Introduction
Epidermal growth factor receptor (EGFR), a member of
the receptor tyrosine kinase (RTK) family, plays an important role
in many fundamental cellular processes, including proliferation,
differentiation, migration, and survival (1–3). Due to
these crucial functions, an activating mutation in EGFR may lead to
malignancies, including non-small cell lung cancer (NSCLC) and
glioblastoma multiforme (GBM, glioma grade IV) (4–6).
Although major activating mutations in EGFR occur in
the intracellular kinase domain in NLCLCs (7), several deletion mutations in the
extracellular domain (ECD) are found in GBM (1,8). An
oncogenic variant III (EGFRvIII), which contains a deletion of 267
amino acids spanning exons 2–7 of the EGFR gene, is the most
common active mutant and is present in 25–33% of all GBM patients
(3,9,10). This
mutation leads to an incapability to bind any known EGFR ligand;
however, it exhibits constitutive tyrosine kinase activity
(3,11–14).
EGFRvIII downstream signaling displays distinct features in signal
strength from wild-type EGFR (EGFRwt) (2,8). It has
less kinase activity than ligand-activated EGFRwt; however, this
constitutive activity is sufficient to provoke downstream signaling
(8). EGFRvIII is generally
co-expressed with wild-type EGFR (EGFRwt); therefore, the
ligand-induced activation of EGFRwt affects the oncogenic potential
of EGFRvIII. Discrepancies have been noted in previous findings on
the synergistic or antagonistic effects of EGFRwt on EGFRvIII
functions (2).
We previously provided evidence for the negative
feedback regulation of EGFR/ErbB family kinases by the
non-canonical phosphorylation of conserved threonine residues in
the juxtamembrane domain (15–17). The
ERK kinase, a main regulator of oncogenic EGFR/ErbB signaling, is
involved in phosphorylation of the threonine residue, which results
in rapid feedback inhibition of the tyrosine kinase activity of
ErbB receptor dimers (15).
In the present study, we attempted to investigate
the role of the feedback loop in the activation of the EGFRvIII
mutant, and found that the ERK-induced phosphorylation of
juxtamembrane Thr-402 (corresponding to conserved Thr-669 of
EGFRwt) reduced the expression level of constitutively
phosphorylated tyrosine in U87MG human glioblastoma cells. We also
demonstrated that the EGF-induced activation of EGFRwt rapidly
induced the activation of EGFRvIII, and then converted it to an
inactivation signal for EGFRvIII via an ERK-mediated feedback
mechanism.
Materials and methods
Antibodies and reagents
Phospho-specific antibodies against EGFR (Tyr-1068
and Thr-669) and ERK (Thr-202/Tyr-204), were purchased from Cell
Signaling Technology. Antibodies against total EGFR (A-10) and
actin (C-11) were obtained from Santa Cruz Biotechnology (Santa
Cruz Biotechnology, Inc.). Recombinant human EGF and trametinib
were obtained from R&D Systems and Cayman Chemical,
respectively. TPA (12-O-tetradecanoylphorbol-13-acetate) and
the Phos-tag ligand were purchased from Wako Pure Chemical
Industries. SCH772984 was purchased from Chemietek. All chemical
inhibitors were dissolved in dimethyl sulfoxide (DMSO), and the
final concentration of DMSO was less than 0.1%.
Cell lines and culture conditions
Human U87MG glioblastoma cells that overexpress
EGFRwt and EGFRvIII were provided by Professors Webster K. Cavenee
(University of California San Diego) and Motoo Nagane (Kyorin
University) (18,19). The original U87MG cells (glioblastoma
of unknown origin) were obtained from the American Type Culture
Collection. 293 cells were obtained from the ATCC. All cells were
cultured in Dulbecco's Modified Eagle's medium (DMEM) supplemented
with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and
100 µg/ml streptomycin at 37°C in 5% CO2.
Cell transfection
Human EGFRvIII cDNA was amplified by reverse
transcription-PCR and inserted into the pcDNA3.1 vector. Plasmid
DNAs were transfected into 293 cells with Lipofectamine 2000
reagent (Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol. The substitution of Thr-669 to Ala was
generated by site-directed mutagenesis with KOD FX Neo Polymerase
(TOYOBO).
Western blotting
Whole cell lysates were prepared in lysis buffer
containing 20 mM β-glycerophosphate, 1 mM dithiothreitol (DTT), 1
mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF),
10 µg/ml aprotinin, and 10 µg/ml leupeptin. Each sample was mixed
with the same volume of sample buffer [100 mM Tris-HCl (pH. 6.8),
2.0% SDS, 70 mM DTT, 10% glycerol, and 0.10% bromophenol blue] and
then heated at 95°C for 5 min. Cell lysates were subjected to
6.5–10% SDS-PAGE and were then transferred to an Immobilon-P
transfer membrane (Merck Millipore Ltd.). The membrane was blocked
with BlockAce (Dainippon Sumitomo Pharmaceutical Co., Ltd.) and
incubated with an appropriate primary antibody at room temperature.
The membrane was then incubated with secondary antibodies, either
anti-rabbit or anti-mouse conjugated to horseradish peroxidase
(DAKO), diluted in PBS containing 0.1% Tween-20 (Wako Pure Chemical
Industries). Signals were detected with an enhanced
chemiluminescence (ECL) system (Thermo Fisher Scientific, Inc.).
