Introduction
Colorectal cancer (CRC) is one of the most common
cancer types and the leading cause of cancer-associated mortality
globally (1,2). The current standard treatment for colon
cancer is surgery combined with chemotherapy. However, a proportion
of patients still suffer from local recurrence and remote
metastasis following surgery (3).
Furthermore, patients with similar clinical or pathological
conditions may exhibit unpredictable and diversified clinical
outcomes, even when treated in the same way. This phenomenon
reveals the limitation of the classic tumor-node-metastasis (TNM)
staging system (4). Reliable and
robust molecular markers, in addition to the current clinical and
pathological factors used for determining the risk of CRC
recurrence, are required to improve personalized therapy for
patients (5).
The development of bioinformatics and gene
expression profiling technologies provides additional opportunities
to characterize the molecular features of cancer. Gene-expression
profiling has been used to develop genomic tests that may provide
better predictions of clinical outcomes in combination with
traditional clinicopathologic factors (6). Although a number of studies have used
this method in CRC, to the best of our knowledge, this has not been
applied clinically (7). Therefore,
establishing a novel and more effective signature for assessing CRC
recurrence is urgently required.
Previously, emerging evidence has suggested that
tumor progression and recurrence are not only regulated by the
abnormal gene expression of cancer cells but also by the tumor
microenvironment (TME) factors, including the infiltration of a
number of immune cell subsets (8).
The expression of TME-associated genes, including chemokine and
inflammatory factors, affects the infiltration of immune cells in
TME, and is associated with the recurrence and survival of patients
with cancer (9).
In the present study, the association between immune
cell subtypes in CRC tumor microenvironment and recurrence-free
survival (RFS) time was evaluated. Subsequently, to explore the hub
genes that are associated with the immune cell subtypes in the TME,
weighted correlation network analysis, protein-protein interaction
network, univariate Cox and multivariate Cox analyses were
sequentially performed. Eventually, the influence of the degree of
DNA methylation of hub genes-associated CpG sites on their
expression was evaluated.
Materials and methods
Collection of gene expression
datasets
GSE39582 were downloaded from the Gene Expression
Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39582)
(10). GSE39582 includes 585
colorectal cancer samples and 19 non-tumoral tissue samples, which
were from a French cohort (11).
GSE71187 data were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71187),
which included a Chinese cohort with the mRNA expression profiles
of 52 human biopsy samples of CRC, and included overall survival
time information (12). The DNA
methylation profile, RNA-seq raw counts and clinical information of
234 patients with CRC was retrieved from The Cancer Genome Atlas
(TCGA) data portal using R package TCGAbiolinks (13).
Immune cell infiltration analysis
CIBERSORT (https://cibersort.stanford.edu/) (14) is an online analytical tool that is
used to provide an estimation of the abundance of a number of
immune cell sub-types in a mixed cell population using gene
expression data. LM22 (downloaded from http://media.nature.com/original/nature-assets/nmeth/journal/v12/n5/extref/nmeth.3337-S2.xls)
was used as the signature gene file for distinguishing 22 immune
cell types in CIBERSORT (14,15). The
GSE39582 expression mixture file was uploaded to the CIBERSORT
website. CIBERSORT was subsequently run online to calculate the
ratio of the 22 immune cell subsets, due to the mRNA expression of
immune signature genes in the cancer tissue of patients with CRC,
and non-tumoral tissue samples. Permutations was set to 1,000,
quantile normalization was used for GSE39582 gene chip data and
disable quantile normalization was used for TCGA CRC RNA-seq data
(14).
Recurrence-associated genes
screening
The genes combined with recurrence clinical traits,
which were significant in univariate Cox analysis, were identified
as recurrence-associated genes based on GSE39582. R package
survival (version 2.42–6; http://cran.r-project.org/web/packages/survival/index.html)
was used for univariate Cox analysis (16). P<0.05 was considered to indicate a
statistically significant difference. Hazard ratios (HRs) were used
to identify protective (HR<1) and risk genes (HR>1) (17).
Weighted correlation network analysis
(WGCNA)
The WGCNA package (version 1.63), in R, was used to
assess correlation patterns among genes, and identify modules of
highly correlated genes (18). WGCNA
was used in the present study to construct the co-expression
network for the recurrence-associated genes that were identified
using univariate Cox analysis, as described previously (17). β was a soft-thresholding parameter
that was able to emphasize strong correlations between genes and
penalize weak correlations (18,19). The
power of β=4 (scale free R2=0.8) was selected as the
soft-threshold to ensure a scale-free network. A cut height of 0.25
and minimum size of 50 were used to identify modules. Pearson's
correlation matrices were subsequently calculated between each
module and immune cell subset.
Enrichment analysis
R package clusterProfiler (version 3.9.1; http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
was used for Gene Ontology (GO) Biological Process and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analysis of the genes in modules that were identified using WGCNA
(20). P<0.05 was considered to
indicate a statistically significant difference.
Protein-protein interaction (PPI)
network construction
STRING (https://string-db.org/) (21) was used to construct the PPI networks
for distance-associated genes using the genes from modules
identified by WGCNA. The PPI networks were then exported from
STRING and imported into Cytoscape (22).
Hub gene identification
To identify the hub genes, all the listed genes were
ranked based on their degrees calculated in the PPI network and
co-expression network. The sum rank (Nsum rank=Nppi
rank+Nco-expression rank) was then used to
identify hub genes.
Gene set enrichment analysis
(GSEA)
Patients in GSE39582 and TCGA Colon Adenocarcinoma
(COAD) were divided into two groups (an IRF1 high-expression group
and an IRF1 low-expression group) based on the median of IRF1
expression in each dataset. The median of IRF1 expression was of
11.6 and 7.23 for TCGA COAD and GSE39582 dataset, respectively. R
package clusterProfiler was then used for GSEA to compare the
different KEGG pathways between the two groups (23).
Methylation analysis
As the GSE39582 dataset lacked methylation data, the
TCGA CRC Level 3 450K DNA methylation dataset was used for the
further analysis of DNA methylation. The acquired methylated sites
were annotated based on the GPL13534 (Illumina HumanMethylation450
BeadChip; Illumina, Inc.) annotation information. Pearson's
correlation coefficient was used to evaluate the relevance between
gene expression and the degree of methylated CpG sites (24). A Wilcoxon's test was used to select
the differentially methylated CpG sites between the two groups
(25). P<0.05 was considered to
indicate a statistically significant difference.
Statistical analysis
Statistical analysis was performed using R (version
3.5.3). The rank-sum t-test was used to test the differences
between two groups. A one-way analysis of variance was used to test
the differences among multiple groups, using the Tukey-honestly
significant difference test as the post-hoc test (26). R package survival (version 2.42–6)
was used for Kaplan-Meier survival analysis and the log-rank test.
R package corrplot (version 0.84) was used to calculate the
Pearson's correlation coefficient between genes and the ratio of
immune cells (27). P<0.05 was
considered to indicate a statistically significant difference
(28). Data are presented as median
± quartile for boxplot.
Results
Influence of immune cell infiltration
on the recurrence of patients with CRC
A previous study demonstrated that the mRNA
expression of immune signature genes may predict the ratio of a
number of immune cell subsets (15).
