Introduction
Thyroid cancer is the most commonly diagnosed
endocrine cancer with ~60,000 new cases diagnosed in the USA in
2018 (1,2). Thyroid cancer is not restricted to a
certain age group; however, its aggressiveness increases
significantly with the age of the patient (2). Anaplastic thyroid cancer (ATC) is the
most aggressive type of thyroid cancer. The incidence rate of ATC
has increased between 1973 and 2014, and is still associated with a
higher risk of tumor progression and cancer metastasis (3).
Normal p53 acts as a tumor suppressor gene
that contributes to the prevention of tumor growth (4). However, mutant p53 proteins may acquire
oncogenic properties that promote tumor invasion, metastasis,
cancer cell proliferation and survival (5). Point mutations in p53 are
frequently detected in 50–80% of ATC tissues (6). Loss of p53 function has been reported
to result in poorly differentiated thyroid tumors (6). Hence, mutant p53 protein can be
considered a crucial therapeutic target in patients with ATC
(6–8).
Several tyrosine kinase inhibitors (TKIs) have been
used to treat advanced thyroid cancer, due to their ability to
block kinases or kinase receptors (9,10). TKIs
can either repress cell proliferation or attenuate the neoplastic
transformation of thyroid cells (11). For example, sorafenib, a multi-kinase
inhibitor that acts predominantly through the inhibition of
Raf-kinase and vascular endothelial growth factor receptor 2, has
been approved for the treatment of metastatic and differentiated
thyroid cancer (12,13). However, although sorafenib may be
effective in the treatment of advanced medullary thyroid cancer, it
is not effective against ATC with a poor survival rate (14,15).
Combination treatments might improve outcomes in ATC (15), which emphasizes that combined
targeted therapy may be highly effective for advanced thyroid
carcinomas (16).
A large number of drug candidates, including small
molecules or chemotherapeutic agents, have been identified or
designed to rescue mutant p53 and reactivate its antitumor
capacity through various mechanisms. For example, PRIMA-1 can
restore active conformation of mutant p53 and PK7088 can
upregulate p21 expression and apoptotic functions (7,17). In
the present study, CP-31398, a stabilizing agent that restores the
wild-type conformation of mutated p53 protein (18), was used to improve the
chemotherapeutic efficacy of sorafenib for p53-mutated ATC
cells. The molecular function of CP-31398 was evaluated using
western blot analysis and a luciferase reporter assay. Sorafenib
and CP-31398 synergistically inhibited the viability of the SW579
ATC cell line. Furthermore, it was demonstrated that the
combination of targeted therapy (with sorafenib) and CP-31398
potentially changed the characteristics of p53-mutant ATC
cells.
Materials and methods
Cell cultures
SW579 cells that harbor p53 mutations were
purchased from the American Type Culture Collection. The cells were
cultured in Dulbecco's modified Eagle's medium (Lonza Group, Ltd.)
supplemented with 10% fetal bovine serum (Greiner Bio-One
International GmbH) and antibiotics (100 U/ml penicillin and 100
µg/ml streptomycin; Lonza Group, Ltd.) at 37°C and 5%
CO2 in a humidified atmosphere.
Western blot analysis
SW579 cells were seeded onto a 15-cm dish (at a
density of 3.5×106) and treated with 0, 0.3 or 3 µM
CP-31398 (cat. no. PZ0115; Merck KGaA) for 24 h and placed at 37°C
in a humidified incubator containing 5% CO2. Total cell
lysates were prepared using radioimmunoprecipitation assay buffer
(cat. no. R0278; Merck KGaA) and protease inhibitors (cat. no.
P8340; Merck KGaA) on ice for 5 min. The protein concentration was
determined using the Bio-Rad protein assay (Bio-Rad Laboratories,
Inc.). Proteins (30 µg) were separated on NuPAGE 4–12% Bis-Tris Gel
(Thermo Fisher Scientific, Inc.) and transferred to polyvinylidene
difluoride membranes (cat. no. IPVH00010; Merck KGaA). The
membranes were incubated with primary antibodies at room
temperature for 30 min for the following proteins: p21 (1:200; cat.
no. 2947; Cell Signaling Technology, Inc.), Noxa (1:100; cat. no.
ab13654; Abcam), p53 (1:200; cat. no. SC-126; Santa Cruz
Biotechnology, Inc.) and glyceraldehyde 3-phosphate dehydrogenase
(GAPDH; 1:6,000; cat. no. AM4300; Thermo Fisher Scientific, Inc.).
The membranes were subsequently incubated with secondary antibody
at room temperature for 30 min. The secondary antibodies were:
Biotinylated anti-rabbit IgG (1:1,000; cat. no. BA-1000; Vector
Laboratories, Inc.; Maravai LifeSciences) for p21; biotinylated
anti-mouse IgG (1:1,000; cat. no. BA-2000; Vector Laboratories,
Inc.; Maravai LifeSciences) for Noxa and p53; and horseradish
peroxidase-conjugated anti-mouse IgG for GAPDH (1:5,000; cat. no.
ab6808; Abcam). The bands were then visualized using: Vectastain
ABC-AmP Chemiluminescence Detection kit (cat. no. AK-6601; Vector
Laboratories, Inc.; Maravai LifeSciences) for p21, Noxa and p53;
and the Western Lightning Ultra-ECL (cat. no. NEL113001EA;
PerkinElmer, Inc.) for GAPDH; both according to the manufacturer's
instructions. Finally, images were acquired and protein bands were
quantified by densitometric analysis using an Alpha Innotech
FluorChem FC2 Imager (version 3.2.2; ProteinSimple). The relative
protein levels were calculated and determined by normalizing their
expression to that of the GAPDH internal control.
