Introduction
Hepatocellular carcinoma (HCC) is the most common
type of primary hepatocellular carcinoma, accounting for 90% of
primary hepatocellular carcinoma cases, and is the leading cause of
cancer-associated death worldwide (1). Globally, ~350 million people are
infected with hepatitis B virus (HBV), and HBV infection accounts
for at least 50% of HCC cases (2).
HBV-associated HCC is therefore frequently observed. Notable
progress has been made in the diagnosis and treatment of HCC, and
overall treatment efficacy has been improved, especially following
the development of multi-modality therapy, such as hepatectomy,
minimally invasive treatment and liver transplantation (3). However, most patients with HCC are
diagnosed during the middle and advanced stages, during which
traditional radiotherapy and chemotherapy have limited clinical
benefit. This is primarily due to high rates of recurrence and
metastasis (4), therefore, it is
important to identify novel diagnostic and prognostic markers to
aid earlier intervention.
The occurrence and development of HCC are caused by
genetic and epigenetic variations, including gene mutations, copy
number variations and abnormal methylation (5). High-throughput technology has
identified a large number of tumor driver gene mutations and
epigenetic alterations associated with the pathogenesis of HCC
(6–10), which may have potential as diagnostic
and prognostic molecular markers (11,12).
Mutations in key driver genes lead to uncontrolled proliferation
and clonal amplification of cancer cells (13–15).
However, differentially expressed genes in HCC have not been
screened and the subsequent signaling pathways involved in the
development of HCC have not been validated.
The present study aimed to identify potentially
novel therapeutic targets for HCC using high-throughput-based gene
expression analysis in human HCC tissues. To prevent false-positive
results from high-throughput screening, immunohistochemical
analysis was also performed to verify differential gene
expression.
Materials and methods
Tissue collection
The samples were collected from eight patients with
HBV-associated HCC at the Department of Hepatobiliary Gland
Extracorporeal Surgery of Guigang People's Hospital (Guigang,
China) between January 2017 and March 2017. The age of the patients
ranged from 38 to 61 years (median age, 48.5 years; male: Female
ratio of 5:3), and the primary HCC tissue specimens and matched
paracancerous liver tissues were collected during hepatectomy.
Clinicopathological characteristics of patients, including age and
serum levels of bilirubin, alkaline phosphatase, albumin, alanine
transaminase (ALT), aspartate transaminase (AST), alphafetoprotein
(AFP), hepatitis B surface antigen (HBsAg), hepatitis B surface
antibody (HBsAb), hepatitis B e antigen (HBeAg), hepatitis B e
antibody (HBeAb) and hepatitis B core antibody (HBcAb) were
collected. HCC was confirmed by a pathological report from Guigang
People's Hospital based on the evaluation of differentiation and
metastasis as previously described (16). The tissues were divided into two
groups: Paracancerous tissue group and hepatocellular carcinoma
tissue group. The inclusion criteria were as follows: i) No
etiological evidence of hepatitis C virus, type 2 diabetes
mellitus, fatty liver, alcoholic liver or aflatoxin exposure
history; ii) no preoperative radiotherapy, digital subtraction
angiography intervention, antineoplastic drug therapy or and
radiofrequency ablation; and iii) no other history of cancer.
Patients who did not meet one of the aforementioned criteria were
excluded. Three tumor tissues and corresponding paracancerous
tissues were analyzed using high-throughput sequencing. Three
differentially expressed genes, stratifin (SFN), cyclin B1
(CCNB1) and cyclin-dependent kinase 1 (CDK1) were
verified in the total 8 tumor tissues and corresponding
paracancerous tissues.
After the specimens were removed, tumors and
paracancerous liver tissues were cut into small pieces (~2 mm) on
ice, then put into 1.5 ml nucleic acid-free centrifugal tubes and
stored in liquid nitrogen. Cancer tissues and paracancerous tissues
were collected from the non-necrotic area of the cancer, and
paracancerous liver tissues were taken from the non-invasive area 2
cm away from the edge of the obvious mass, avoiding the burning
site of the scalpel and preventing the loss of DNA and RNA due to
high temperature. Patients provided informed written consent before
organizing the collection, and the study was approved by the Ethics
Committee of Guigang City People's Hospital.
Construction of the Illumina
sequencing library
Total RNA was extracted from the samples using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
The concentration and purity of the RNA were detected using
Nanodrop 2000 (Thermo Fisher Scientific, Inc.), the integrity of
RNA was detected by agarose gel electrophoresis (0.5% gel; ethidium
bromide staining and visualization at UV light), and the value of
RNA integrity was determined using Agilent 2100 (Agilent
Technologies, Inc.). The total amount of RNA required for single
library construction was >5 µg, the concentration was >200
ng/µl, and optical density 260/280 was between 1.8 and 2.2. Then, a
Ribo-Zero Magnetic kit (Epicentre; Illumina, Inc.) was used to
remove ribosomal RNA, RNase R (Epicentre; Illumina, Inc.) was used
to remove linear RNA and a TruSeq™ Stranded Total RNA Library Prep
kit (Illumina, Inc.) was used to construct Paired-End sequencing
library. The analysis of mRNA was performed using the Hiseq4000
sequencing platform (Illumina, Inc.). Principal component analysis
(PCA) was used to analyze multidimensional data.