Some antibody reactions were performed in Can Get Signal solution
(TOYOBO).
Zn2+ Phos-tag SDS-PAGE
Whole cell lysates were prepared with RIPA buffer as
described previously (20,21). Samples were mixed with a half volume
of SDS-PAGE sample buffer [195 mM Tris-HCl (pH. 6.8), 30% glycerol,
15% 2-mercaptoethanol, 3% SDS, and 0.10% bromophenol blue], and
then heated at 95°C for 5 min. The acrylamide pendant Phos-tag
ligand and two equivalents of ZnCl2 were added to the
separating gel before polymerization. The running buffer for
Zn2+ Phos-tag electrophoresis consisted of 100 mM Tris
and 100 mM MOPS containing 0.1% SDS and 5.0 mM sodium bisulfite.
Furthermore, the gel was washed in solution containing 25 mM Tris,
192 mM glycine, 10% methanol, and 1.0 mM EDTA for 20 min, then
washed using a solution containing 25 mM Tris, 192 mM glycine, and
10% methanol for 20 min.
Results
TPA-induced feedback phosphorylation
of EGFR via the MEK-ERK pathway
We initially investigated whether feedback
regulation occurs in EGFRwt-overexpressing U87MG human glioma
cells. A Phos-tag immunoblot analysis detected a shift in EGFR
bands by the TPA stimulation, indicating the strong phosphorylation
of EGFR (Fig. 1A). In normal
immunoblot analyses, ERK activation and Thr-669 phosphorylation
(EGFR phosphorylated on Thr-669 is described as pT-EGFR) were
up-regulated in a similar time course (Fig. 1B). Conversely, tyrosine
phosphorylation on Tyr-1068 (EGFR phosphorylated on Tyr-1068 is
described as pY-EGFR) was gradually decreased (Fig. 1B). In addition, trametinib, a MEK
inhibitor (22), completely blocked
the negative feedback loop, maintaining pY-EGFR, even in the
presence of TPA (Fig. 1C). These
results confirmed that the MEK-ERK pathway controlled the negative
feedback regulation of EGFRwt by juxtamembrane phosphorylation in
glioblastoma cells.
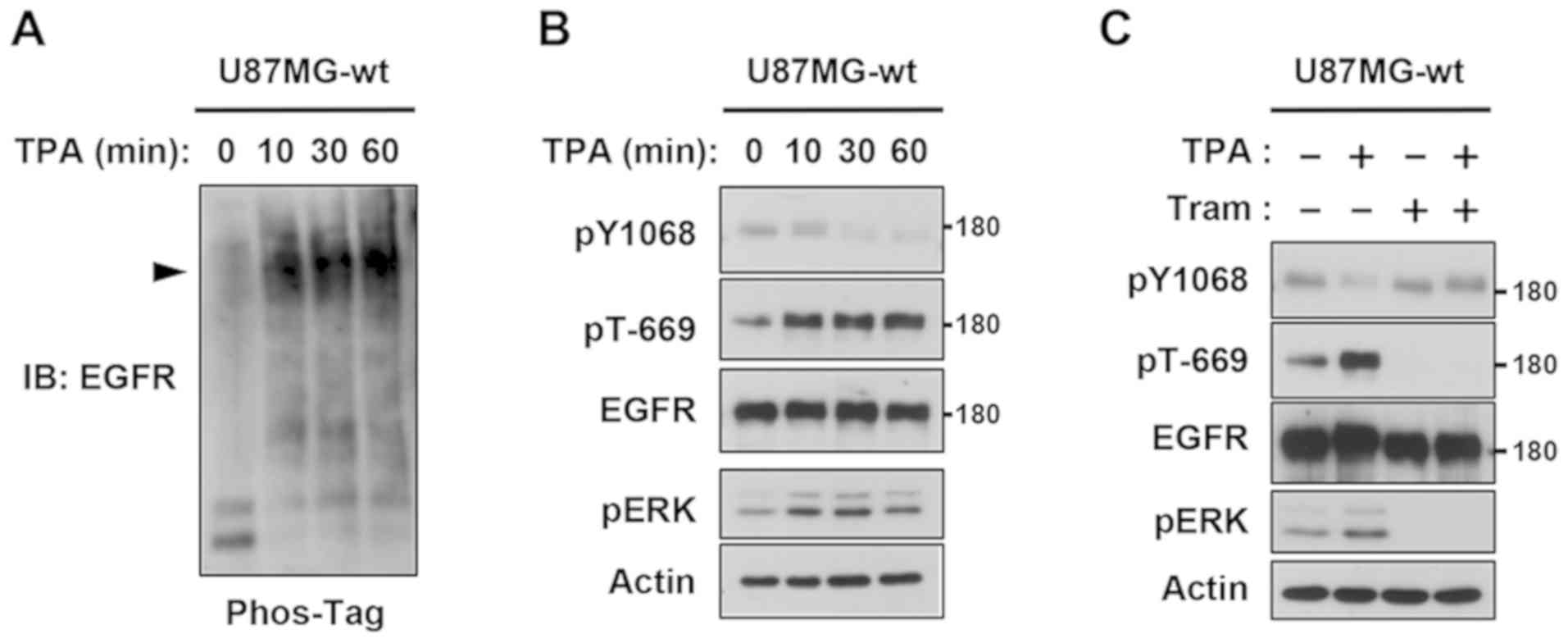 | Figure 1.TPA-induced feedback phosphorylation
of EGFR via the ERK pathway. (A) Whole cell lysates from U87MG-wt
cells stimulated with 100 ng/ml TPA for the indicated periods were
separated by Zn2+ Phos-tag SDS-PAGE, followed by
immunoblotting (IB) with EGFR antibody. (B) Cells were stimulated
with 100 ng/ml TPA then the whole-cell lysates were electrophoresed
and immunoblotted with phospho-EGFR (Thr-669 and Tyr-1068), EGFR,
phospho-ERK, and actin antibodies. (C) Cells were pretreated with
0.03 µM trametinib (Tram) for 30 min, and then stimulated with TPA
for another 10 min. Whole cell lysates were separated by normal
SDS-PAGE followed by immunoblotting with primary antibodies against
phospho-EGFR (Thr-669 and Tyr-1068), EGFR, phospho-ERK, and actin.