Change in viability of SW579 cells
following CP-31398 treatment
The growth of SW579 cells was evaluated using MTT
assay (cat. no. M5655; Merck KGaA). Firstly, SW579 cells were
cultured at 1×104 cells/well in 96-well plates for 24 h.
To calculate the half-maximal inhibitory concentration
(IC50) of CP-31398, the chemosensitivity of cells was
determined following incubation for 24 h at 0, 1, 1.5, 2, 2.5, 3,
3.5 and 4 µM. To evaluate the efficiency of adjuvant therapy, SW579
cells were treated with 1 µM sorafenib (cat. no. 284461-73-0;
ApexBio Technology) and 3 µM CP-31398 alone or combined for 72 h
and cell viability was calculated using MTT assay. The cells were
then treated with 10 µl of the MTT reagent, and incubated in the
dark for 4 h. Dimethyl sulfoxide (100 µl) was subsequently added to
dissolve the purple precipitate formed by the viable cells. The
absorbance of each well at 540 nm was read by a Synergy HT
Multi-Mode microplate reader (BioTek Instruments, Inc.). Data were
obtained from three independent experiments.
Cell cycle analysis through image
cytometry
The cell cycle analysis of SW579 cells was performed
using a fluorescence image cytometer (NucleoCounter NC-3000;
ChemoMetec A/S). SW579 cells were seeded in a 10-cm dish at a
density of 1.1×106 cells per dish. Treatment with
CP-31398, at concentrations of 0, 0.3 or 3 µM, was initiated 16 h
following seeding. After a 72-h incubation, cells were harvested by
trypsinization, loaded into a Via-1-Casette (ChemoMetec), and
counted according to the manufacturer's instructions. Approximately
1.5×106 cells were suspended in 0.5 ml of
phosphate-buffered saline (PBS), and fixed with 4.5 ml of 70% cold
ethanol for at least 2 h. Subsequently, ethanol was removed, and
the cells were resuspended in PBS. Cell pellets were harvested by
centrifugation at 500 × g for 5 min at 4°C and incubated with 0.5
ml DAPI solution [0.1% Triton X-100 and
4′,6-diamidino-2-phenylindole (DAPI)] for 5 min at 37°C. The
stained cells were loaded into an NC-Slide A8 (ChemoMetec) and
evaluated using a Fixed Cell Cycle-DAPI/DNA fragmentation assay
protocol in the NucleoCounter NC-3000 image cytometer (ChemoMetec)
(19). The acquired DNA content
histograms were used to distinguish cells at different stages of
the cell cycle with the NucleoView NC-3000 software (version
2.1.25.12; ChemoMetec).
Luciferase reporter assay
The p53-dependent reporter plasmid, PG13-luc, which
carries the firefly luciferase gene driven by a promoter containing
13 p53-binding elements, was provided by Dr Shih-Ming Huang
(Department and Graduate Institute of Biochemistry, National
Defense Medical Center, Taiwan). Clones of SW579 cells expressing
luciferase were obtained after transfecting the cells with
PG13-luc, using the T-Pro NTRIII transfection reagent (Jifeng
Biotechnology) for 5 h according to the manufacturer's protocol.
The background levels of relative luciferase units (RLUs) were
detected from SW579 cells without PG13-luc plasmid. Luciferase
activity was measured 24 h after the cells were treated with or
without CP-31398 (3 µM). The SW579/PG13-luc cell lysate (5 µg in 20
µl) was added to 100 µl of the luciferase assay reagent (Luciferase
Assay System with Reporter Lysis Buffer; Promega Corporation) in
white clear-bottomed 96-well plates (Greiner Bio-One International
GmbH), in duplicate or triplicate for each experimental condition.
Bioluminescence produced for 10 sec was automatically measured
under the Centro LB 960 Microplate Luminometer (Berthold
Technologies GmbH).
