Introduction
Diffuse large B-cell lymphoma (DLBCL) is an
aggressive non-Hodgkin lymphoma (1).
The incidence rate is 6.3%, with an estimated 25,380 new cases in
the United States in 2016 (2). DLBCL
can be classified into three molecular subtypes; the germinal
center B cell-like subtype, the activated B cell-like subtype and
primary mediastinal B cell lymphoma (1). Cyclophosphamide, doxorubicin,
vincristine and prednisolone (CHOP) are the standard treatment for
non-Hodgkin lymphoma (3). The 3-year
overall survival rate is ~60% following CHOP treatment in patients
with DLBCL (4).
Large genomics studies have been conducted to
characterize the genetic alterations in DLBCL genomes, which
provide an unbiased view of the landscape of mutations and the
pathogenesis of the disease (5–7). Lohr
et al (5) performed exome
sequencing on 55 paired tumor and normal samples of primary DLBCL
and identified 58 significantly mutated genes, such as CD79b
molecule (CD79B), Tumor Protein P53 (TP53), caspase
recruitment domain family member 11 (CARD11), MYD88 innate
immune signal transduction adaptor (MYD88) and enhancer of
zeste 2 polycomb repressive complex 2 subunit (EZH2). Reddy
et al (6) identified 150
genetic drivers by performing an integrative analysis of whole
exome sequencing in a cohort of 1,001 patients with DLBCL. In the
previously mentioned study, CRISPR-based knockout of 35 driver
genes resulted in the decreased viability of DLBCL cells,
suggesting these driver genes serve an oncogenic function. MYC
proto-oncogene (MYC), CD79B and zinc finger and
AT-hook domain containing (ZFAT) mutations were
significantly associated with poor overall survival rate of
patients with DLBCL. While, mutations in neurofibromin 1 and
serum/glucocorticoid regulated kinase 1 were associated with
favorable overall survival rate of patients with DLBCL (5). Chapuy et al (7) integrated genetic drivers using
consensus clustering and identified 5 distinct DLBCL subgroups
associated with distinct pathogenic mechanisms and clinical
outcomes. For example, tumors in the activated B cell (ABC) and
germinal center B cell (GCB)-independent group are characterized by
biallelic inactivation of TP53, cyclin dependent kinase
inhibitor 2A loss, and associated with genomic instability
(7).
Driver genes that are recurrently mutated in a large
cohort of cancer samples have consistently been a focus of the
previously published studies of DLBCL; however, the mutation
frequency of numerous driver genes may remain relatively low (e.g.
<1%) in tumors (8). Few studies
have been conducted on the driver genes with low mutation frequency
in DLBCL (5,6). In the present study, four computational
tools were used to identify driver genes and conduct integrative
analyses on these genes in 48 DLBCL samples. The study aimed to
detect novel driver genes, driver pathways and their association
with clinical characteristics of patients with DLBCL; enhancing the
understanding of this disease and providing potential therapeutic
targets in DLBCL.
Materials and methods
Data for analysis of somatic mutations
in DLBCL
In total, 16,918 somatic mutations of 8,672 genes
were identified in 48 patients with DLBCL (22 men and 26 women; age
range, 23–82 years; mean age, 56.27 years) and obtained from The
Cancer Genome Atlas (TCGA) database (9). The functional impact of somatic
mutations was evaluated using Ensembl Variant Effect Predictor
(10) and the mutations were
classified into 9 categories according to their functional impact,
including frame shift insertions and deletions (indels), in frame
indels, missense mutation, nonsense mutation, RNA, silent and
splice site. RNA denotes somatic mutations that are located in the
5′-untranslated region (UTR) or 3′-UTR and may be functional, but
probably act by impacting RNA levels.
Prediction of driver genes and
pathways
Driver genes were predicted using 4 distinct
computational tools, including OncodriveCLUST v.0.4.1 (11), OncodriveFM v.0.0.1 (12), The integrated Cancer Genome Score
(iCAGES,) (13) and Drivers Genes
and Pathways (DrGaP v.0.1.0) (14).
The parameters were set to default values. Driver genes were
determined based on the following criteria: Genes with q-values
<0.05 were considered as driver genes using OncodriveCLUST and
OncodriveFM; genes with iCAGES gene scores >0.5 were determined
as drivers using iCAGES and genes or pathways with P-values
<0.05 were regarded as driver genes or pathways using DrGaP. To
further annotate the driver genes, the list of driver genes were
compared with curated ONGene (15)
and TSGene (16) databases.
Gene Ontology (GO) term and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analyses
In order to characterize the functional enrichment
of all driver genes, GO (17)
biological process terms and KEGG pathway enrichment analyses
(18) were performed with The
Database for Annotation, Visualization and Integrated Discovery
(DAVID) (19). Driver genes were
considered to be significantly enriched in GO terms or KEGG
pathways using a cut-off of Benjamini adjusted P-value of
<0.05.
Co-expression network analysis in
patients with DLBCL
Normalized read counts of driver genes of 48
patients with DLBCL were downloaded from TCGA database (http://firebrowse.org/?cohort=DLBC&download_dialog=true).
Co-expression networks were constructed with the R package of
weighted gene co-expression network analysis (WGCNA version 1.67)
using normalized read counts of driver genes (20). The softpower and minimum number of
genes were set as 7 and 10 respectively, all other parameters were
set to the default values. Identification of gene co-expression
modules was conducted using hierarchical average-linkage
clustering. The dynamic tree-cut algorithm was used to identify
modules and genes in the same branch that could be assigned to
different modules (20). Genes with
high intramodular connectivity were considered as intramodular hub
genes. The clinicopathologic characteristics investigated in the
study are patient age, sex, clinical stage [Ann Arbor staging
system; (21)], radiation therapy,
ethnicity, survival status and follow-up time and were obtained
from the TCGA database. Module-trait associations were estimated
using the correlation between the module eigengene and clinical
traits, which enables the identification of modules highly
correlated with clinical features.
Protein-protein interaction (PPI)
network construction and analysis
A PPI network was constructed using the Search Tool
for the Retrieval of Interacting Genes/Proteins (STRING) database
(22). Visualization and calculation
of degree value for each node was performed using Cytoscape
software v3.7.2 (23). Degree
centrality was defined as the number of connections one node has
and was also analyzed using Cytoscape software. Hub nodes which
have the highest degree of centrality connect most adjacent
proteins in the PPI network (22).
Furthermore, Molecular Complex Detection (MCODE) (24) was used to detect hub clustering
modules in the PPI network with default parameters in Cytoscape. GO
and KEGG pathway enrichment analyses were also conducted for genes
in significant modules.
Copy number variation (CNV)
analyses
Focal CNVs and gene-level CNVs of 48 DLBCL samples
were detected using the GISTIC algorithm (25) and were downloaded from TCGA database
(9). Focal CNVs were considered
statistically significant at the cut-off value of q<0.25. The
gene-level copy-number alterations of the top 20 frequently altered
driver genes were clustered using the heatmap.2 function of gplots
package in R (26).
