Introduction
Hepatocellular carcinoma (HCC) is one of the most
challenging health problems worldwide. Currently, HCC is the sixth
most commonly diagnosed cancer and the fourth leading cause of
cancer-related death globally (1).
Although HCC is more prevalent in Asian and African nations,
important evidence indicates that the incidence of HCC is rising in
developed countries (1). The main
risk factors for HCC development are chronic infection with
hepatitis B virus (HBV) or hepatitis C virus (HCV), alcohol abuse,
and metabolic syndrome, including type 2 diabetes and non-alcoholic
steatohepatitis (NASH) (2). Other
cofactors, such as smoking and aflatoxin-contaminated food supplies
are well-characterized contributors to HCC (1–3). Over
the past decade, advances in genomic research have increased our
knowledge of HCC molecular pathogenesis. However, the exact
molecular mechanisms underlying the development of HCC are still
unclear. Each HCC appears to be characterized by a distinctive
pattern of somatic mutations in passenger and driver genes that
accumulate overtime (4). Recent
studies performing whole-genome or whole-exome sequencing have
identified specific somatic mutations in driver genes that appear
to contribute to tumor initiation and progression (4–6). The
most frequently detected mutations affect the catalytic subunit of
telomerase holoenzyme, the telomerase reverse transcriptase (TERT).
These mutations are detected in 44–65% of HCC (7,8),
representing the most frequent somatic genetic alterations in human
HCC (9). TERT promoter
mutations are associated with an increased expression of
telomerase, which allow cells to acquire the ability to overcome
senescence and to become immortal (10). TERT mutations can be found in
preneoplastic lesions and in early-stage HCCs (11,12).
Thus, TERT promoter mutations correlate with tumor
initiation, while mutations in other genes, such as TP53,
CTNNB1, are associated with later stages of HCC progression,
causing further genomic modification (12). It is of note that HBV and HCV
infections have different impacts on the TERT gene (8,9).
TERT promoter mutations have been more frequently found in
HCV-related and alcohol intake-related HCC than in HBV-related HCC
(9,13,14),
where induction of telomerase overexpression is in some cases due
to integration of HBV DNA sequences into the TERT promoter
(8,15). Other driver genes frequently mutated
in HCC belong to key signaling pathways of oncogenesis as the
WNT/β-catenin pathway and the P53 cell cycle pathway (5,6). Indeed,
somatic mutations in CTNNB1 (coding for β-catenin) and in
TP53 tumor suppressor gene are recurrently detected in HCCs.
Mutations in the TP53 gene are detected in 12–48% of HCC,
with high frequency in advanced tumors (6,16). The
mutational spectrum in this gene has shown a strong molecular
association between environmental carcinogen exposure and cancer.
In Asia and Africa the increased risk of HCC is particularly
related to the dietary intake of aflatoxin B1 (AFB1), a fungal
mycotoxin, which contaminates foods that may act in synergy with
chronic HBV infection. Exposure to AFB1 induces G: C to T: A
transversions at the third base in codon 249 of TP53 leading to the
R249S substitution (17,18). No other specific recurrent TP53
hotspot somatic mutations outside the R249S mutation have been
identified in HCC (16). Concerning
CTNNB1, somatic mutations in this gene have been found in
11–37% of HCC samples (6,16,19).
CTNNB1 mutations can be nucleotide substitutions or in-frame
deletions in the consensus site targeted by the APC/AXIN1/GSK3B
inhibitory complex (6,19,20). As
a consequence this leads to the impairment of β-catenin degradation
and Wnt signaling activation, which promotes cell motility,
de-differentiation, and proliferation (20). CTNNB1 and TP53
mutations are frequently mutually exclusive, whereas CTNNB1
mutations are associated with TERT mutations (11,21).
Though recurrent somatic mutations in the TERT promoter
region, in the exon 3 of CTNNB1 gene, and in TP53
gene have been recognized as common events in HCC they show
variable frequencies in different geographic areas, depending on
liver disease etiology and environmental factors (6,9,14,16,18,22).
In this study, we investigated the presence of
CTNNB1, TP53, and TERT promoter mutations in paired
tumor and non-tumor liver specimens from a cohort of HCC patients
from Southern Italy, a geographic region with a high incidence of
liver cancer (23–25).
Patients and methods
Patients
Frozen tumor and non-tumor liver specimens from 67
HCC patients (47 males and 20 females; mean age, 66.4±9 years) were
analysed. Additionally, we studied frozen liver specimens from 41
control patients (19 males and 22 females; mean age, 49.2±13.1
years) with morbid obesity that underwent bariatric surgery and
whose liver histology showed the presence of non-alcoholic fatty
liver (NAFL) with no sign of steatohepatitis and fibrosis (26). The choice of a control group, which
included people with simple hepatic steatosis and no sign of
hepatic injury was due to the fact that patients who usually
undergo liver biopsy are those that frequently show severe chronic
liver disease, and as demonstrated by Nault et al (11), these patients may have already
developed TERT promoter mutations in the liver. Indeed, TERT is
considered as the earliest genomic event currently identified in
the multistep process of liver carcinogenesis on cirrhosis. Forty
(59.7%) of the 67 patients with HCC had HCV- and 3 (4.5%) had
HBV-related liver diseases. Among the other 24 patients, 2 had
alcohol-related liver disease and 22 had cryptogenic liver disease.
