Introduction
Since the introduction of paclitaxel >20 years
ago for the treatment of epithelial ovarian cancer (EOC), only a
few phase III trials testing other therapeutic agents have
demonstrated notable data in terms of clinical outcome (1). Two studies demonstrated an increase in
progression-free survival (PFS) time and, in a selected subgroup of
patients, overall survival (OS) time (2) or only in PFS time (3), with the introduction of the
anti-vascular endothelial growth factor (VEGF) monoclonal antibody,
bevacizumab, using a therapeutic schedule based on paclitaxel
(2–5).
More recently, poly(ADP-ribose) polymerase (PARP)
inhibitors have begun to be used as a new therapeutic approach in
the management of EOC (6),
particularly for patients with assessed defects in the homologous
recombination (HR) DNA repair process, which is strictly linked to
BRCA1/2 gene mutations (7).
Considering the few available therapeutic options for EOC
treatment, this important discovery has addressed scientific
research into novel strategies exploiting DNA repair deficiencies
and PARP inhibitors are the first drugs with this peculiar
mechanism of action and are active in patients with recurrent EOC
with HR deficiencies (6,7). In spite of this potentially
revolutionary evidence, these molecules have been demonstrated to
be also active in patients without HR deficiencies (8). Three PARP inhibitors, olaparib,
rucaparib and niraparib, are commercially available and approved by
the Food and Drug Administration (FDA) for the treatment of
patients with recurrent EOC, with different clinical indications
and toxicity profiles. In addition, two other molecules, veliparib
and talazoparib, are still under clinical investigation. In the
literature, to the best of our knowledge, no comparisons among the
three commercial drugs have been made so far; however, ongoing
trials are now focusing their attention on new clinical indications
and on additional therapeutic strategies in combination with
conventional antineoplastic drugs.
The aim of the present review was to discuss the
current status of PARP inhibitors in terms of the mechanisms of
action, molecular activity and clinical applications, as well as to
evaluate their future prospective in oncological therapy.
BRCA mutations and cancer risk
Previous studies have demonstrated the association
between germline mutations of BRCA1 and BRCA2 genes
and the early development of both breast and ovarian cancer
(9), as well as other neoplasms
caused by either germline or somatic mutations (10).
The techniques used for the detection of BRCA
gene mutations depend on DNA sequencing procedures, which are,
however, susceptible to yielding false-positive results. In fact,
these genes can also be affected by certain benign non-pathogenic
variations, termed variants of unknown significance, which
represent ~13% of BRCA1 and BRCA2 mutations,
suggesting clinical uncertainty and ambiguity in the risk
assessment of patients undergoing the analysis (11,12). As
a consequence, different polymorphisms of these genes complicate
the identification of BRCA mutations.
Breast cancer-related to BRCA1 mutation is
more likely estrogen receptor (ER)-negative when compared with
BRCA2 and non-BRCA1 tumors (13). This evidence is substantial as
estrogens influence certain genes controlling growth regulation;
therefore, both breast and ovarian cancer are assessed for ER
status to predict prognosis, future treatment or preventive and
curative measures in both BRCA and non-BRCA tumors.
The failure of BRCA function and estrogen signaling,
together with other subcellular mechanisms, causes tumor growth due
to the lack of appropriate DNA surveillance. Silencing the
BRCA1 gene leads to increased expression of the gene
codifying the aromatase enzyme that is responsible for the
conversion of steroids into active estrogens, promoting their
synthesis and biological activity (14).