EGFR, phospho-ERK and actin antibodies. TPA,
12-O-tetradecanoylphorbol-13-acetate; EGFR, epidermal growth
factor receptor; wt, wild-type; Tram, trametinib; IB,
immunoblotting. |
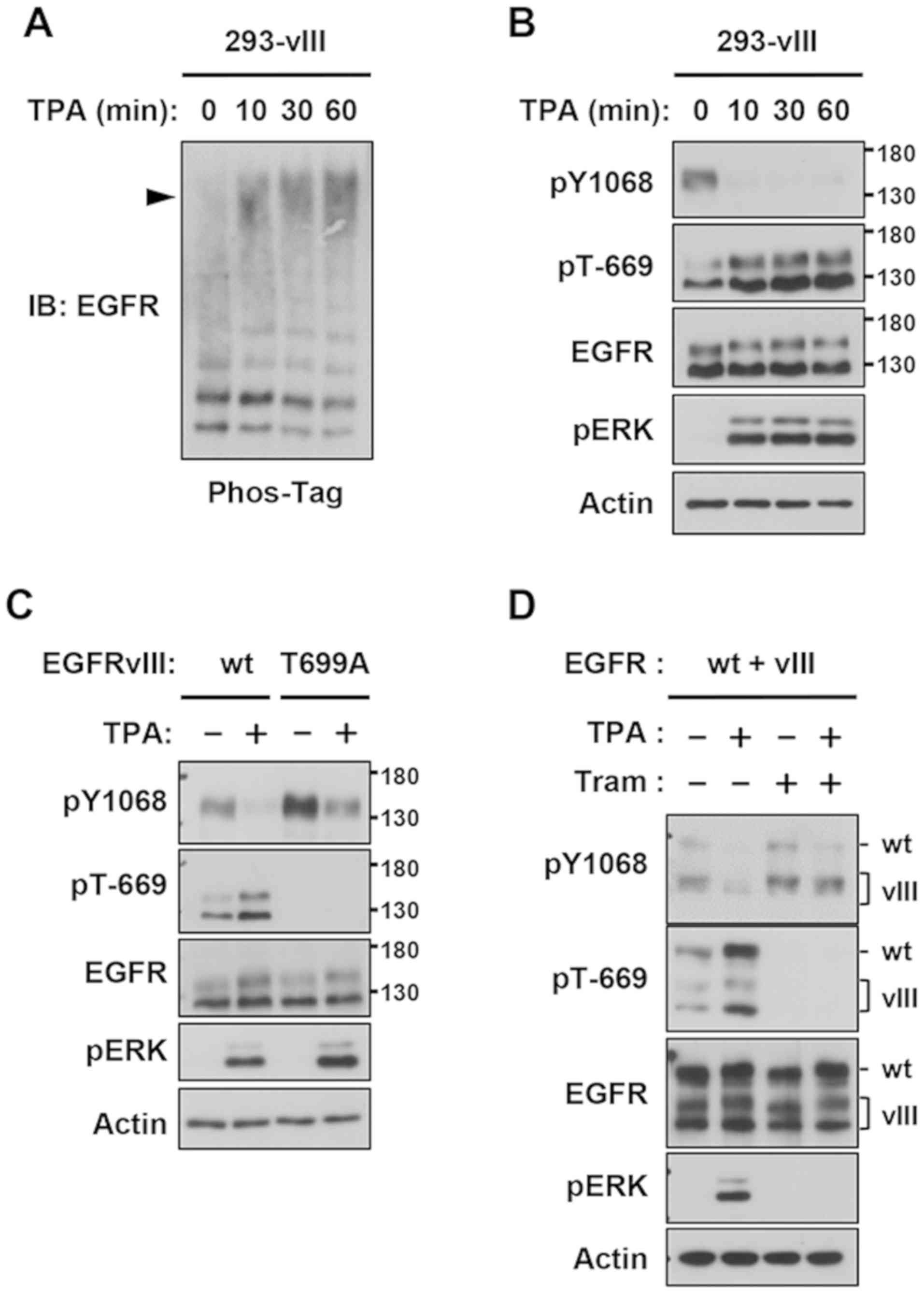
Down-regulation of the constitutive
tyrosine phosphorylation of EGFRvIII in 293 cells
We performed transient transfection experiments
using 293 cells to characterize the feedback regulation of
EGFRvIII. Thr-402 of EGFRvIII is the residue corresponding to
feedback Thr-669 of EGFRwt; therefore, Thr-402 of EGFRvIII was also
indicated as Thr-669 in the present study. Similar to the results
obtained using U87MG-wt cells (Fig.