Bootstrap model validation of survival
analyses
In order to confirm the associations of driver gene
expression with overall survival rate in patients with DLBCL,
bootstrap methodology (27) was used
for validation. Bootstrapping methodology randomly selected 80% of
samples with replacement from the original dataset as a ‘training’
set to determine the median values for driver genes. The original
dataset was then used as a ‘testing’ set in which the patients were
divided into high and low-expression groups according to the median
values. The log-rank test was used to compare the difference in
survival rates between the high- and low-expression groups using
the R package of survival V3.1–11 (28). This process was repeated 1,000 times,
generating 1,000 P-values for each driver gene and the frequency of
P<0.05 was counted for each driver gene.
Statistical analysis
The difference in mutation density between groups
were compared by Wilcoxon rank-sum test. Linear regression model
was used to characterize the associations between clinical
features, driver genes and overall survival rate. To establish the
association of driver gene expression with overall survival rate of
patients with DLBCL, patients were assigned to the
‘high-expression’ group if they exhibited gene expression levels
greater than the median values of driver gene expression and to the
‘low-expression’ group if they exhibited expression levels lower
than the median value. Kaplan-Meier survival analysis was
performed, and survival curves generated. The log-rank test was
used to compare the difference in survival rates between the high-
and low-expression groups using the R package of survival v3.1–11
(28). P<0.05 was considered to
indicate a statistically significant difference.
Results
Somatic mutations in patients with
DLBCL
In total, 16,918 somatic mutations were detected in
patients with DLBCL (n=48). Somatic mutations were comprised of
9,623 missense, 6,230 silent, 188 splice-site, 353 nonsense, 9 RNA
and 515 indels. Of the 515 indels, 332 caused reading frame shifts,
and 135 deletions and 48 insertions were located in open reading
frames (Fig. S1A). C>T/G>A,
A>G/T>C and C>A/G>T were the 3 predominant transitions,
with mutation rates of 52.9, 17.5 and 8.5% respectively (Fig. S1B). Indels accounted for 3.2% of
variants in DLBCL (Fig. S1B). The
somatic mutation density ranged between 0.68–131.29
mutations/megabase (Mb) with an average mutation density of 9.64
mutations/Mb (data not shown). To understand the cause of the
mutation density variation, mutation statuses in the DNA
mismatch-repair (MMR) pathway genes mutL homolog 1 (MLH1),
mutL homolog 3 (MLH3), mutS homolog 2 (MSH2), mutS
homolog 3 (MSH3), mutS homolog 6 (MSH6) and PMS1
homolog 2 (PMS2) were analyzed. This revealed that 11
patients with DLBCL had mutations in one of the MMR genes and the
average mutation density in patients harboring an MMR mutation was
significantly higher compared with wild-type MMR (23.97
mutations/Mb vs. 5.40 mutations/Mb; P<0.01; Fig. S1C). Notably, the DLBCL with the
highest mutation density (131.29 mutations/Mb) had 1 missense
mutation in PMS2 (data not shown).
Prediction of driver genes and
pathways
Overall, 8,672 genes were mutated in ≥ one DLBCL
sample. There were 12, 47, 109 and 59 driver genes predicted by
OncodriveCLUST, OncodriveFM, iCAGES and DrGaP respectively
(Tables SI–IV). Combining these 4 sets of driver
genes, a total of 208 unique driver genes were detected using all 4
tools. Zinc finger protein 814 (ZNF814), major
histocompatibility complex, class I, C (HLA-C),
CD79B, rhophilin Rho GTPase binding protein 2
(RHPN2), MYD88 and EZH2 were the common genes
identified by OncodriveCLUST and OncodriveFM. Suppressor of
cytokine signaling 1 (SOCS1), TP53, signal transducer
and activator of transcription 6 (STAT6), actin beta
(ACTB), protein tyrosine phosphatase non-receptor type 6
(PTPN6) and LYN proto-oncogene (LYN) were common to
OncodriveFM and iCAGES. Fas cell surface death receptor
(FAS), inhibitor of nuclear factor kappa B kinase subunit
beta (IKBKB), tumor necrosis factor (TNF) and
TP53 were common driver genes detected by iCAGES and DrGaP.
TP53, BTG anti-proliferation factor 2 (BTG2) and
ubiquitin conjugating enzyme E2 A (UBE2A) were common to
OncodriveFM and DrGaP, MLH1 was the overlapping driver gene
found between OncodriveCLUST and iCAGES. TP53 was the only
driver gene predicted by OncodriveFM, iCAGES and DrGaP. Among the
208 driver genes; immunoglobulin lambda like polypeptide 5
(IGLL5), myeloid/lymphoid or mixed-lineage leukemia 2
(MLL2), BTG anti-proliferation factor 2 (BTG2),
beta-2-microglobulin (B2M) and Pim-1 proto-oncogene,
serine/threonine kinase (PIM1) were the top five
recurrently-mutated genes in patients with DLBCL with mutation
rates of 41.7, 35.4, 33.3, 27.1, 25.0, respectively (Fig. 1; Table
SV). The majority of driver genes were mutated at a low
frequency in DLBCL with an average mutation rate of 6.1% (Table SV). By comparing the list of driver
genes with curated ONGene and TSGene databases, numerous known
oncogenes were identified in the current study, such as signal
transducer and activator of transcription 3 (STAT3),
epidermal growth factor receptor (EGFR), as well as tumor
suppressor genes, such as ATM serine/threonine kinase (ATM),
phosphatase and tensin homolog (PTEN). In addition to the
list of driver genes, DrGaP also identified 31 driver pathways in
DLBCL, including the MAPK signalling pathway, cytokine-cytokine
receptor interaction, cell cycle, apoptosis, p53 signalling
pathway, pathways in cancer, pancreatic cancer, the Wnt signalling
pathway and chronic myeloid leukemia (data not shown).