Patients' characteristics are shown in Table I. Tumor and non-tumor liver tissues
were obtained by surgical resection. Similarly, tissue specimens
from obese subjects were obtained by bariatric surgery. Each liver
specimen was stored in an appropriate volume of RNAlater RNA
stabilization reagent (Ambion) at −80°C. The study protocol was
approved by the Ethics Committee of the Messina University
Hospital, and written informed consent was obtained from all
patients.
 | Table I.Demographic, clinical, and genetic
characteristics of the studied patients with HCC and the control
subjects. |
Table I.
Demographic, clinical, and genetic
characteristics of the studied patients with HCC and the control
subjects.
|
Characteristics | Patients with HCC
(n=67) | Control subjects
(n=41) | P-value |
|---|
| Age, years | 66.4 (±9) | 49.2 (±13.11) | <0.0001 |
| Sex,
Male/Female | 47/20 | 19/22 | 0.013 |
| Etiology |
|
|
|
|
HCV | 39/67 (58.2%) | 0/41 (0%) |
|
|
HBV | 3/67 (4.5%) | 0/41 (0%) |
|
| HCV +
HBV | 1/67 (1.49%) | 0/41 (0%) |
|
|
Alchool | 2/67 (3%) | 0/41 (0%) |
|
|
Unknown | 22/67 (32.8%) | 0/41 (0%) |
|
| CTNNB1
mutations, exon 3 mutated | 0/67 (0%) | 0/41 (0%) |
|
| TP53
mutations |
|
|
|
|
R249S | 0/67 (0%) | 0/41 (0%) |
|
|
R72P | 10/67 (14.9%) | 0/41 (0%) | 0.009 |
| TERT
promoter mutations |
|
|
|
| −124
(G>A) | 28/67 (41.8%) | 0/41 (0%) | <0.0001 |
| −245
(G>A) | 47/67 (70.1%) | 22/41 (53.7%) | 0.08 |
| −297
(C>T) | 5/67 (7.5%) | 0/41 (0%) | 0.007 |
PCR amplification and sequencing
analysis
Exon 3 of CTNNB1, exons 4 and 7 of
TP53, and the TERT promoter region were analysed by
PCR amplification and Sanger's direct sequencing. DNA was extracted
from the frozen liver specimens of each patient by standard
procedures. In brief, tissue specimens were homogenized by
digestion in 150 mmol/l NaCl, 50 mmol/l Tris-HCl (pH 7.4), 10
mmol/l EDTA, 1% SDS and proteinase K (800 µg/ml) overnight at 37°C.
After extraction with phenol/chloroform, nucleic acids were
precipitated in 2 volumes of pure, cold ethanol. Nucleic acids were
then resuspended and digested with pancreatic ribonuclease (100
µg/ml), followed by extraction with phenol/chloroform and
reprecipitation in pure cold ethanol. DNA was resuspended in 10
mmol/l Tris-HCl (pH 7.4), 1 mmol/l EDTA. DNA concentration was
measured using the ND-1000 spectrophotometer (NanoDrop
Technologies) at 260 nm.
PCR amplification of the TERT promoter was carried
out using additives (dimethylsulphoxide 5% and glycerol 5%) under
the following conditions: 95°C for 2 min and then 35 cycles of 95°C
for 30 sec, 62°C for 40 sec, 72°C for 50 sec. Amplification of
CTNNB1 exon 3 and TP53 exons 4 and 7 were carried out
under the following conditions: 95°C for 5 min and then 35 cycles
of 94°C for 15 sec, 55°C for 20 sec, 72°C for 60 sec.
Nucleotide sequences of PCR products were determined
using the BigDye Terminator Cycle Sequencing Ready Reaction kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The sequencing products were resolved in an automatic
DNA sequencer (ABI PRISM 3500 Dx Genetic Analyzer; Thermo Fisher
Scientific, Inc.). The oligonucleotide primers used for PCR
amplification and sequencing of TERT promoter, of
TP53 exons 4 and 7, and of CTNNB1 exon 3 are reported
in Table II. Mutations detected in
each genomic region analysed were confirmed by sequencing both DNA
strands of a second independent PCR amplification product. The
somatic or germline status of the mutations was assessed by
sequencing the corresponding non-tumor liver tissue.
| Table II.Oligonucleotide primers used for PCR
amplification and sequencing of TERT promoter, of
TP53 exons 4 and 7, and of CTNNB1 exon 3. |
Table II.