1A), TPA caused a Phos-tag band shift of EGFRvIII (Fig. 2A). The immunoblot analysis
demonstrated that constitutive pY-EGFRvIII completely disappeared
with TPA, which inversely correlated with the up-regulation of the
ERK-mediated Thr-669 phosphorylation of EGFRvIII (Fig. 2B). To elucidate the role of Thr-669
in feedback inhibition, Thr-669 was substituted to alanine (T669A).
The lack of Thr-669 resulted in increased pY-EGFRvIII in the
absence of TPA. In addition, although ERK was strongly activated,
the down-regulation of pY-EGFRvIII was not observed in the
EGFRvIII-T669A mutant (Fig. 2C).
Moreover, to elucidate the role of Thr-669 in the EGFRwt/EGFRvIII
heterodimer, 293 cells were co-transfected with these EGFR
plasmids. TPA efficiently induced the phosphorylation of Thr-669
and dephosphorylation of Tyr-1068 in EGFRwt and EGFRvIII, and
trametinib completely blocked these feedback reactions (Fig. 2D). Collectively, these results
clearly demonstrated that negative feedback occurred either in the
EGFRvIII homodimer or EGFRwt/EGFRvIII heterodimer through
ERK-mediated Thr-669 phosphorylation.
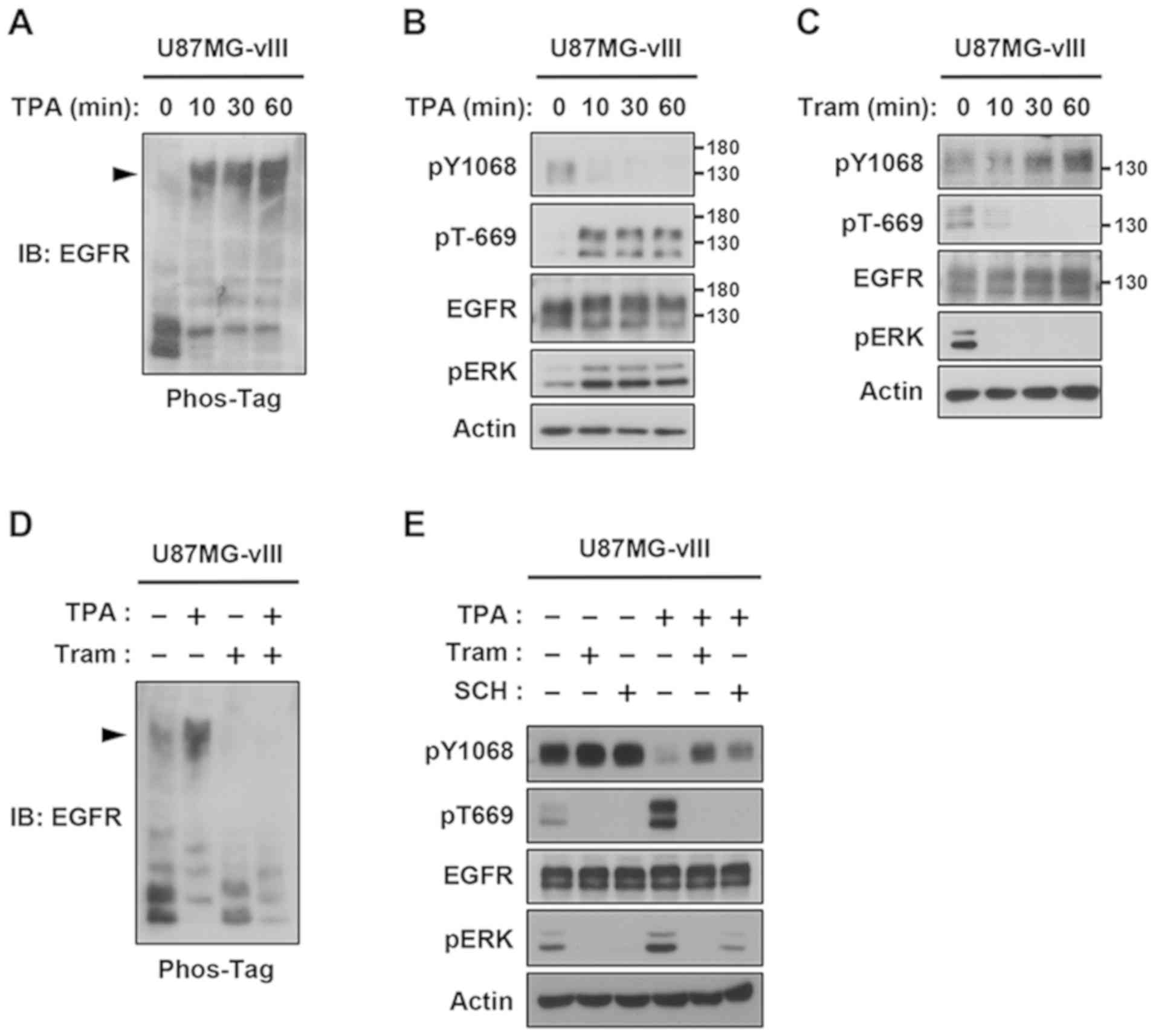
Negative feedback regulation of
EGFRvIII in glioblastoma cells
We attempted to provide direct evidence of negative
feedback regulation in glioma cells using U87MG cells
overexpressing EGFRvIII. Similar to the results obtained using 293
cells (Fig. 2), TPA induced a
Phos-tag band shift and negative feedback reactions of EGFRvIII on
Tyr-1068 and Thr-669 in U87MG-vIII cells (Fig. 3A and B). Since U87MG-vIII cells
constitutively express a basal level of active ERK, we examined the
effects of selective inhibitor of MEK1/2, trametinib (22,23), on
the phosphorylation of Tyr-1068 and Thr-669. In parallel to the
rapid reduction in ERK activation and pT-EGFRvIII, pY-EGFRvIII
gradually increased, indicating that basal EGFRvIII activity was
also controlled by the phosphorylation of Thr-669 (Fig. 3C). Furthermore, trametinib
effectively canceled the TPA-induced feedback regulation of
EGFRvIII (Fig. 3D and E). In
addition, we investigated the effect of a new selective ERK1/2
inhibitor, SCH772984, on the phosphorylation of Tyr-1068 and
Thr-669 (23). This inhibitor also
canceled the TPA-induced inhibition of EGFRvIII (Fig. 3E). Overall, these results revealed
that ERK activates the Thr-669-dependent EGFRvIII feedback pathway
in glioma cells.
