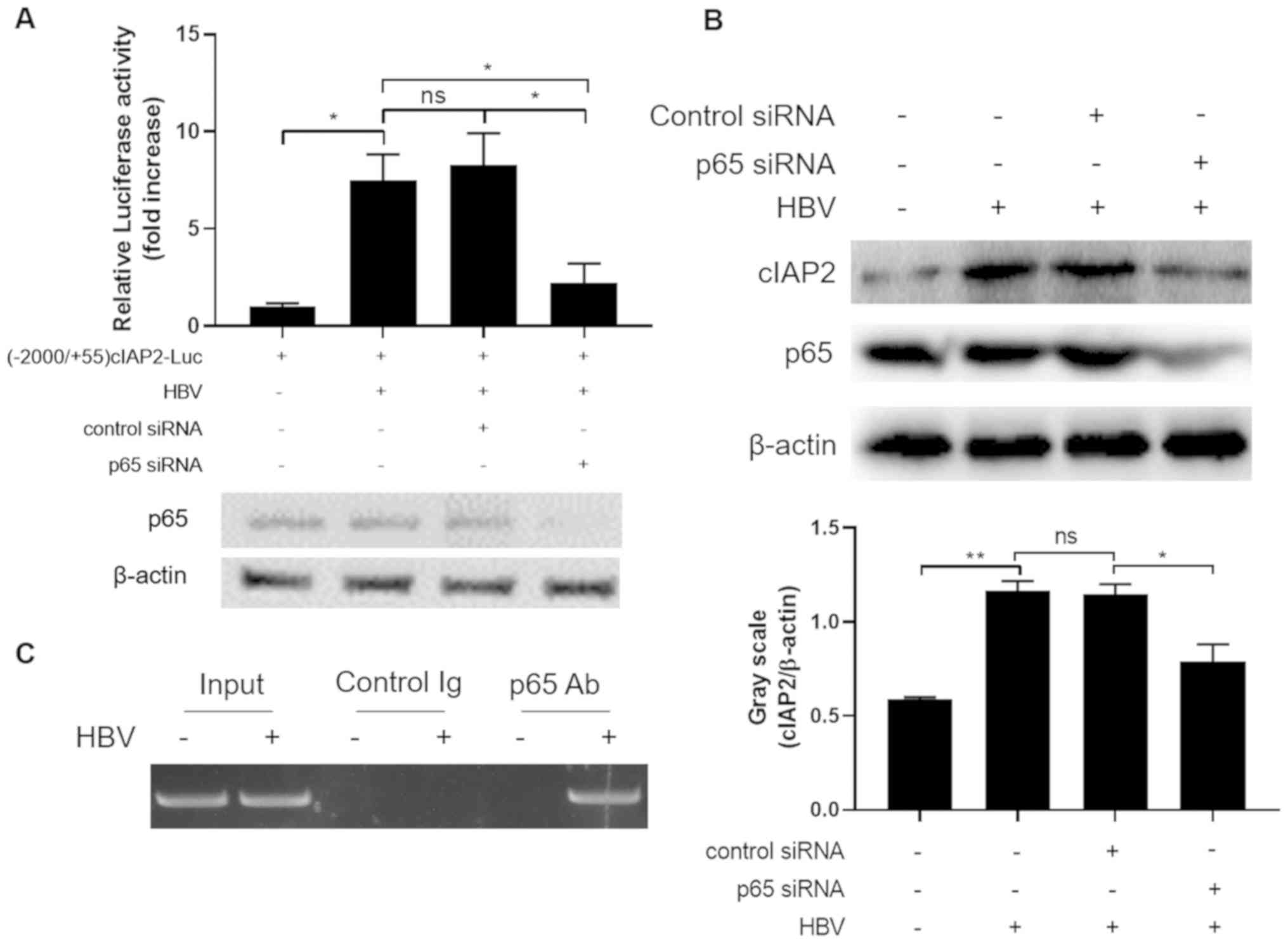 | Figure 3.HBV-infection induces NF-κB binding
onto cIAP2 promoter. (A) THLE-3 cells co-transfected with
(−2,000/+55)cIAP2-Luc and p65 siRNA or control siRNA, were infected
with HBV. Cells were lysed, and luciferase activity was measured 24
h after transfection. Data are presented as the mean ± standard
deviation of three independent experiments. *P<0.05. (B) THLE-3
cells transfected with p65 siRNA or control siRNA were infected
with HBV and cIAP2 protein expression was determined using western
blot analysis. ns, statistically not significant; *P<0.05;
**P<0.01. (C) Chromatin immunoprecipitation was performed
following THLE-3 cells mock infected or infected with HBV, with
either p65 or a control Ab, to pull down DNA segment containing the
NF-κB binding site in the cIAP2 promoter. For western blot
analysis, one representative result out of three is shown. cIAP2,
cellular inhibitor of apoptosis protein 2; si, small interfering;
HBV; hepatitis B virus; Ab, antibody; ns; not statistically
significant. |
To further confirm that NF-κB binds to cIAP2
promoter, ChIP assay was performed with cells mock infected with
medium or infected with HBV. As presented in Fig. 3C, p65 antibody pulled down cIAP2
promoter sequence only when cells were infected with HBV,
indicating that upon HBV infection, NF-κB could bind to cIAP2
promoter and may assist the transcription of this gene.