Sequencing data analysis
SepPrep and Silkle software (linux-64 v1.3.2;
Anaconda Clound) were used to examine the data quality and the data
obtained after quality control (Phred quality score) with the
reference genome data were compared using Bowtie version 2. KNIEF
and CIRCexplorer2 were used to predict the expression of RNA, and
then levels of RNA in the samples were calculated.
Differentially expressed gene
enrichment analysis
Gene Ontology software was used for enrichment
analysis of GO functions, including molecular function, cell
components, and biological processes, and Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway enrichment analysis was carried
out using the KEGG Orthology-Based Annotation System and KEGG
annotation (https://www.genome.jp/kegg-bin/get_htext?ko00001).
Prediction of target genes
Differential analysis of gene expression was
performed using DESeq version 2 (Bioconductor) to predict target
genes of RNA.
Quantitative (q)PCR
According to the results of high-throughput
sequencing, three differentially expressed RNAs (SFN, CCNB1
and CDK1) were analyzed using fluorescence qPCR. The primer
sequences used are listed in Table
I. Total mRNA was amplified using a one-step RT-PCR kit
(00081405, CWBio). The primers were added into a 25-µl ULtraSYBR
mixture (01170, CWBio) PCR reaction system according to the
manufacturer's protocol. The thermocycling conditions were as
follows: Pre-denaturation at 95°C for 10 min, 95°C denaturation for
10 sec, 58.5°C annealing for 30 sec and 72°C extension for 30 sec
(40 cycles). The Cq value for each gene was determined and
expression levels of target genes were calculated using the
2−ΔΔCq method (17). mRNA
expression of target genes were normalized to GAPDH.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Genes | Primers, 5′-3′ | Primer length,
bp | Product length,
bp | Annealing, °C |
|---|
| SFN |
|
| 116 | 61.4 |
|
Forward |
GGTGACTACTACCGCTACCTGG | 22 |
|
|
|
Reverse |
GGCATCTCCTTCTTGCTGACG | 21 |
|
|
| CCNB1 |
|
| 157 | 56.9 |
|
Forward |
TTGAGGAAGAGCAAGCAGTC | 20 |
|
|
|
Reverse |
AACCGATCAATAATGGAGACAG | 22 |
|
|
| CDK1 |
|
| 110 | 57.5 |
|
Forward |
AGGATGTGCTTATGCAGGATTC | 22 |
|
|
|
Reverse |
CATGTACTGACCAGGAGGG | 19 |
|
|
| GAPDH |
|
| 106 | 57.2 |
|
Forward |
CAATGACCCCTTCATTGACC | 20 |
|
|
|
Reverse |
GAGAAGCTTCCCGTTCTCAG | 20 |
|
|
Immunohistochemistry
Cancer tissues were fixed with 4% paraformaldehyde
overnight at 4°C. The tissues were then dehydrated using 70, 80 and
90% ethanol, and mixed with anhydrous ethanol and xylene for 15
min, xylene I for 15 min and xylene II for 15 min (until
transparent). Tissues were embedded in paraffin and sliced into
10-µm sections. The paraffin slices were dewaxed and hydrated in
70, 75, 80, 85 and 95% alcohol. Then, 3% (v/v)
H2O2 was used to block endogenous peroxidase
activity for 5 min at room temperature. Immunostaining of the
slides was performed using the following antibodies overnight at
4°C: Rabbit polyclonal anti-SFN (1:50, bs-20373R, BIOSS), rabbit
polyclonal anti-CCNB1 (1:400, bs-20373R, BIOSS) and rabbit
polyclonal anti-CDK1 (1:100, bs-20373R, BIOSS). The slides were
then washed with PBS and incubated with horseradish
peroxidase-labeled goat anti-rabbit IgG secondary antibody
(1:10,000; A16104SAMPLE; Thermo Fisher Scientific, Inc.) for 30 min
at room temperature, and visualized with 3,3′-diaminobenzidine
chromogen for 3 min at room temperature. At least four fields were
taken from each image using a light microscope (magnification,
×200; BX51, Olympus Corporation). Staining intensity was analyzed
by Image-Pro Plus software (National Institutes of Health). The
relative expressions of target proteins were normalized to the
negative control (without primary antibody).
Statistical analysis
All data were analyzed using SPSS version 19.0 (IBM
Corp.) The differences between groups were analyzed using unpaired
Student's t-tests and P<0.05 was considered to indicate a
statistically significant difference.
Results
Basic information of the patients
Eight patients with HCC were enrolled in the present
study. The age of the patients ranged from 38 to 61 years. The mean
values of bilirubin, alkaline phosphatase, albumin, ALT, AST and
AFP serum levels were 15.0 µM, 92.1 U/l, 44.6 g/l, 31.5 U/l, 33.6
U/l and 785.3 ng/ml, respectively. HBsAg, HBeAb and HBcAb were
positive in all patients. The patients were in a stage of medium-
and low-differentiation without lymph node involvement and distal
metastasis. All patients were diagnosed with cirrhosis and portal
vein embolism, but without ascites (data not shown).
Quality analysis of the data
The quality of the data is presented in Table II. The sequence numbers obtained in
paracancerous tissues and cancer tissues were 43,704,194,
41,440,988, 42,235,972 and 4,092,974, 44,150,718 and 42,218,858,
respectively. Phred bases with a value >20 accounted for 97% of
the total number of bases, indicating that the original sequencing
data were of good quality and could be used for subsequent data
analysis.
| Table II.Quality analysis of the original
data. |
Table II.
