Introduction
Colorectal cancer (CRC) is the third most prominent
type of cancer worldwide, which is commonly diagnosed in patients
above the age of 50 years (1–3). The
incidence of CRC among adolescents and young adults has exhibited
an increase over the past decades (1–3),
particularly in patients aged 20–39 years (4,5).
Early-onset CRC usually presents at an advanced stage at the time
of diagnosis with a more aggressive biological behavior, which is
unique to this subset of CRC (6–8). A study
indicated that the prevalence of hereditary cancer syndromes in CRC
patients aged 35 years or younger was ~35% (7). This group of patients (aged 35 years or
younger) has comparatively more concerns in various aspects that
require to be addressed (including the impact on fertility)
(9), which raises a compelling need
for the identification of prognostic markers and optimization of
treatment strategies. However, studies focusing on the genetic
features of early-onset CRC in patients aged 35 or younger are
limited (7).
The carcinogenesis of CRC is described by a genetic
model of cancer comprising the sequential accumulation of genetic
alterations. There are two distinct genetic pathways: The
adenomatous polyposis coli (APC)/β-catenin pathway, which exhibits
sequential alterations of genes including APC, K-RAS proto-oncogene
(KRAS) and tumor protein 53 (TP53), and the microsatellite
instability (MSI) pathway, which comprises a deficiency in DNA
mismatch repair (MMR) genes (10,11). In
addition, the methylation of CpG islands, an epigenetic alteration,
has been recognized as an early event involved in the development
of CRC (12). The genetic
alterations of non-polyposis CRC in younger patients manifest as
Lynch syndrome (LS), which exhibits germline mutations in MMR
genes; and sporadic CRC, which presents as more complex and diverse
genetic alterations (13).
Certain genetic test panels have been reported to be
a useful tool with which to identify multiple genetic mutations
(14). However, the genetic
alterations that were identified from these pathways or panels have
limited clinical utility at present. The major molecular
alterations validated as significant markers are high levels of
MSI/defective MMR (MSI-H/dmmr; good prognosis, insensitive to
5-fluorouracil chemotherapy), mutation in the B-Raf proto-oncogene
(BRAF) [poor prognosis, resistance to anti-epidermal growth factor
receptor (EGFR) antibodies] and mutations in KRAS/N-RAS
proto-oncogene (NRAS) genes (resistance to anti-EGFR antibodies)
(15,16).
More genes, particularly prognosis- and
treatment-associated genes, should be identified in order to
enhance the current understanding of early-onset CRC. The
identification of these genes may aid in the development of more
specific and suitable genetic analyses and may also help to improve
management strategies. There are certain therapies that were
initially used to target specified molecular alterations in a
particular tumor type and have been successfully utilized in other
cancer types. A successful example of this is the status regarding
anaplastic lymphoma kinase (ALK) gene rearrangement, which was
initially identified in anaplastic large-cell lymphomas, and is
currently an important indicator for ALK inhibitor treatment in
lung cancer (17). It would be of
interest to determine whether CRC exhibits certain genetic
mutations that are already considered as prognostic and/or
predictive markers in other cancer types and also to determine what
the frequencies of these genetic mutations are. Thus, after
reviewing the literature and searching certain databases (COSMIC,
https://cancer.sanger.ac.uk/cosmic
and My Cancer Genome, http://www.mycancergenome.org), the present study
selected 44 exons of 17 genes, which have been reported to be
prognostic and/or predictive markers in CRC and other malignancies
(Table SI) for analysis. The aim of
the present study was to use next-generation sequencing (NGS) to
identify these genetic mutations in a cohort of patients with
early-onset non-polyposis CRC. Of note, a set of genetic mutations
was identified, which was rarely reported previously, as well as
other features, which may indicate that this subset of CRC has
unique genetic features.
Materials and methods
Patient selection
A total of 40 patients aged 35 years or younger who
had undergone surgery for CRC at West China Hospital (Sichuan
University, Chengdu, Sichuan, China) between November 2014 and May
2015 were selected for the present study, which was performed
according to protocols approved by the West China Hospital
Institutional Review Board (Chengdu, China). They primarily
presented with solitary tumors without polyposis or any clinical
history of inflammatory bowel disease. Information regarding
medical history and family history and pathological data were
collected.
Immunohistochemistry (IHC) of MMR
protein
Tissue sections (4-µm-thick) were cut from the
formalin-fixed, paraffin-embedded (FFPE) tumors. The slides had
undergone deparaffinization, rehydration and antigen retrieval, and
endogenous peroxidase activity was blocked by incubation with 3%
H2O2 at room temperature for 25 min. The
sections were then mounted on a DAKO Autostainer Link 48 and then
exposed to the ready-to-use mutL homolog 1 (MLH1; cat. no.
IR07961), mutS homolog 2 (MSH2; cat. no. IR08561), mutS homolog 6
(MSH6; cat. no. IR08661), PMS1 homolog 2 (PMS2) (cat. no. IR08761)
rabbit and mouse monoclonal antibodies (Dako; Agilent Technologies,
Inc.) at 37°C for 1 h. The antigen-antibody reaction was visualized
with the Envision kit (Dako; Agilent Technologies, Inc). Finally,
the slides were counterstained with hematoxylin.
The tumors were considered to be devoid of MLH1,
MSH2, MSH6 and PMS2 expression if no nuclear staining of tumor
cells was detected. However, tumor cells that were strongly and
weakly stained were considered as positive. The surrounding stromal
cells, lymphocytes served as an internal positive control (18).
Genomic DNA extraction
The FFPE colorectal tumors obtained during surgery
were analyzed and for this, biopsy samples were extracted from
them. The sample blocks which harbored tumors that covered >50%
of the area were selected by a pathologist (DJ). The genomic DNA
was extracted from the FFPE samples using the QIAamp DNA FFPE
Tissue kit (Qiagen GmbH) as per the manufacturer's protocol. The
DNA concentration was measured using a Nanodrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.) and normalized
to 20–50 ng/µl. The DNA samples were then stored at −20°C until
use.