To investigate the function of each TME immune cell subset
infiltration in recurrence, CIBERSORT was used to calculate the
ratio of 22 immune cell subsets in TME, for each patient with CRC,
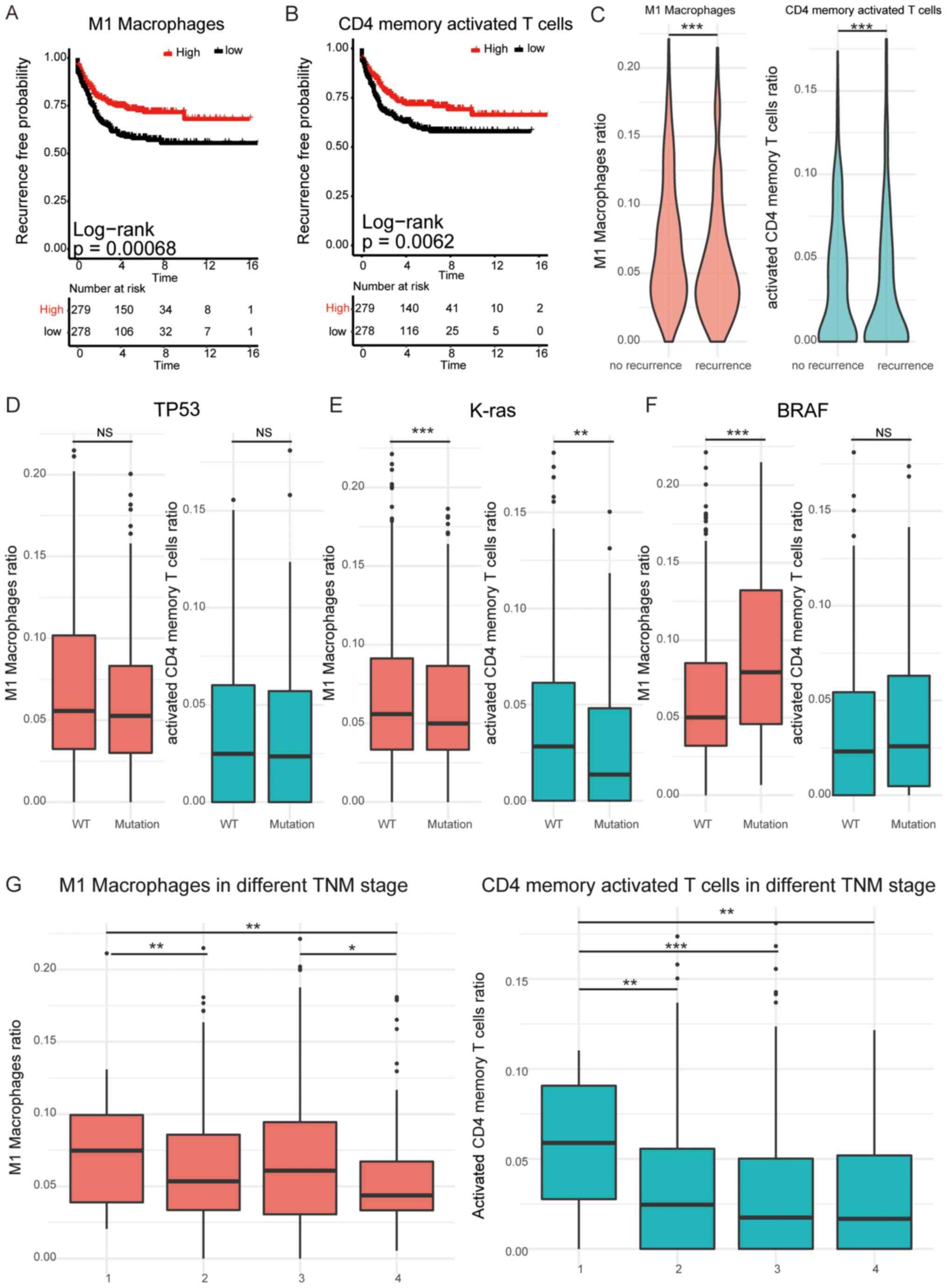
based on the mRNA expression data of immune signature genes. M1
macrophages and activated memory CD4+ T cells were
indicated to be protective factors and indicative of improved RFS
time (P<0.01; Fig. 1A and B). Due
to the LM22, the molecular pattern of M1 macrophages identification
included ADAMDEC1, AIF1, ALOX15, CCL13, CCL14, CCL18, CCL23, CCL8,
CD209, CD4, CD68, CFP, CHI3L1, CLEC10A, CLEC4A, CLIC2, CRYBB1,
EBI3, FAM198B, FES, FRMD4A, FZD2, GGT5, GSTT1, HRH1, HTR2B, MS4A6A,
NME8, NPL, P2RY13, PDCD1LG2, RENBP, SIGLEC1, SLC15A3, TLR8, TREM2
and WNT5B. The molecular pattern of activated memory
CD4+ T cells included CCL20, CD2, CD247, CD28, CD3D,
CD3G, CD40LG, CD6, CD7, CDC25A, CSF2, CTLA4, CXCL13, DPP4, GPR171,
GPR19, GZMB, ICOS, IFNG, IL12RB2, IL17A, IL26, IL2RA, IL3, IL4,
IL9, LAG3, LCK, LTA, NKG7, ORC1, PMCH, RRP9, SH2D1A, SKA1, TNFRSF4,
TNIP3, TRAC, TRAT1 and UBASH3A. The ratio of M1 macrophages and
activated CD4+ memory T cells was significantly lower in
patients with recurrence compared with patients with no recurrence
(P<0.001; Fig. 1C). Further
analysis revealed that the ratio of M1 macrophages and activated
CD4+ memory T cells demonstrated no significant
difference between patients with a TP53 mutation and wild-type (WT)
patients, but was significantly reduced in patients with the K-ras
mutation compared with WT patients (P<0.01; Fig. 1D and E). Additionally, the ratio of
M1 macrophages and activated CD4+ memory T cells was
significantly increased in patients with the B-Raf proto-oncogene,
serine/threonine kinase mutation compared with WT patients
(P<0.001; Fig. 1F). In comparison
with patients with advanced CRC, early-stage patients exhibited a
significantly higher ratio of M1 macrophages and activated memory
CD4+ T cells (P<0.05; Fig.
1G). These results revealed that the ratio of M1 macrophages
and activated memory CD4+ T cells in TME serve a vital
function in the recurrence of patients with CRC.
Identification of key modules
associated with immune cell infiltration
To investigate the factors that influenced the ratio
of M1 macrophages and activated memory CD4+ T cells, and
the response of recurrence in patients with CRC, 3,530
recurrence-associated genes were identified using univariate Cox
analysis, which may be used as independent factors for CRC.