Quality analysis of the original
data.
| Sample | Reads no. | Bases, bp | Q30, bp | N, % | Q20, % | Q30, % |
|---|
| Control 1 | 43,704,194 | 6,599,333,294 | 6,226,745,352 | 0.001709 | 97.73 | 94.35 |
| Control 2 | 41,440,988 | 6,257,589,188 | 5,914,658,053 | 0.001422 | 97.81 | 94.51 |
| Control 3 | 42,235,972 | 6,377,631,772 | 6,099,059,319 | 0.001376 | 98.28 | 95.63 |
| Cancer 1 | 40,926,974 | 6,179,973,074 | 5,744,415,761 | 0.000725 | 97.11 | 92.95 |
| Cancer 2 | 44,150,718 | 6,666,758,418 | 6,218,604,371 | 0.001765 | 97.24 | 93.27 |
| Cancer 3 | 42,218,858 | 6,375,047,558 | 5,979,826,518 | 0.001859 | 97.5 | 93.82 |
Quality control analysis of sequencing
data
The data quality control analysis showed that the
total number of reference genome sequences was >90% (Table III), indicating that the quality
control of sequencing data was good and could be used in subsequent
data analysis.
| Table III.Statistical analysis of RNASeq
map. |
Table III.
Statistical analysis of RNASeq
map.
| Sample | Clean_Reads | Total_Mapped, n
(%) | Multiple_Mapped, n
(%) | Uniquely_Mapped, n
(%) |
|---|
| Control 1 | 43,594,562 | 40,504,709
(92.91) | 2,163,607
(5.34) | 38,341,102
(94.66) |
| Control 2 | 41,339,382 | 38,683,072
(93.57) | 1,788,575
(4.62) | 36,894,497
(95.38) |
| Control 3 | 42,162,810 | 39,597,473
(93.92) | 1,290,633
(3.26) | 38,306,840
(96.74) |
| Cancer 1 | 40,695,892 | 37,637,436
(92.48) | 1,630,956
(4.33) | 36,006,480
(95.67) |
| Cancer 2 | 44,010,224 | 40,362,249
(91.71) | 4,109,173
(10.18) | 36,253,076
(89.82) |
| Cancer 3 | 42,106,446 | 38,830,579
(92.22) | 4,455,543
(11.47) | 34,375,036
(88.53) |
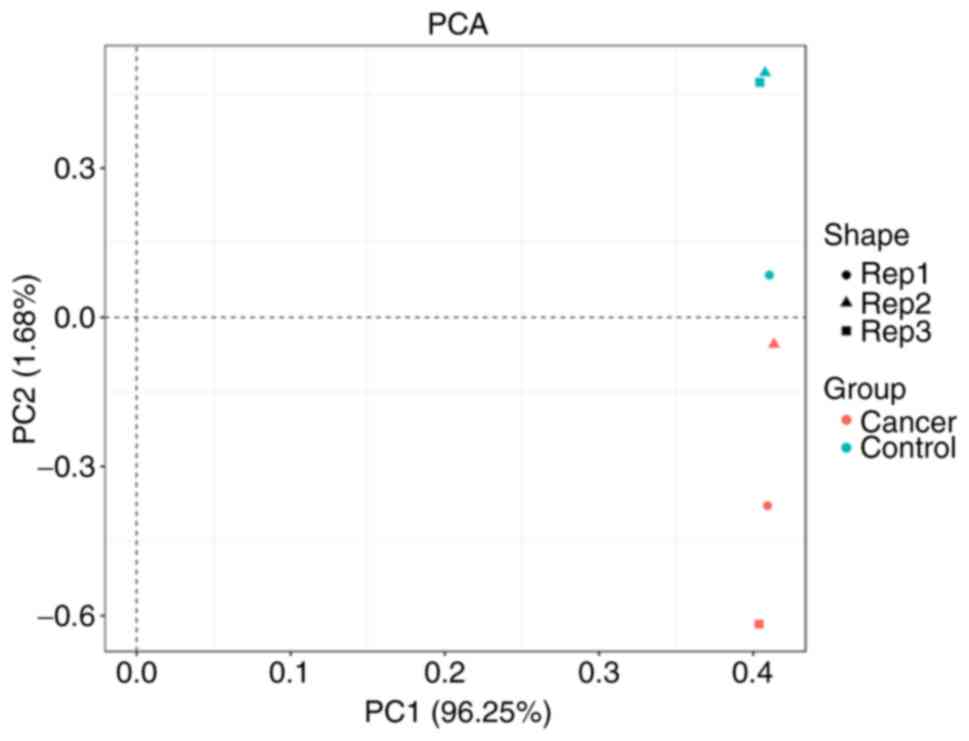
PCA analysis
PCA analysis showed that there were obvious
differences between the control group and cancer group. By
contrast, there was no inner difference among the data in each
group. Thus, the data could be used in the subsequent analysis
(Fig. 1).