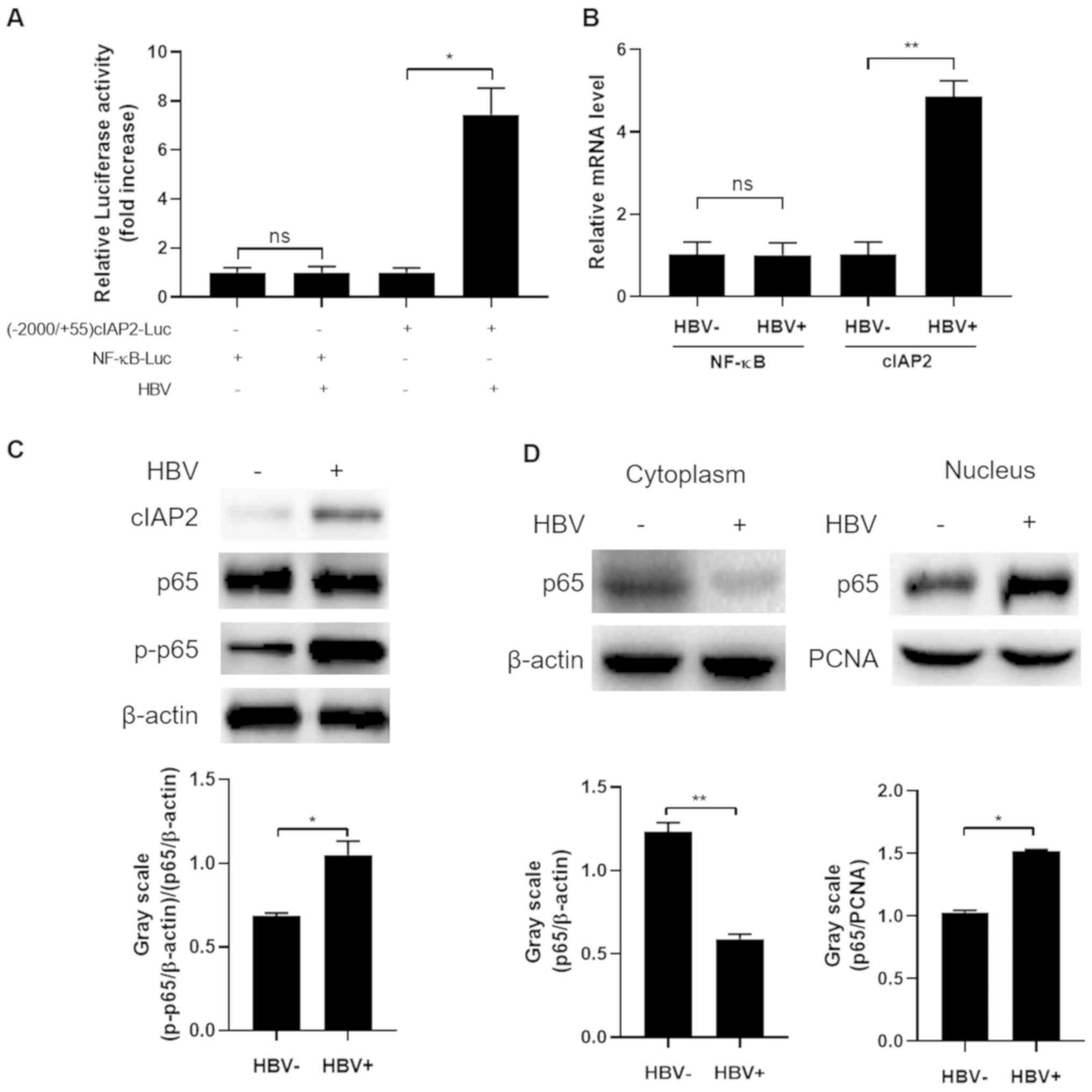
HBV infection increases NF-κB
phosphorylation and nuclear translocation
The aforementioned data revealed that NF-κB
responded to HBV infection and played an important role in
HBV-induced cIAP2 elevation. Subsequently, it was further
investigated whether HBV infection would change NF-κB expression
and/or function. A plasmid with a firefly luciferase reporter gene
under the control of NF-κB p65 promoter was constructed and its
response to HBV infection was measured. As shown in Fig. 4A, HBV infection showed no apparent
activation of the p65 promoter, as luciferase activity was not
altered in THLE-3 cells transfected with NF-κB-Luc prior to and
following HBV infection. As a control, HBV infection significantly
enhanced luciferase activity in cells transfected with (−2000/+55)
cIAP2-Luc, an indication of the activation of cIAP2 promoter.
Similar results were observed when p65 mRNA level was determined.
HBV infection did not enhance NF-κB mRNA level, however did
significantly enhance that of cIAP2 (P<0.01; Fig. 4B). Western blot analysis of p65
expression further confirmed that HBV infection did not affect p65
expression, however, considerably increased phosphorylation of this
protein was detected when cells were infected with HBV, indicating
an enhanced NF-κB activation (Fig.
4C). Since activated NF-κB needs to translocate from the
cytoplasm into the nucleus in order to exert its function, it was
subsequently determined whether a translocation of NF-κB was