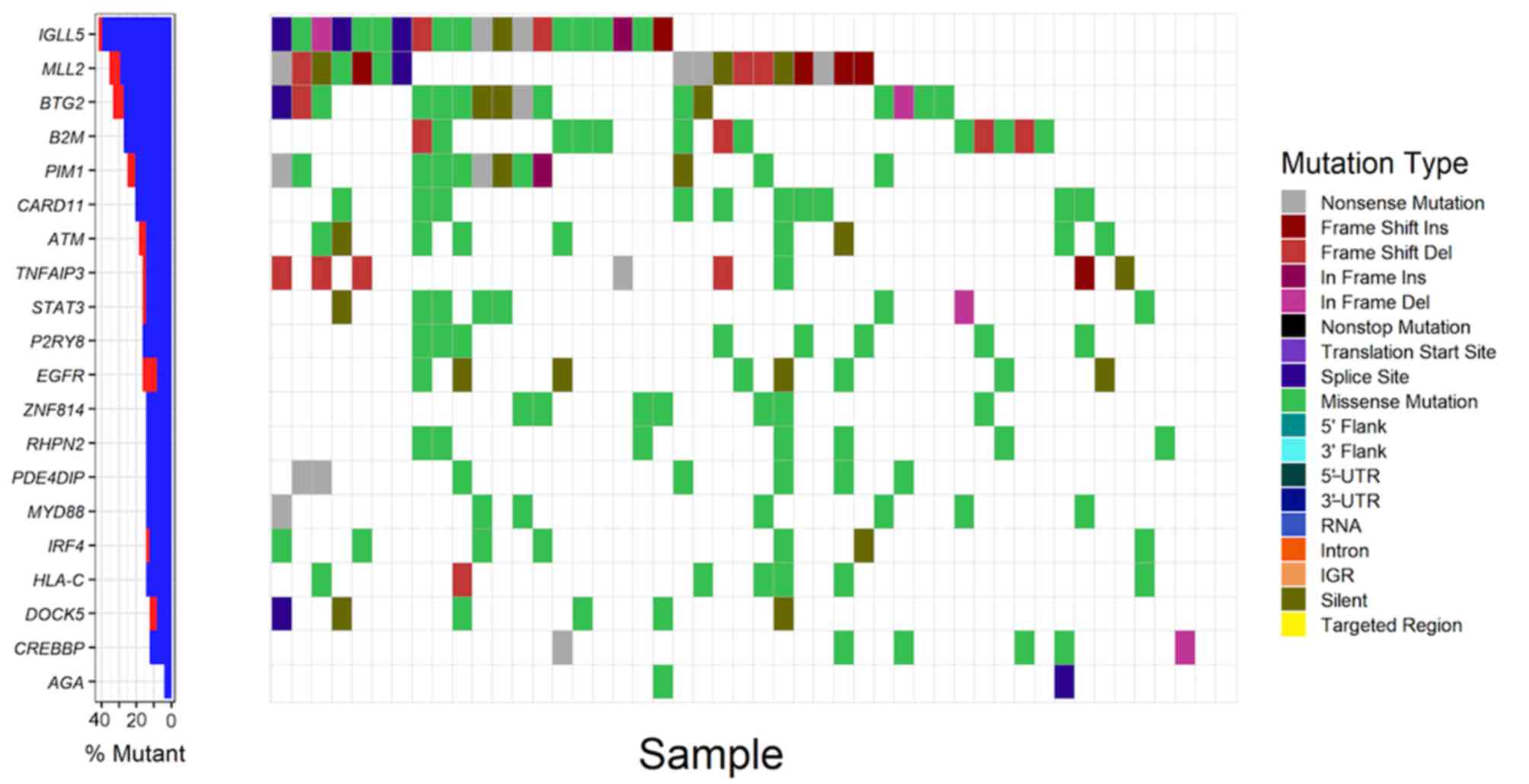 | Figure 1.Analysis of somatic mutations of the
top 20 frequently mutated driver genes in patients with DLBCL
(n=48). The left panel demonstrates the mutation rates of the 20
driver genes, blue and red denote synonymous and non-synonymous
mutations respectively. The right panel demonstrates the
distribution of mutations with different classes of functions in
patient samples. UTR, untranslated region; IGR, intergenic region;
DLBCL, diffuse large B cell lymphoma; Ins, insertion; Del,
deletion; IGLL5, Immunoglobulin lambda like polypeptide 5; MLL2,
myeloid/lymphoid or mixed-lineage leukemia 2; BTG2, BTG
anti-proliferation factor 2; B2M, beta-2-microglobulin; PIM1, Pim-1
proto-oncogene; CARD11, caspase recruitment domain family member
11; ATM, ATM serine/threonine kinase; TNFAIP3, TNF alpha induced
protein 3; STAT3, signal transducer and activator of transcription
3; P2YR8, P2Y receptor family member 8; EGFR, epidermal growth
factor receptor; ZNF184, zinc finger protein 184; RHPN2, rhophilin
Rho GTPase binding protein 2; PDE4DIP, phosphodiesterase 4D
interacting protein; MYD88, MYD88 innate immune signal transduction
adaptor; IRF4, interferon regulatory factor 4; HLA-C, major
histocompatibility complex class I C; DOCK5, dedicator of
cytokinesis 5; CREBBP, CREB binding protein; AGA,
aspartylglucosaminidase. |
GO term and KEGG pathway enrichment
analyses
GO term and KEGG pathway enrichment analyses were
performed for 208 driver genes using DAVID. GO enrichment analysis
indicated that driver genes were significantly overrepresented in
35 biological processes (Benjamini-adjusted P-value <0.05;
Table SVI). The main GO biological
processes exhibited a wide spectrum of functional processes,
including ‘IκB kinase/NF-κB signaling’, ‘extracellular matrix
organization’ and ‘regulation of phosphatidylinositol 3-kinase
signaling’ DAVID also revealed driver genes were significantly
enriched in 88 KEGG pathways, including ‘acute myeloid leukemia’,
‘melanoma’, ‘colorectal cancer’, ‘non-small cell lung cancer’, ‘T
cell receptor signaling pathway’, ‘antigen processing and
presentation’, ‘the mTOR signaling pathway’, ‘apoptosis’ and
‘cell cycle’ (Benjamini-adjusted P-value <0.05; Table SVII).