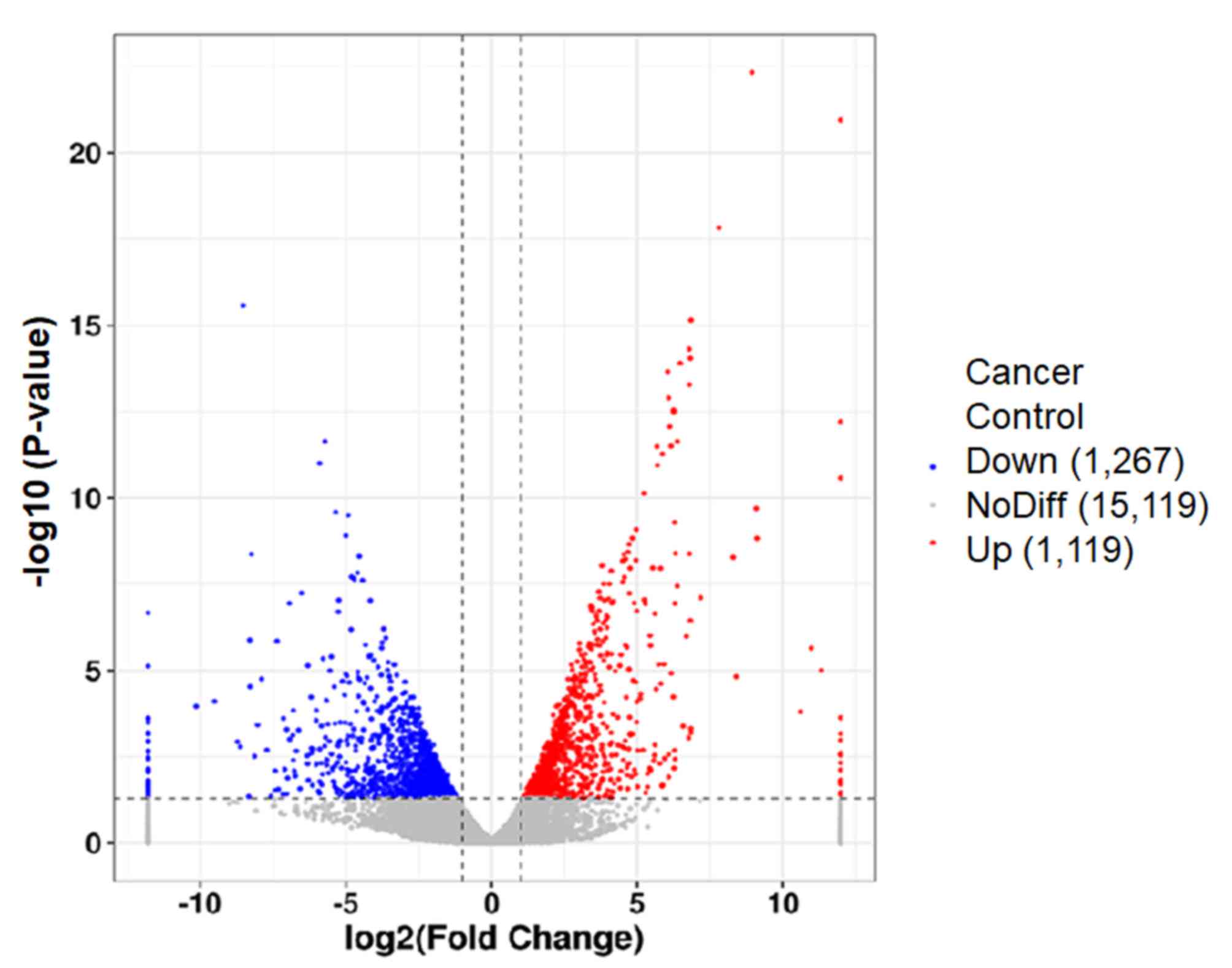
Differential gene expression
analysis
Differential gene expression analysis is presented
in Fig. 2. 2,386 differentially
expressed genes were screened (multiple of differentially expressed
genes between samples was >1, and the value of differentially
expressed genes in samples was <0.05). There were 1119
downregulated and 1,267 upregulated genes in cancer tissues
compared with paracancerous tissues.
GO functional enrichment analysis
GO functional enrichment analysis of differentially
expressed genes was used to identify GO functional items. Firstly,
all genes were mapped to each term in GO database and the number of
differentially expressed genes was calculated. Then, significantly
enriched differentially expressed genes with the whole genome were
analyzed. The GO analysis showed that differentially expressed
genes were enriched in the following GO molecular functions: GO:
0046395 carboxylic acid catabolic process, GO: 0032787
monocarboxylic acid metabolic process and GO: 1901605 α-amino acid
metabolic processes. Genes were also enriched in the following
cellular component functions: GO: 0070062 extracellular exosome,
GO: 0044444 cytoplasmic part and GO: 0044459 plasma membrane part.
Differentially expressed genes were enriched in the following
biological processes: GO: 0016614 oxidoreductase activity, acting
on CH-OH group of donors, GO: 0042802 identical protein binding,
GO: 0043168 anion binding, GO: 0050662 coenzyme binding and GO:
0008028 monocarboxylic acid transmembrane transporter activity
(data not shown).