induced by HBV infection. As expected, upon HBV infection, there
was a marked decrease of p65 in the cytoplasm while a significant
increase in the nucleus was detected (P<0.05; Fig. 4D). Together, these data here indicate
that HBV infection does not affect NF-κB expression but promotes
its activation and translocation from the cytoplasm into the
nucleus.
HBV infection-induced cIAP2 is
mediated by the PI3K/AKT/NF-κB signaling pathway
To further explore the signaling pathways HBV
employs to increase cIAP2 expression, signaling pathway inhibitors
specifically targeting NF-κB, MAPKK, PI3K and p38 were analyzed for
their ability to block HBV-infection induced cIAP2 transactivation.
Of the inhibitors investigated, only NF-κB and PI3K pathway
inhibitors significantly suppressed cIAP2 transactivation included
by HBV infection (Fig. 5A). Further
western blot analysis revealed that HBV infection did not affect
PI3K and AKT expression, but markedly increased the p-AKT level
(Fig. 5B). To further confirm the
importance of AKT in HBV-induced cIAP2 expression, HBV activation
of cIAP2 promoter in the presence or absence of AKT siRNA was
analyzed. The data showed that AKT-knockdown by siRNA significantly
reduced the level of cIAP2 promoter activation induced by HBV
infection (Fig. 5C). Western blot
results further confirmed the involvement of AKT in HBV-induced
cIAP2 expression, as AKT-knockdown reduced the level of cIAP2 and
p-p65, which was enhanced by HBV infection (Fig. 5D).