Statistical analysis
Statistical analysis was performed using the SPSS
software (version 13.0; SPSS Inc.). Numerical data are expressed as
mean ± standard deviation. The Student's t-test was used for the
analysis of two groups, and one-way analysis of variance with least
significant difference for multiple comparisons was performed for
determining the significance of differences between the various
experimental groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Determination of the IC50
of CP-31398 in p53-mutated ATC cells
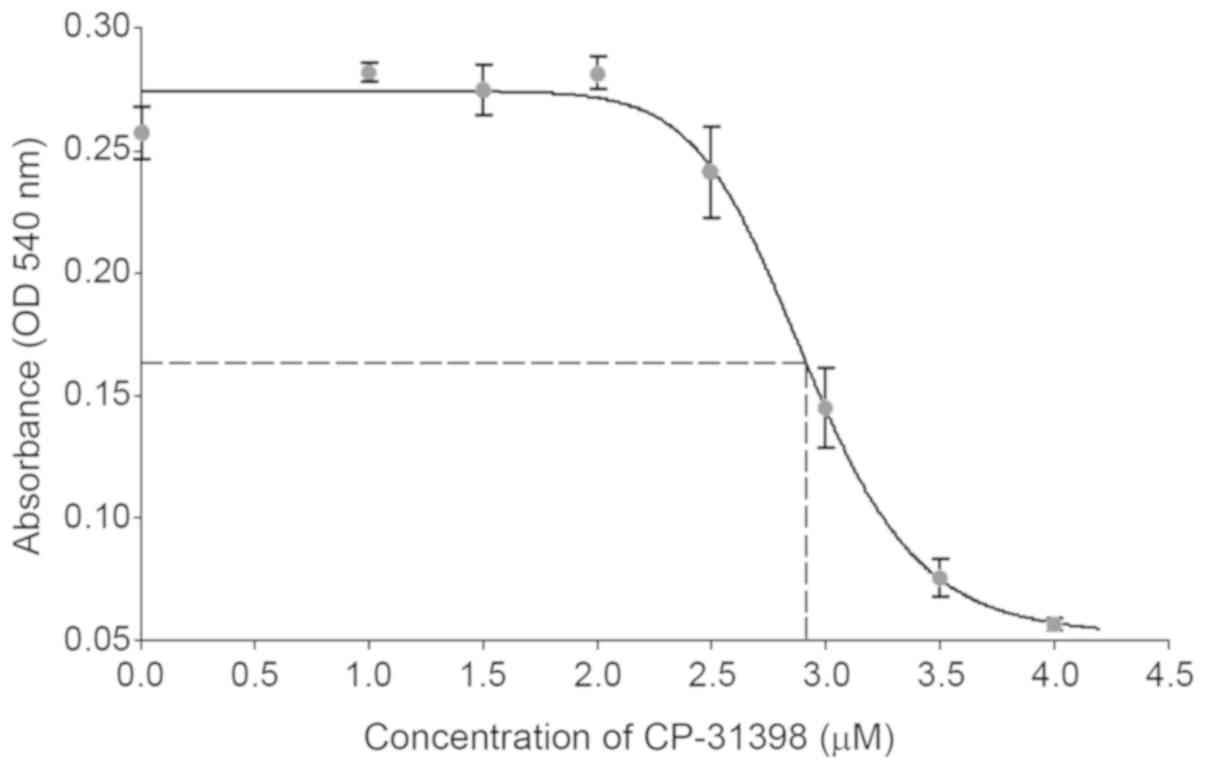
CP-31398 inhibited the growth of SW579 thyroid
cancer cells, an ATC cell line harboring p53 mutations
(20,21). The IC50 of CP-31398 in
SW579 cells was determined 24 h after treatment with doses ranging
from 0–4 µM. The potential inhibitory effect of CP-31398 on SW579
cells is shown in Fig. 1. The
concentration of 3 µM was chosen for the following experiments
since it was closed to the IC50 (~2.9 µM). Recent
studies reported that CP-31398 restores the function of p53
and could be considered as a therapeutic strategy for
p53-dysfunctional cancers, including cervical cancer or
esophageal cancer (22,23). The present study demonstrated that
CP-31398 restored the function of mutated p53 in SW579
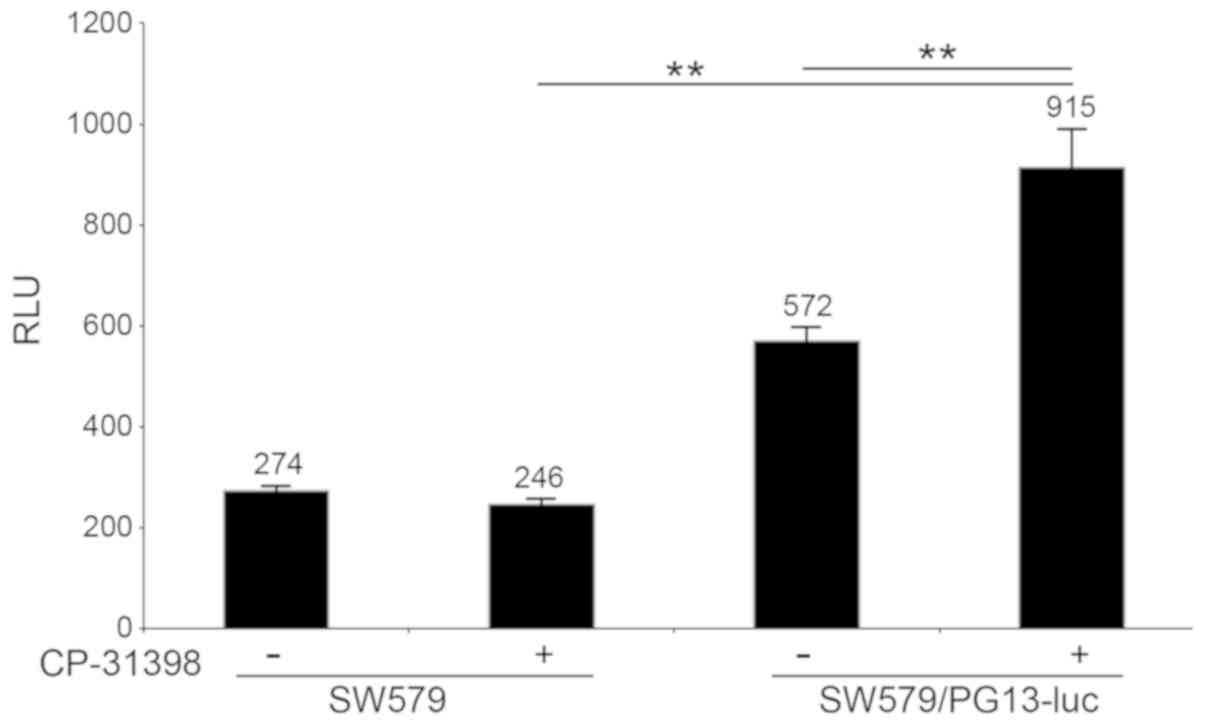
cells. Firstly, 3 µM CP-31398 induced the promoter activity of the
plasmid PG13-luc that requires wild-type p53 binding sites.