Subsequently, based on the mRNA expression of recurrence-associated
genes, a co-expression network was constructed and key modules were
identified using WGCNA, which were used to aggregate genes with the
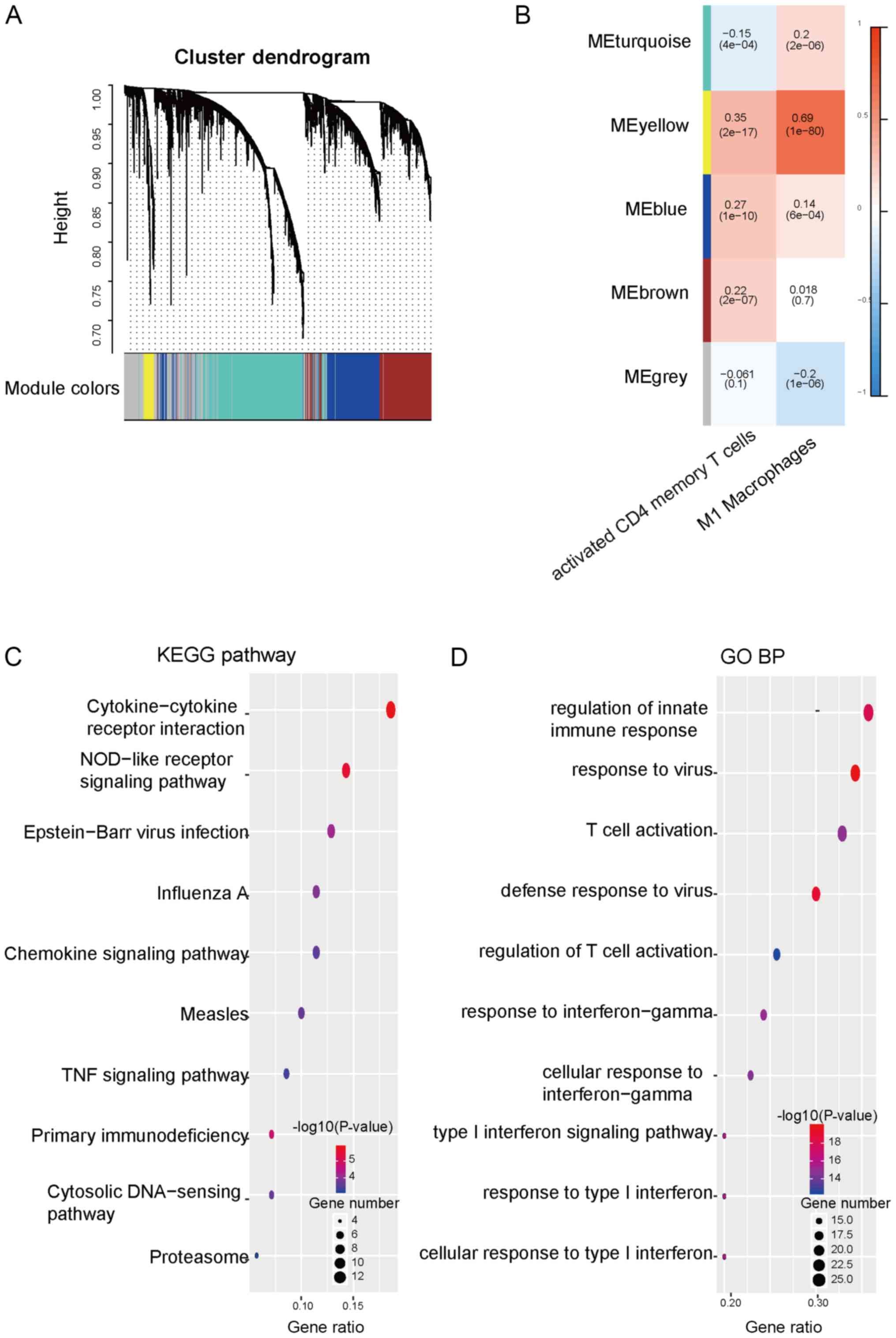
same expression pattern. The results revealed that the
recurrence-associated genes may be grouped into 4 major modules,
which were identified as blue, brown, turquoise and yellow modules
(Fig. 2A). The association between
the infiltration of every immune cell subset and the modules was
then assessed. The data revealed that the yellow module exhibited a
positive correlation with the ratio of M1 macrophages and the ratio
of activated memory CD4+ T cells (Fig. 2B). KEGG pathway and GO enrichment
analysis were then performed for the yellow module. KEGG pathway
enrichment analysis revealed that influenza A, Epstein-Bar virus
infection, NOD-like receptor signaling pathway and
cytokine-cytokine receptor interaction were enriched in the yellow
module (Fig. 2C). The results of the
GO enrichment analysis revealed that the yellow module was mainly
enriched in the cellular response to type I interferon, regulation
of innate immune response, T cell activation, defense response to
virus and response to virus (Fig.
2D). These data indicated that recurrence-associated genes
enriched in the yellow module may be involved in regulating the
infiltration or function of immune cells in TME.
Identified hub genes in the yellow
module influenced the RFS time of patients with CRC
Hub genes, which exhibited the highest degree and
greatest number of associations with other genes, serve a key
function in the yellow module (29).
To further investigate the biological functions of the yellow
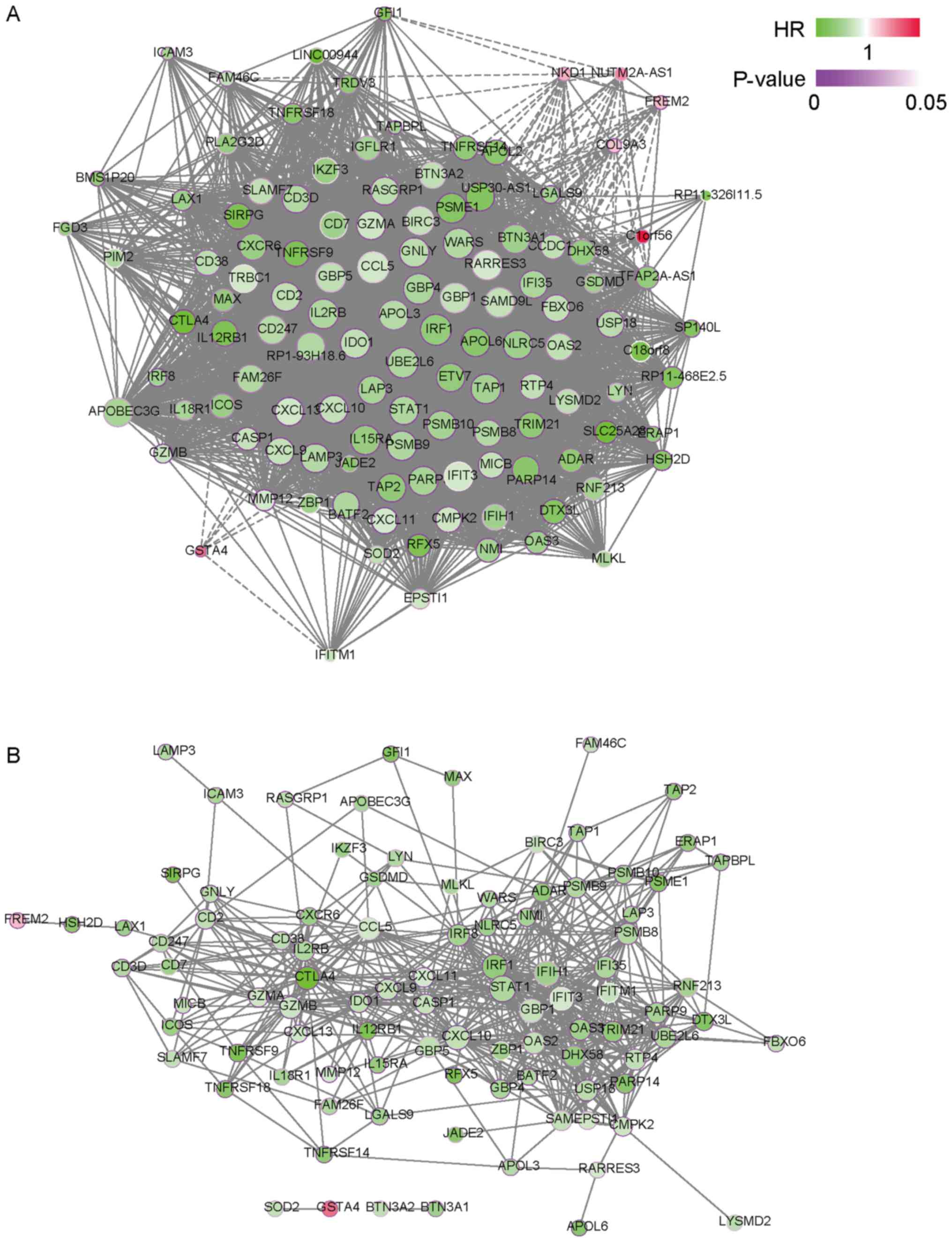
module, co-expression networks that were based on the WGCNA and PPI