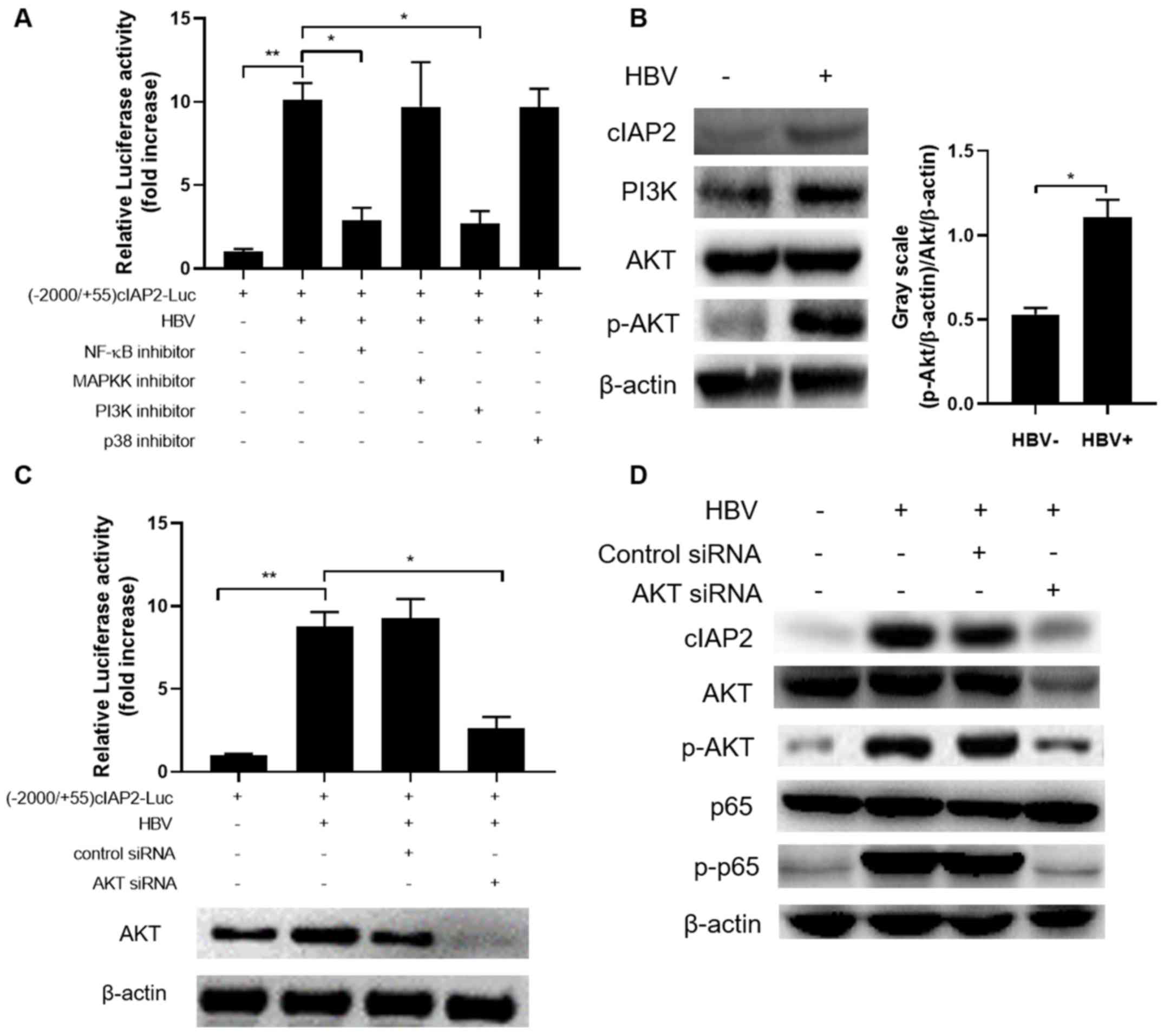 | Figure 5.HBV induces cIAP2 expression by
activating PI3K/AKT signaling pathway. (A) THLE-3 cells transfected
with (−2,000/+55)cIAP2-Luc were mock-infected or infected with HBV,
in the presence or absence of signaling pathway inhibitors. Cells
were lysed, and luciferase activity was measured 24 h later. (B)
THLE-3 cells were mock-infected or infected with HBV, then cIAP2,
PI3K, AKT and p-AKT expression was determined using western blot
analysis. (C) THLE-3 cells co-transfected with
(−2,000/+55)cIAP2-Luc and AKT siRNA or control siRNA, were infected
with HBV. Cells were lysed, and luciferase activity was measured 24
h later. (D) THLE-3 cells transfected with AKT siRNA or control
siRNA were infected with HBV and cIAP2, AKT, p-AKT, NF-κB and
p-NF-κB expression was determined using western blot analysis. For
western blot analysis, one representative result out of three is
shown. Data are presented as the mean ± standard deviation of three
independent experiments. *P<0.05; **P<0.01. HBV; hepatits B
virus; cIAP2 cellular inhibitor of apoptosis protein 2; si, small
interfering; p, phosphorylated. |
Discussion
It has been well documented that chronic HBV
infection has a role in liver cancer pathogenesis, however,
mechanisms underlying HBV-induced liver cancer seem to be
multifaceted and cannot be exhaustive (10,20,21).