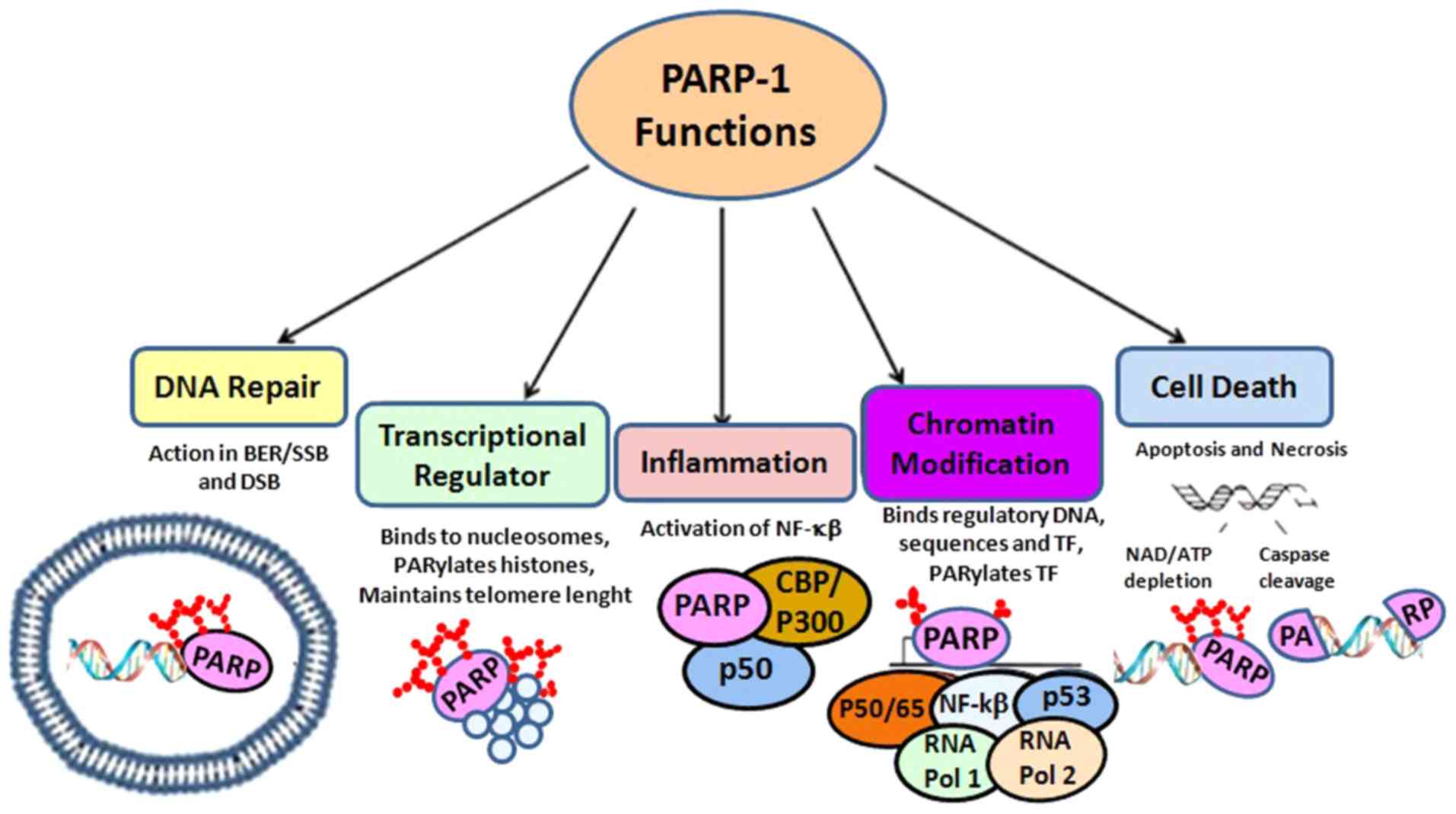
Molecular mechanisms of PARP enzymes
Tumor genomic instability results in DNA aberrations
consisting of point mutations, tandem duplications and
translocations, which induce carcinogenesis and tumor progression
(15,16). The integrity of chromosomal
structure, transcription, replication, recombination and DNA repair
are under the control of a pool of 17 enzymes, constituting the
PARP family of proteins (17,18)
(Fig. 1).
Human cells have at least five primary mechanisms of
DNA repair (19), such as mismatch
repair (MMR), base excision repair (BER), nucleotide excision
repair (NER) and double-strand break (DSB) recombination repair,
including both non-homologous end-joining (NHEJ) and homologous
recombination repair. The dysfunction, reduction or absence of
proteins involved in these pathways may lead to dangerous cellular
implications, determining mutagenesis and toxicity (20).
Different insults can affect DNA, although single
alterations are the most recurrent and are repaired by a
combination of BER, NER and MMR pathways using the undamaged DNA
strand as a template. The predominant mechanism of single-strand
break (SSB) repair is BER with the activity of PARP enzymes
(21).