Ion Torrent Personal Genome Machine
(PGM) library preparation and sequencing
Libraries were prepared using the CureSeq PAN-Cancer
Panel (ACCB Biotech Ltd.) on the Ion Torrent™ System according to
the manufacturer's protocol. The genomic regions of selected genes
(Table I) were amplified using
pooled primer pairs, followed by ligation with adaptors and
barcodes. The hot points and exons that were tested in the 17 genes
are listed in Table I. Following
purification, the libraries were quantified using a Qubit dsDNA HS
Assay kit on a Qubit 2.0 fluorimeter (Life Technologies; Thermo
Fisher Scientific, Inc.) diluted to a concentration of 3 ng/ml and
pooled in equal volumes. The library pool was clonally amplified in
an emulsion PCR reaction using Ion Sphere Particles on the OneTouch
2 instrument (Life Technologies; Thermo Fisher Scientific, Inc.).
Subsequently, template-positive ion sphere particles were enriched
on the Ion OneTouch ES (Life Technologies; Thermo Fisher
Scientific, Inc.). Following enrichment, sequencing primers and
polymerase were added from the Ion PGM™ Sequencing Supplies 200 v2
kit (Life Technologies; Thermo Fisher Scientific, Inc.). The
mentioned procedures were performed according to the manufacturer's
protocol. The libraries were loaded onto an Ion 318 chip (Life
Technologies; Thermo Fisher Scientific, Inc.) and sequenced on an
Ion Torrent PGM (Life Technologies; Thermo Fisher Scientific, Inc.)
instrument to generate the sequencing data.
 | Table I.Detected gene panel and mutation
sites used in the present study. |
Table I.
Detected gene panel and mutation
sites used in the present study.
| Gene name | Detected mutation
sites |
|---|
| KRAS | Exons 2 and 3 |
| NRAS | Exons 2 and 3 |
| BRAF | Exons 11 and
15 |
| PIK3CA | Exons 9 and 20 |
| KIT | Exons 9, 11, 13, 14
and 17 |
| PDGFRA | Exons 12, 14 and
18 |
| EGFR | Exons 18, 19, 20
and 21 |
| ERBB2 | Exon 20 |
| DDR2 | Exon 18 |
| ALK | Codon 1196, 1202
and 1206 at exon 23, and codon 1269 at exon 25 |
| RET | Codon 634 at exon
11, codon 918 at exon 16 |
| SMO | Codon 473 at exon
8 |
| TSC1 | Exon 15 |
| FLT3 | Codon 835 at exon
20, exon 14 and 15 |
| NPM1 | Exon 11 |
| DNMT3A | Codon 882 at exon
23, exons 15–22 |
| ABL1 | Codons 253–255 at
exon 4, codons 299 and 317 at exon 5, and codons 351–359 at exon
6 |
Variant calling
The generated data were initially processed using
the Ion Torrent pipeline software Torrent Suite (Life Technologies;
Thermo Fisher Scientific, Inc.) to generate sequence reads, trim
adapter sequences and filter and remove poor signal-profile reads.
Initial variant calling from the sequencing data was generated
using Torrent Suite Software v3.4 with the plug-in ‘variant
caller’. In order to eliminate erroneous base calling and generate
the final variant calling, the variants in amplicon AMPL339432
(PIK3CA, exon13, ch3: 178938822-178938906) were eliminated as
previously described (19). The
sequence read distribution across the 67 amplicons generated from
the 40 FFPE specimens was normalized to 300,000 reads per sample
(Fig. S1).
Somatic mutations and bioinformatic
validation
The detected mutations were compared with variants
in the 1000 Genomes Project (https://www.internationalgenome.org) and 6500 exomes
of National Heart, Lung, and Blood Institute Exome Sequencing
Project (https://www.nhlbi.nih.gov), COSMIC
database (http://cancer.sanger.ac.uk/cosmic), MyCancerGenome
database (https://www.mycancergenome.org) and certain pieces of
published literature to assess the mutations in CRC in PubMed
database using the gene names as keywords.
Statistical analysis
Categorical variables were examined using the
Chi-squared or Fisher's exact test. A two-sided P<0.05 was
considered to indicate a statistically significant difference. All
the statistical analyses were performed using SPSS software
(version 24.0 for Windows; IBM Corp.).
Results
Clinicopathological
characteristics
The clinicopathological features of the patients of
the present study are summarized in Table II. The cohort consisted of 22 males
and 18 females with a mean age of 30 years (range, 18–35 years).
The rectum was the most common tumor site, followed by the right
colon and the left colon. Half of the patients presented with stage
III carcinoma at the time of diagnosis, followed by stage II, stage
I and stage IV. In total, 6 patients had poorly differentiated
tumors, including 4 cases of signet ring cell carcinoma. In
addition, 15 of the patients had relatives within three generations
who had been diagnosed with carcinoma and 9 patients had relatives
who were diagnosed with CRC.
| Table II.Patient demographics and
clinicopathological characteristics. |
Table II.
Patient demographics and
clinicopathological characteristics.