Therefore, identifying new mechanisms is not only important for
understanding the pathogenesis of this disease, but also beneficial
for the development of new diagnostic and treatment targets. In the
present study, it was identified that HBV-infection can enhance
anti-apoptotic protein cIAP2 expression by activating its promoter
via the PI3K/AKT/NF-κB signaling pathway. These findings have
proposed a novel mechanism underlying HBV-induced liver cancer and
may offer valuable information for the development against liver
cancer.
cIAP2 belongs to the IAP protein family, which is a
highly conserved protein family that regulate cell survival,
immunity, inflammation and cell division (22). cIAP2 and other members of this family
are frequently dysregulated in numerous types of cancer, including
colorectal cancer, lung cancer and prostate cancer, and high
expression of this protein is also associated with poor outcome in
these cancers (22–24). In liver cancer, to the best of our
knowledge, the importance of cIAP2 has not been well studied. In
the present study, it was revealed that cIAP2 is upregulated by HBV
infection in both clinical tissues and cell lines. However, due to
the limited number of clinical samples obtained, a more in-depth
investigation on the relationship between cIAP2 expression and
HBV-induced liver cancer severity and prognosis could not be
performed. Further investigation with a higher number of clinical
samples to further access this relationship would give a better
understanding of cIAP2 in liver cancer and may also be beneficial
for the evaluation of cIAP2 as a diagnostic and/or treatment
target.
Drug development targeting cIAP2 and other IAP
family members, albeit in very limited number, has been studied at
both the pre-clinical and clinical levels. Up to now, several
strategies have been evaluated, including small chemical compounds,
Smac mimetics and antisense oligonucleotides. For example, YM155, a
novel molecule targeting and inhibiting IAP member survivin, has
shown good anticancer activity against a variety of tumors
including esophageal cancer and colorectal cancer in a Phase I
clinical trial (30). Now this small
molecule is being studied in combination with other anticancer
treatment drugs in various cancer clinical studies (31,32).
Birinapant, a bivalent Smac mimetic targeting TNF receptor
associated factor-2-associated cIAPs, is another anticancer drug
targeting cIAPs that has shown good efficacy in suppressing tumor
growth in a range of solid tumors, including esophageal, thymic and
colorectal tumors, and lymphoma when used alone or in combination
with other drugs, such as elrotnib and rituximab, in Phase I and II
clinical studies (33–35). Since this study reveals cIAP2 is
associated with HBV-infection induced liver cancer, further
investigation is required to explore the efficacy of IAP-targeting
anticancer drugs on liver cancer treatment.
HBV, a DNA virus with a 3.2 kb genome, contains four
overlapping open reading frames coding for the surface (HBs), Core
(HBc), X and the polymerase genes. Several viral genes have been
identified as cancer related. Among them, HBx is the mostly studied
one (13–16). HBx is associated with a wide range of
cancer-related biological pathways, including the cell cycle, cell
growth, apoptosis, inflammation, genome stability and metastasis
(36,37). HBs can also modulate inflammation and
the cell cycle by various pathways, including downregulation of
TLR9 and inhibition of interferon production, which can be
contributors to liver cancer pathogenesis (38). Although still controversial, HBc can
reportedly suppress cell apoptosis by downregulation of Fas, Fas
ligand and TP53 (39). In the
present study, the identification of one of more viral genes which
may trigger cIAP2 upregulation was not determined. However, it
would be of interest to investigate the viral components in cIAP2
expression, in a further study.