Oligonucleotide primers used for PCR
amplification and sequencing of TERT promoter, of
TP53 exons 4 and 7, and of CTNNB1 exon 3.
| Gene | Primer | Sequence
5′->3′ |
|---|
| hTERT | hTERT FWD |
ACGAACGTGGCCAGCGGCAG |
|
| hTERT REV |
CTGGCGTCCCTGCACCCTGG |
| TP53 exon 7 | TP53 exon 7
FWD |
GCGCACTGGCCTCATCTTG |
|
| TP53 exon 7
REV |
GGGTCAGCGGCAAGCAGAG |
| TP53 exon 4 | TP53 exon 4
FWD |
CTGGTCCTCTGACTGCTCTT |
|
| TP53 exon 4
REV |
AGGCATTGAAGTCTCATGGA |
| CTNNB1 exon 3 | CTNNB1 exon 3
FWD |
GGTATTTGAAGTATACCATAC |
|
| CTNNB1 exon 3
REV |
CTGGTCCTCGTCATTTAGCAG |
RNA extraction and TERT reverse
transcription-quantitative PCR (qPCR)
To evaluate the effect of the TERT promoter
mutations on gene transcription, real-time PCR quantification of
TERT transcripts was performed in all tumor and non-tumor liver
specimens as well as in liver tissue specimens from 20 patients
with NAFL. RNA extraction was performed using the QIAzol reagent
(Qiagen) following the manufacturer's instructions. Total liver RNA
was resuspended in nuclease-free water and concentration was
determined by spectrophotometry at 260 nm. To eliminate DNA
contamination each sample was treated with RQ1 RNase-Free DNase
(Promega Corporation) for 30 min, at 37°C. Then RNA was reverse
transcribed for first-strand cDNA synthesis by using AffinityScript
Multi-Temp cDNA Synthesis kit (Agilent Technologies) and random
examers. RNA reverse transcription was performed under the
following conditions: 65°C for 5 min, 42°C for 5 min, 55°C for 60
min and 70°C for 15 min.
TERT expression was assessed using TaqMan
Applied Biosystems gene expression assay (Hs00972656_m1) (Thermo
Fisher Scientific, Inc.). The relative amount of RNA was calculated
with the 2−∆∆Cq method (27). TERT gene expression was normalized to
internal control ribosomal 18S RNA (Hs99999901_s1), and the
expression level in the tumor specimens was compared with the mean
level of the gene expression in liver tissues from subjects with
NAFL and expressed as an n-fold ratio.
Statistical analysis
All statistical analyses were performed using the
SPSS 22.0 software package (SPSS Inc.) for Windows. Numerical data
were expressed as mean and standard deviation (SD) and categorical
variables as number and percentage. χ2 test was used for comparison
of categorical data. Analyses by the Kolmogorov-Smirnov test showed
that TERT gene expression did not followed a normal distribution.
Therefore, a non-parametric Kruskal-Wallis test was used for
comparisons of mean values among the different groups, followed by
post-hoc testing using un-paired Mann-Whitney U tests with a
Bonferroni-adjusted alpha level of 0.017. All tests were
two-tailed. P<0.05 was considered to indicate a statistically
significant difference.
Results
Sequencing analysis of CTNNB1, TP53,
and the TERT promoter region
Sequencing analysis showed the absence of mutations
in CTNNB1 exon 3 as well as the absence of the R249S
substitution in TP53 gene both in patients with HCC and in
the control cases. Interestingly, the functionally significant
215G>C polymorphism at codon 72 (R72P, rs1042522) of TP53
gene, which has been associated with a higher risk of developing
several different types of cancers (including oral, lung, thyroid,
bladder, and liver cancer) (28–33), was
found in 10/67 (14.9%) tumors, in 0/67 (0%) non-tumor tissues
(P=0.001), and in 0/41 (0%) controls (P=0.009) analysed. The
homozygous 215 G>C mutation leading to R72P was identified in 2
of 67 cases (2.9%). Heterozygosity of 215 G>C was revealed in 8
of 67 cases (11.9%).