As shown in Fig. 2, CP-31398
significantly increased the luciferase activity from 572±26 RLUs in
non-treated cells to 915±76 RLUs in treated SW579/PG13-luc cells
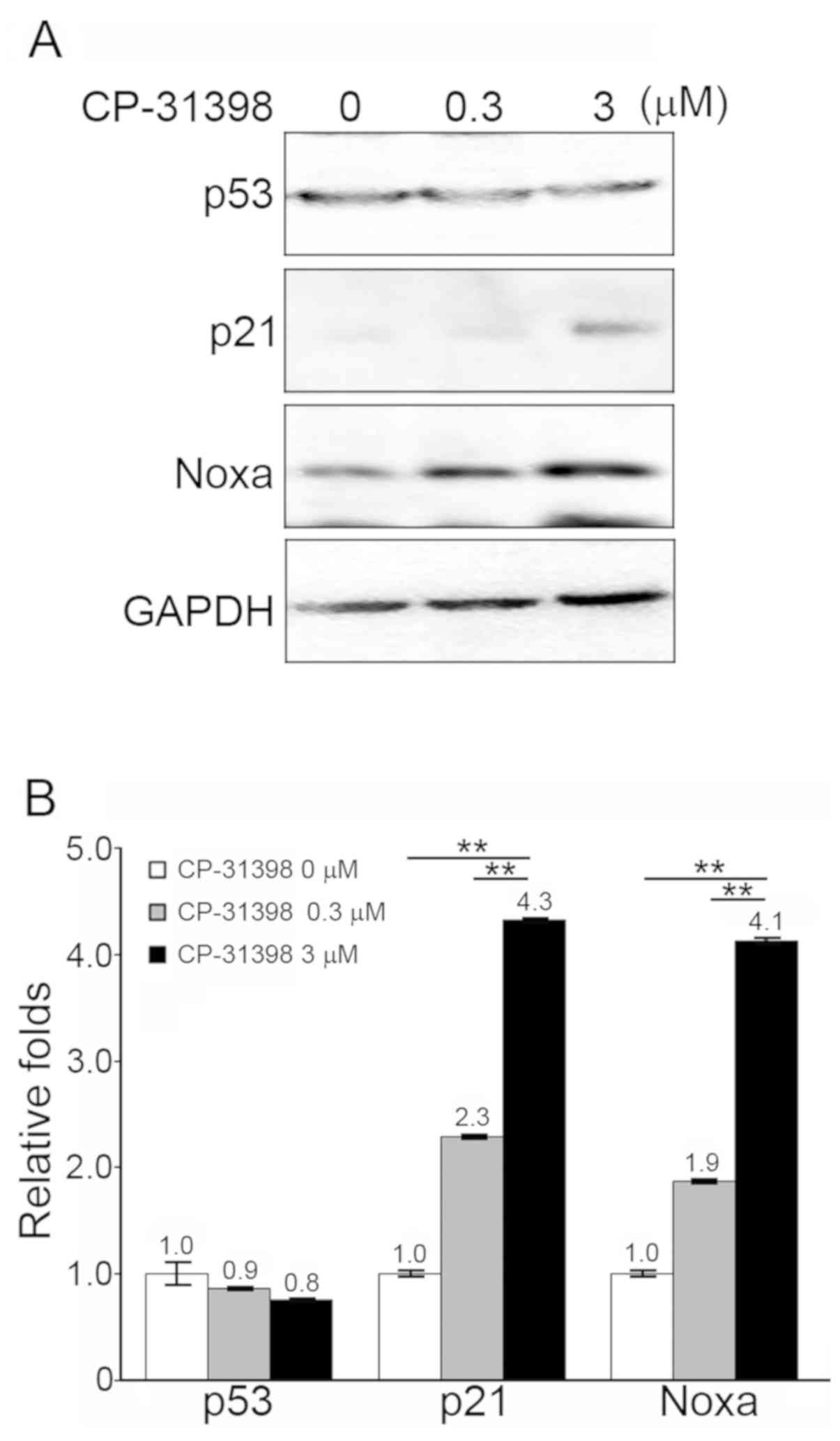
(P<0.01). Secondly, SW579 cells treated with CP-31398 (3 µM)
exhibited significantly increased protein levels of p21 and Noxa,
which are p53-dependent proteins (Fig.
3A and B).