There are three subtypes of AKT proteins in humans
(namely AKT1, 2 and 3) and the AKT antibody used in the present
study could detect all three subtypes of AKT, therefore it was not
possible to directly identify which subtype may play a major role.
However, previous publications indicate that AKT1 is involved in
cell survival pathways by inhibiting apoptotic processes (40,41),
AKT2 is an important molecule in insulin signaling pathway
(42), and AKT3 with a less clear
role is predominantly expressed in the brain (43). Therefore, it is likely that AKT1 was
the subtype identified in the present study and might play a role
in HBV-induced cIAP2 expression.
Taken together, the current study has identified
that HBV-infection can enhance anti-apoptotic protein cIAP2
expression by activating its promoter via the PI3K/AKT/NF-κB
signaling pathway. These findings have proposed a novel mechanism
underlying HBV-induced liver cancer and may offer valuable
information for the development against liver cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
JL and LC designed the experiments. JL, YZ, LH and
HC performed the experiments. JC, YZ, LH, KH and JZ analyzed the
data. JL wrote the paper. KH, JZ and LC checked and finalized the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All protocols involving human samples were reviewed
and approved by the Ethical Review Board at The Affiliated Hospital
of Jinggangshan University (Jiangxi, China; ref. JGSU-20170071),
and written informed consent was provided from all the
participating patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huang J, Ye X, Guan J, Chen B, Li Q, Zheng
X, Liu L, Wang S, Ding Y, Ding Y and Chen L: Tiam1 is associated
with hepatocellular carcinoma metastasis. Int J Cancer. 132:90–100.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Balogh J, Victor D III, Asham EH,
Burroughs SG, Boktour M, Saharia A, Li X, Ghobrial RM and Monsour
HP Jr: Hepatocellular carcinoma: A review. J Hepatocell Carcinoma.
3:41–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ringelhan M, McKeating JA and Protzer U:
Viral hepatitis and liver cancer. Philos Trans R Soc Lond B Biol
Sci. 372:20160274. 2017.PubMed/NCBI
|
|
4
|
Fujiwara N, Friedman SL, Goossens N and
Hoshida Y: Risk factors and prevention of hepatocellular carcinoma
in the era of precision medicine. J Hepatol. 68:526–549. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levrero M and Zucman-Rossi J: Mechanisms
of HBV-induced hepatocellular carcinoma. J Hepatol. 64:S84–S101.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang S, Chen N, Chen Y, Sun L, Li L and
Liu H: Elevated GPC3 level promotes cell proliferation in liver
cancer. Oncol Lett. 16:970–976. 2018.PubMed/NCBI
|
|
7
|
Tian Y and Ou JH: Genetic and epigenetic
alterations in hepatitis B virus-associated hepatocellular
carcinoma. Virol Sin. 30:85–91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Trepo C, Chan HL and Lok A: Hepatitis B
virus infection. Lancet. 384:2053–2063. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sung WK, Zheng H, Li S, Chen R, Liu X, Li
Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al: Genome-wide
survey of recurrent HBV integration in hepatocellular carcinoma.
Nat Genet. 44:765–769. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buendia MA and Neuveut C: Hepatocellular
carcinoma. Cold Spring Harb Perspect Med. 5:a0214442015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murakami Y, Saigo K, Takashima H, Minami
M, Okanoue T, Bréchot C and Paterlini-Bréchot P: Large scaled
analysis of hepatitis B virus (HBV) DNA integration in HBV related
hepatocellular carcinomas. Gut. 54:1162–1168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lau CC, Sun T, Ching AK, He M, Li JW, Wong
AM, Co NN, Chan AW, Li PS, Lung RW, et al: Viral-human chimeric
transcript predisposes risk to liver cancer development and
progression. Cancer Cell. 25:335–349. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tropberger P, Mercier A, Robinson M, Zhong
W, Ganem DE and Holdorf M: Mapping of histone modifications in
episomal HBV cccDNA uncovers an unusual chromatin organization
amenable to epigenetic manipulation. Proc Natl Acad Sci USA.