Concerning the TERT promoter region, 28/67
(41.8%) tumors, 0/67 (0%) non-tumor tissues (P<0.0001), and 0/41
(0%) control tissues (P<0.0001) showed the recurrent somatic
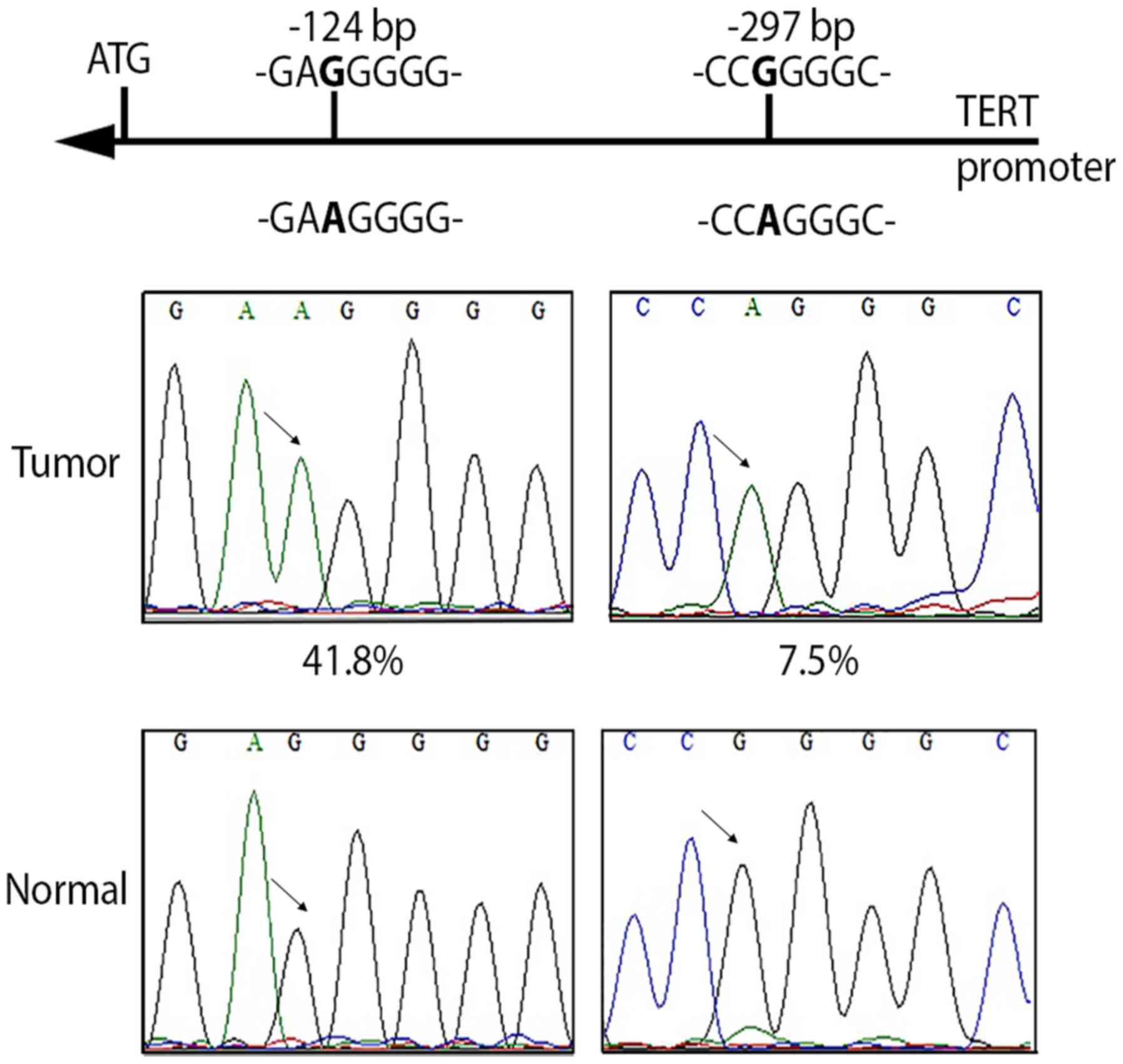
mutation at the previously described hot spot located at −124 bp
(−124G>A) from the ATG start site of TERT gene (11,34–36)
(Fig. 1), whereas the other
described hot spot located at −146 bp (−146 bp G>A) from the ATG
start site (11,34–36) was
not detected in any of the HCC cases nor in the controls.
Analogously, neither the HCC or control cases showed the mutation
at −57 bp (−57 bp A>C) previously described in melanoma and in
bladder cancer (35,37) or the tandem GG>AA mutation, a
hallmark of ultraviolet-induced mutagenesis, described in melanoma
(34,35). The rs2853669 A>G single nucleotide
polymorphism (SNP) located at −245 bp from the TERT ATG
start site-able to modify the TERT expression levels induced
by TERT promoter somatic mutations (38,39)-was
found in 47/67 (70.1%) tumors, in 44/67 (65.7%) non-tumors (P=0.6),
and in 22/41 (53.7%) controls (P=0.08).
Interestingly, in 5/67 (7.5%) tumors, in 0/67 (0%)
non-tumor tissues (P<0.0001), and in 0/41 (0%) controls (P=0.07)
analysed, a new mutation was identified in the TERT
promoter, located at −297 bp (−297 C>T; G>A on opposite
strand) from the ATG start site (Fig.
1). This mutation creates an activating protein 2 (AP2)
consensus sequence (CCGGGGC>CCAGGGC) (40,41) and
was found alone (1 tumor tissue) or in combination (4 tumor
tissues) with the −124 bp mutation.