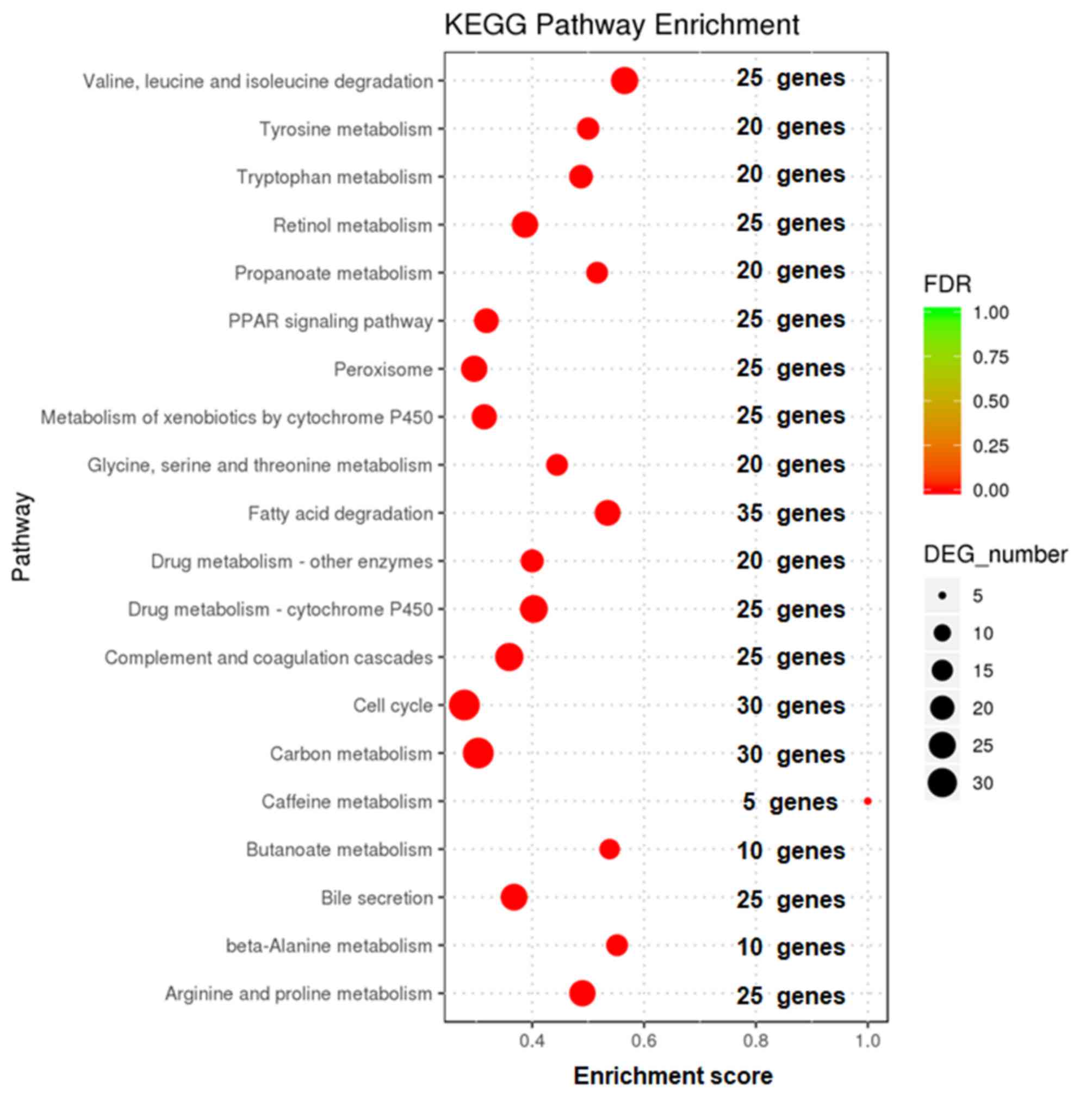
Enrichment analysis of KEGG signaling
pathways of differentially expressed genes
Enrichment analysis of KEGG signaling pathways of
differentially expressed genes was based on the whole genome and is
shown in Fig. 3. The metabolic
pathways and signaling pathways in which differentially expressed
genes functioned were determined. The results showed that the
differentially expressed genes were mainly concentrated in 20
signaling pathways, including degradation of valine, leucine and
isoleucine, retinol metabolism and the cell cycle.
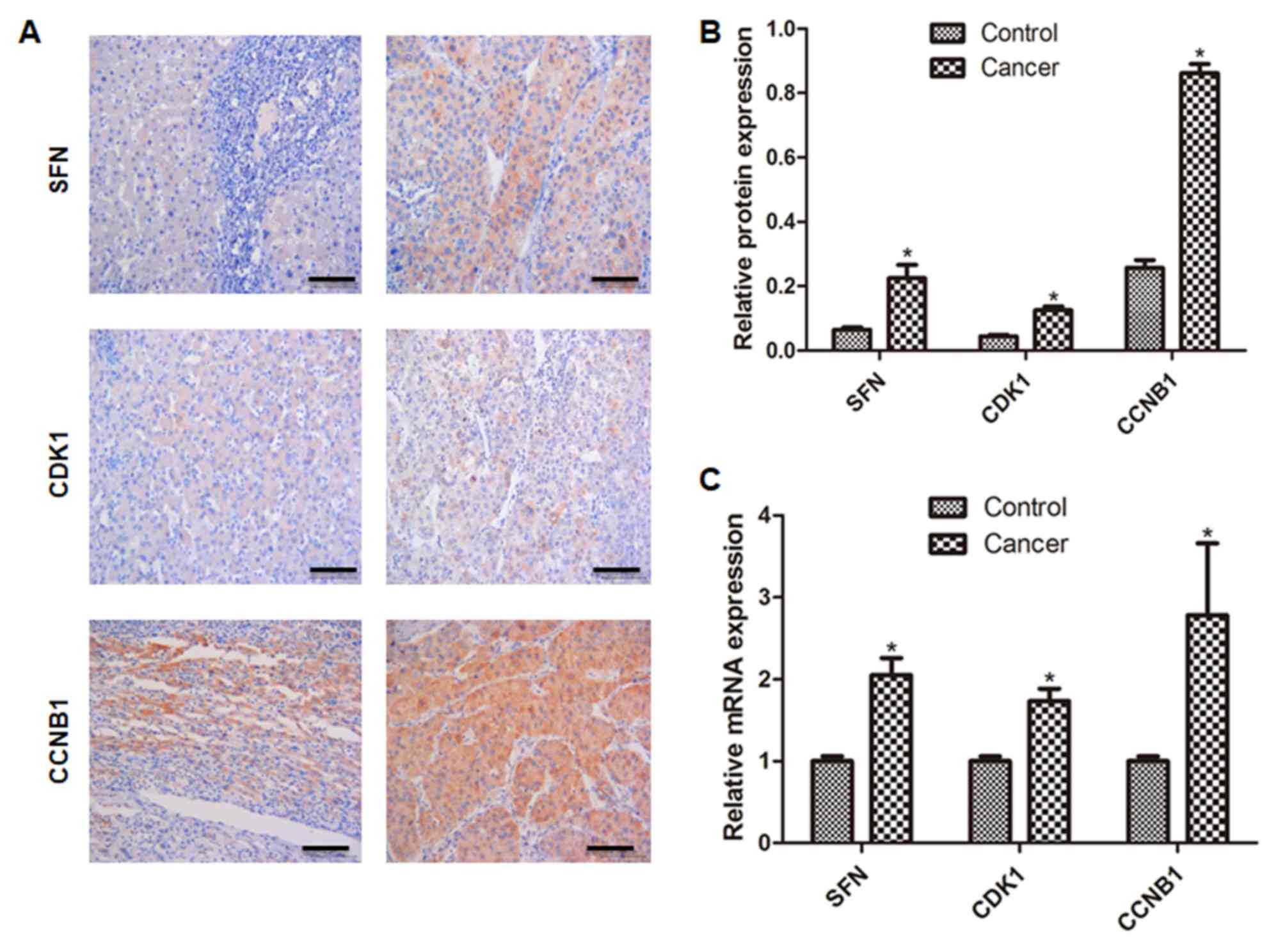
Validation of differentially expressed
genes
The validation of three differentially expressed
genes is shown in Fig. 4. The
results showed that the expression of SFN, CCNB1 and
CDK1 genes in tumors were upregulated in cancer tissues
compared with control (P<0.05; Figs.
4A and B, S1), which was
consistent with the results of high-throughput sequencing. The
results were further confirmed using qPCR and a consistent result
was obtained (vs. control; P<0.05; Fig. 4C).
Discussion
In the present study, differential gene expression
between HCC and paracancerous tissues was analyzed. The
differentially expressed genes were mainly concentrated in 20
signaling pathways, such as valine, leucine and isoleucine
degradation, retinol metabolism and the cell cycle. It was then
further confirmed that cell cycle-associated genes (SFN,
CCNB1 and CDK1) were abnormally expressed in HCC
tissues. The present study may provide novel therapeutic targets
for HCC.
Due to the lack of early diagnostic measures, the
incidence of recurrence and metastasis after operation in HCC is
high, and drug treatments have limited therapeutic benefit
(18,19). Therefore, early diagnosis and
prediction are important to improve the treatment of HCC. Similar
to other malignant tumors, HCC is often caused by the activation of
oncogenes (myc, ras) or the inactivation of tumor suppressor
genes (e.g., p53) (20,21). The