network were constructed to identify hub genes in the yellow module
(Fig. 3A and B; Table SI). By ranking the degree of these
networks, IRF1, C-C motif chemokine ligand 5 (CCL5), ubiquitin
conjugating enzyme E2 L6 (UBE2L6), guanylate binding protein 1
(GBP1) and interleukin 2 receptor subunit beta (IL2RB) were
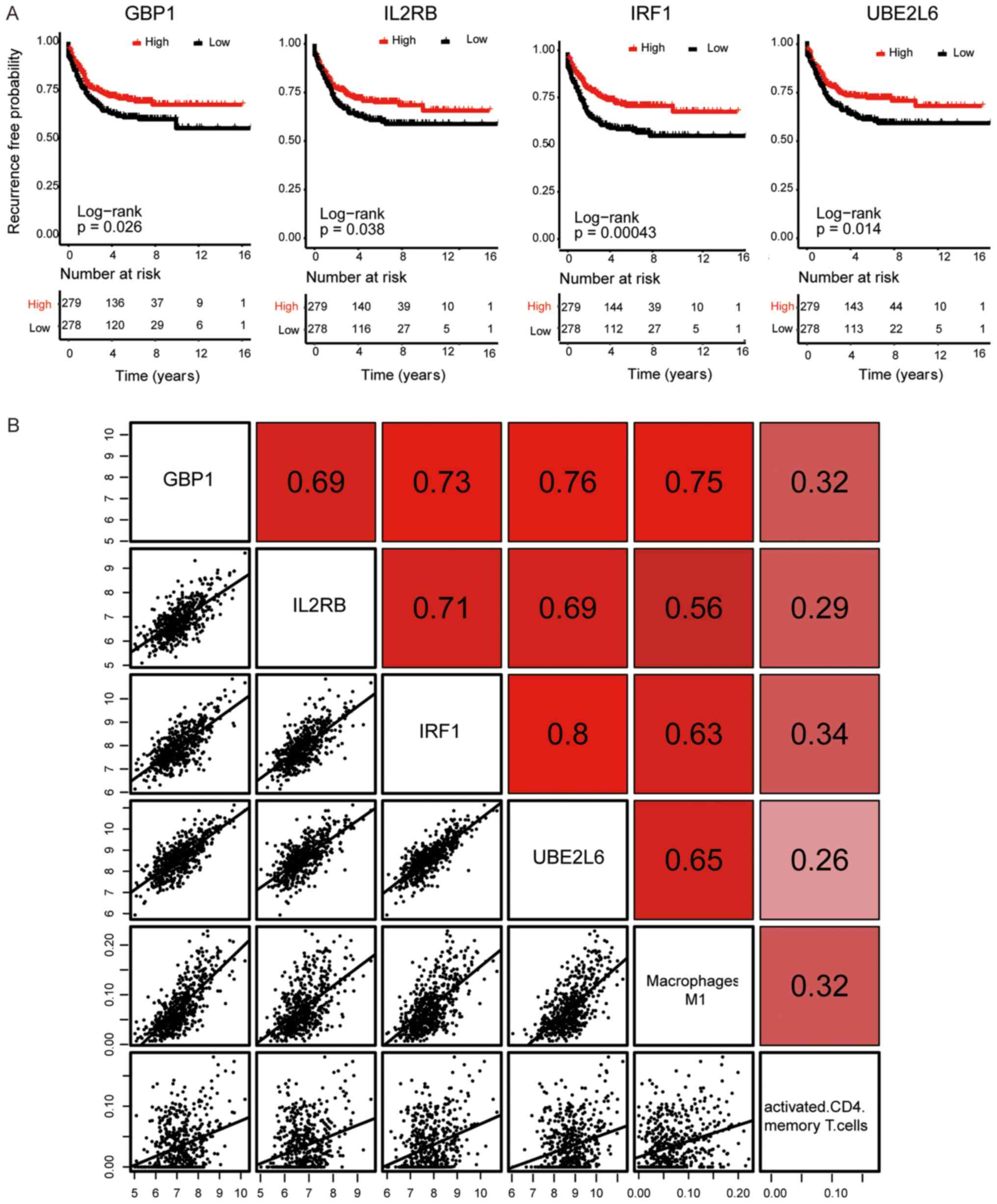
identified as the hub genes in the yellow module. Kaplan-Meier
curve and Log-rank analysis revealed that the expression of IRF1,
UBE2L6, GBP1 and IL2RB significantly influenced the RFS time of
patients with CRC (P<0.05; Fig.
4A). All 4 hub genes indicated a significant positive
correlation with each other, in addition to the ratio of M1
macrophages and activated memory CD4+ T cells (Fig. 4B).
IRF1 was able to predict the RFS time
of patients with CRC
Although all hub genes exhibited the potential to be
independent diagnostic biomarkers for the RFS time of patients with
CRC, IRF1 most significantly influenced this and was indicated to
be a potential diagnostic biomarker for RFS in patients with CRC
(P=0.001; Table I). TCGA data was
then used for further validation, and the results revealed that
patients with CRC and a lower expression of IRF1 exhibited a worse
prognosis compared with those with a higher expression of IRF1,
which is consistent with the results of the GSE39582 dataset
(P<0.01; Fig. 5A). Based on TCGA
data, IRF1 expression was also significantly positively correlated
with the ratio of M1 macrophages and activated memory
CD4+ T cells (P<0.01; Fig.
5B and C). An additional dataset from a Chinese cohort was also
used, which contained the mRNA expression profiles of 52 human
biopsy samples of CRC, and included overall survival time
information (12). The data revealed
that IRF1 expression in Chinese patients with CRC exhibited similar
results compared with those identified in the French CRC cohort.