Expression levels of TERT gene in
tumor and non-tumor liver tissues
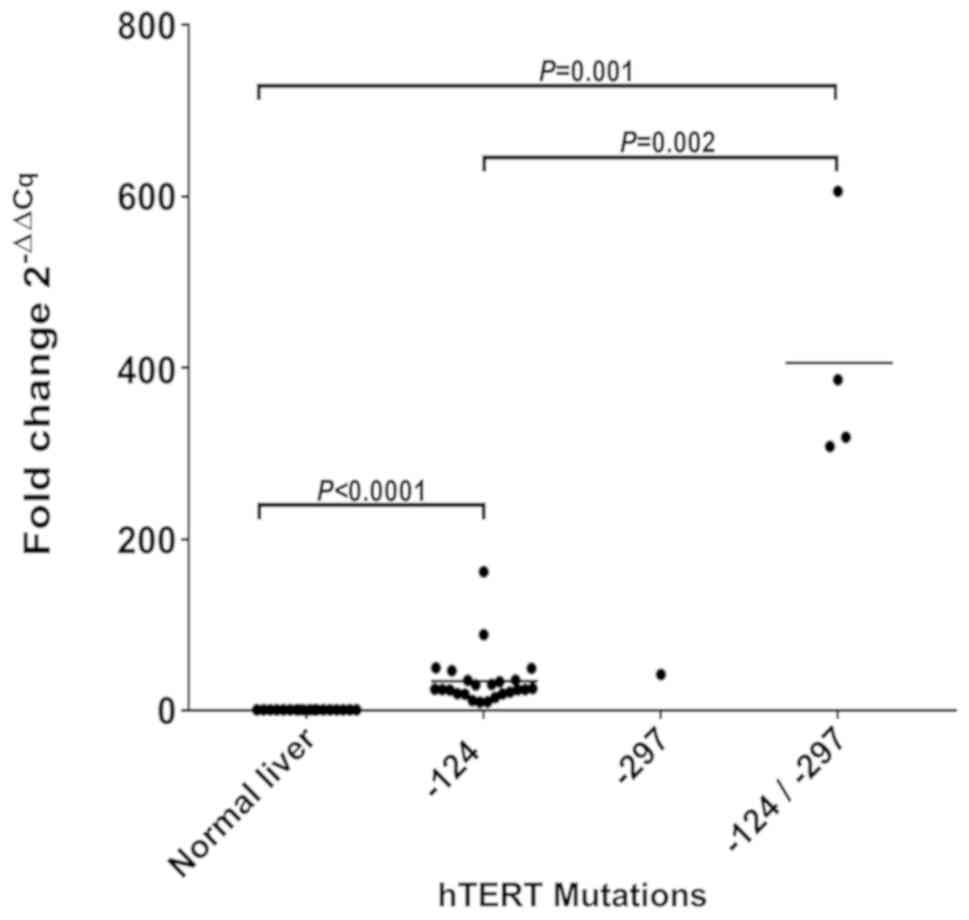
Real-time qPCR quantifications of TERT
transcripts confirmed that within the tumors harboring the −124 bp
mutation, TERT expression levels were significantly
upregulated compared with control liver tissues (P<0.0001, fold
change tumors/control livers=33). Interestingly, significantly
higher levels of TERT expression were also found in tumors
harbouring the −297 bp somatic mutation. In particular, the tumor
specimen with the single mutation at −297 bp showed TERT
expression levels 40-fold higher compared with control liver
tissues. The 4 tumor specimens harbouring both the −124 and the
−297 bp mutations showed that TERT gene expression was
significantly further increased compared with control tissues
(P=0.001; fold change tumors/control livers >300) and even when
compared with tumors harboring the −124 bp mutation alone (P=0.002;
fold change tumors with both −124 and −297 bp mutations/tumors with
−124 bp mutation alone >11) (Fig.
2). In these 4 tumors TERT transcripts were
significantly increased also when compared with tumors not mutated
in the TERT promoter (P=0.001; fold change
tumors/non-mutated tumors >8). The SNP rs2853669 associated
modulatory effect on TERT expression was not detectable in
tumors with or without TERT promoter somatic mutations.
Summarising, 29 (43.3%) of the 67 patients with HCC
harboured the −124 bp and/or the −297 bp somatic mutation in tumor
tissue. The underlying liver disease of the 29 HCC patients, was
HCV-related in 18 (62%) cases, HBV-related in 2 (6.9%) cases, and
cryptogenic in 9 (31%) cases (P=0.59). Therefore, TERT
mutations were observed at similar frequencies in viral-related
HCCs and in HCCs related to other causes of liver disease.
Amongst the other variables tested, there were
significant differences in age distribution between patients with
and without the −124 bp mutation. Patients with the −124 bp
mutation were older (70.7±7.5 years) than those without the
mutation (63.4±8.4 years, P=0.0008). No other variable was
associated with the TERT somatic mutations.