CP-31398 induces apoptosis and G2/M
cell cycle arrest in SW579 cells
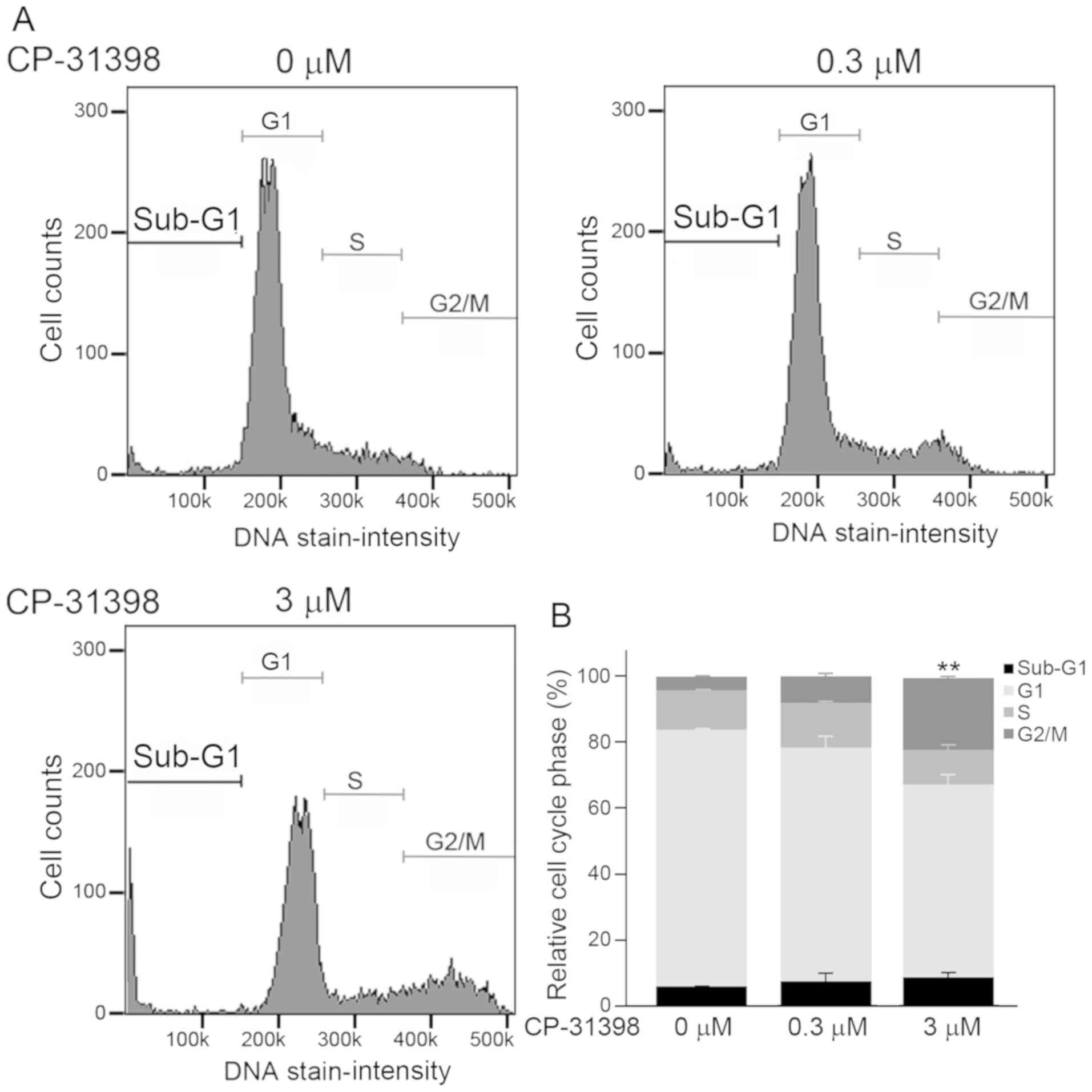
To determine whether the increased expression of p21
and Noxa in SW579 cells, following treatment with CP-31398, matched
with the induction of apoptosis, the percentages of cells at each
stage of the cell cycle were determined. The cell cycle
distribution in SW579 cells was examined after treatment with
CP-31398 at 0, 0.3 or 3 µM for 72 h. Treatment with 3 µM CP-31398
resulted in changes in cell cycle phases (Fig. 4A). Results from Fig. 4B demonstrated an increased percentage
of cells with sub-G1 DNA content (0 to 0.3 µM, +1.5%; 0.3 to 3 µM,
+1.0%), a decreased percentage of cells at G1 phase (0 to 0.3 µM,
−7.5%; 0.3 to 3 µM, −12.5%), and arrested cell cycle at the G2/M
phase with statistical significance (0 to 3 µM, +3.5%; 0.3 to 3 µM,
+13.5%; P<0.01).