Overall survival analysis indicated that Chinese patients with CRC
and high IRF1 expression exhibited a high survival probability,
although this was not significant. Patients with CRC and higher
IRF1 expression exhibited better survival time compared with
patients with lower IRF1 expression, and IRF1 expression
demonstrated a positive association with the ratio of M1
macrophages and activated memory CD4+ T cells, though
this was not statistically significant as it was a smaller sample
size (Fig. S1). Although age
exhibited a significant difference, there was no difference of all
other clinical variables between these two cohorts, including sex,
TNM stage, overall survival time and survival state (Table SII). This indicated that the
expression of IRF1 may predict the RFS of patients with CRC in
different cohorts, including Europeans, Americans and Asians.
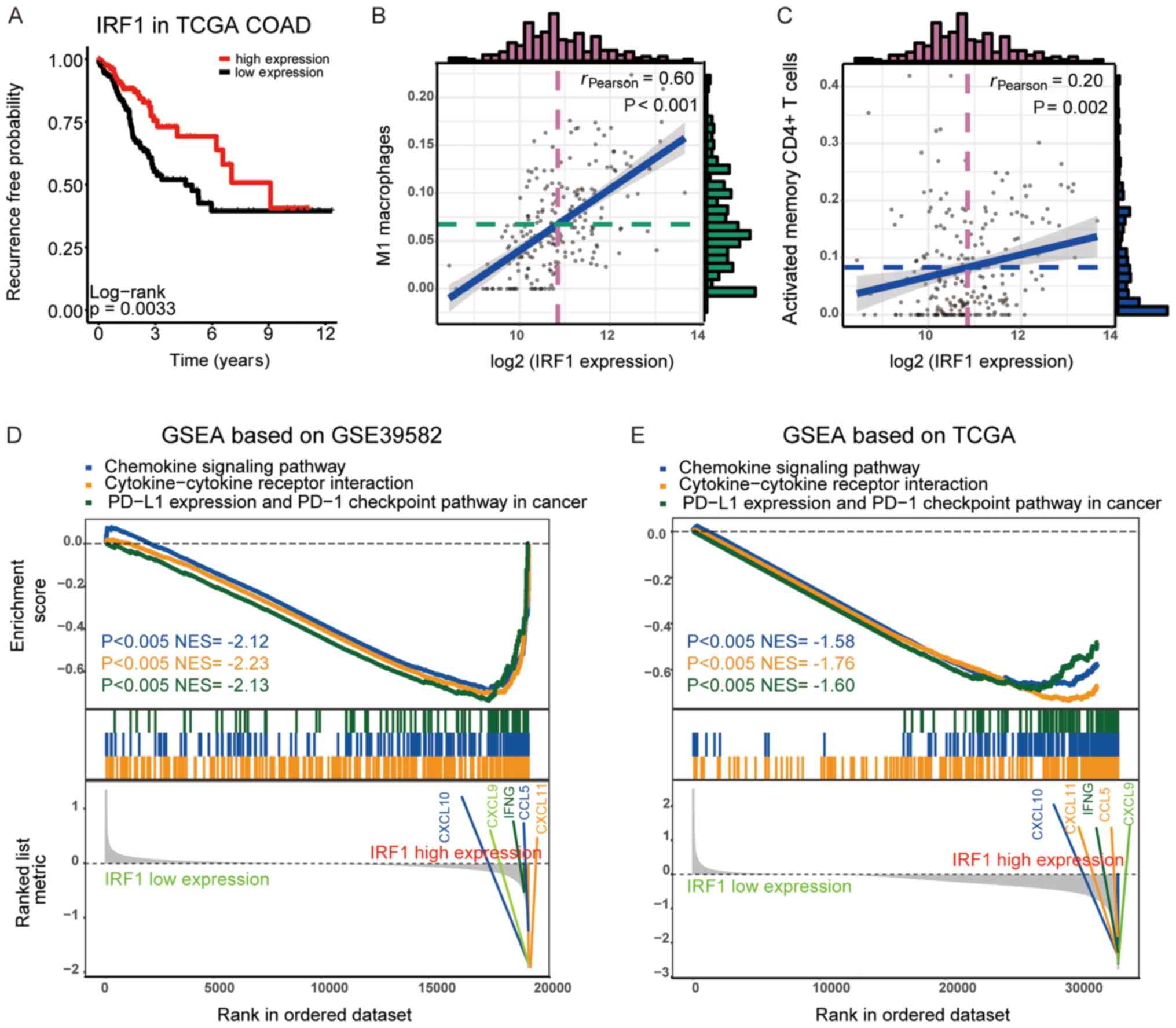 | Figure 5.Verification of IRF1 prognostic
signature using TCGA datasets and the GSEA of the DEGs between the
high- and low-expression IRF1 groups. (A) Kaplan Meier survival
plots of the association between recurrence-free survival and IRF1
expression in TCGA datasets. The association between IRF1
expression and the ratio of (B) M1 macrophages and (C) activated
CD4+ memory T cells in TCGA datasets. The histogram
exhibited the frequency in the value. The violet dotted line
represents the median value of log2 (IRF1 expression)
and the green dotted line represents the median value of the ratio
of (B) M1 macrophages or (C) activated CD4+ memory T
cells. The blue line revealed the line of best fit of the
association between the log2 (IRF1 expression) and cell
ratio of (B) M1 macrophages or (C) activated CD4+ memory
T cells. GSEA of the DEGs between the high- and low-expression IRF1
groups using (D) GSE39582 and (E) TCGA datasets. The P-value and
NES of different pathways were indicated by different colors,
respectively. IRF1, interferon regulatory factor 1; TCGA, The
Cancer Genome Atlas; GSEA, Gene set enrichment analysis; PD-1,
programmed cell death 1; PD-L1, programmed death ligand 1; CXCL,
C-X-C motif chemokine ligand; IFNG, interferon γ; CCL5, C-C motif
chemokine ligand 5; DEG, differentially expressed gene; NES,
normalized enrichment score; COAD, colon adenocarcinoma. |
 | Table I.Univariate Cox analyses and
multivariate Cox analysis of 4 hub genes. |
Table I.
Univariate Cox analyses and
multivariate Cox analysis of 4 hub genes.
|
| Univariate Cox
analysis | Multivariate Cox
analysis |
|---|
|
|
|
|
|---|
| Gene | 95% CI | P-value | HR | 95% CI | P-value | HR |
|---|
| GBP1 | 0.821 | 0.683–0.987 | 0.035a |
|
| >0.05 |
| IL2RB | 0.741 | 0.59–0.93 | 0.01a |
|
| >0.05 |
| UBE2L6 | 0.73 | 0.604–0.881 | 0.001a |
|
| >0.05 |
| IRF1 | 0.618 | 0.497–0.769 |
<0.001a | 0.527 | 0.359–0.772 | 0.001a |
Patients from GSE39582 and TCGA were divided into
two groups according to the median IRF1 expression. GSEA was used
to investigate the different KEGG pathways between these groups,
and the results demonstrated that pathways associated with immune
cell recruitment and activation, including the chemokine signaling
pathway, cytokine-cytokine receptor interaction and programmed
death ligand 1 expression and programmed cell death 1 checkpoint
pathways in cancer, were upregulated in the IRF1 high-expression
group. A number of chemokines, including C-X-C motif chemokine
ligand (CXCL) 9–11 and CCL5, were highly expressed in the IRF1
high-expression group. These results indicated that a higher
expression of IRF may reduce the risk of recurrence through
influencing the recruitment and activation of immune cells,
particularly M1 macrophages and activated memory CD4+ T
cells.