aim of the present study was to detect differentially expressed
genes in HCC and paracancerous tissues using high-throughput
sequencing.
Tumor development is usually associated with
multiple signaling pathways (22).
KEGG analysis showed that the differentially expressed genes were
mainly concentrated in 20 signaling pathways, such as valine,
leucine and isoleucine degradation, retinol metabolism and the cell
cycle. In addition, 2,386 differentially expressed genes were
screened. The differentially expressed genes of SFN, CCNB1
and CDK1 were selected from the high throughput results for
further verification. SFN, CCNB1 and CDK1 genes
expressed differently in high-throughput results, were associated
to the cell cycle (23). Experiments
have shown that knocking out the SFN gene can lead to cell
failure to maintain stable G2/M cycle after DNA damage
and sensitizes cells to DNA damage. This method is therefore used
in the treatment of neuroblastic tumor (24). SFN can arrest cell cycle and induce
apoptosis of human tumor cells (25). Increased expression of SFN results in
the chelation of CDK1/cyclinB1 in cytoplasm, thus blocking the
interaction between cell division cycle 2 (CDC2) gene and
CDK1 and preventing cells from entering into cell mitosis (26–28).
CDK1 is a member of the protein kinase family and is
encoded by the CDC2 gene. At the late stage of
G2, CDK1 and cyclin B1 lotus root synthesize complex,
forming mitotic promoter factor, which can promote cell cycling
from G2 to M (29). CDK1
is an important regulator of mitotic initiation, cell cycling and
metastasis. High expression of active CDK1 promotes G2/M
expression and accelerates cancer cell growth (30). Some studies have shown that specific
inhibitors of CDK1 can induce reversible dormancy of human cells in
G2/M phase, leading to the apoptosis of cancer cells,
suggesting that selective inhibitors of CDK1 may play a pivotal
role in the treatment of cancer (30,31).
CCNB1 is an important regulator of the cell cycle
associated with the detection point of G2/M phase.