| Case no. | Age (years) | Gender | Family history
(number of relatives with cancer) | Tumor site |
Differentiationa | Perineural
invasion | AJCC stage |
|---|
| C1 | 28 | F | 1, FDR with
CRC | Right colon | 2 | No | IIIb |
| C2 | 34 | M | 1, FDR with
CRC | Right colon | 2 | No | IIa |
| C3 | 31 | F | 1, FDR with
CRC | Right colon | 1 | No | IIa |
| C4 | 26 | F | 1, FDR with
CRC | Right colon | 3 | No | IIIb |
| C5 | 31 | F | 1, FDR with
CRC | Rectum | 2 | No | IIb |
| C6 | 33 | M | 2, FDR with
CRC | Right colon | 1 | No | IIa |
| C7 | 33 | M | 1, SDR with
CRC | Rectum | 3 | No | IIIc |
| C8 | 34 | F | 1, SDR with
CRC | Rectum | 1 | No | I |
| C9 | 34 | M | 2, SDR with CRC; 1,
TDR with CRC | Rectum | 2 | No | IIIc |
| C10 | 34 | M | 1, FDR with other
cancer | Right colon | 2 | Yes | IIIb |
| C11 | 33 | F | 1, FDR with other
cancer | Right colon | 2 | No | IIIb |
| C12 | 33 | M | 1, FDR with other
cancer | Right colon | 2 | No | IIa |
| C13 | 31 | M | 1, FDR with other
cancer; 1, SDR with other cancer | Rectum | 2 | No | IVa |
| C14 | 31 | M | 1, SDR with other
cancer | Right colon | 2 | No | IIIb |
| C15 | 25 | M | 1, SDR with other
cancer | Rectum | 2 | No | I |
| C16 | 27 | M | None | Right colon | 1 | No | IIc |
| C17 | 26 | M | None | Left colon | 2 | No | IIb |
| C18 | 27 | F | None | Rectum | 2 | No | IIIb |
| C19 | 31 | M | None | Rectum | 3 | No | IIIb |
| C20 | 31 | M | None | Rectum | 2 | No | IIIa |
| C21 | 27 | M | None | Rectum | 1 | No | I |
| C22 | 18 | M | None | Right colon | 3 | No | IIIc |
| C23 | 23 | M | None | Right colon | 3 | No | IIIb |
| C24 | 30 | F | None | Left colon | 2 | No | IIIb |
| C25 | 27 | F | None | Rectum | 3 | No | IIa |
| C26 | 33 | F | None | Rectum | 2 | No | IIIb |
| C27 | 29 | M | None | Left colon | 2 | No | IIIc |
| C28 | 28 | M | None | Left colon | 1 | No | IIb |
| C29 | 34 | F | None | Right colon | 2 | No | IIIa |
| C30 | 28 | F | None | Right colon | 2 | No | IIIa |
| C31 | 35 | F | None | Right colon | 2 | No | IIIb |
| C32 | 35 | F | None | Right colon | 2 | No | IIb |
| C33 | 30 | F | None | Rectum | 2 | No | IV |
| C34 | 26 | F | None | Rectum | 2 | Yes | IV |
| C35 | 34 | M | None | Rectum | 1 | No | Ib |
| C36 | 33 | M | None | Left colon | 2 | No | IIIb |
| C37 | 35 | F | None | Right colon | 2 | No | IIIc |
| C38 | 27 | F | None | Rectum | 2 | No | IIb |
| C39 | 32 | M | None | Rectum | 2 | No | IIb |
| C40 | 35 | M | None | Rectum | 2 | No | IIa |
MMR protein expression
Representative IHC images for MMR are presented in
Fig. 1. The results regarding MMR
are summarized in Table III. The
absence of at least one MMR protein was observed in 11 tumors. No
solitary MLH1 absence was observed. Solitary MSH2 or PMS2 absence
was observed in three and one of the cases, respectively.
Furthermore, five of the tumors presented with both MSH2 and MSH6
absence and two of the tumors exhibited both MLH1 and PMS2 protein
absence. In total, 7 out of the 17 cancers that were located in the
right colon presented with MMR protein absence and among the 18
rectal cancers, only 4 samples exhibited MMR protein absence. MMR
proteins were observed in all of the 5 cases of cancer of the left
colon.
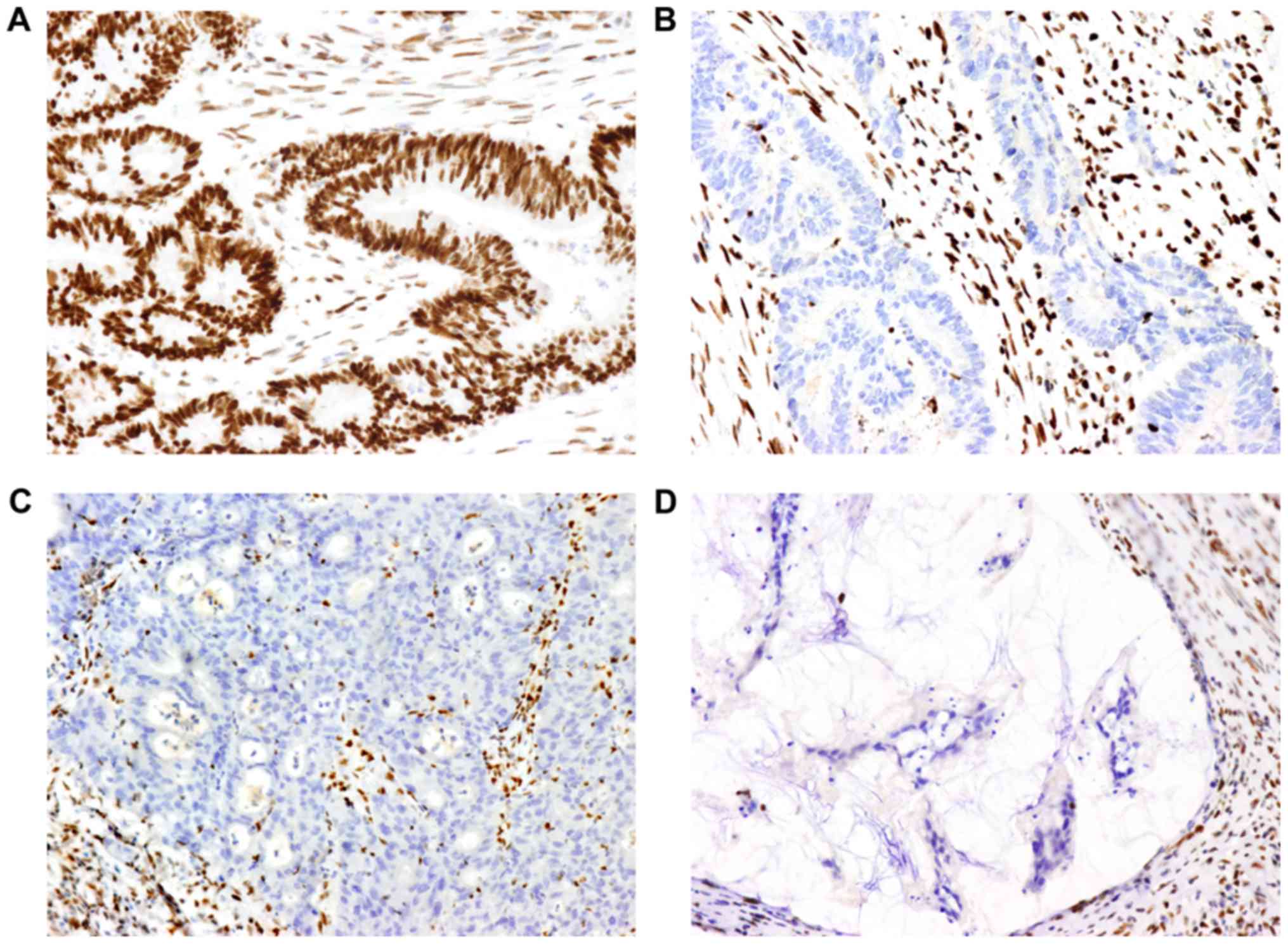 | Figure 1.Images of MMR proteins assessed by
immunohistochemistry on paraffin-embedded colorectal cancer
specimens. (A) Positive expression of MLH1 protein (magnification,
×200). (B, C and D) Lack of expression of (B) MSH2, (C) MSH6 and
(D) PMS2 protein (magnification, ×200). Positive expression is
defined as the MMR protein being expressed in the tumor cell
nucleus. A negative result is defined as the absence of nuclear
staining detected in tumor cells, while the surrounding stromal
cells and lymphocytes present with staining and serve as an
internal positive control. MMR, mismatch repair; MLH1 mutL homolog
1, DNA mismatch repair protein MLH, putative; MSH2, mutS homolog 2;
MSH6, mutS homolog 6; PMS2, PMS1 homolog 2, mismatch repair system
component. |
| Table III.Mismatch repair status and genetic
mutations present in this group of early-onset nonpolyposis
colorectal cancers. |
Table III.