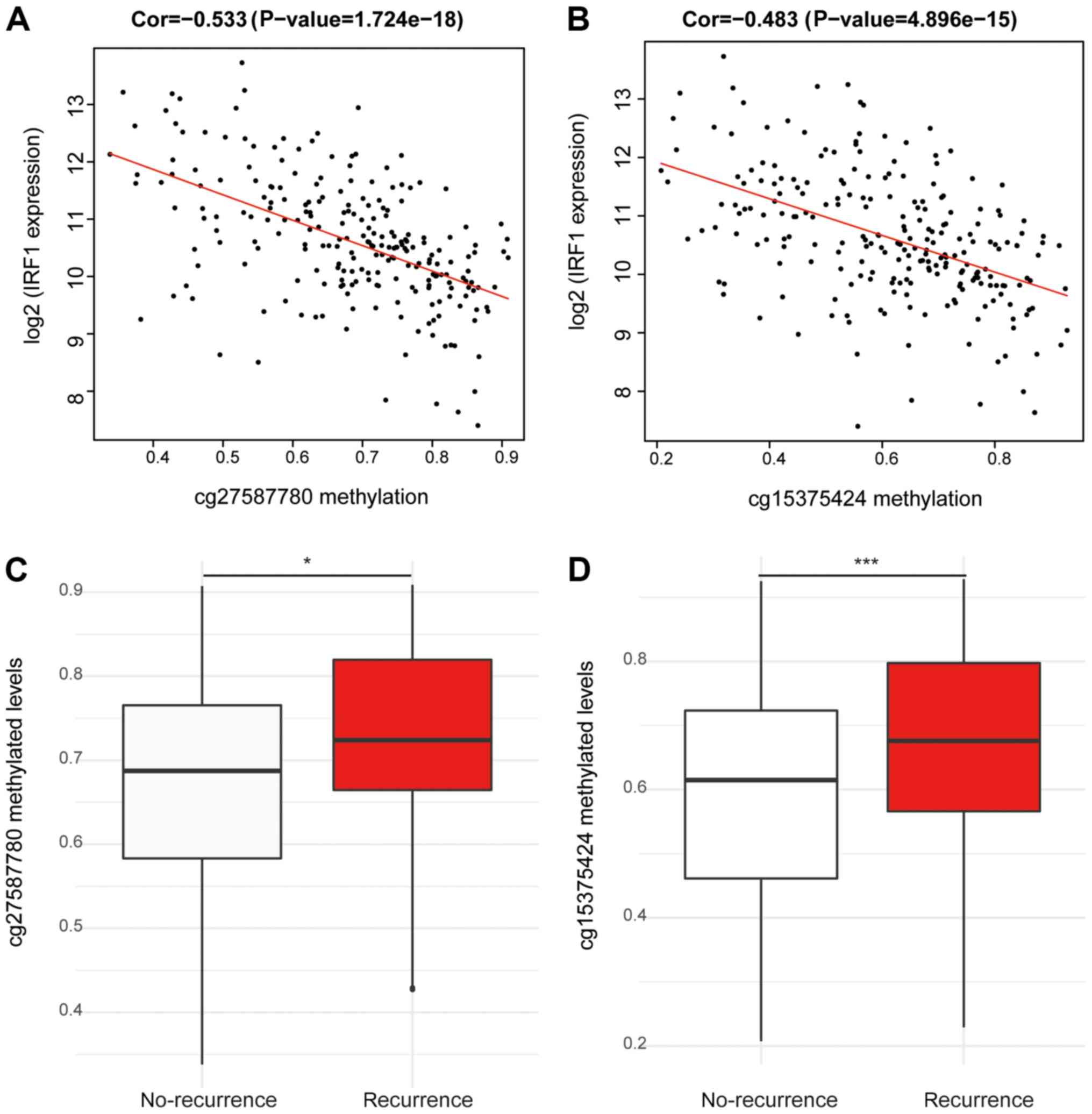
DNA methylation levels of two CpG
sites (cg27587780 and cg15375424) are negatively correlated with
the expression of IRF1 and positively correlated with the
recurrence of patients with CRC
To investigate the aberrant expression of IRF1, the
association between the degree of DNA methylation of
IRF1-associated methylated CpG sites and the expression of IRF1
were analyzed based on TCGA dataset. The results indicated that two
CpG sites, cg27587780 and cg15375424, were significant negatively
correlated with IRF1 expression (P<0.001; Fig. 6A and B). These two methylated CpG
sites were hypermethylated in patients with CRC recurrence compared
with patients with CRC without recurrence (P<0.05; Fig. 6C and D).
Discussion
Colorectal cancer is the third leading cause of
cancer-associated mortality globally (1,2).
Although the majority of patients with CRC are treated using
surgery combined with chemotherapy, local recurrence and remote
metastases following therapy often influence their survival rate
(30). At present, there is a lack
of effective biomarkers for CRC recurrence. TME, particularly
immune cell infiltration, serves an important function in affecting
the metastasis and recurrence of patients with CRC (31). Therefore, investigating the
mechanisms of TME and TME-associated factors may be useful to
identify novel biomarkers for CRC prognosis and may provide more
effective, target-specific or personalized therapeutic strategies
for patients with CRC.
In the present study, based on the data from
GSE39582, M1 macrophages and activated memory CD4+ T
cells were indicated to be protective factors for the survival of
patients with CRC. Patients with a high score of M1 macrophages and
activated memory CD4+ T cells exhibited a lower risk of
recurrence. M1 macrophages and activated memory CD4+ T
cells have been confirmed as tumor-preventing cells in CRC, but
factors affecting the distribution of these cells in TME remain
unclear (32,33). The results of the present study
revealed that a module, which contained 120 genes from
recurrence-associated genes in CRC, was positively correlated with
these two immune cell subsets in TME. In this module, the top 5 hub
genes were identified to be IRF1, CCL5, UBE2L6, GBP1 and IL2RB.
Further analysis using a Kaplan-Meier curve and
Log-rank test revealed that all top 5 hub genes, except CCL5,
influenced the RFS of patients with CRC. IRF1, which belongs to the
IRF family (34), is weakly
expressed in resting dendritic cells and macrophages, but is
induced by interferon-γ (IFN-γ) in M1 polarized macrophages
(35,36). Previous studies have revealed that
IRF1 inhibited the proliferation and metastasis of CRC (37,38).
UBE2L6, which is also known as UBCH8, promotes apoptosis in
cervical cancer cells (39). In the
present study, the low expression of UBE2L6 was associated with a
poor prognosis in patients with CRC. GBP-1 is highly induced by
IFN-γ in a number of different cell types, and functions as a tumor
suppressor, which arrests tumor evasion in CRC (40,41).