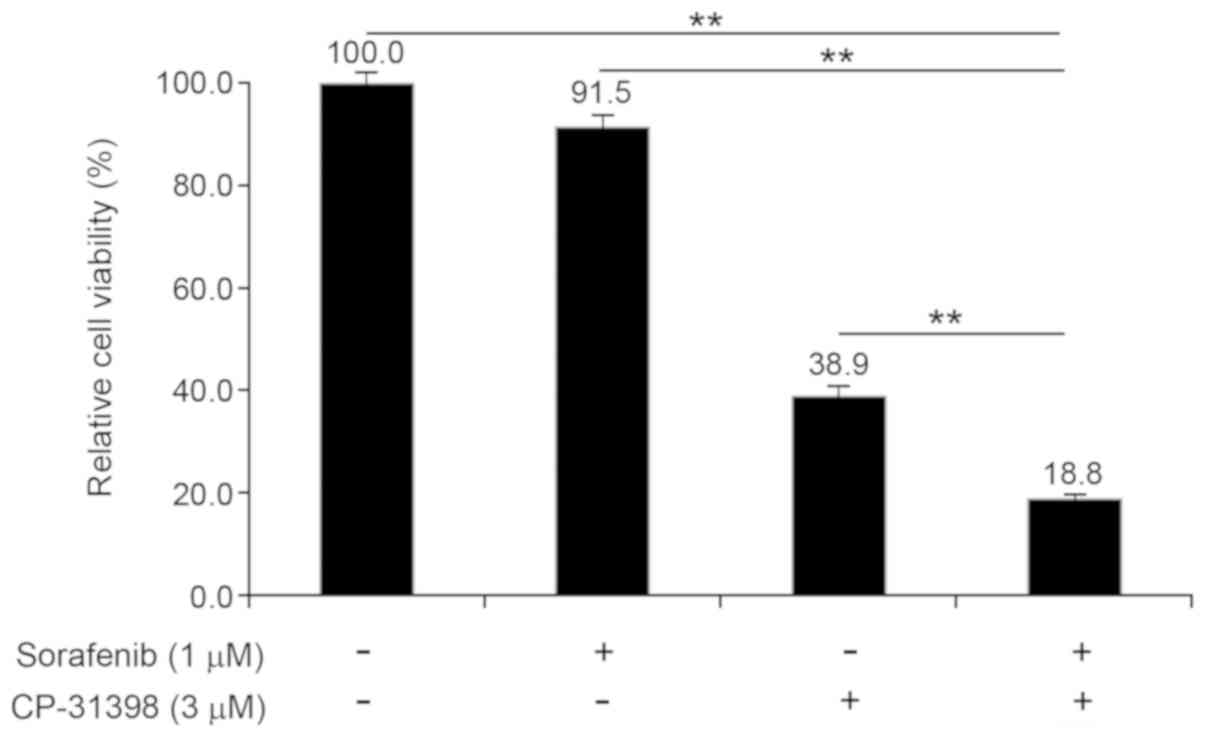
Adjuvant therapy with sorafenib for
thyroid cancer in the presence of CP-31398
Sorafenib has been used to treat different types of
thyroid cancer (14,24). However, previous studies indicated
that sorafenib has an IC50 value of 1.7±0.2 µM against
SW579 cells (25). In the present
study, 1 µM sorafenib did not effectively inhibit the viability of
SW579 cells, however treatment with 3 µM CP-31398 inhibited their
viability after 72 h incubation (Fig.
5). Notably, combination treatment with 1 µM sorafenib and 3 µM
CP-31398 for 72 h resulted in a significantly greater decrease in
cell viability (18.8±0.9 of untreated control cells; P<0.01)
compared with single-agent treatments (1 µM sorafenib, 91.5±2.4; 3
µM CP-31398, 38.9±1.8; both P<0.01).
Discussion
Although rare, ATC is one of the most aggressive
human malignancies, and most patients with ATC have a poor
prognosis, with a rate of thyroid cancer-associated
associated-mortality of almost 50% (26). In addition, Liu et al also
reported that the 2-year overall survival rate of patients with ATC
was low (26.0%) in Asia (27). In
clinical settings, sorafenib provides sufficient tumor reduction to
allow for thyroidectomy and radioactive iodine therapy in most
types of thyroid cancer (12,28).
However, Ito et al (14)
reported that sorafenib appeared to be effective in the treatment
of advanced medullary thyroid carcinoma but not ATC. Therefore,
targeted therapy with sorafenib in combination with other drugs
could be clinically effective for patients with ATC (15).
In the present study, SW579 cells (carrying
p53 mutations) were treated with a small-molecule agent
CP-31398, which induces the desired phenotypic change (apoptosis
and proliferation inhibition) in cancer cells with p53
mutations (29,30). CP-31398 caused notably decreased
viability of SW579 cells (31).
Mutated p53 (mutations at codons 151, 152, 248 and 255) is
frequently detected in endocrine tumors, including ATC (21), and the DNA-binding domain of
p53 in SW579 cells was defective due to a point mutation at
codon 255 (Ile to Ser) (31). In
general, the DNA-binding ability and molecular function of p53 in
SW579 cells improved when they were treated with CP-31398 (31). Xu et al (32) reported that CP-31398 treatment
stabilizes the levels of mutant p53 and enhances p21 protein
level in rhabdomyosarcoma. In the present study, the plasmid,
PG13-luc, containing a p53-binding site in its promoter was
reactivated following treatment with CP-31398 in SW579 cells.
Secondly the protein expression levels of two p53-dependent
proteins, p21 and Noxa, were increased. However, the level of
mutant p53 did not increase in the CP-31938-treated SW579
cells. These data demonstrated that the molecular function of p53
was restored when SW579 cells were treated with the optimal
concentration (3 µM) of CP-31398. It was hypothesized that the
increased levels of p21 and Noxa contributed to the reduction in
cell viability, by causing cells to arrest at the G2/M phase and
inducing apoptosis (33,34).