Mismatch repair status and genetic
mutations present in this group of early-onset nonpolyposis
colorectal cancers.
| A, Family history
of CRC |
|---|
|
|---|
| Case no. | MMR
expressiona | Gene 1 | Mutation 1 | Gene 2 | Mutation 2 | Gene 3 | Mutation 3 | Gene 4 | Mutation 4 |
|---|
| C1 | MSH2(−) | PIK3CA | p.E545G |
|
|
|
|
|
|
| C2 | MLH1(−),
PMS2(−) | KRAS | p.G12D | PIK3CA | p.H1047R | KIT | p.L682fs | DDR2 | p.V797F |
| C3 | MSH2(−),
MSH6(−) | KRAS | p.G13D |
|
|
|
|
|
|
| C4 | MSH2(−),
MSH6(−) | ERBB2 | p.V777M | KRAS | p.G12D |
|
|
|
|
| C5 | MSH2(−),
MSH6(−) | PIK3CA | p.H1047R |
|
|
|
|
|
|
| C6 | MSH2(−) | PIK3CA | p.H1047Y | DNMT3A | p.R882H | DNMT3A | p.A741V | DNMT3A | p.A572G |
| C7 | PMS2(−) | KRAS | p.G13D | BRAF | p.V600E |
|
|
|
|
| C8 | MSH2(−),
MSH6(−) | PIK3CA | p.E545K |
|
|
|
|
|
|
| C9 | MSH2(−),
MSH6(−) | WT |
|
|
|
|
|
|
|
|
| B, Family
history of other cancer types |
|
| Case
no. | MMR
expressiona | Gene 1 | Mutation
1 |
|
|
|
|
|
|
|
| C10 | MLH1(−),
PMS2(−) | WT |
|
|
|
|
|
|
|
| C11 | (+) | WT |
|
|
|
|
|
|
|
| C12 | (+) | ERBB2 | p.C805S |
|
|
|
|
|
|
| C13 | (+) | PIK3CA | p.E545K |
|
|
|
|
|
|
| C14 | (+) | WT |
|
|
|
|
|
|
|
| C15 | (+) | DDR2 | p.V797L |
|
|
|
|
|
|
|
| C, No family
history of cancer |
|
| Case
no. | MMR
expressiona | Gene 1 | Mutation
1 | Gene 2 | Mutation
2 |
|
|
|
|
|
| C16 | MSH2(−) | KRAS | p.G12D |
|
|
|
|
|
|
| C17 | (+) | WT |
|
|
|
|
|
|
|
| C18 | (+) | WT |
|
|
|
|
|
|
|
| C19 | (+) | WT |
|
|
|
|
|
|
|
| C20 | (+) | NRAS | p.G12D |
|
|
|
|
|
|
| C21 | (+) | ABL1 | p.V323F |
|
|
|
|
|
|
| C22 | (+) | WT |
|
|
|
|
|
|
|
| C23 | (+) | WT |
|
|
|
|
|
|
|
| C24 | (+) | KRAS | p.G12D |
|
|
|
|
|
|
| C25 | (+) | KRAS | p.G13D | PIK3CA | p.E545K |
|
|
|
|
| C26 | (+) | WT |
|
|
|
|
|
|
|
| C27 | (+) | BRAF | p.V600E |
|
|
|
|
|
|
| C28 | (+) | KRAS | p.Q61L |
|
|
|
|
|
|
| C29 | (+) | KRAS | p.G12V |
|
|
|
|
|
|
| C30 | (+) | KRAS | p.G12D |
|
|
|
|
|
|
| C31 | (+) | PIK3CA | p.T1025S |
|
|
|
|
|
|
| C32 | (+) | WT |
|
|
|
|
|
|
|
| C33 | (+) | KRAS | p.G12D |
|
|
|
|
|
|
| C34 | (+) | WT |
|
|
|
|
|
|
|
| C35 | (+) | WT |
|
|
|
|
|
|
|
| C36 | (+) | WT |
|
|
|
|
|
|
|
| C37 | (+) | KRAS | p.G12V |
|
|
|
|
|
|
| C38 | (+) | KRAS | p.G12V |
|
|
|
|
|
|
| C39 | (+) | TSC1 | p.Q654E |
|
|
|
|
|
|
| C40 | (+) | KRAS | p.G12V |
|
|
|
|
|
|
Genetic mutations detected by NGS
The results regarding genetic mutation are
summarized in Table III and
Fig. 2. In total, 26 of the tumors
(26/40, 65%) carried one or more of the tested genetic mutations.
Single mutations were observed in 21 of the tumors, double
mutations in 3 tumors and three or more gene mutations in 2 tumors.
The most common mutated gene was KRAS, followed by
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha (PIK3CA), BRAF, erb-b2 receptor tyrosine kinase 2 (ERBB2),
discoidin domain receptor tyrosine kinase 2 (DDR2), NRAS, KIT
proto-oncogene (KIT), TSC complex subunit 1 (TSC1), DNA
methyltransferase 3 alpha (DNMT3A) and ABL proto-oncogene 1 (ABL1).
The specific codon data are presented in Table III.