Interaction of CCNB1 and CDK1 phosphorylates the substrate cell
division homologous protein cyclin 25, initiates cell cycle
progression from G1/S phase to G2/M phase and
promotes mitosis (32). A previous
study reported that the abnormal expression of CCNB1 is associated
with abnormal cell proliferation and tumorigenesis (33). A number of studies have shown that
CCNB1 is highly expressed in breast, lung and gastrointestinal
cancer (34–36). In the present study, the results of
qPCR and immunohistochemistry demonstrated that the expression of
SFN, CCNB1 and CDK1 was elevated in cancer tissues
and that these results were consistent with the high-throughput
results.
There were still limitations of the present study.
First, only three tumor tissues and corresponding paracancerous
tissues were analyzed using high-throughput sequencing. Three
differentially expressed genes, SFN, CCNB1 and CDK1
were verified in the total 8 tumor tissues and corresponding
paracancerous tissues. Consistent results were obtained from
high-throughput sequencing, PCR and western blotting; however, the
differentially expressed genes and their proteins need external
validation in a larger cohort of patients with cirrhosis and
different stages of HCC. Second, although differentially expressed
genes in HCC were screened, the potential value of these genes in
the treatment and prognosis of HCC were not confirmed. Third, there
were still some discrepancies regarding the differentially
expressed genes reported in the present study and previous studies.
These might be caused by different patient ethnicities, types of
HCC and disease stages. However, larger sample size is crucial and
laboratory experiments should be carried out to validate these
findings and disclose potential mechanisms.
In conclusion, differentially expressed genes were
screened using high-throughput technology and enriched in
tumor-associated signaling pathways and functions. The next step is
to investigate the functions of these differentially expressed
genes.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Construction Funding of Key Clinical Specialties
Guangxi Zhuang Autonomous Region (Z20190320).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
HZ, YH, WQ, PC, LH, WZ, LL and HL performed the
experiments and analyzed the data. HZ and XQ designed the study and
wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All patients provided informed written consent
before tissue collection, and the study was approved by the Ethics
Committee of Guigang City People's Hospital (approval no.
013142017).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang X, Zhang D, Liu S, Li X, Hu W and Han
C: KLF4 suppresses the migration of hepatocellular carcinoma by
transcriptionally upregulating monoglyceride lipase. Am J Cancer
Res. 8:1019–1029. 2018.PubMed/NCBI
|
|
2
|
Xie Y: Hepatitis B virus-associated
hepatocellular carcinoma. Adv Exp Med Biol. 1018:11–21. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xing H, Qiu H, Ding X, Han J, Li Z, Wu H,
Yan C, Li H, Han R, Zhang H, et al: Clinical performance of
alpha-L-fucosidase for early detection of hepatocellular carcinoma.
Biomark Med. 13:545–555. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiao D, Li Y, Yang F, Han D, Wu J, Shi S,
Tian F, Guo Z, Xi W, Li G, et al: Expression of prostate-specific
membrane antigen in tumor-associated vasculature predicts poor
prognosis in hepatocellular carcinoma. Clin Transl Gastroenterol.
May 15–2019.doi: 10.14309/ctg.0000000000000041 (Online ahead of
print). View Article : Google Scholar
|
|
5
|
Xiao Z, Yan Y, Zhou Q, Liu H, Huang P,
Zhou Q, Lai C, Zhang J, Wang J and Mao K: Development and external
validation of prognostic nomograms in hepatocellular carcinoma
patients: A population based study. Cancer Manag Res. 11:2691–2708.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ziats MN and Rennert OM: Identification of
differentially expressed microRNAs across the developing human
brain. Mol Psychiatry. 19:848–852. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang ZY, Ye SL, Liu YK, Qin LX, Sun HC, Ye
QH, Wang L, Zhou J, Qiu SJ, Li Y, et al: A decade's studies on
metastasis of hepatocellular carcinoma. J Cancer Res Clin Oncol.
130:187–196. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sells MA, Chen ML and Acs G: Production of
hepatitis B virus particles in Hep G2 cells transfected with cloned
hepatitis B virus DNA. Proc Natl Acad Sci USA. 84:1005–1009. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakabayashi H, Taketa K, Miyano K, Yamane
T and Sato J: Growth of human hepatoma cells lines with
differentiated functions in chemically defined medium. Cancer Res.
42:3858–3863. 1982.PubMed/NCBI
|
|
11
|
Schulze K, Nault JC and Villanueva A:
Genetic profiling of hepatocellular carcinoma using next-generation
sequencing. J Hepatol. 65:1031–1042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin Y, Lee WY, Toh ST, Tennakoon C, Toh
HC, Chow PK, Chung AY, Chong SS, Ooi LL, Sung WK and Lee CG:
Comprehensive analysis of transcriptome profiles in hepatocellular
carcinoma. J Transl Med. 17:2732019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tian J, Tang ZY, Ye SL, Liu YK, Lin ZY,
Chen J and Xue Q: New human hepatocellular carcinoma (HCC) cell
line with highly metastatic potential (MHCC97) and its expressions
of the factors associated with metastasis. Br J Cancer. 81:814–821.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Y, Tang ZY, Ye SL, Liu YK, Chen J, Xue
Q, Chen J, Gao DM and Bao WH: Establishment of cell clones with
different metastatic potential from the metastatic hepatocellular
carcinoma cell line MHCC97. World J Gastroenterol. 7:630–636. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kan Z, Zheng H, Liu X, Li S, Barber TD,
Gong Z, Gao H, Hao K, Willard MD, Xu J, et al: Whole-genome
sequencing identifies recurrent mutations in hepatocellular
carcinoma. Genome Res. 23:1422–1433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen D, Li Z, Song Q, Qian L, Xie B and
Zhu J: Clinicopathological features and differential diagnosis of
hepatocellular carcinoma in extrahepatic metastases. Medicine
(Baltimore). 97:e133562018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He ZX, Xiang P, Gong JP, Cheng NS and
Zhang W: Radiofrequency ablation versus resection for Barcelona
clinic liver cancer very early/early stage hepatocellular
carcinoma: A systematic review. Ther Clin Risk Manag. 12:295–303.