Furthermore, the present study demonstrated the
reduction in the viability of SW579 cells by CP-31398 treatment to
be augmented by the addition of the targeted therapeutic agent
sorafenib. Nevertheless, the antitumor activity of combined
targeted therapy with sorafenib and CP-31398 must be further
investigated using an in vivo xenograft model or a real-time
image of orthotopic model (35,36).
In summary, the present study demonstrated that
CP-31398 induces apoptosis and arrests the cell cycle at the G2/M
phase in SW579 cells, by restoring the molecular function of p53.
Notably, CP-31398 increased the antimitogenic effect of sorafenib,
and the combination of these two agents synergistically reduced the
viability of SW579 cells. Overall, the present study indicates a
potential clinical application of CP-31398 for treating patients
with ATC harboring p53 mutations, who generally respond
poorly to sorafenib-only treatment.
Acknowledgements
The authors would like to thank Dr Shih-Ming Huang
(Department of Biochemistry, National Defense Medical Center,
Taipei, Taiwan R.O.C) for providing the plasmid.
Funding
The present study was supported by the Cathay
General Hospital (Taipei, Taiwan; grant. no. CGH-MR-A10809; awarded
to YS).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CLL, JTW and YCS designed the study. CLL, JTW and
CJH wrote the initial version of the manuscript. CJH and CCC
conducted the cell studies. CCC performed the statistical analyses.
YCC performed western blotting. All authors discussed, modified and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATC
|
anaplastic thyroid cancer
|
|
TKIs
|
tyrosine kinase inhibitors
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blackburn BE, Ganz PA, Rowe K, Snyder J,
Wan Y, Deshmukh V, Newman M, Fraser A, Smith K, Herget K, et al:
Aging-related disease risks among young thyroid cancer survivors.
Cancer Epidemiol Biomarkers Prev. 26:1695–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Janz TA, Neskey DM, Nguyen SA and Lentsch
EJ: Is the incidence of anaplastic thyroid cancer increasing: A
population based epidemiology study. World J Otorhinolaryngol Head
Neck Surg. 5:34–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Labuschagne CF, Zani F and Vousden KH:
Control of metabolism by p53-cancer and beyond. Biochim Biophys
Acta Rev Cancer. 1870:32–42. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Manzella L, Stella S, Pennisi MS, Tirro E,
Massimino M, Romano C, Puma A, Tavarelli M and Vigneri P: New
insights in thyroid cancer and p53 family proteins. Int J Mol Sci.
18:E13252017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Y, Wang Z, Chen Y, Petersen RB, Zheng L
and Huang K: Salvation of the fallen angel: Reactivating mutant
p53. Br J Pharmacol. 176:817–831. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu L, Li D, Chen Z, Yang J, Ma Y, Cai H,
Shan C, Lv Z and Zhang X: Wild-type p53 induces sodium/iodide
symporter expression allowing radioiodide therapy in anaplastic
thyroid cancer. Cell Physiol Biochem. 43:905–914. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Date E, Okamoto K, Fumita S and Kaneda H:
Gastrointestinal perforation related to lenvatinib, an
anti-angiogenic inhibitor that targets multiple receptor tyrosine
kinases, in a patient with metastatic thyroid cancer. Invest New
Drugs. 36:350–353. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saito Y, Sugino K, Takami H, Matsuzu K,
Uruno T, Ohkuwa K, Kitagawa W, Nagahama M, Kawakubo H, Ito K and
Kitagawa Y: Clinical status and treatment of liver metastasis of
differentiated thyroid cancer using tyrosine kinase inhibitors.
World J Surg. 42:3632–3637. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cabanillas ME, McFadden DG and Durante C:
Thyroid cancer. Lancet. 388:2783–2795. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Corrado A, Ferrari SM, Politti U, Mazzi V,
Miccoli M, Materazzi G, Antonelli A, Ulisse S, Fallahi P and
Miccoli P: Aggressive thyroid cancer: Targeted therapy with
sorafenib. Minerva Endocrinol. 42:64–76. 2017.PubMed/NCBI
|
|
13
|
Ramakrishnan V, Timm M, Haug JL, Kimlinger
TK, Wellik LE, Witzig TE, Rajkumar SV, Adjei AA and Kumar S:
Sorafenib, a dual Raf kinase/vascular endothelial growth factor
receptor inhibitor has significant anti-myeloma activity and
synergizes with common anti-myeloma drugs. Oncogene. 29:1190–1202.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ito Y, Onoda N, Ito KI, Sugitani I,
Takahashi S, Yamaguchi I, Kabu K and Tsukada K: Sorafenib in
Japanese patients with locally advanced or metastatic medullary
thyroid carcinoma and anaplastic thyroid carcinoma. Thyroid.
27:1142–1148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen G, Nicula D, Renko K and Derwahl M:
Synergistic anti-proliferative effect of metformin and sorafenib on
growth of anaplastic thyroid cancer cells and their stem cells.