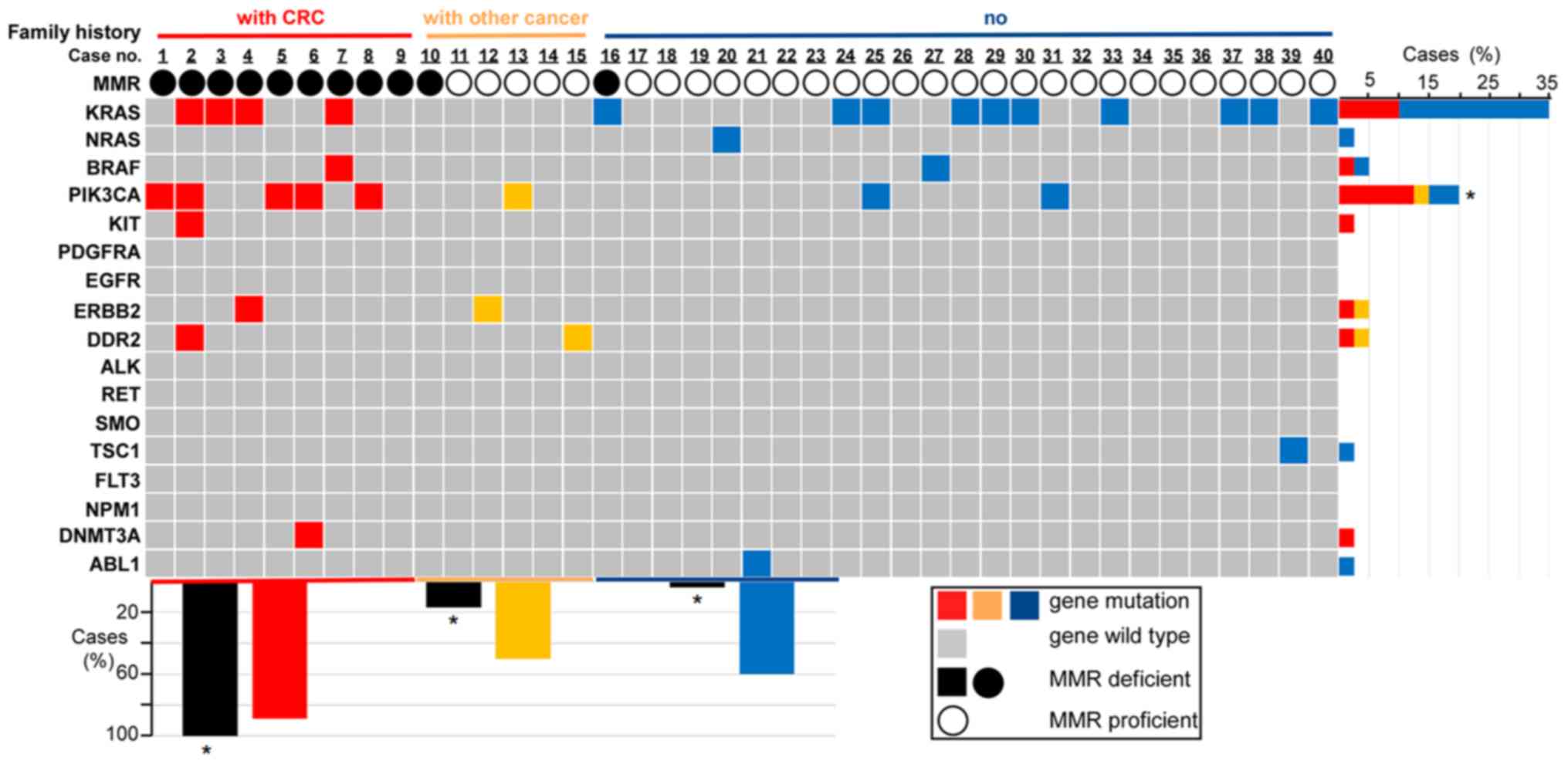 | Figure 2.Family history, MMR status and
genetic mutations for each sample. The association between genetic
mutation and family history or the MMR status is provided. Family
history and MMR status are represented by differently colored bars
or circles. For family history (first row in the figure), red
represents the cases (cases from 1 to 9) with a family history of
CRC, yellow represents the cases (cases from 10 to 15) with a
family history of other types of cancer and blue represents the
cases (cases from 16 to 40) without any family history of cancer.
For the MMR status (third row in the figure), the solid black
circle represents the dMMR status (loss of expression of at least
one MMR protein), the hollow black circles represent the pMMR
status (case exhibits positivity for MLH, MSH2, MSH6 and PMS2
proteins). The grid figure shows the genetic mutations of each
sample (grey grids represent the cases without genetic mutations
tested in the experiment, while the genetic mutational cases are
colored differently according to their family history status) and
all the tested genes in the study are listed on the left. The bar
chart on the right illustrates the percentages of mutational cases
with a different family history (differently colored bars indicate
a different family history status) for each gene. For instance, the
top bar indicates that 35% of the cases in this group of patients
had a KRAS gene mutation, of which 10% of total cases had a family
history of CRC (red bar), while the other 25% of total cases had no
family history of cancer (blue bar). The lower bar chart
illustrates the percentages of dMMR cases (black bars) and the
percentages of genetic mutational cases (differently colored bars
based on different family history) classified by three different
family history statuses (colored differently on the horizontal
axis). For instance, for the cases with a family history of CRC
(red horizontal axis), 100% showed dMMR (black bar on the left) and
88.9% of these cases (patients with CRC family history) had genetic
mutations (red bar). *P<0.05 (patients with CRC family history
vs. patients with other cancer history and vs. patients with no
family history). CRC, colorectal cancer; MMR, mismatch repair;
d/pMMR, MMR deficient/proficient; KRAS, KRAS proto-oncogene,
GTPase; NRAS, NRAS proto-oncogene; BRAF, B-Raf proto-oncogene,
serine/threonine kinase; PIK3CA,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha; KIT, KIT proto-oncogene, receptor tyrosine kinase; PDGFRA,
platelet derived growth factor receptor alpha; EGFR, epidermal
growth factor receptor; ERBB2, erb-b2 receptor tyrosine kinase 2;
DDR2, discoidin domain receptor tyrosine kinase 2; GTPase; ALK, ALK
receptor tyrosine kinase; RET, ret proto-oncogene; SMO, smoothened,
frizzled class receptor; TSC1, TSC complex subunit 1; FLT3, fms
related receptor tyrosine kinase 3; NPM1, nucleophosmin 1; DNMT3A,
DNA methyltransferase 3 alpha; ABL1, ABL proto-oncogene 1. |
Correlation between MMR status and
genetic mutations
Out of the 11 dMMR tumors, 9 samples (9/11, 81.8%)
exhibited genetic mutations in the genes tested in the present
study, while only 17 samples of the 29 MMR-proficient (pMMR) tumors
exhibited genetic mutations (P=0.158). KRAS/NRAS was mutated in 5
dMMR tumors and in 10 pMMR tumors (P=0.469). PI3KCA was mutated in
5 dMMR tumors and 3 pMMR tumors (P=0.025). KIT and DNMT3A mutations
were only detected in dMMR right colon cancers (but not in all dMMR
right colon cancers). TSC1 and ABL1 mutations were only observed in
pMMR rectal cancers (but not in all pMMR rectal cancers). BRAF,
ERBB2 and DDR2 mutations were identified both in dMMR and pMMR
cancers (not all cancers). The results are presented in Fig. 2.