Discussion
In this study, we analysed the mutational pattern of
TP53, CTNNB1, and TERT promoter in tumor and
non-tumor liver tissue specimens from patients with HCC and from
control obese patients, all from Southern Italy. Interestingly, we
detected no CTNNB1 and TP53 R249S somatic mutations in any
patients. CTNNB1 mutations have been identified in about
20–40% of HCCs in previous studies (19,42–44), and
this prevalence was shown to be higher in individuals with
HCV-related HCC than in those infected by HBV (21). In our study population, HCC was
related to HCV in 59.7% of the patients. Given the absence of
CTNNB1 somatic mutations in the studied HCCs it is possible
that Wnt/β-catenin activation in our patients is induced
independently of the CTNNB1 genetic background as it has
been shown for adrenal aldosterone producing adenomas (45,46).
Concerning the TP53 gene, the absence of
R249S somatic mutation in the analysed HCCs could be explained by
the fact that all the studied patients were from a geographic area
where there is no dietary exposure to the human liver carcinogen
AFB1, and where the general prevalence of HBV infection is low
(less than 1%). In TP53 gene we detected the 215G>C
polymorphism at codon 72 (R72P, rs1042522). It was found in 10% of
the HCC specimens and in none of the non-tumor liver tissues and
the control livers. Thus, the prevalence of this polymorphism was
significantly higher in HCCs than in the control tissues. Our
results are in accordance with previous studies showing that
carriers of both the heterozygous and homozygous TP53-SNP72
genotypes are at a high risk of HCC development (31,47–49).
Interestingly, it has been hypothesized that tumorigenesis, relying
on TP53 R72P, might play a role only in selected populations
of patients living in low incidence geographic areas (50). Indeed, in areas of high HCC incidence
(Far East and Southern Africa) the presence of potent risk factors
(HBV infection and AFB1) able to induce major genomic alterations
in the exposed populations may likely further reduce the weak
oncogenic impact of the R72P SNP (50).
Concerning the TERT gene promoter, in this
study we report for the first time the identification of a somatic
mutation located at −297 bp (−297 G>A) from the ATG start site
of the TERT gene. This mutation was detected in 7.5% of the
studied tumor specimens but in none of either the paired non-tumor
or control liver tissues. The nucleotide change in the sequence
generates a de novo consensus binding motif for AP2
transcription factor (40,41) and real time PCR quantification
revealed that TERT transcripts in the tumors harboring the
−297 bp mutation were 1.2-fold higher than those expressed in
tumors showing the −124 bp mutation, known to create novel
consensus binding motifs for E-twenty-six (ETS) and ternary complex
factor (TCF) transcription factors (35). Interestingly, tumors harboring both
−124 and −297 bp mutations had more than 300-fold increase in
TERT transcript levels compared with control tissues, thus
clearly indicating that the combination of the 2 somatic mutations
has a strong impact on the promoter activation and the
up-regulation of TERT gene expression.
AP2 family genes have been implicated in a large
number of tumors in various stages of tumorigenesis (51). Our data demonstrating the creation of
a de novo AP2 binding site in TERT promoter indicate
that TERT may become an AP-2 target gene, and
this-possibly-not only in HCC but also in other cancer types,
strengthening the crucial role of TERT promoter mutations
and telomerase activation in the carcinogenetic process, and
confirming them as excellent candidate biomarkers for early tumor
detection or monitoring.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TP, GR, CS and GN conceived and designed the study.
DL, GC, CM, VC and FCdT performed all the analyses. TP, DL, CS, DG
and MSF were involved in the interpretation of all data. DL was
involved in the preparation of the figures and tables. AA performed
statistical analysis. TP wrote the manuscript and GR revised the
manuscript. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of the Messina University Hospital (reference no. 65-15),
and written informed consent was obtained from all patients, parent
or guardian. In addition, the procedures of this manuscript were in
accordance with the Helsinki declaration of 1964 and its later
amendments.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kulik L and El-Serag HB: Epidemiology and
management of hepatocellular carcinoma. Gastroenterology.
156:477–491.e1. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Laursen L: A preventable cancer. Nature.
516:S2–S3. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schulze K, Nault JC and Villanueva A:
Genetic profiling of hepatocellular carcinoma using next-generation
sequencing. J Hepatol. 65:1031–1042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Watson IR, Takahashi K, Futreal PA and
Chin L: Emerging patterns of somatic mutations in cancer. Nat Rev
Genet. 14:703–18. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, Zucman-Rossi J, Pikarsky E,
Sangro B, Schwartz M, Sherman M and Gores G: Hepatocellular
carcinoma. Nat Rev Dis Primers. 2:160182016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Quaas A, Oldopp T, Tharun L, Klingenfeld
C, Krech T, Sauter G and Grob TJ: Frequency of TERT promoter
mutations in primary tumors of the liver. Virchows Arch.
465:673–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Totoki Y, Tatsuno K, Covington KR, Ueda H,
Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H,
et al: Trans-ancestry mutational landscape of hepatocellular
carcinoma genomes. Nat Genet. 46:1267–1273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nault JC and Zucman-Rossi J: TERT promoter
mutations in primary liver tumors. Clin Res Hepatol Gastroenterol.