Oncol Rep. 33:1994–2000. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Orlandi F, Caraci P, Berruti A, Puligheddu
B, Pivano G, Dogliotti L and Angeli A: Chemotherapy with
dacarbazine and 5-fluorouracil in advanced medullary thyroid
cancer. Ann Oncol. 5:763–765. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Binayke A, Mishra S, Suman P, Das S and
Chander H: Awakening the ‘guardian of genome’: Reactivation of
mutant p53. Cancer Chemother Pharmacol. 83:1–15. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mularski J, Malarz K, Pacholczyk M and
Musiol R: The p53 stabilizing agent CP-31398 and multi-kinase
inhibitors. Designing, synthesizing and screening of
styrylquinazoline series. Eur J Med Chem. 163:610–625. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Beberok A, Rzepka Z, Respondek M, Rok J,
Stradowski M and Wrześniok D: Moxifloxacin as an inducer of
apoptosis in melanoma cells: A study at the cellular and molecular
level. Toxicol In Vitro. 55:75–92. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang LC, Tam KW, Liu WN, Lin CY, Hsu KW,
Hsieh WS, Chi WM, Lee AW, Yang JM, Lin CL and Lee CH: CRISPR/Cas9
genome editing of epidermal growth factor receptor sufficiently
abolished oncogenicity in anaplastic thyroid cancer. Dis Markers.
2018:38357832018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoshimoto K, Iwahana H, Fukuda A, Sano T,
Saito S and Itakura M: Role of p53 mutations in endocrine
tumorigenesis: Mutation detection by polymerase chain
reaction-single strand conformation polymorphism. Cancer Res.
52:5061–5064. 1992.PubMed/NCBI
|
|
22
|
Liu L, Yu TT, Ren CC, Yang L, Cui SH and
Zhang XA: CP-31398 inhibits the progression of cervical cancer
through reversing the epithelial mesenchymal transition via the
downregulation of PAX2s. J Cell Physiol. 234:2929–2942. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhong B, Shingyoji M, Hanazono M, Nguyen
TTT, Morinaga T, Tada Y, Hiroshima K, Shimada H and Tagawa M: A
p53-stabilizing agent, CP-31398, induces p21 expression with
increased G2/M phase through the YY1 transcription factor in
esophageal carcinoma defective of the p53 pathway. Am J Cancer Res.
9:79–93. 2019.PubMed/NCBI
|
|
24
|
Krajewska J, Handkiewicz-Junak D and
Jarzab B: Sorafenib for the treatment of thyroid cancer: An updated
review. Expert Opin Pharmacother. 16:573–583. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang L, Zhang Y, Zhang Q, Zhu G, Zhang Z,
Duan C, Lu T and Tang W: Discovery of potent Pan-Raf inhibitors
with increased solubility to overcome drug resistance. Eur J Med
Chem. 163:243–255. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Salehian B, Liem SY, Mojazi Amiri H and
Maghami E: Clinical trials in management of anaplastic thyroid
carcinoma; progressions and set backs: A systematic review. Int J
Endocrinol Metab. 17:e677592019.PubMed/NCBI
|
|
27
|
Liu TR, Xiao ZW, Xu HN, Long Z, Wei FQ,
Zhuang SM, Sun XM, Xie LE, Mu JS, Yang AK, et al: Treatment and
prognosis of anaplastic thyroid carcinoma: A clinical study of 50
cases. PLoS One. 11:e01648402016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Danilovic DLS, Castro G Jr, Roitberg FSR,
Vanderlei FAB, Bonani FA, Freitas RMC, Coura-Filho GB, Camargo RY,
Kulcsar MA, Marui S and Hoff AO: Potential role of sorafenib as
neoadjuvant therapy in unresectable papillary thyroid cancer. Arch
Endocrinol Metab. 62:370–375. 2018.PubMed/NCBI
|
|
29
|
Arihara Y, Takada K, Kamihara Y, Hayasaka
N, Nakamura H, Murase K, Ikeda H, Iyama S, Sato T, Miyanishi K, et
al: Small molecule CP-31398 induces reactive oxygen
species-dependent apoptosis in human multiple myeloma. Oncotarget.
8:65889–65899. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He XX, Zhang YN, Yan JW, Yan JJ, Wu Q and
Song YH: CP-31398 inhibits the growth of p53-mutated liver cancer
cells in vitro and in vivo. Tumour Biol. 37:807–815. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grassi ES, Vezzoli V, Negri I, Labadi A,
Fugazzola L, Vitale G and Persani L: SP600125 has a remarkable
anticancer potential against undifferentiated thyroid cancer
through selective action on ROCK and p53 pathways. Oncotarget.
6:36383–36399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu J, Timares L, Heilpern C, Weng Z, Li C,
Xu H, Pressey JG, Elmets CA, Kopelovich L and Athar M: Targeting
wild-type and mutant p53 with small molecule CP-31398 blocks the
growth of rhabdomyosarcoma by inducing reactive oxygen
species-dependent apoptosis. Cancer Res. 70:6566–6576. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shibue T, Takeda K, Oda E, Tanaka H,
Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T and Tanaka
N: Integral role of Noxa in p53-mediated apoptotic response. Genes
Dev. 17:2233–2238. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tran Cao HS, Kaushal S, Snyder CS, Ongkeko
WM, Hoffman RM and Bouvet M: Real-time imaging of tumor progression
in a fluorescent orthotopic mouse model of thyroid cancer.
Anticancer Res. 30:4415–4422. 2010.PubMed/NCBI
|
|
36
|
Kim S, Yazici YD, Calzada G, Wang ZY,
Younes MN, Jasser SA, El-Naggar AK and Myers JN: Sorafenib inhibits
the angiogenesis and growth of orthotopic anaplastic thyroid
carcinoma xenografts in nude mice. Mol Cancer Ther. 6:1785–1792.
2007. View Article : Google Scholar : PubMed/NCBI
|