112:E5715–E5724. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ringelhan M, O'Connor T, Protzer U and
Heikenwalder M: The direct and indirect roles of HBV in liver
cancer: Prospective markers for HCC screening and potential
therapeutic targets. J Pathol. 235:355–367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Menolfi D, Delamarre A, Lengronne A,
Pasero P and Branzei D: Essential roles of the Smc5/6 complex in
replication through natural pausing sites and endogenous DNA damage
tolerance. Mol Cell. 60:835–846. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang HC, Huang W, Lai MD and Su IJ:
Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and
hepatocarcinogenesis. Cancer Sci. 97:683–688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weber A, Boege Y, Reisinger F and
Heikenwälder M: Chronic liver inflammation and hepatocellular
carcinoma: Persistence matters. Swiss Med Wkly.
141:w131972011.PubMed/NCBI
|
|
18
|
Chao CC: Inhibition of apoptosis by
oncogenic hepatitis B virus X protein: Implications for the
treatment of hepatocellular carcinoma. World J Hepatol.
8:1061–1066. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang CS, Huang SM, Lin HH, Wu CC and Wang
CJ: Different expression of apoptotic proteins between HBV-
infected and non-HBV-infected hepatocellular carcinoma.
Hepatogastroenterology. 54:2061–2068. 2007.PubMed/NCBI
|
|
20
|
Fattovich G, Stroffolini T, Zagni I and
Donato F: Hepatocellular carcinoma in cirrhosis: Incidence and risk
factors. Gastroenterology. 127:S35–S50. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sunami Y, Ringelhan M, Kokai E, Lu M,
O'Connor T, Lorentzen A, Weber A, Rodewald AK, Müllhaupt B,
Terracciano L, et al: Canonical NF-κB signaling in hepatocytes acts
as a tumor-suppressor in hepatitis B virus surface antigen-driven
hepatocellular carcinoma by controlling the unfolded protein
response. Hepatology. 63:1592–1607. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Q, Wang X and Evers BM: Induction of
cIAP-2 in human colon cancer cells through PKC delta/NF-kappa B. J
Biol Chem. 278:51091–51099. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gill C, Dowling C, O'Neill AJ and Watson
RW: Effects of cIAP-1, cIAP-2 and XIAP triple knockdown on prostate
cancer cell susceptibility to apoptosis, cell survival and
proliferation. Mol Cancer. 8:392009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu HH, Wu JY, Cheng YW, Chen CY, Lee MC,
Goan YG and Lee H: cIAP2 upregulated by E6 oncoprotein via
epidermal growth factor receptor/phosphatidylinositol 3-kinase/AKT
pathway confers resistance to cisplatin in human papillomavirus
16/18-infected lung cancer. Clin Cancer Res. 16:5200–5210. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miura K, Karasawa H and Sasaki I: cIAP2 as
a therapeutic target in colorectal cancer and other malignancies.
Expert Opin Ther Targets. 13:1333–1345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang W, Hu K, Luo S, Zhang M, Li C, Jin
W, Liu Y, Griffin GE, Shattock RJ and Hu Q: Herpes simplex virus
type 2 infection of human epithelial cells induces CXCL9 expression
and CD4+ T cell migration via activation of
p38-CCAAT/enhancer-binding protein-β pathway. J Immunol.
188:6247–6257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yoneda M, Hyun J, Jakubski S, Saito S,
Nakajima A, Schiff ER and Thomas E: Hepatitis B virus and DNA
stimulation trigger a rapid innate immune response through NF-κB. J
Immunol. 197:630–643. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mrani S, Chemin I, Menouar K, Guillaud O,
Pradat P, Borghi G, Trabaud MA, Chevallier P, Chevallier M, Zoulim
F and Trépo C: Occult HBV infection may represent a major risk
factor of non-response to antiviral therapy of chronic hepatitis C.