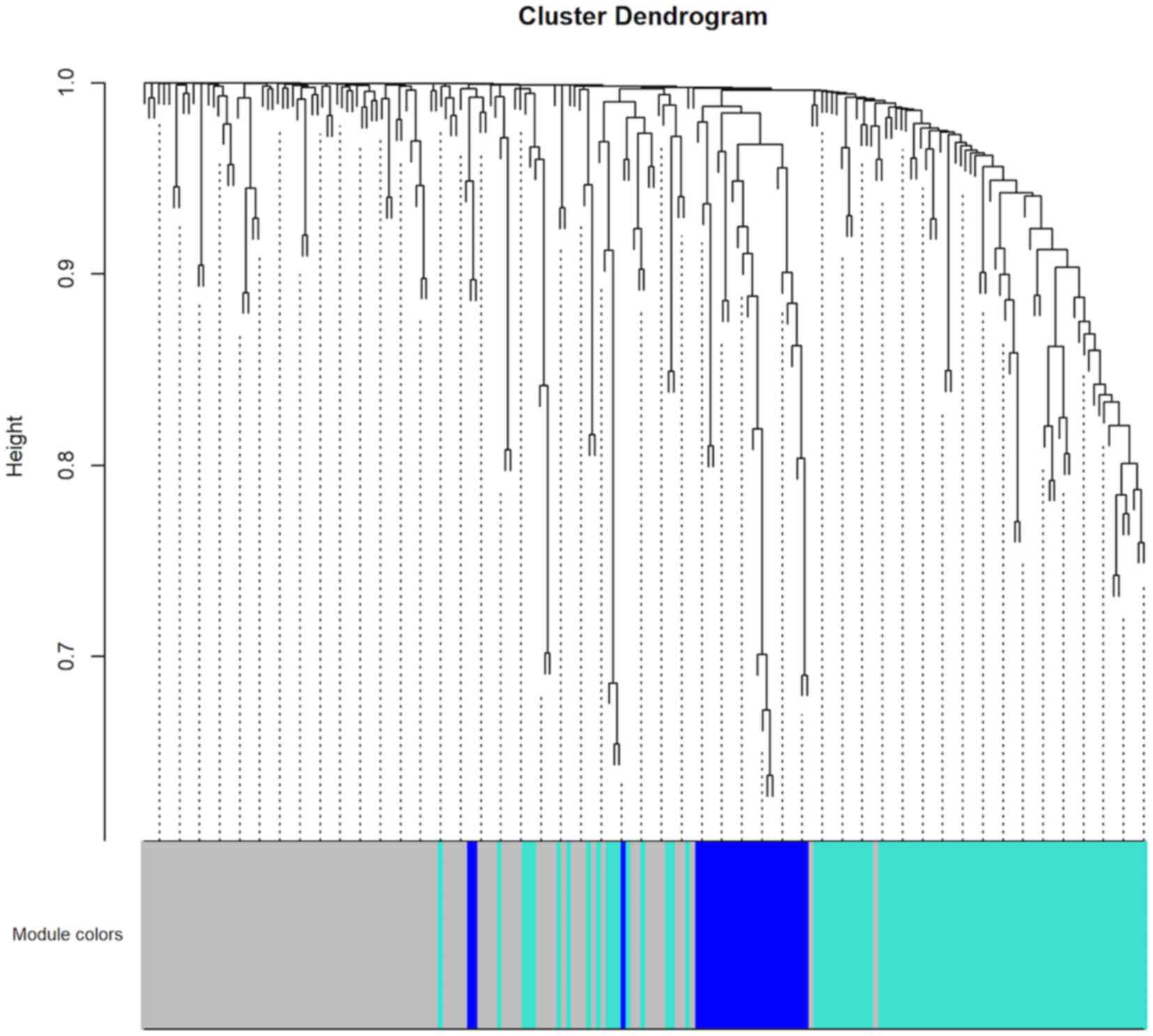
Co-expression network analysis
To construct the co-expression network of the 208
driver genes, WGCNA was used based on the expression correlation
between driver genes in 48 DLBCL samples. WGCNA analysis identified
3 distinct co-expression modules in DLBCL. These co-expression
modules are demonstrated in different colors with 94, 83 and 26
genes in the grey, turquoise and blue modules respectively
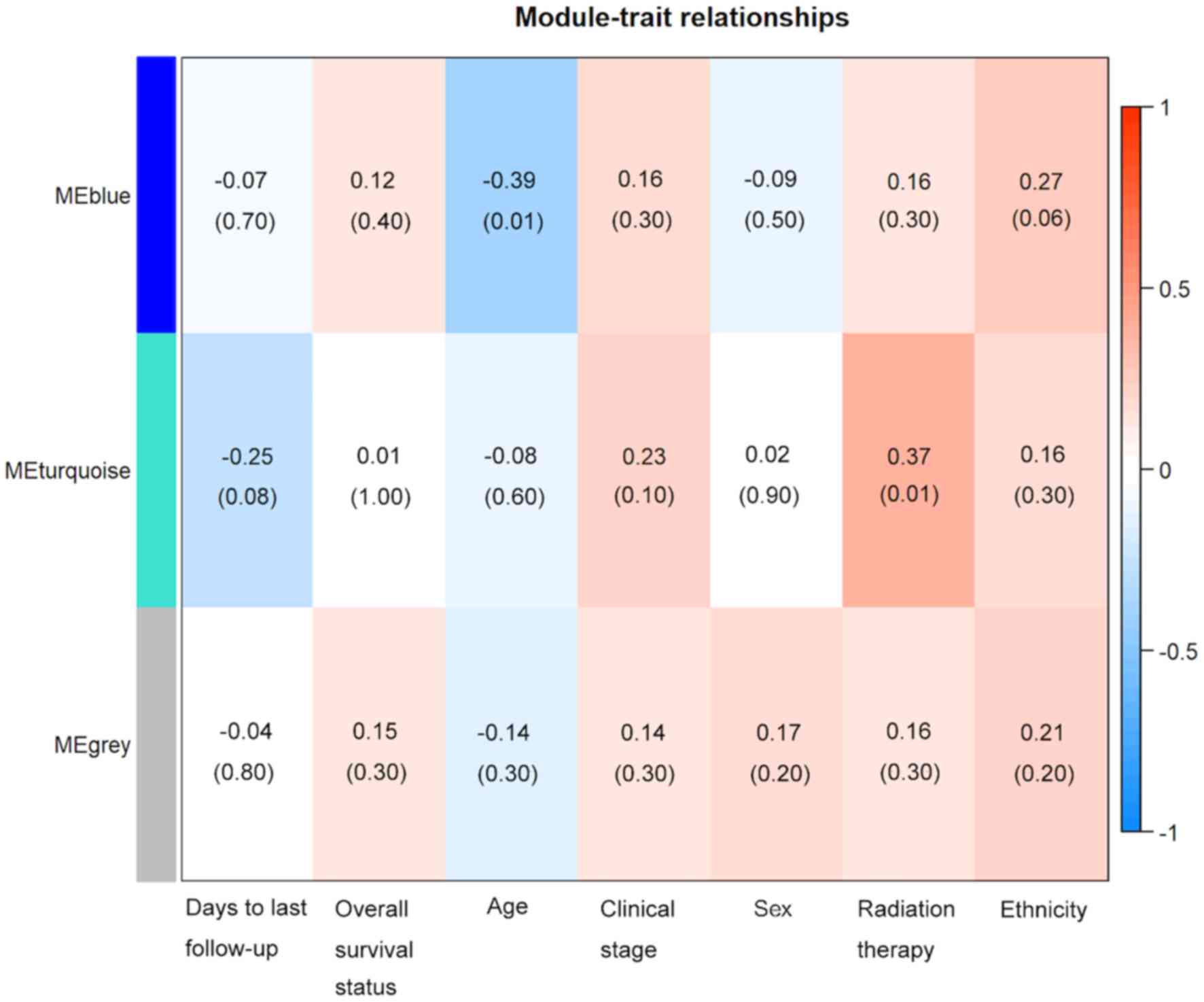
(Fig. 2). The module-trait
association analysis indicated that the turquoise module was
positively correlated with radiation therapy and the blue module
was negatively correlated with patient age (P<0.05; Fig. 3). Mitogen-activated protein kinase 8
(MAPK8) was the hub gene in the turquoise module. Notch
receptor 3 (NOTCH3) was the hub gene in the blue module.