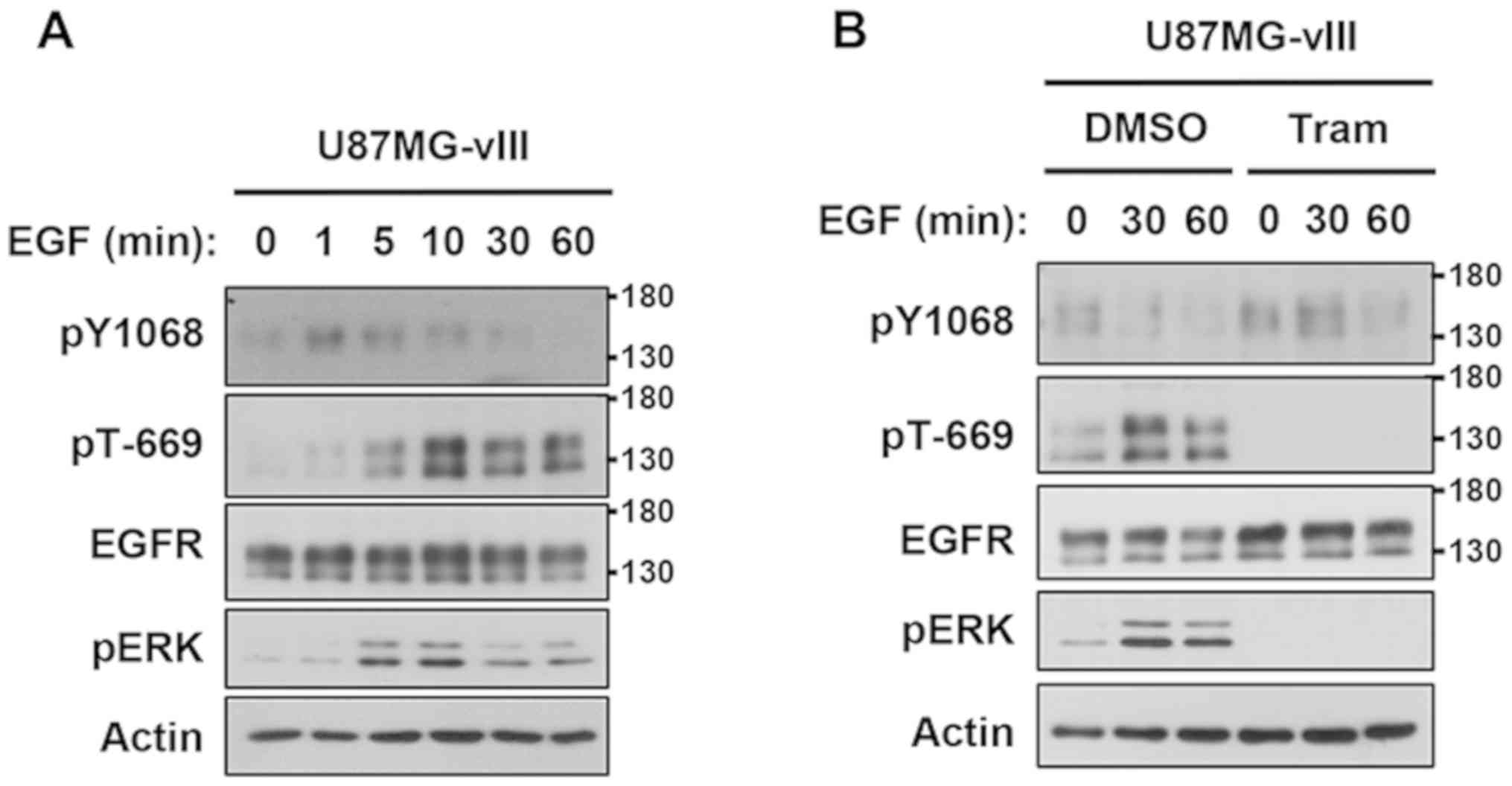
Ligand-induced negative feedback
inhibition of EGFRvIII
EGFRwt is an important counterpart for EGFRvIII to
promote or antagonize EGFRvIII activity (24–26).