MMR status and genetic mutations
classified by family history
The results regarding MMR status and genetic
mutations classified by family history are illustrated in Fig. 2. All of the 9 patients who had
relatives with CRC presented with tumors with dMMR, the prevalence
of which differed significantly from the other groups (100 vs.
16.7% of patients with a family history of other cancer types vs.
4% of patients without a family history of cancer; P<0.001). In
total, 8 out of the 9 patients with a family history of CRC
presented with mutations in genes that were assessed in the present
study. The mutation of PIK3CA was more frequently detected in the
group with a family history of CRC compared to that in the other
two groups (P=0.01), while the other genetic features exhibited no
statistically significant differences (Fig. 2).
The coincidence of dMMR and genetic mutations
occurring simultaneously was not observed in the 6 patients who had
relatives with other types of carcinoma. There were genetic
mutations in only 3 samples, while dMMR was observed in another
single case separately. ERBB2, DDR2, KIT and DNMT3A mutations were
only detected in patients who presented with a family history of
carcinoma. For the other 25 patients who did not have any family
history of cancer, 15 cases exhibited genetic mutations together
with pMMR.
Discussion
CRC is recognized as a highly heterogeneous type of
tumor at the molecular level (20).
The most important characteristic of young patients with CRC is
considered to be the high prevalence of hereditary cancer syndromes
(7). However, the results of the
present study revealed the genetic features of this subgroup of CRC
from a different perspective. The present study focused on the
genetic mutations, which are already recognized as prognostic
and/or predictive markers in multiple malignancies and various
noteworthy results were obtained. These results may aid in the
understanding of the genetic features associated with this subset
of patients with CRC. Most importantly, these results may serve as
a basis to investigate these newly identified mutations in future
research.
Early-onset non-polyposis CRC is a distinct entity
with unique genetic features. Accumulating data suggest that
early-onset CRC may have distinct clinicopathological features,
including a more frequent occurrence in the rectum (32-57.7%),
manifesting as mucinous (12.6%) or signet ring (10.8%) carcinoma
and exhibiting poor differentiation features (20.4%) on
histological analysis, and that diagnosis is mostly made at stages
III and IV (8,21–23). All
of these features were also determined in the present study.
In the cohort of the present study, the frequency of
loss of MMR function was 27.5%, which may be close to that of CRC
in general (15-20%) (24). The
mutation frequencies in the RAS signaling pathway (KRAS, 35%; NRAS,
2.5%; and BRAF, 5%) and the PI3K/AKT pathway (PIK3CA, 20%) were
similar to the general CRC series (KRAS, 35%; NRAS, 1–6%; BRAF,
5–15%; and PIK3CA, 10–30%) (25–27).
However, certain distinct molecular features were still recognized.
First, mutations were identified in certain receptor tyrosine
kinases (ERBB, 2.5%; DDR, 2.5%; KIT, 2.5%; and ABL1, 2.5%) which
have been rarely studied in CRC. This result indicates that these
receptor tyrosine kinases, which participate in several signaling
pathways, may be involved in the development of CRC. Furthermore,
these tested genes are mutated more frequently in dMMR (81.8%)
tumors than in pMMR (58.6%) tumors. This result is in accordance
with the observations of a previous study, which suggests that
multiple genes, including transforming growth factor beta receptor
2 (TGFBR2; ~90%, TGFB signaling), activin A receptor type 2A (~86%,
activin signaling), BAX (~50%, apoptosis) and phosphatase and
tensin homolog (~30%, growth regulation) are frequently mutated in
MSI-H/dmmr CRC (28). Finally, the
association of the MMR status with genetic mutations may be
classified based on family history. Patients with a family history
of CRC exhibited a higher mutational load in the present study.
Another gene that should be mentioned is DNMT3A, a
DNA methyltransferase and an epigenetic modifier. This gene has
mutations that may be detected in ~20% of acute myeloid leukemia
and is considered to be a marker of poor prognosis (29,30).
However, DNMT3A has not been identified in CRC (31,32). In
the present study, a tumor sample from a 33-year-old patient
exhibited a DNMT3A mutation and the absence of MSH2 protein
expression. Of note, both parents of this patient had CRC at the
age of 45 and 56 years, respectively. Further research is required
on this particular gene, since there may be a noteworthy biological
association.
Certain recommendations may be made regarding
stratified genetic analysis for early-onset non-polyposis CRC. The
genetic features discovered in the present study may aid clinicians
in the development of more effective strategies with which to
analyse genetic mutations in early-onset CRC. First, this involves
testing for more RAS gene mutations. The RAS gene status is crucial
for patients who are being considered for anti-EGFR antibody
therapy (33). For early-onset CRC,
it may be necessary to test for multiple RAS gene mutations, as the
mutation may occur at rare locations that render treatment
ineffective. This result is consistent with the findings of a
previous study, which suggested testing for broad RAS genes
(34). Furthermore, it is advisable
to provide a more extensive genetic analysis for patients with
early-onset CRC with dMMR. It has been observed that dMMR tumors
presented with more genetic mutations in comparison to pMMR
cancers. In addition, it is advised that more extensive gene
testing is considered for patients with a family history of CRC,
even in cases in which the MSI-H/dMMR status is fully known. In the
present study, almost all patients who were related to individuals
with CRC had both dMMR status and genetic mutations. Finally, for
patients with no family history of cancer, or those with a family
history of other cancer types, it is important to further
understand the status of other genes, even though the tumors do not
have dMMR status or may not be LS, since 50–60% of the patients
presented with other genetic mutations in the present study. On the
other hand, this suggests that 40–50% of tumors from these two
groups exhibited genetic wild-type mutations based on the testing
results from the current 17-gene panel. Thus, a larger number of
such cases require to be investigated and other types of genetic
alterations, other genes or even epigenetic modifications require
to be explored in future research.