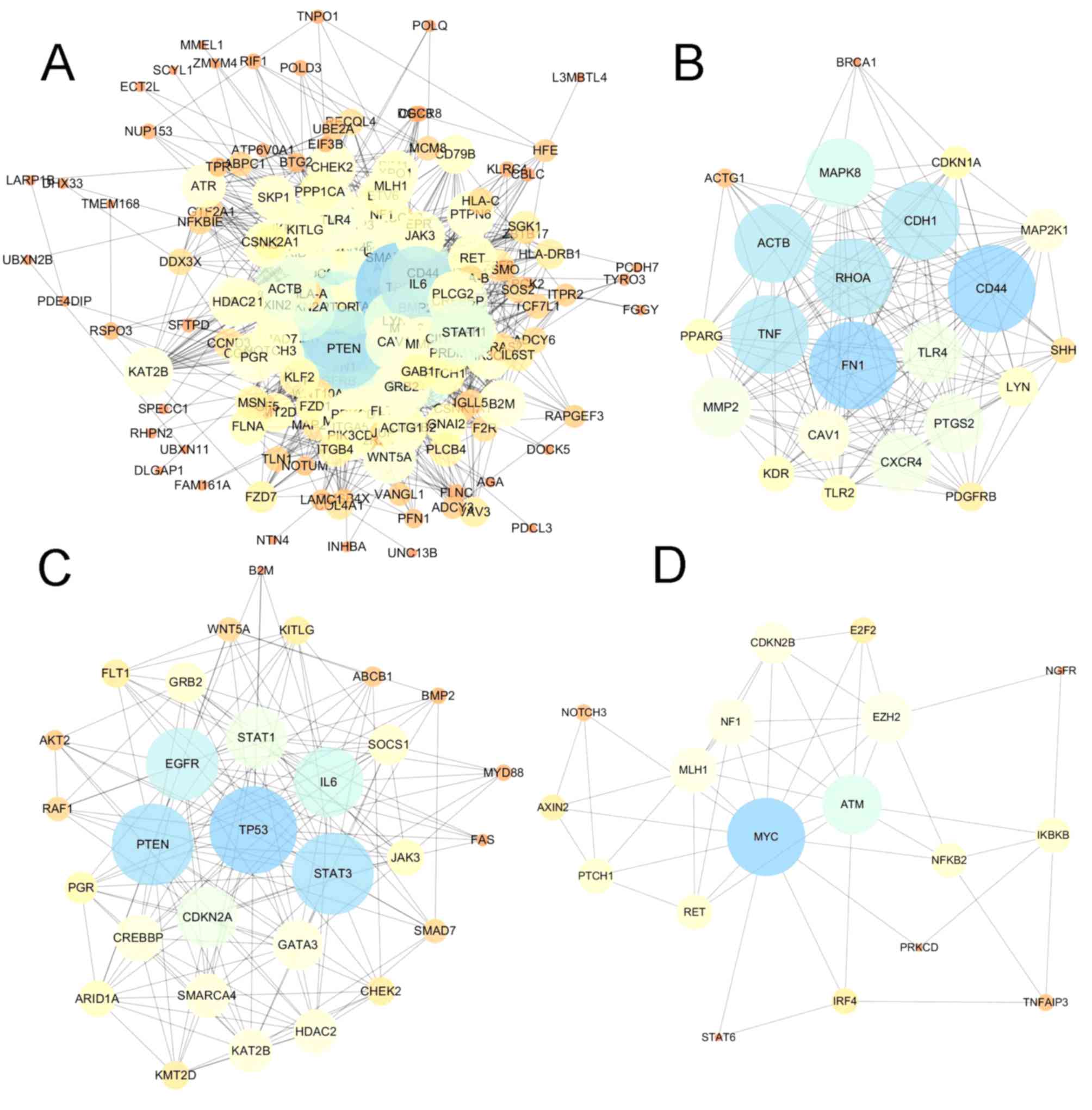
Protein-protein interaction (PPI)
network construction and analysis
In addition to the co-expression network of driver
genes, the present study also characterized the interactions of
driver genes at the protein level. STRING was used to construct a
PPI network for driver genes. The PPI network comprised 208 nodes
and 2,041 edges, with an average node degree of 19.6 (Fig. 4A). The PPI network had significantly
more interactions than expected for a random set of proteins of
similar size (PPI enrichment; P<0.0001). The nodes which have
high degrees possess intensive interactions with other nodes and
may serve as key nodes in the PPI network. A total of 9 candidate
hub nodes, the degree of which was >4 times the corresponding
median values were identified, namely, TP53, MYC, EGFR,
PTEN, interleukin 6 (IL6), signal transducer and
activator of transcription 3 (STAT3), mitogen-activated
protein kinase 8 (MAPK8), tumor necrosis factor (TNF)
and cadherin 1 (CDH1) (Fig.
4A). Furthermore, module analysis was performed to obtain the
top 3 modules with high scores using MCODE (Fig. 4B-D). The 9 candidate hub nodes were
contained in the three modules. In relation to GO enrichment
analysis, genes in module 1 (Fig.
4B) were significantly correlated with 208 GO terms, including
‘positive regulation of apoptosis’, ‘programmed cell death’, ‘cell
migration’ and ‘response to hypoxia.’ Genes in module 2 (Fig. 4C) were primarily enriched in
‘regulation of apoptotic process’, ‘cell proliferation’, ‘MAPK
cascade’ and ‘phosphatidylinositol-mediated signaling’. Genes in
module 3 (Fig. 4D) were not
significantly enriched in any GO terms. With respect to KEGG
pathway enrichment analysis, the genes in module 1 were enriched in
‘leukocyte transendothelial migration’, ‘melanoma’ and ‘Toll-like
receptor signaling pathway’. The genes in module 2 mainly were
predominantly enriched in ‘chronic myeloid leukemia’, ‘acute
myeloid leukemia’, ‘p53 signaling pathway’ and the
‘hypoxia-inducible factor 1 signaling pathway’. The genes in module
3 were significantly implicated in the ‘cell cycle’, ‘microRNAs in
cancer’, ‘small-cell lung cancer’ and the ‘NF-κB signaling
pathway’.
Copy number variation (CNV)
analyses
Focal CNVs of 48 patients with DLBCL were obtained
from TCGA. Significant focal gains and deletions (q<0.25) were
identified at 40 loci (14 amplifications and 26 deletions) in 93.8%
(45/48) of DLBCL samples. Among them, deletions at 6q14.1 and
9p21.3, and amplifications at 1q24.2, 2p16.1 and 7p22.3 were the
top 5 most frequent CNVs in DLBCL, with occurrence rates of 35.4
(17/48), 35.4 (17/48), 33.3 (16/48), 33.3 (16/48) and 33.3%
(16/48), respectively (Fig. S2).