Since EGFRvIII is frequently co-expressed with EGFRwt in GBM
(2,6), we examined the effects of EGF on the
negative feedback regulation of EGFRvIII. The tyrosine
phosphorylation of EGFRvIII increased within 1 min, gradually
decreased, and was completely suppressed at 60 min in accordance
with the inverse increase in the phosphorylation of ERK and Thr-669
(Fig. 4A). Since EGFRvIII is unable
to bind ligands (3,11,13),
these inducible events were evoked by endogenous EGFRwt. Thus, the
status of EGFRvIII was rapidly changed from the activated to
silenced form by the EGF-induced activation of EGFRwt. In addition,
the EGF stimulation failed to promote the feedback regulation of
EFGRvIII when cells were pretreated with trametinib (Fig. 4B). Collectively, these results
demonstrated the physiological feedback inhibition of EGFRvIII as
an outcome of ligand stimulation.
Discussion
Cancer cells may attenuate negative feedback to
promote oncogenic growth factor signaling and establish acquired
resistance to anti-cancer agents. We previously reported that the
conserved juxtamembrane threonine residue plays a major role in the
feedback mechanism of EGFR/ErbB receptor via ERK activation
(15–17). Recent study by Soo-Jung Kwon and
colleagues demonstrated MEK partner-1 that encoded by LAMTOR3 gene
contributes to the neural cancer stemness by control ERK activity
in glioma cells expressing EGFRvIII (27). Considering that MEK/ERK pathway is
one of important downstream signaling in EGFR (28), in the present study we investigated
the role of conserved juxtamembrane threonine in
EGFRvIII-expressing glioblastoma cells.
There are two opposing observations related to the
co-expression of EGFRwt and EGFRvIII in human glioblastoma, namely,
synergistic and antagonistic interactions (2). Fan and colleagues reported that EGFR
and EGFRvIII collaborated to activate STAT transcription factors
(25). In contrast, Li and
colleagues found an antagonistic interaction, in which
ligand-induced EGFRwt signaling caused the inactivation of c-Met by
inducing the dissociation of a c-Met-EGFRvIII complex (29). A similar antagonistic role was found
in the EGFR-mediated activation of NF-kB in glioma cells (30). In order to provide further evidence
for this controversial issue, we attempted to elucidate the role of
negative feedback phosphorylation in the functional interaction
between EGFRwt and EGFRvIII. Although EGFRvIII is incapable of
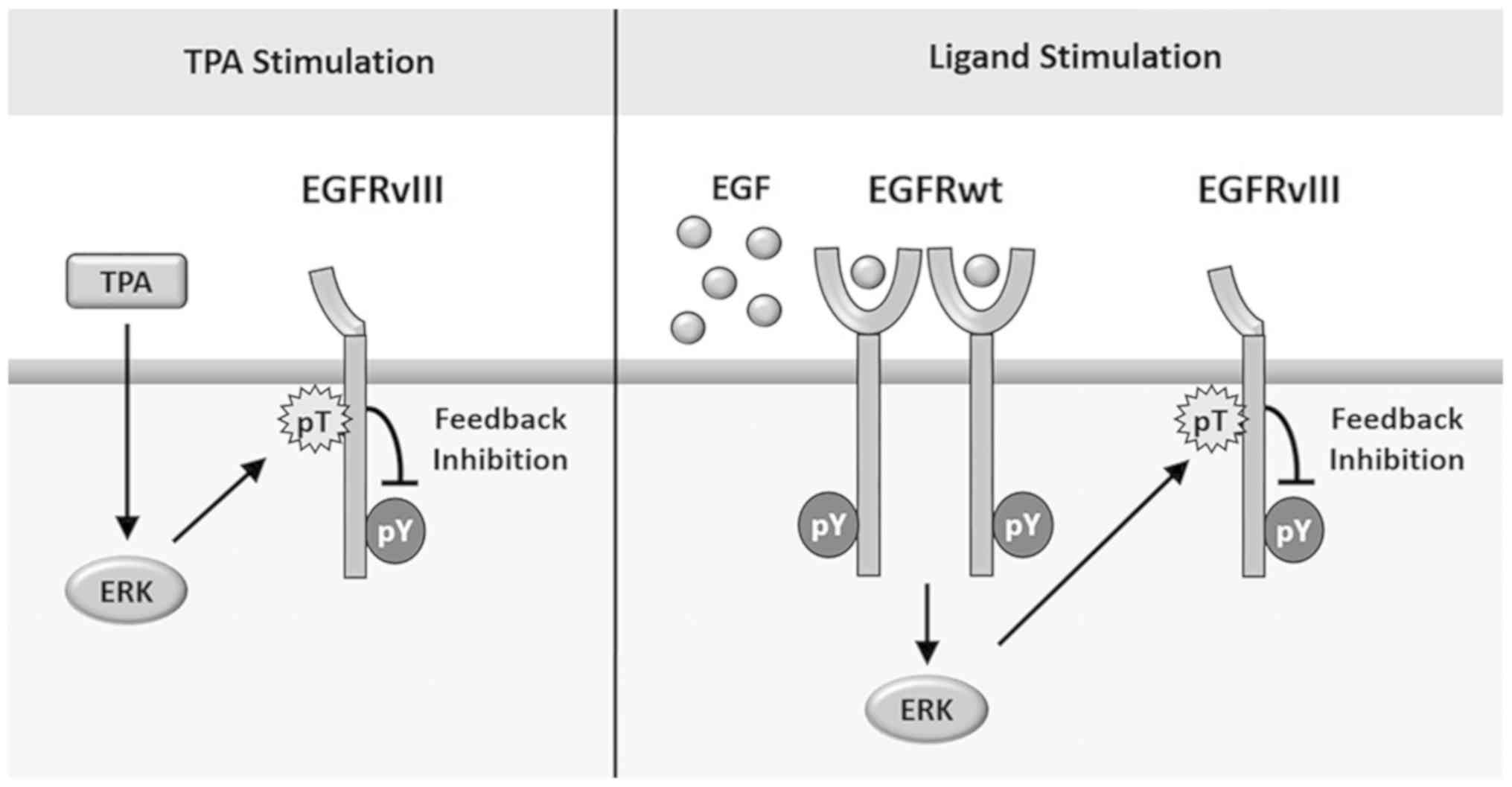
binding ligands, exogenous EGF caused the trans-phosphorylation of
EGFRvIII by EGFRwt within one minute (Fig. 4A). More importantly, after the early
trans-activation of EGFRvIII, EGF-induced ERK activation caused the
feedback suppression of EGFRvIII within 60 min, indicating that the
feedback loop is involved in the antagonistic role of EGFRwt in
EGFRvIII activation (Fig. 5).
The EGFR homodimer, which is composed of an
activator and receiver, has been shown to have an asymmetric dimer
structure. We previously demonstrated that Thr-669 in only the
receiver EGFR was involved in negative feedback. Due to the weak
expression of EGFRwt in U87MG-vIII cells, feedback inactivation may
occur in EGFRvIII homodimers via EGFRwt-dependent ERK activation.
Therefore, the stoichiometric ratio of wild-type and mutant EGFR,
which influences the dimer status, will affect the balance between
the synergistic and antagonistic activities of EGFRwt signaling in
the oncogenic functions of EGFRvIII.
Collectively, these results provide insights into
the existence of negative feedback regulation in an oncogenic
EGFRvIII deletion mutant, suggesting that the loss of this function