40:9–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rudolph KL, Hartmann D and Opitz OG:
Telomere dysfunction and DNA damage checkpoints in diseases and
cancer of the gastrointestinal tract. Gastroenterology.
137:754–762. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nault JC, Mallet M, Pilati C, Calderaro J,
Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C and
Zucman- Rossi J: High frequency of telomerase reverse-transcriptase
promoter somatic mutations in hepatocellular carcinoma and
preneoplastic lesions. Nat Commun. 4:22182013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nault JC, Calderaro J, Di Tommaso L,
Balabaud C, Zafrani ES, Bioulac-Sage P, Roncalli M and Zucman-Rossi
J: Telomerase reverse transcriptase promoter mutation is an early
somatic genetic alteration in the transformation of premalignant
nodules in hepatocellular carcinoma on cirrhosis. Hepatology.
60:1983–1992. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee SE, Chang SH, Kim WY, Lim SD, Kim WS,
Hwang TS and Han HS: Frequent somatic TERT promoter mutations and
CTNNB1 mutations in hepatocellular carcinoma. Oncotarget.
7:69267–69275. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pezzuto F, Buonaguro L, Buonaguro FM and
Tornesello ML: Frequency and geographic distribution of TERT
promoter mutations in primary hepatocellular carcinoma. Infect
Agent Cancer. 12:272017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kawai-Kitahata F, Asahina Y, Tanaka S,
Kakinuma S, Murakawa M, Nitta S, Watanabe T, Otani S, Taniguchi M,
Goto F, et al: Comprehensive analyses of mutations and hepatitis B
virus integration in hepatocellular carcinoma with
clinicopathological features. J Gastroenterol. 51:473–486. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zucman-Rossi J, Villanueva A, Nault JC and
Llovet JM: Genetic landscape and biomarkers of hepatocellular
carcinoma. Gastroenterology. 149:1226–1239 e4. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hussain SP, Schwank J, Staib F, Wang XW
and Harris CC: TP53 mutations and hepatocellular carcinoma:
Insights into the etiology and pathogenesis of liver cancer.
Oncogene. 26:2166–2176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tornesello ML, Buonaguro L, Tatangelo F,
Botti G, Izzo F and Buonaguro FM: Mutations in TP53, CTNNB1 and
PIK3CA genes in hepatocellular carcinoma associated with hepatitis
B and hepatitis C virus infections. Genomics. 102:74–83. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
de La Coste A, Romagnolo B, Billuart P,
Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C,
Kahn A and Perret C: Somatic mutations of the beta-catenin gene are
frequent in mouse and human hepatocellular carcinomas. Proc Natl
Acad Sci USA. 95:8847–8851. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saitta C, Lanza M, Bertuccio A, Lazzara S,
Navarra G, Raimondo G and Pollicino T: Evaluation of CTNNB1 and
TP53 variability in patients with hepatocellular carcinoma and
occult hepatitis B virus infection. Cancer Genet. 208:513–516.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Statistics INIo. http://dati.istat.it/?lang=en2018
|
|
24
|
Fusco M, Girardi E, Piselli P, Palombino
R, Polesel J, Maione C, Scognamiglio P, Pisanti FA, Solmone M, Di
Cicco P, et al: Epidemiology of viral hepatitis infections in an
area of southern Italy with high incidence rates of liver cancer.
Eur J Cancer. 44:847–853. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dal Maso L, Lise M, Zambon P, Crocetti E,
Serraino D, Ricceri F, Vercelli M, De Lisi V, Tagliabue G, Federico
M, et al: Incidence of primary liver cancer in Italy between 1988
and 2002: An age-period-cohort analysis. Eur J Cancer. 44:285–292.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chalasani N, Younossi Z, Lavine JE,
Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM and Sanyal AJ:
The diagnosis and management of nonalcoholic fatty liver disease:
Practice guidance from the American association for the study of
liver diseases. Hepatology. 67:328–357. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mostaid MS, Ahmed MU, Islam MS, Bin Sayeed
MS and Hasnat A: Lung cancer risk in relation to TP53 codon 47 and
codon 72 polymorphism in Bangladeshi population. Tumour Biol.
35:10309–10317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu B, Guo D and Guo Y: Association between
p53 Arg72Pro polymorphism and thyroid cancer risk: A meta-analysis.
Tumour Biol. 35:561–565. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang YC, Chen CY, Chen SK, Chang YY and
Lin P: p53 codon 72 polymorphism in Taiwanese lung cancer patients:
Association with lung cancer susceptibility and prognosis. Clin
Cancer Res. 5:129–134. 1999.PubMed/NCBI
|
|
31
|
Jia S, Tang W and Luo Y: p53 codon 72
polymorphism and hepatocellular carcinoma: A meta-analysis. Hepatol
Int. 7:669–675. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeng XT, Luo W, Geng PL, Guo Y, Niu YM and
Leng WD: Association between the TP53 codon 72 polymorphism and
risk of oral squamous cell carcinoma in Asians: A meta-analysis.