There are various challenges and opportunities in
treating non-polyposis early-onset CRC. Emerging evidence suggests
that early-onset CRC has a more progressive biological behavior and
a worse prognosis (6,35). This subset of patients has
comparatively more concerns in a number of aspects, including the
effects of treatment on fertility (8). These features raise a compelling
concern as to the identification of prognostic markers and optimal
treatment strategies, which may differ from the older-aged subset
of patients.
The present study not only identified certain
mutations in various pathways that may be considered as prognostic
and predictive markers, but also provided insight into therapy to
be identified and developed in future research. Due to the
mutations detected in the present study, it is suggested that these
receptor tyrosine kinases and certain rarely reported genes should
be investigated for non-polyposis early-onset CRC. Furthermore, it
is crucial that further research determines whether these targeted
agents may also be efficient in early-onset CRC due to the fact
that there are established treatment strategies available for these
kinases that are normally utilized in other cancer types (36–38).
Continued focus on this tumor entity may provide an opportunity to
identify more targetable genetic changes that may improve the
current understanding and treatment of this disease.
It should be noted that the present study still has
certain limitations. One limitation is the relatively small sample
size in the present study. Although unique genetic features of
patients with CRC aged ≤35 years were identified, only 40 patients
were included. However, the incidence of early-onset CRC is
relatively low, particularly in patients aged ≤35 years (~1.5% in
all CRC patients) (5). Furthermore,
only the MMR IHC analysis rather than both MMR and MSI analysis was
performed in the present study. However, according to the high
consistency (>95%) between MMR IHC and MSI PCR results reported
(39), the MMR status may precisely
represent the MSI status and vice versa. Another limitation is that
a 17-gene panel with testing in hot mutational spots on 44 exons
was applied, which do not include intron mutations. Finally, the
number of selected genes was limited, while the major goal of the
present study was to reveal the genetic mutations that are relevant
to treatment and prognosis. The panel used in the present study
included genes that are biomarkers for the treatment and prognosis
in multiple cancer types and certain genes which have targeted
therapeutic agents in clinical practice or other studies.
Therefore, further studies using larger sample sizes and
investigating more genetic alterations are required in order to
clarify the genetic alterations occurring in young patients with
CRC.
In conclusion, the present pilot study tested
genetic mutations in 40 patients with early-onset non-polyposis CRC
using the NGS technique. The results reveal genetic mutations at
different frequencies and a possible association between the tested
genes with the MMR status or with family history. The present study
provides evidence that may aid in the understanding of the genetic
features of patients with early-onset CRC and provides insight into
potential gene-targeted therapeutic agents for early-onset
non-polyposis CRC. However, investigation of a greater number of
such cases is required in order to obtain further information and
to confirm the present findings.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dana Philips for her
help revising the language of this manuscript.
Funding
This work was financially supported by the National
Natural Science Foundation of China (grant no. 81401990).
Availability of data and materials
All of the analyzed results are included in this
article. The original sequencing data were not submitted to a
database, as a further study is underway. Datasets used and or
analyzed during the current study are available from the
corresponding author on reasonable request.
Authors' contributions
DJ and CS designed the study; CL, YW and LS
performed experiments; DJ and CS analyzed and interpreted the data;
DJ and CS wrote the manuscript. All authors have read and approved
the manuscript.
Ethics approval and consent to
participate
This study was approved by the West China Hospital
Institutional Review Board, Sichuan University (Chengdu, China; no.
K2016031). Informed consent of patients was obtained at the time of
tissue collection.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stigliano V, Sanchez-Mete L, Martayan A
and Anti M: Early-onset colorectal cancer: A sporadic or inherited
disease? World J Gastroenterol. 20:12420–12430. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Singh KE, Taylor TH, Pan CG, Stamos MJ and
Zell JA: Colorectal cancer incidence among young adults in
California. J Adolesc Young Adult Oncol. 3:176–184. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bailey CE, Hu CY, You YN, Bednarski BK,
Rodriguez-Bigas MA, Skibber JM, Cantor SB and Chang GJ: Increasing
disparities in the age-related incidences of colon and rectal
cancers in the United States, 1975–2010. JAMA Surg. 150:17–22.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ,
Meester RGS, Barzi A and Jemal A: Colorectal cancer statistics,
2017. CA Cancer J Clin. 67:177–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vuik FE, Nieuwenburg SA, Bardou M,
Lansdorp-Vogelaar I, Dinis-Ribeiro M, Bento MJ, Zadnik V, Pellisé
M, Esteban L, Kaminski MF, et al: Increasing incidence of
colorectal cancer in young adults in Europe over the last 25 years.
Gut. 68:1820–1826. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Al-Barrak J and Gill S: Presentation and
outcomes of patients aged 30 years and younger with colorectal
cancer: A 20-year retrospective review. Med Oncol. 28:1058–1061.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mork ME, You YN, Ying J, Bannon SA, Lynch
PM, Rodriguez-Bigas MA and Vilar E: High prevalence of hereditary
cancer syndromes in adolescents and young adults with colorectal
cancer. J Clin Oncol. 33:3544–3549. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siegel RL, Jakubowski CD, Fedewa SA, Davis
A and Azad NS: Colorectal Cancer in the Young: Epidemiology,
prevention, management. Am Soc Clin Oncol Educ Book. 40:1–14.
2020.PubMed/NCBI
|
|
9
|
Yantiss RK, Goodarzi M, Zhou XK, Rennert
H, Pirog EC, Banner BF and Chen YT: Clinical, pathologic, and
molecular features of early-onset colorectal carcinoma. Am J Surg
Pathol. 33:572–582. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith G, Carey FA, Beattie J, Wilkie MJ,
Lightfoot TJ, Coxhead J, Garner RC, Steele RJ and Wolf CR:
Mutations in APC, Kirsten-ras, and p53-alternative genetic pathways
to colorectal cancer. Proc Natl Acad Sci USA. 99:9433–9438. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos
JL: Genetic alterations during colorectal-tumor development. N Engl
J Med. 319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okugawa Y, Grady WM and Goel A: Epigenetic
alterations in colorectal cancer: Emerging biomarkers.