PR/SET domain 1 (PRDM1), cyclin dependent kinase inhibitor
2A (CDKN2A), cyclin dependent kinase inhibitor 2B
(CDKN2B), TNF alpha induced protein 3 (TNFAIP3) and
R-spondin 3 (RSPO3) were the top five most frequently
deleted driver genes in DLBCL, while actin beta (ACTB), BTG
anti-proliferation factor 2 (BTG2), placenta expressed
transcript 1 (PLET1), CARD11 and DIX domain
containing 1 (DIXDC1) were the top five most frequently
amplified driver genes in DLBCL (Fig.
S3).
Prognosis of patients with DLBCL
Linear regression model analysis demonstrated
overall survival rate was not significantly associated with patient
age, clinical stage, radiation therapy, sex and ethnicity in DLBCL
(all P>0.05; Table SVIII). To
evaluate the association of driver gene expression with patient
survival, patients with DLBCL were divided into low- and
high-expression groups based on the median expression values of the
driver genes. Kaplan-Meier survival analysis indicated that
patients with high eukaryotic translation initiation factor 3
subunit B (EIF3B), mutL homolog 1(MLH1), protein
phosphatase 1 catalytic subunit alpha (PPP1CA) and RecQ like
helicase 4 (RECQL4) expression levels exhibited improved
overall survival rate compared with those with low EIF3B, MLH1,
PPP1CA and RECQL4 expression levels (Fig. 5). Patients with high exportin 1
(XPO1) and LYN expression exhibited a less favorable
prognosis compared with patients with low XPO1 and
LYN expression (all P<0.05; Fig. 5). To further verify the
aforementioned findings, driver genes were evaluated for their
associations with overall survival rate using Kaplan-Meier survival
analysis with 1,000 bootstrap resampling. EIF3B, MLH1, PPP1CA,
RECQL4, XPO1 and LYN were significantly associated with
overall survival rate in patients with DLBCL across the 1,000
bootstrapped samples. EIF3B, MLH1, PPP1CA, RECQL4 and
XPO1 exhibited a P-value <0.05 in >60% of the 1,000
testing datasets (EIF3B, 82.6%; MLH1, 94.1%;
PPP1CA, 83.2%; RECQL4, 93%; and XPO1, 64.8%),
while LYN had a P-value <0.05 in only 29.2% of the
testing datasets (data not shown). The current results indicate
that EIF3B, MLH1, PPP1CA, RECQL4 and XPO1 may
represent potential prognostic biomarkers for patients with DLBCL
in the future.
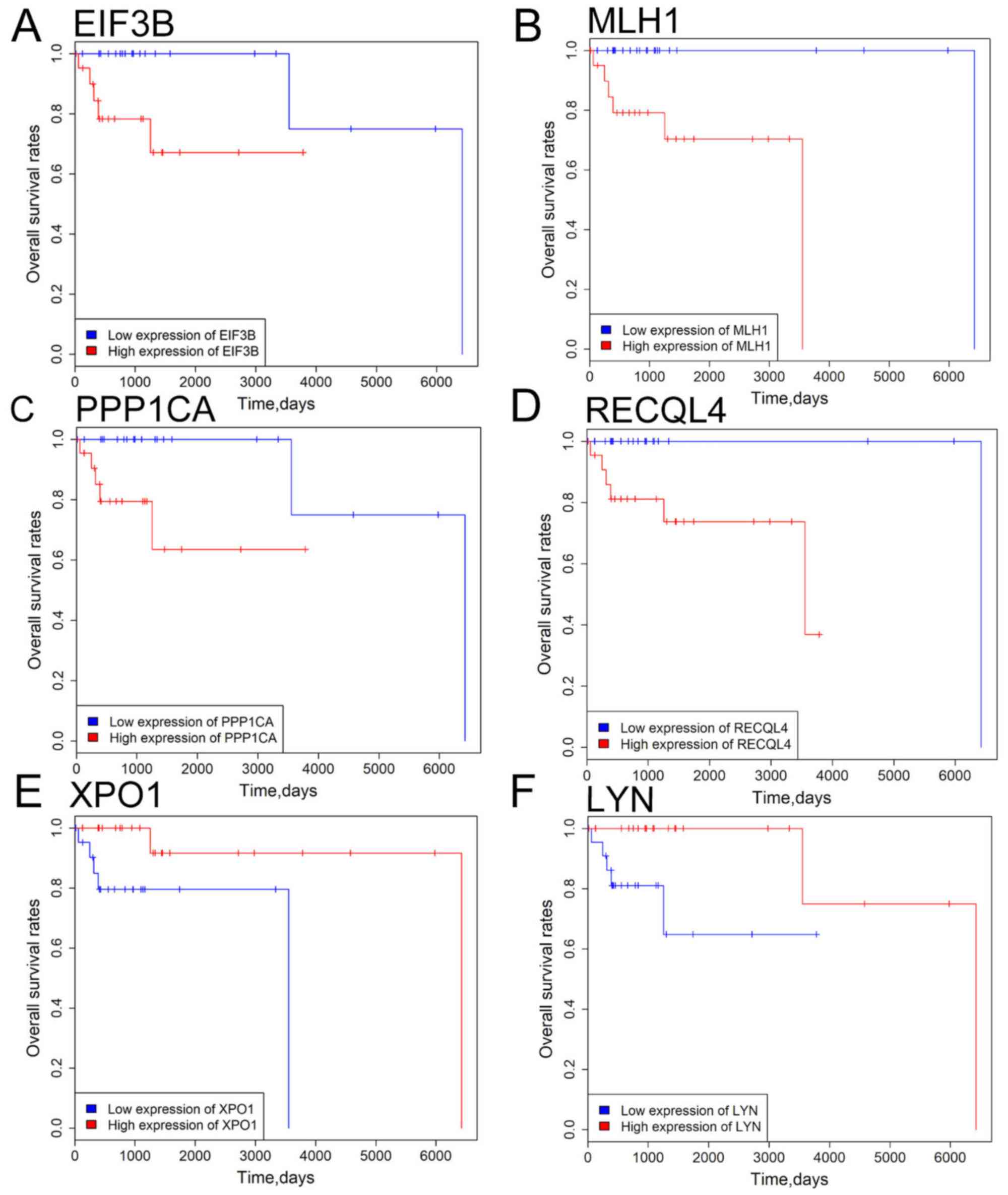 | Figure 5.Survival analysis of 6 driver genes
in patients with DLBCL. (A) EIF3B, (B) MLH1, (C)
PPP1CA, (D) RECQL4 expression, (E) XPO1 and
(F) LYN expression levels were significantly associated with
overall survival rate of DLBCL patients. Red and blue curves
represent high- and low-expression groups, respectively. P<0.05
was considered to be statistically significant. EIF3B, eukaryotic
translation initiation factor 3 subunit B; MLH1, mutL homolog 1;
PPP1CA, protein phosphatase 1 catalytic subunit alpha; RECQL4, RecQ
like helicase 4; XPO1, exportin 1; LYN, LYN proto-oncogene. |
Discussion
Cancer is initiated by the accumulation of driver
mutations in cancer genes, which confers a proliferation advantage
to cancer cells (29). The average
mutation rate is 9.64 mutations/Mb in DLBCL, which is higher
compared with the mutation rate in other hematopoietic
malignancies, such as chronic lymphocytic leukemia and other
leukemias (29,30), and multiple myeloma (31). In the present study, the mutation
density varied considerably across DLBCL samples with increased
mutation rates in MMR-mutant samples. Moreover, the DLBCL sample
with the highest mutation density had one missense mutation in
PMS2, which is a key component of the mismatch repair system to
correct DNA mismatches and small indels that occur during DNA
replication and homologous recombination (32). The results of the present study
suggested that mutation density variation is, to a large extent,
attributable to mutations in the DNA mismatch repair genes in
DLBCL.