2016.PubMed/NCBI
|
|
19
|
Tejeda-Maldonado J, García-Juárez I,
Aguirre-Valadez J, González-Aguirre A, Vilatobá-Chapa M,
Armengol-Alonso A, Escobar-Penagos F, Torre A, Sánchez-Ávila JF and
Carrillo-Pérez DL: Diagnosis and treatment of hepatocellular
carcinoma: An update. World J Hepatol. 7:362–376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khordadmehr M, Jigari-Asl F, Ezzati H,
Shahbazi R, Sadreddini S, Safaei S and Baradaran B: A comprehensive
review on miR-451: A promising cancer biomarker with therapeutic
potential. J Cell Physiol. 234:21716–21731. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mirza AZ: Advancement in the development
of heterocyclic nucleosides for the treatment of cancer-A review.
Nucleosides Nucleotides Nucleic Acids. 38:836–857. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nwabo Kamdje AH, Takam Kamga P, Tagne Simo
R, Vecchio L, Seke Etet PF, Muller JM, Bassi G, Lukong E, Kumar
Goel R, Mbo Amvene J and Krampera M: Developmental pathways
associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer
Biol Med. 14:109–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Diaz-Moralli S, Tarrado-Castellarnau M,
Miranda A and Cascante M: Targeting cell cycle regulation in cancer
therapy. Pharmacol Ther. 138:255–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Banelli B, Bonassi S, Casciano I, Mazzocco
K, Di Vinci A, Scaruffi P, Brigati C, Allemanni G, Borzì L, Tonini
GP and Romani M: Outcome prediction and risk assessment by
quantitative pyrosequencing methylation analysis of the SFN gene in
advanced stage, high-risk, neuroblastic tumor patients. Int J
Cancer. 126:656–668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clarke JD, Dashwood RH and Ho E:
Multi-targeted prevention of cancer by sulforaphane. Cancer Lett.
269:291–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Singh SV, Herman-Antosiewicz A, Singh AV,
Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L and Baskaran R:
Sulforaphane-induced G2/M phase cell cycle arrest involves
checkpoint kinase 2-mediated phosphorylation of cell division cycle
25C. J Biol Chem. 279:25813–25822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Steiner M, Clark B, Tang JZ, Zhu T and
Lobie PE: 14-3-3σ mediates G2-M arrest produced by
5-aza-2′-deoxycytidine and possesses a tumor suppressor role in
endometrial carcinoma cells. Gynecol Oncol. 127:231–240. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thangapandiyan S, Ramesh M, Hema T,
Miltonprabu S, Uddin MS, Nandhini V and Bavithra Jothi G:
Sulforaphane potentially ameliorates arsenic induced hepatotoxicity
in albino Wistar rats: Implication of PI3K/Akt/Nrf2 signaling
pathway. Cell Physiol Biochem. 52:1203–1222. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang Y, Xue K, Li Z, Zheng W, Dong W, Song
J, Sun S, Ma T and Li W: c-Myc regulates the CDK1/cyclin B1
dependentG2/M cell cycle progression by histone H4 acetylation in
Raji cells. Int J Mol Med. 41:3366–3378. 2018.PubMed/NCBI
|
|
30
|
Wang J, Chang L, Lai X, Li X, Wang Z,
Huang Z, Huang J and Zhang G: Tetrandrine enhances radiosensitivity
through the CDC25C/CDK1/cyclin B1 pathway in nasopharyngeal
carcinoma cells. Cell Cycle. 17:671–680. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jang SH, Kim AR, Park NH, Park JW and Han
IS: DRG2 regulates G2/M progression via the cyclin B1-cdk1 Complex.
Mol Cells. 39:699–704. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fang Y, Yu H, Liang X, Xu J and Cai X:
Chk1-induced CCNB1 overexpression promotes cell proliferation and
tumor growth in human colorectal cancer. Cancer Biol Ther.
15:1268–1279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Chen YL, Xie YT, Zheng LY, Han JY,
Wang H, Tian XX and Fang WG: Association study of germline variants
in CCNB1 and CDK1 with breast cancer susceptibility, progression,
and survival among Chinese Han women. PLoS One. 8:e844892013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Song GQ and Zhao Y: MicroRNA-211, a direct
negative regulator of CDC25B expression, inhibits triple-negative
breast cancer cells' growth and migration. Tumour Biol.
36:5001–5009. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun Q, Shi R, Wang X, Li D, Wu H and Ren
B: Overexpression of ZIC5 promotes proliferation in non-small cell
lung cancer. Biochem Biophys Res Commun. 479:502–509. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shi Q, Wang W, Jia Z, Chen P, Ma K and
Zhou C: ISL1, a novel regulator of CCNB1, CCNB2 and c-MYC genes,
promotes gastric cancer cell proliferation and tumor growth.
Oncotarget. 7:36489–36500. 2016. View Article : Google Scholar : PubMed/NCBI
|