contributes to the progression of GBMs. Moreover, MEK inhibition by
trametinib counteracted the basal level of the negative feedback
regulation of EGFRvIII, resulting in the up-regulation of EGFRvIII
activity. Therefore, negative feedback events need to be considered
when applying precision medicine to EGFRvIII-expressing
glioblastoma.
Acknowledgements
The authors would like to thank Professor Webster K.
Cavenee (University of California San Diego) and Professor Motoo
Nagane (Kyorin University) for providing U87MG glioblastoma
cells.
Funding
The present study was supported in part by JSPS
KAKENHI (grant nos. 16H04694 and 19H03368) and the Research and
Innovation in Science and Technology Project (RISET-Pro) of the
Ministry of Research, Technology, and Higher Education of Republic
of Indonesia (World Bank Loan no. 8245-ID).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
RDH wrote the manuscript and conducted experiments.
TT, YZ and SY designed and helped conduct the experiments. HS
designed the study and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huang PH, Xu AM and White FM: Oncogenic
EGFR signaling networks in glioma. Sci Signal. 2:re62009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guo G, Gong K, Wohlfeld B, Hatanpaa KJ,
Zhao D and Habib AA: Ligand-independent EGFR signaling. Cancer Res.
75:3436–3442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gan HK, Cvrljevic AN and Johns TG: The
epidermal growth factor receptor variant III (EGFRvIII): Where wild
things are altered. FEBS J. 280:5350–5370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gazdar AF: Activating and resistance
mutations of EGFR in non-small-cell lung cancer: Role in clinical
response to EGFR tyrosine kinase inhibitors. Oncogene. 28 (Suppl
1):S24–S31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taylor TE, Furnari FB and Cavenee WK:
Targeting EGFR for treatment of glioblastoma: Molecular basis to
overcome resistance. Curr Cancer Drug Targets. 12:197–209. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Padfield E, Hayley P and Kurian KM:
Current therapeutic advances targeting EGFR and EGFRvIII in
glioblastoma. Front Oncol. 5:52015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bethune G, Bethune D, Ridgway N and Xu Z:
Epidermal growth factor receptor (EGFR) in lung cancer: An overview
and update. J Thorac Dis. 2:48–51. 2010.PubMed/NCBI
|
|
8
|
An Z, Aksoy O, Zheng T, Fan QW and Weiss
WA: Epidermal growth factor receptor and EGFRvIII in glioblastoma:
Signaling pathways and targeted therapies. Oncogene. 37:1561–1575.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang J, Yan J and Liu B: Targeting
EGFRvIII for glioblastoma multiforme. Cancer Lett. 403:224–23.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arteaga CL and Engelman JA: ERBB
receptors: From oncogene discovery to basic science to
mechanism-based cancer therapeutics. Cancer Cell. 25:282–303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gan HK, Kaye AH and Luwor RB: The EGFRvIII
variant in glioblastoma multiforme. J Clin Neurosci. 16:748–754.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tomas A, Futter CE and Eden ER: EGF
receptor trafficking: Consequences for signaling and cancer. Trends
Cell Biol. 24:26–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chistiakov DA, Chekhonin IV and Chekhonin
VP: The EGFR variant III mutant as a target for immunotherapy of
glioblastoma multiforme. Eur J Pharmacol. 810:70–82. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Keller S and Schmidt MHH: EGFR and
EGFRvIII promote angiogenesis and cell invasion in glioblastoma:
Combination therapies for an effective treatment. Int J Mol Sci.