The widely applied approach for the detection of
driver genes identifies significantly mutated genes in a cohort of
cancer samples as compared with the background mutation rate
(33,34). In the present study, 4 computational
tools, OncodriveCLUST, OncodriveFM, iCAGES and DrGaP were used to
detect driver genes using somatic mutations of patients with DLBCL
(n=48). MLL2, TP53, CD79B, B2M, CARD11 and EZH2 were
predicted as driver genes in DLBCL in the present study, which is
consistent with previously published reports (5,6). By
comparing the list of driver genes with curated oncogene (15) and tumor suppressor gene (16) databases, numerous known oncogenes
were identified in the current study, such as EGFR, STAT3,
as well as tumor suppressor genes, such as ATM, PTEN.
Notably, in the present study a large fraction of driver genes had
low mutation frequencies and were first reported as driver genes in
DLBCL, such as CSNK2A1, RECQL4, LARP1B and GAB1.
Therefore, the combination of the 4 tools enabled the detection of
recurrently and rarely mutated driver genes. For instance, DrGap
detected a new driver gene IGLL5, which was not identified
via the other 3 computational tools. This may be due to DrGaP
predicting driver genes and driver signaling pathways according to
a different algorithm. DrGaP integrates biological knowledge of the
mutational process in tumors, including the length of
protein-coding regions, transcript isoforms, variation in mutation
types, differences in background mutation rates, redundancy of the
genetic code and multiple mutations in one gene. DrGaP use a
Poisson process to model the random nature of somatic mutations, a
Bayesian model to estimate background mutation rates and a
likelihood ratio test to test the significance of driver genes and
pathways. The newly identified driver genes in the present study
provide promising candidates for functional validation in future
studies.
By performing WGCNA analysis in the present study, 3
co-expression modules were detected, the turquoise module was
positively associated with radiation therapy and the blue module
was negatively associated with patient age. MAPK8 was the
hub gene in the turquoise module. NOTCH3 was the hub gene
indicating that these genes were correlated with other genes at the
mRNA expression level. Therefore, they may have key roles in the
co-expression network. The PPI network analysis also identified 9
candidate hub nodes, namely, TP53, MYC, EGFR, PTEN, IL6, STAT3,
MAPK8, TNF and CDH1, and 3 modules. The 9 nodes
identified in the present study were major hub nodes and the 3
modules may represent the key biological characteristics in the PPI
network.
Finally, in the present study 5 driver genes were
significantly associated with the overall survival rate of patients
with DLBCL, including EIF3B, MLH1, PPP1CA, RECQL4 and
XPO1. Of the five genes, XPO1 has been reported to be
oncogene and a negative prognostic factor in mantle cell lymphoma
(35), lung adenocarcinoma (36) and gastric cancer (37). XPO1 encodes a protein which
functions as the trafficker of a wide range of proteins, including
tumor suppressors, growth regulatory, proinflammatory and
antiapoptotic proteins (38).
XPO1 serves oncogenic and anti-apoptotic roles in
transformed cells and is upregulated in mantle cell lymphoma
(35), lung adenocarcinoma (36) and gastric cancer (37). In concordance with the findings of
the present study, upregulated expression of XPO1 is
associated with poor prognosis in gastric carcinoma (37), acute myeloid leukemia (39), pancreatic cancer (40) and lung adenocarcinoma (31). The results obtained in the present
study, combined with previously published reports (35–39),
indicate that XPO1 may exert oncogenic functions and
represent a negative prognostic factor in cancers.
Expression analysis of EIF3B, MLH1, PPP1CA,
RECQL4 and XPO1 may be valuable in clinical settings.
Cytological or surgical specimens of DLBCL exhibiting high
expression of EIF3B, MLH1, PPP1CA, RECQL4 and low expression
of XPO1 may be associated with a favorable clinical outcome,
which needs to be verified in large-scale and more vigorous future
studies.
Despite enhancing the understanding of pathogenesis
of DLBCL, the present study is not without limitations. For
example, there was a lack of functional validation for the novel
driver genes. Furthermore, the overall survival rate-associated
genes were not validated in an independent DLBCL dataset, due to
lack of publicly available DLBCL data. Overall, the present study
discovered a set of driver genes and driver pathways in DLBCL, and
demonstrated that the driver genes, such as EIF3B, MLH1, PPP1CA,
RECQL4 and XPO1 may serve as potential prognostic
biomarkers for patients with DLBCL.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Science and
Technology Bureau of Yinzhou (grant no. 20191JCGY020007).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available in the figshare repository (https://figshare.com/s/268d9f525ceb5d172b60).
Authors' contributions
YL designed the study. RP, KS and LC downloaded
somatic mutation, RNA-seq data, CNVs and clinical data from the
TCGA database. KS, LC, ZF, TW and RP predicted driver genes and
pathways, performed WGCNA co-expression, PPI network, CNV and
survival analyses. LC, ZF, RP and KS participated in the writing
and revision of the manuscript. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DLBCL
|
diffuse large B cell lymphoma
|
|
TCGA
|
The Cancer Genome Atlas
|
|
iCAGES
|
Integrated Cancer Genome Score
|
|
GO
|
Gene ontology
|
|
WGCNA
|
weighted gene correlation network
analysis
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
STRING
|
Search Tool for the Retrieval of
Interacting Genes and Proteins
|
|
DAVID
|
Database for Annotation,
Visualization and Integrated Discovery
|
|
CNV
|
copy number variation
|
References
|
1
|
Lenz G and Staudt LM: Aggressive
lymphomas. N Engl J Med. 362:1417–1429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Teras LR, DeSantis CE, Cerhan JR, Morton
LM, Jemal A and Flowers CR: 2016 US lymphoid malignancy statistics
by world health organization subtypes. CA Cancer J Clin.