Gastroenterology. 149:1204–1225 e12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peltomaki P and de la Chapelle A:
Mutations predisposing to hereditary nonpolyposis colorectal
cancer. Adv Cancer Res. 71:93–119. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haraldsdottir S, Hampel H, Tomsic J,
Frankel WL, Pearlman R, de la Chapelle A and Pritchard CC: Colon
and endometrial cancers with mismatch repair deficiency can arise
from somatic, rather than germline, mutations. Gastroenterology.
147:1308–1316 e1. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
National Comprehensive Cancer Network, .
Colon cancer treatment guidelines. October
19–2018
|
|
16
|
National Comprehensive Cancer Network, .
Rectal cancer treatment guidelines. August
7–2018
|
|
17
|
Shaw AT and Engelman JA: ALK in lung
cancer: Past, present, and future. J Clin Oncol. 31:1105–1111.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Perez-Carbonell L, Ruiz-Ponte C, Guarinos
C, Alenda C, Paya A, Brea A, Egoavil CM, Castillejo A, Barbera VM,
Bessa X, et al: Comparison between universal molecular screening
for Lynch syndrome and revised Bethesda guidelines in a large
population-based cohort of patients with colorectal cancer. Gut.
61:865–872. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu Z, Huo X, Ye H, Tang C, Nandakumar V,
Lou F, Zhang D, Dong H, Sun H, Jiang S, et al: Genetic mutation
analysis of human gastric adenocarcinomas using ion torrent
sequencing platform. PLoS One. 9:e1004422014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang DT, Pai RK, Rybicki LA, Dimaio MA,
Limaye M, Jayachandran P, Koong AC, Kunz PA, Fisher GA, Ford JM, et
al: Clinicopathologic and molecular features of sporadic
early-onset colorectal adenocarcinoma: An adenocarcinoma with
frequent signet ring cell differentiation, rectal and sigmoid
involvement, and adverse morphologic features. Mod Pathol.
25:1128–1139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Connell JB, Maggard MA, Liu JH, Etzioni
DA, Livingston EH and Ko CY: Do young colon cancer patients have
worse outcomes? World J Surg. 28:558–562. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ballester V, Rashtak S and Boardman L:
Clinical and molecular features of young-onset colorectal cancer.
World J Gastroenterol. 22:1736–1744. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li SKH and Martin A: Mismatch repair and
colon cancer: Mechanisms and therapies explored. Trends Mol Med.
22:274–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Douillard JY, Oliner KS, Siena S,
Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham
D, Jassem J, et al: Panitumumab-FOLFOX4 treatment and RAS mutations
in colorectal cancer. N Engl J Med. 369:1023–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen Y, Wang J, Han X, Yang H, Wang S, Lin
D and Shi Y: Effectors of epidermal growth factor receptor pathway:
The genetic profiling of KRAS, BRAF, PIK3CA, NRAS mutations in
colorectal cancer characteristics and personalized medicine. PLoS
One. 8:e816282013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Samuels Y, Wang Z, Bardelli A, Silliman N,
Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al:
High frequency of mutations of the PIK3CA gene in human cancers.
Science. 304:5542004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grady WM and Carethers JM: Genomic and
epigenetic instability in colorectal cancer pathogenesis.
Gastroenterology. 135:1079–1099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ley TJ, Ding L, Walter MJ, McLellan MD,
Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et
al: DNMT3A mutations in acute myeloid leukemia. N Engl J Med.
363:2424–2433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y,
Shi JY, Zhu YM, Tang L, Zhang XW, et al: Exome sequencing
identifies somatic mutations of DNA methyltransferase gene DNMT3A
in acute monocytic leukemia. Nat Genet. 43:309–315. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim MS, Kim YR, Yoo NJ and Lee SH:
Mutational analysis of DNMT3A gene in acute leukemias and common
solid cancers. APMIS. 121:85–94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li WL, Xiao MS, Zhang DF, Yu D, Yang RX,
Li XY and Yao YG: Mutation and expression analysis of the IDH1,
IDH2, DNMT3A, and MYD88 genes in colorectal cancer. Gene.
546:263–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Van Cutsem E, Cervantes A, Adam R, Sobrero
A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson
A, Bodoky G, et al: ESMO consensus guidelines for the management of
patients with metastatic colorectal cancer. Ann Oncol.
27:1386–1422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Al-Shamsi HO, Alhazzani W and Wolff RA:
Extended RAS testing in metastatic colorectal cancer-Refining the
predictive molecular biomarkers. J Gastrointest Oncol. 6:314–321.
2015.PubMed/NCBI
|
|
35
|
Chou CL, Chang SC, Lin TC, Chen WS, Jiang
JK, Wang HS, Yang SH, Liang WY and Lin JK: Differences in
clinicopathological characteristics of colorectal cancer between
younger and elderly patients: An analysis of 322 patients from a
single institution. Am J Surg. 202:574–582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bai Y, Kim JY, Watters JM, Fang B, Kinose
F, Song L, Koomen JM, Teer JK, Fisher K, Chen YA, et al: Adaptive
responses to dasatinib-treated lung squamous cell cancer cells
harboring DDR2 mutations. Cancer Res. 74:7217–7228. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Heinrich MC, Corless CL, Demetri GD,
Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den
Abbeele AD, Druker BJ, et al: Kinase mutations and imatinib
response in patients with metastatic gastrointestinal stromal
tumor. J Clin Oncol. 21:4342–4349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Soverini S, Hochhaus A, Nicolini FE,
Gruber F, Lange T, Saglio G, Pane F, Muller MC, Ernst T, Rosti G,
et al: BCR-ABL kinase domain mutation analysis in chronic myeloid
leukemia patients treated with tyrosine kinase inhibitors:
Recommendations from an expert panel on behalf of European Leukemia
Net. Blood. 118:1208–1215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
McConechy MK, Talhouk A, Li-Chang HH,
Leung S, Huntsman DG, Gilks CB and McAlpine JN: Detection of DNA
mismatch repair (MMR) deficiencies by immunohistochemistry can
effectively diagnose the microsatellite instability (MSI) phenotype
in endometrial carcinomas. Gynecol Oncol. 137:306–310. 2015.
View Article : Google Scholar : PubMed/NCBI
|