18:12952017. View Article : Google Scholar
|
|
15
|
Sato K, Shin MS, Sakimura A, Zhou Y,
Tanaka T, Kawanishi M, Kawasaki Y, Yokoyama S, Koizumi K, Saiki I
and Sakurai H: Inverse correlation between Thr-669 and constitutive
tyrosine phosphorylation in the asymmetric epidermal growth factor
receptor dimer conformation. Cancer Sci. 104:1315–1322. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kawasaki Y, Sakimura A, Park CM, Tomaru R,
Tanaka T, Ozawa T, Zhou Y, Narita K, Kishi H, Muraguchi A and
Sakurai H: Feedback control of ErbB2 via ERK-mediated
phosphorylation of a conserved threonine in the juxtamembrane
domain. Sci Rep. 6:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haryuni RD, Watabe S, Yamaguchi A, Fukushi
Y, Tanaka T, Kawasaki Y, Zhou Y, Yokoyama S and Sakurai H: Negative
feedback regulation of ErbB4 tyrosine kinase activity by
ERK-mediated non-canonical phosphorylation. Biochem Biophys Res
Commun. 514:456–461. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishikawa R, Ji XD, Harmon RC, Lazar CS,
Gill GN, Cavenee WK and Huang HJ: A mutant epidermal growth factor
receptor common in human glioma confers enhanced tumorigenicity.
Proc Natl Acad Sci USA. 91:7727–7731. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nitta Y, Shimizu S, Shishido-Hara Y,
Suzuki K, Shiokawa Y and Nagane M: Nimotuzumab enhances
temozolomide-induced growth suppression of glioma cells expressing
mutant EGFR in vivo. Cancer Med. 5:486–499. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou Y, Yamada N, Tanaka T, Hori T,
Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I and
Sakurai H: Crucial roles of RSK in cell motility by catalysing
serine phosphorylation of EphA2. Nat Commun. 6:76792015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kinoshita E and Kinoshita-Kikuta E:
Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced
protein phosphorylation profiling. Proteomics. 11:319–323. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lugowska I, Koseła-Paterczyk H, Kozak K
and Rutkowski P: Trametinib: A MEK inhibitor for management of
metastatic melanoma. Onco Targets Ther. 8:2251–2259.
2015.PubMed/NCBI
|
|
23
|
Najem A, Krayem M, Perdrix A, Kerger J,
Awada A, Journe F and Ghanem G: New drug combination strategies in
melanoma: Current status and future direction. Anticancer Res.
37:5941–5953. 2017.PubMed/NCBI
|
|
24
|
Li L, Chakraborty S, Yang CR, Hatanpaa KJ,
Cipher DJ, Puliyappadamba VT, Rehman A, Jiwani AJ, Mickey B, Madden
C, et al: An EGFR wild type-EGFRvIII-HB-EGF feed-forward loop
regulates the activation of EGFRvIII. Oncogene. 33:4253–4264. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fan QW, Cheng C, Gustafson WC, Charron E,
Zipper P, Wong RA, Chen J, Lau J, Knobbe-Thomsen C, Weller M, et
al: EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and
progression in glioblastoma. Cancer Cell. 24:438–449. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luwor RB, Zhu HJ, Walker F, Vitali AA,
Perera RM, Burgess AW, Scott AM and Johns TG: The tumor-specific
de2-7 epidermal growth factor receptor (EGFR) promotes cells
survival and heterodimerizes with the wild-type EGFR. Oncogene.
23:6095–6104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kwon SJ, Kwon OS, Kim KT, Go YH, Yu SI,
Lee BH, Miyoshi H, Oh E, Cho SJ and Cha HJ: Role of MEK partner-1
in cancer stemness through MEK/ERK pathway in cancerous neural stem
cells, expressing EGFRviii. Mol Cancer. 16:1402017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wee P and Wang Z: Epidermal growth factor
receptor cell proliferation. Cancers (Basel). 9:522017. View Article : Google Scholar
|
|
29
|
Li L, Puliyappadamba VT, Chakraborty S,
Rehman A, Vemireddy V, Saha D, Souza RF, Hatanpaa KJ, Koduru P,
Burma S, et al: EGFR wild type antagonizes EGFRvIII-mediated
activation of Met in glioblastoma. Oncogene. 34:129–134. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Puliyappadamba VT, Chakraborty S, Chauncey
SS, Li L, Hatanpaa KJ, Mickey B, Noorani S, Shu HK, Burma S,
Boothman DA and Habib AA: Opposing effect of EGFRWT on
EGFRvIII-mediated NF-κB activation with RIP1 as a cell death
switch. Cell Rep. 4:764–775. 2013. View Article : Google Scholar : PubMed/NCBI
|