IL2RB, which is a receptor on numerous different effector immune
cells of interleukin-2, promotes antitumor immunity (42). All aforementioned reported results
are consistent with the results of the present study, which
indicated that patients with CRC and a higher expression of IRF1,
UBE2L6, GBP1 or IL2RB exhibited better RFS time.
Multivariate Cox analysis revealed that IRF1 may be
a diagnostic biomarker for RFS in patients with CRC among these
genes, which was also validated using TCGA datasets and a Chinese
cohort. In aggressive neuroblastoma, a previous study revealed that
IRF1 and nuclear factor-κB restored MHC class I-restricted tumor
antigen processing and presentation to cytotoxic T cells (43). A tumor-derived exosome, which was
induced by IRF-1 overexpression, enhanced the anti-tumor immune
response (44). IRF1 expression in
tumor cells was also reported to be critical for the immune
response to adoptive T cell therapy (45) and the antitumor immunity of
cyclophosphamide (46).
Consistently, the results of the present study revealed that IRF1
expression was correlated with the expression of CXCL9, CXCL10,
CXCL11 and IFNG, which have been demonstrated to influence
macrophage infiltration and memory CD4+ T cell
activation (47,48). The increased predictive ability of
IRF1 mRNA expression may be due to the fact IRF1 is able to
regulate immune subsets infiltration into the TME of CRC.
Epigenetic gene silencing that is caused by DNA methylation has
been widely accepted as a major mechanism of tumor recurrence
(49). IRF4, IRF5 and IRF8 have been
reported to be frequently suppressed in gastric cancer due to DNA
methylation (50). In the present
study, two CpG sites, cg27587780 and cg15375424, in the IRF1 DNA
region, were demonstrated to be significantly negatively correlated
with IRF1 expression. This result may explain the abnormal IRF1
expression.
Collectively, the expression of IRF1 may predict the
RFS of patients with CRC in different cohorts, which may be due to
IRF1 serving a function in regulating the ratio of M1 macrophages
and activated memory CD4+ T cells. This result may be
considered useful information for use in treatment or immunotherapy
in clinical practice.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant. no. 81372272) and the
Provincial Natural Science Foundation of Tibet (grant. no.
XZ2019ZRG-135).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YW devised the concept, designed experiments,
analyzed data, and wrote the manuscript; SZ analyzed data; JY
devised the concept, designed the research, supervised the study,
and revised the paper. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This research abides by the International and
National regulations in accordance with the Declaration of
Helsinki. It was approved by the Ethics Committee of the Fourth
People's Hospital of Shaanxi (Shanxi, China).
Patient consent for publication
Not applicable.
Competing interests
The authors have declared that they have no
competing interests.
Glossary
Abbreviations
Abbreviations:
|
CRC
|
colorectal cancer
|
|
WGCNA
|
weighted gene co-expression network
analysis
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
COAD
|
Colon Adenocarcinoma
|
|
GEO
|
Gene Expression Omnibus
|
|
BP
|
biological process
|
|
FDR
|
false discovery rate
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TNM
|
Tumor-Node-Metastasis
|
|
RFS
|
recurrence-free survival
|
|
TME
|
tumor microenvironment
|
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baca B, Beart RW Jr and Etzioni DA:
Surveillance after colorectal cancer resection: A systematic
review. Dis Colon Rectum. 54:1036–1048. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Inadomi JM: Screening for colorectal
neoplasia. N Engl J Med. 376:1599–1600. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee JS: Exploring cancer genomic data from
the cancer genome atlas project. BMB Rep. 49:607–611. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yip S, Christofides A, Banerji S, Downes
MR, Izevbaye I, Lo B, MacMillan A, McCuaig J, Stockley T, Yousef GM
and Spatz A: A Canadian guideline on the use of next-generation
sequencing in oncology. Curr Oncol. 26:e241–e254. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun D, Chen J, Liu L, Zhao G, Dong P, Wu
B, Wang J and Dong L: Establishment of a 12-gene expression
signature to predict colon cancer prognosis. PeerJ. 6:e49422018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee JH, Jung S, Park WS, Choe EK, Kim E,
Shin R, Heo SC, Lee JH, Kim K and Chai YJ: Prognostic nomogram of
hypoxia-related genes predicting overall survival of colorectal
cancer-analysis of TCGA database. Sci Rep. 9:18032019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yan X, Jiao SC, Zhang GQ, Guan Y and Wang
JL: Tumor-associated immune factors are associated with recurrence
and metastasis in non-small cell lung cancer. Cancer Gene Ther.
24:57–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marisa L, de Reynies A, Duval A, Selves J,
Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D,
Ayadi M, et al: Gene expression classification of colon cancer into
molecular subtypes: Characterization, validation, and prognostic
value. PLoS Med. 10:e10014532013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
An N, Shi X, Zhang Y, Lv N, Feng L, Di X,
Han N, Wang G, Cheng S and Zhang K: Discovery of a novel immune
gene signature with profound prognostic value in colorectal cancer:
A model of cooperativity disorientation created in the process from
development to cancer. PLoS One. 10:e01371712015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mounir M, Lucchetta M, Silva TC, Olsen C,
Bontempi G, Chen X, Noushmehr H, Colaprico A and Papaleo E: New
functionalities in the TCGAbiolinks package for the study and
integration of cancer data from GDC and GTEx. PLoS Comput Biol.
15:e10067012019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen B, Khodadoust MS, Liu CL, Newman AM
and Alizadeh AA: Profiling tumor infiltrating immune cells with
CIBERSORT. Methods Mol Biol. 1711:243–259. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Q, Su YL and Shen WX: A novel
prognostic signature of seven genes for the prediction in patients
with thymoma. J Cancer Res Clin Oncol. 145:109–116. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Y, Lu Q, Shao X, Mo B, Nie X, Liu W,
Chen X, Tang Y, Deng Y and Yan J: Development of A three-gene
prognostic signature for hepatitis B virus associated
hepatocellular carcinoma based on integrated transcriptomic
analysis. J Cancer. 9:1989–2002. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan L, Zeng G, Chen L, Wang G and Wang X,
Cao X, Lu M, Liu X, Qian G, Xiao Y and Wang X: Identification of
key genes and pathways in human clear cell renal cell carcinoma
(ccRCC) by co-expression analysis. Int J Biol Sci. 14:266–279.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47((D1)): D607–D613.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu G, Wang LG, Han Y and He QY:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu Z, Jin Z, Deng Y, Wei L, Yuan X, Zhang
M and Sun D: Co-expression network analysis identifies four hub
genes associated with prognosis in soft tissue sarcoma. Front
Genet. 10:372019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Zhou S, Li S, Jiang Y, Wan Y, Ma X
and Cheng W: Eleven genes associated with progression and prognosis
of endometrial cancer (EC) identified by comprehensive
bioinformatics analysis. Cancer Cell Int. 19:1362019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gu S, Lin S, Ye D, Qian S, Jiang D, Zhang
X, Li Q, Yang J, Ying X, Li Z, et al: Genome-wide methylation
profiling identified novel differentially hypermethylated biomarker
MPPED2 in colorectal cancer. Clin Epigenetics. 11:412019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Turcan S, Rohle D, Goenka A, Walsh LA,
Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al: IDH1
mutation is sufficient to establish the glioma hypermethylator
phenotype. Nature. 483:479–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun X, Han Q, Luo H, Pan X, Ji Y, Yang Y,
Chen H, Wang F, Lai W, Guan X, et al: Profiling analysis of long
non-coding RNAs in early postnatal mouse hearts. Sci Rep.