J Med Virol. 79:1075–1081. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu K, He S, Xiao J, Li M, Luo S, Zhang M
and Hu Q: Interaction between herpesvirus entry mediator and HSV-2
glycoproteins mediates HIV-1 entry of HSV-2-infected epithelial
cells. J Gen Virol. 98:2351–2361. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu D, Zhu Y, Pang J, Weng X, Feng X and
Guo Y: Knockdown of long non-coding RNA MALAT1 inhibits growth and
motility of human hepatoma cells via modulation of miR-195. J Cell
Biochem. 119:1368–1380. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hong SY, Yoon WH, Park JH, Kang SG, Ahn JH
and Lee TH: Involvement of two NF-kappa B binding elements in tumor
necrosis factor alpha-, CD40-, and epstein-barr virus latent
membrane protein 1-mediated induction of the cellular inhibitor of
apoptosis protein 2 gene. J Biol Chem. 275:18022–18028. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Satoh T, Okamoto I, Miyazaki M, Morinaga
R, Tsuya A, Hasegawa Y, Terashima M, Ueda S, Fukuoka M, Ariyoshi Y,
et al: Phase I study of YM155, a novel survivin suppressant, in
patients with advanced solid tumors. Clin Cancer Res. 15:3872–3880.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shimizu T, Nishio K, Sakai K, Hayashi H,
Okamoto K, Takeda M, Iwasa T, Tanaka K, Aoyama K, Morishita M and
Nakagawa K: Phase I study of YM155, selective survivin suppressant
in combination with elrotnib in patients with EGFR-mutant advanced
non-small cell lung cancer. J Clin Oncol. 34:e205852016. View Article : Google Scholar
|
|
35
|
Papadopoulos KP, Lopez-Jimenez J, Smith
SE, Steinberg J, Keating A, Sasse C, Jie F and Thyss A: A
multicenter phase II study of sepantronium bromide (YM155) plus
rituximab in patients with relapsed aggressive B-cell Non-Hodgkin
lymphoma. Leuk Lymphoma. 57:1848–1855. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Benetatos CA, Mitsuuchi Y, Burns JM,
Neiman EM, Condon SM, Yu G, Seipel ME, Kapoor GS, Laporte MG,
Rippin SR, et al: Birinapant (TL32711), a bivalent SMAC mimetic,
targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB
activation, and is active in patient-derived xenograft models. Mol
Cancer Ther. 13:867–879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Amaravadi RK, Schilder RJ, Martin LP,
Levin M, Graham MA, Weng DE and Adjei AA: A phase I study of the
SMAC-mimetic birinapant in adults with refractory solid tumors or
lymphoma. Mol Cancer Ther. 14:2569–2575. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Crawford N, Salvucci M, Hellwig CT,
Lincoln FA, Mooney RE, O'Connor CL, Prehn JH, Longley DB and Rehm
M: Simulating and predicting cellular and in vivo responses of
colon cancer to combined treatment with chemotherapy and IAP
antagonist Birinapant/TL32711. Cell Death Differ. 25:1952–1966.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang SQ and Wu JG: Biological effects of
HBV X protein on hepatocellular carcinogenesis in association with
cellular factors. Virol Sin. 23:146–151. 2008. View Article : Google Scholar
|
|
40
|
Lee WY, Bachtiar M, Choo CCS and Lee CG:
Comprehensive review of Hepatitis B Virus-associated hepatocellular
carcinoma research through text mining and big data analytics. Biol
Rev Camb Philos Soc. 94:353–367. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li YW, Yang FC, Lu HQ and Zhang JS:
Hepatocellular carcinoma and hepatitis B surface protein. World J
Gastroenterol. 22:1943–1952. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu W, Lin YT, Yan XL, Ding YL, Wu YL,
Chen WN and Lin X: Hepatitis B virus core protein inhibits
Fas-mediated apoptosis of hepatoma cells via regulation of
mFas/FasL and sFas expression. FASEB J. 29:1113–1123. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tuttle RL, Gill NS, Pugh W, Lee JP,
Koeberlein B, Furth EE, Polonsky KS, Naji A and Birnbaum MJ:
Regulation of pancreatic beta-cell growth and survival by the
serine/threonine protein kinase Akt1/PKBalpha. Nat Med.
7:1133–1137. 2001. View Article : Google Scholar : PubMed/NCBI
|