BMC Cancer. 14:4692014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vinagre J, Almeida A, Populo H, Batista R,
Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, et al:
Frequency of TERT promoter mutations in human cancers. Nat Commun.
4:21852013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin
L and Garraway LA: Highly recurrent TERT promoter mutations in
human melanoma. Science. 339:957–959. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Horn S, Figl A, Rachakonda PS, Fischer C,
Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al:
TERT promoter mutations in familial and sporadic melanoma. Science.
339:959–961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda
C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL,
Giovanella BC, et al: TERT promoter mutations occur frequently in
gliomas and a subset of tumors derived from cells with low rates of
self-renewal. Proc Natl Acad Sci USA. 110:6021–6026. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Giedl J, Rogler A, Wild A, Riener MO,
Filbeck T, Burger M, Rummele P, Hurst C, Knowles M, Hartmann A, et
al: TERT core promotor mutations in early-onset bladder cancer. J
Cancer. 7:915–920. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park CK, Lee SH, Kim JY, Kim JE, Kim TM,
Lee ST, Choi SH, Park SH and Kim IH: Expression level of hTERT is
regulated by somatic mutation and common single nucleotide
polymorphism at promoter region in glioblastoma. Oncotarget.
5:3399–3407. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ko E, Seo HW, Jung ES, Kim BH and Jung G:
The TERT promoter SNP rs2853669 decreases E2F1 transcription factor
binding and increases mortality and recurrence risks in liver
cancer. Oncotarget. 7:684–699. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Eckert D, Buhl S, Weber S, Jäger R and
Schorle H: The AP-2 family of transcription factors. Genome Biol.
6:2462005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Williams T and Tjian R: Analysis of the
DNA-binding and activation properties of the human transcription
factor AP-2. Genes Dev. 5:670–682. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang H, Fujii H, Sankila A, Mahler-Araujo
BM, Matsuda M, Cathomas G and Ohgaki H: Beta-catenin mutations are
frequent in human hepatocellular carcinomas associated with
hepatitis C virus infection. Am J Pathol. 155:1795–1801. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zucman-Rossi J, Benhamouche S, Godard C,
Boyault S, Grimber G, Balabaud C, Cunha AS, Bioulac-Sage P and
Perret C: Differential effects of inactivated Axin1 and activated
beta-catenin mutations in human hepatocellular carcinomas.
Oncogene. 26:774–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pezzuto F, Izzo F, Buonaguro L, Annunziata
C, Tatangelo F, Botti G, Buonaguro FM and Tornesello ML: Tumor
specific mutations in TERT promoter and CTNNB1 gene in hepatitis B
and hepatitis C related hepatocellular carcinoma. Oncotarget.
7:54253–54262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Berthon A, Drelon C, Ragazzon B, Boulkroun
S, Tissier F, Amar L, Samson-Couterie B, Zennaro MC, Plouin PF,
Skah S, et al: WNT/β-catenin signalling is activated in
aldosterone-producing adenomas and controls aldosterone production.
Hum Mol Genet. 23:889–905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Boulkroun S, Samson-Couterie B, Golib-Dzib
JF, Amar L, Plouin PF, Sibony M, Lefebvre H, Louiset E, Jeunemaitre
X, Meatchi T, et al: Aldosterone-producing adenoma formation in the
adrenal cortex involves expression of stem/progenitor cell markers.
Endocrinology. 152:4753–4763. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yoon YJ, Chang HY, Ahn SH, Kim JK, Park
YK, Kang DR, Park JY, Myoung SM, Kim DY, Chon CY and Han KH: MDM2
and p53 polymorphisms are associated with the development of
hepatocellular carcinoma in patients with chronic hepatitis B virus
infection. Carcinogenesis. 29:1192–1196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhu ZZ, Cong WM, Liu SF, Xian ZH, Wu WQ,
Wu MC, Gao B, Hou LF and Zhu GS: A p53 polymorphism modifies the
risk of hepatocellular carcinoma among non-carriers but not
carriers of chronic hepatitis B virus infection. Cancer Lett.
229:77–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhu ZZ, Cong WM, Liu SF, Dong H, Zhu GS
and Wu MC: Homozygosity for Pro of p53 Arg72Pro as a potential risk
factor for hepatocellular carcinoma in Chinese population. World J
Gastroenterol. 11:289–292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rebbani K, Marchio A, Ezzikouri S, Afifi
R, Kandil M, Bahri O, Triki H, El Feydi AE, Dejean A, Benjelloun S
and Pineau P: TP53 R72P polymorphism modulates DNA methylation in
hepatocellular carcinoma. Mol Cancer. 14:742015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kołat D, Kałuzińska Ż, Bednarek AK and
Płuciennik E: The biological characteristics of transcription
factors AP-2α and AP-2γ and their importance in various types of
cancers. Biosci Rep. 39(pii): BSR201819282019. View Article : Google Scholar : PubMed/NCBI
|