7:434852017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pesenti C, Navone SE, Guarnaccia L,
Terrasi A, Costanza J, Silipigni R, Guarneri S, Fusco N, Fontana L,
Locatelli M, et al: The genetic landscape of human glioblastoma and
matched primary cancer stem cells reveals intratumour similarity
and intertumour heterogeneity. Stem Cells Int. 2019:26170302019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wen Q, Yang Y, Chen XH, Pan XD, Han Q,
Wang D, Deng YC, Li XH, Yan J and Zhou JH: Competing endogenous RNA
screening based on long noncoding RNA-messenger RNA co-expression
profile in Hepatitis B virus-associated hepatocarcinogenesis. J
Tradit Chin Med. 37:510–521. 2017. View Article : Google Scholar
|
|
29
|
Chen P, Wang F, Feng J, Zhou R, Chang Y,
Liu J and Zhao Q: Co-expression network analysis identified six hub
genes in association with metastasis risk and prognosis in
hepatocellular carcinoma. Oncotarget. 8:48948–48958.
2017.PubMed/NCBI
|
|
30
|
Zhang Y, Chen Z and Li J: The current
status of treatment for colorectal cancer in China: A systematic
review. Medicine (Baltimore). 96:e82422017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tosolini M, Kirilovsky A, Mlecnik B,
Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH,
Pages F and Galon J: Clinical impact of different classes of
infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in
patients with colorectal cancer. Cancer Res. 71:1263–1271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deschoolmeester V, Baay M, Lardon F,
Pauwels P and Peeters M: immune cells in colorectal cancer:
Prognostic relevance and role of MSI. Cancer Microenviron.
4:377–392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Edin S, Wikberg ML, Dahlin AM, Rutegard J,
Oberg A, Oldenborg PA and Palmqvist R: The distribution of
macrophages with a M1 or M2 phenotype in relation to prognosis and
the molecular characteristics of colorectal cancer. PLoS One.
7:e470452012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alsamman K and El-Masry OS: Interferon
regulatory factor 1 inactivation in human cancer. Biosci Rep.
38(pii): BSR201716722018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xie C, Liu C, Wu B, Lin Y, Ma T, Xiong H,
Wang Q, Li Z, Ma C and Tu Z: Effects of IRF1 and IFN-β interaction
on the M1 polarization of macrophages and its antitumor function.
Int J Mol Med. 38:148–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang C, Lewis C, Borg NA, Canals M, Diep
H, Drummond GR, Goode RJ, Schittenhelm RB, Vinh A, Zhu M, et al:
Proteomic identification of interferon-induced proteins with
tetratricopeptide repeats as markers of M1 macrophage polarization.
J Proteome Res. 17:1485–1499. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gunthner R and Anders HJ:
Interferon-regulatory factors determine macrophage phenotype
polarization. Mediators Inflamm. 2013:7310232013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hong M, Zhang Z, Chen Q, Lu Y, Zhang J,
Lin C, Zhang F, Zhang W and Li X, Zhang W and Li X: IRF1 inhibits
the proliferation and metastasis of colorectal cancer by
suppressing the RAS-RAC1 pathway. Cancer Manag Res. 11:369–378.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Q, Qiao L, Wang X, Ding C and Chen
JJ: UHRF1 epigenetically down-regulates UbcH8 to inhibit apoptosis
in cervical cancer cells. Cell Cycle. 17:300–308. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Britzen-Laurent N, Lipnik K, Ocker M,
Naschberger E, Schellerer VS, Croner RS, Vieth M, Waldner M,
Steinberg P, Hohenadl C and Stürzl M: GBP-1 acts as a tumor
suppressor in colorectal cancer cells. Carcinogenesis. 34:153–162.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Britzen-Laurent N, Herrmann C, Naschberger
E, Croner RS and Sturzl M: Pathophysiological role of
guanylate-binding proteins in gastrointestinal diseases. World J
Gastroenterol. 22:6434–6443. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Malek TR and Castro I: Interleukin-2
receptor signaling: At the interface between tolerance and
immunity. Immunity. 33:153–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lorenzi S, Forloni M, Cifaldi L, Antonucci
C, Citti A, Boldrini R, Pezzullo M, Castellano A, Russo V, van der
Bruggen P, et al: IRF1 and NF-kB restore MHC class I-restricted
tumor antigen processing and presentation to cytotoxic T cells in
aggressive neuroblastoma. PLoS One. 7:e469282012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang MQ, Du Q, Varley PR, Goswami J, Liang
Z, Wang R, Li H, Stolz DB and Geller DA: Interferon regulatory
factor 1 priming of tumour-derived exosomes enhances the antitumour
immune response. Br J Cancer. 118:62–71. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cascone T, McKenzie JA, Mbofung RM, Punt
S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al:
Increased tumor glycolysis characterizes immune resistance to
adoptive t cell therapy. Cell Metab. 27:977–987.e4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Buccione C, Fragale A, Polverino F,
Ziccheddu G, Arico E, Belardelli F, Proietti E, Battistini A and
Moschella F: Role of interferon regulatory factor 1 in governing
Treg depletion, Th1 polarization, inflammasome activation and
antitumor efficacy of cyclophosphamide. Int J Cancer. 142:976–987.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhuang J, Shan Z, Ma T, Li C, Qiu S, Zhou
X, Lin L and Qi Z: CXCL9 and CXCL10 accelerate acute transplant
rejection mediated by alloreactive memory T cells in a mouse
retransplantation model. Exp Ther Med. 8:237–242. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Corbera-Bellalta M, Planas-Rigol E, Lozano
E, Terrades-García N, Alba MA, Prieto-González S, García-Martínez
A, Albero R, Enjuanes A, Espígol-Frigolé G, et al: Blocking
interferon γ reduces expression of chemokines CXCL9, CXCL10 and
CXCL11 and decreases macrophage infiltration in ex vivo cultured
arteries from patients with giant cell arteritis. Ann Rheum Dis.
75:1177–1186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nassiri F, Mamatjan Y, Suppiah S,
Badhiwala JH, Mansouri S, Karimi S, Saarela O, Poisson L,
Gepfner-Tuma I, Schittenhelm J, et al: DNA methylation profiling to
predict recurrence risk in meningioma: Development and validation
of a nomogram to optimize clinical management. Neuro Oncol. Jan
3–2019.(Epub ahead of print). View Article : Google Scholar
|
|
50
|
Yamashita M, Toyota M, Suzuki H, Nojima M,
Yamamoto E, Kamimae S, Watanabe Y, Kai M, Akashi H, Maruyama R, et
al: DNA methylation of interferon regulatory factors in gastric
cancer and noncancerous gastric mucosae. Cancer Sci. 101:1708–1716.
2010. View Article : Google Scholar : PubMed/NCBI
|