66:443–459. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fisher RI, Gaynor ER, Dahlberg S, Oken MM,
Grogan TM, Mize EM, Glick JH, Coltman CA Jr and Miller TP:
Comparison of a standard regimen (CHOP) with three intensive
chemotherapy regimens for advanced non-Hodgkin's lymphoma. N Engl J
Med. 328:1002–1006. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pfreundschuh M, Schubert J, Ziepert M,
Schmits R, Mohren M, Lengfelder E, Reiser M, Nickenig C, Clemens M,
Peter N, et al: Six versus eight cycles of bi-weekly CHOP-14 with
or without rituximab in elderly patients with aggressive CD20+
B-cell lymphomas: A randomised controlled trial (RICOVER-60).
Lancet Oncol. 9:105–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lohr JG, Stojanov P, Lawrence MS, Auclair
D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW,
Slager SL, et al: Discovery and prioritization of somatic mutations
in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing.
Proc Natl Acad Sci. 109:3879–3884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reddy A, Zhang J, Davis NS, Moffitt AB,
Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-
Lindsberg ML, et al: Genetic and functional drivers of diffuse
large B cell lymphoma. Cell. 171:481–494.e15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chapuy B, Stewart C, Dunford AJ, Kim J,
Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et
al: Molecular subtypes of diffuse large B cell lymphoma are
associated with distinct pathogenic mechanisms and outcomes. Nat
Med. 24:679–690. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ld W, Dw P, Jones S, Lin J, Sjöblom T,
Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The genomic
landscapes of human breast and colorecta l cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hoadley KA, Yau C, Hinoue T, Wolf DM,
Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et
al: Cell-of- origin patterns dominate the molecular classification
of 10,000 tumors from 33 types of cancer. Cell. 173:291–304.e6.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Y, Cunningham F, Rios D, McLaren WM,
Smith J, Pritchard B, Spudich GM, Brent S, Kulesha E, Marin- Garcia
P, et al: Ensembl variation resources. BMC Genomics. 11:2932010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tamborero D, Gonzalez-Perez A and
Lopez-Bigas N: OncodriveCLUST: Exploiting the positional clustering
of somatic mutations to identify cancer genes. Bioinformatics.
29:2238–2244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gonzalez-Perez A and Lopez-Bigas N:
Functional impact bias reveals cancer drivers. Nucleic Acids Res.
40:e1692012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong C, Guo Y, Yang H, He Z, Liu X and
Wang K: iCAGES: Integrated CAncer GEnome score for comprehensively
prioritizing driver genes in personal cancer genomes. Genome Med.
8:1352016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hua X, Xu H, Yang Y, Zhu J, Liu P and Lu
Y: DrGaP: A powerful tool for identifying driver genes and pathways
in cancer sequencing studies. Am J Hum Genet. 93:439–451. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu Y, Sun J and Zhao M: ONGene: A
literature-based database for human oncogenes. J Genet Genomics.
44:2016–2018. 2016.
|
|
16
|
Zhao M, Sun J and Zhao Z: TSGene: A web
resource for tumor suppressor genes. Nucleic Acids Res. 41:970–976.
2013. View Article : Google Scholar
|
|
17
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ogata H, Goto S, Sato K, Fujibuchi W, Bono
H and Kanehisa M: KEGG: Kyoto encyclopedia of genes and genomes.
Nucleic Acids Res. 27:29–34. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang DW, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carbone PP, Kaplan HS, Musshoff K,
Smithers DW and Tubiana M: Report of the committee on Hodgkin's
disease staging classification. Cancer Res. 31:1860–1861.
1971.PubMed/NCBI
|
|
22
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45((D1)): D362–D368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mermel CH, Schumacher SE, Hill B, Meyerson
ML, Beroukhim R and Getz G: GISTIC2.0 facilitates sensitive and
confident localization of the targets of focal somatic copy-number
alteration in human cancers. Genome Biol. 12:R412011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Warnes G, Bolker B, Bonebakker L,
Gentleman R, Huber W, Liaw A, Lumley T, Mächler MM and Steffen AM:
gplots: Various R programming tools for plotting data. R package
version. 2:https://www.researchgate.net/publication/303186599_gplots_Various_R_programming_tools_for_plotting_dataMay–2005
|
|
27
|
Stine R: An introduction to bootstrap
methods: Examples and ideas. Sociol Methods Res. 18:243–291. 1989.
View Article : Google Scholar
|
|
28
|
Fox J: Cox proportional-hazards regression
for survival data the cox proportional-hazards model. An R and
S-PLUS companion to applied regression. 2002:1–18. 2002.
|
|
29
|
Greenman C, Stephens P, Smith R, Dalgliesh
GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C,
et al: Patterns of somatic mutation in human cancer genomes.
Nature. 446:153–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Puente XS, Pinyol M, Quesada V, Conde L,
Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz
M, et al: Whole-genome sequencing identifies recurrent mutations in
chronic lymphocytic leukaemia. Nature. 475:101–105. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chapman MA, Lawrence MS, Keats JJ,
Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann
GJ, Adli M, et al: Initial genome sequencing and analysis of
multiple myeloma. Nature. 471:467–472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kolodner RD and Marsischky GT: Eukaryotic
DNA mismatch repair. Curr Opin Genet Dev. 9:89–96. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dees ND, Zhang Q, Kandoth C, Wendl MC,
Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis
ER, et al: MuSiC: Identifying mutational significance in cancer
genomes. Genome Res. 22:1589–1598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshimura M, Ishizawa J, Ruvolo V, Dilip
A, Quintás-Cardama A, McDonnell TJ, Neelapu SS, Kwak LW, Shacham S,
Kauffman M, et al: Induction of p53-mediated transcription and
apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma.
Cancer Sci. 105:795–801. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao W, Lu C, Chen L and Keohavong P:
Overexpression of CRM1: A characteristic feature in a transformed
phenotype of lung carcinogenesis and a molecular target for lung
cancer adjuvant therapy. J Thorac Oncol. 10:815–825. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou F, Qiu W, Yao R, Xiang J, Sun X, Liu
S, Lv J and Yue L: CRM1 is a novel independent prognostic factor
for the poor prognosis of gastric carcinomas. Med Oncol.
30:7262013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishizawa J, Kojima K, Hail N Jr, Tabe Y
and Andreeff M: Expression, function, and targeting of the nuclear
exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol
Ther. 153:25–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kojima K, Kornblau SM, Ruvolo V, Dilip A,
Duvvuri S, Davis RE, Zhang M, Wang Z, Coombes KR, Zhang N, et al:
Prognostic impact and targeting of CRM1 in acute myeloid leukemia.
Blood. 121:4166–4174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang WY, Yue L, Qiu WS, Wang LW, Zhou XH
and Sun YJ: Prognostic value of CRM1 in pancreas cancer. Clin
Invest Med. 32:E3152009. View Article : Google Scholar : PubMed/NCBI
|