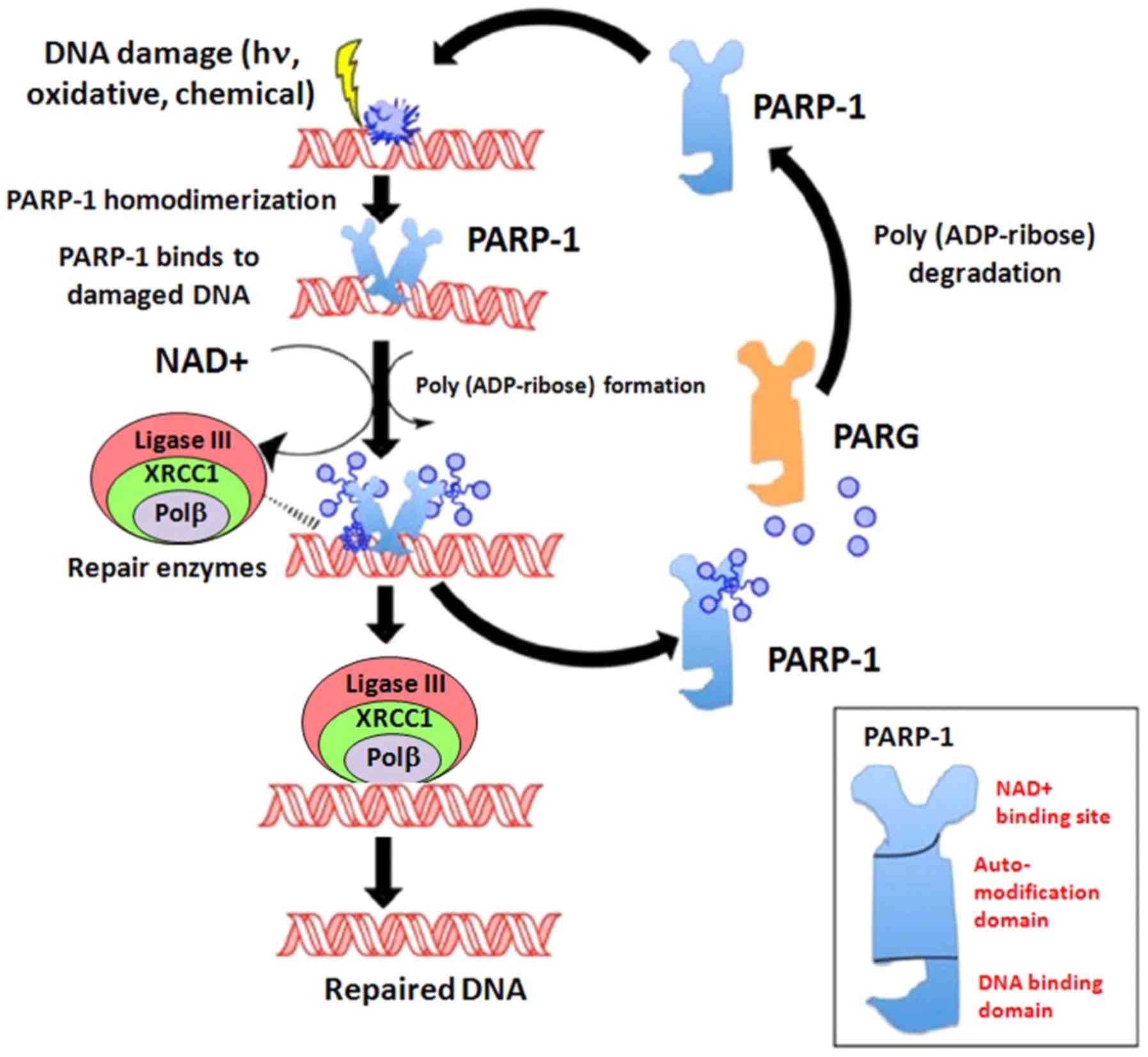
PARP-1 and PARP-2 are activated by DNA damage. In
particular, PARP-1 functions as a molecular sensor binding the
N-terminal zinc finger domains to DNA SSBs and subsequently, by
increasing its activity, catalyzes the transfer of ADP-ribose (poly
ADP-ribosylation) to target proteins through their C-terminal
catalytic domain. Following the activation of the
nicotinamide-adenine-dinucleotide (NAD+), PARPs form PAR
polymer chains that play an essential role for recruiting
intermediates for the DNA repair pathway (20) (Fig.
2). PARP-1 covalently attaches PAR chains to several different
proteins, in the process known as PARylation. Due to its role in
DNA repair, PARP inhibition results in genomic instability and the
accumulation of damaged cells in cell cycle arrest (22–26).
If PARP activity is lacking, more deleterious DSBs
can multiply, beginning from damaged SSBs, which require other
different pathways for repair (19).
Biological link between PARP and
angiogenesis inhibition
Ample experimental data have indicated a link
between PARP enzymes and angiogenesis. Angiogenesis is an important
driver of EOC development and progression and it is a main target
of antitumor therapy (27). In this
regard, since 2011, anti-VEGF therapy with bevacizumab combined
with paclitaxel and carboplatin has been the backbone of treatment
with monoclonal antibodies in patients with locally advanced and
metastatic EOC (FIGO classification stage III and IV) (28).
The PARP-1 pathway is able to regulate gene
expression, controlling angiogenesis through hypoxia-inducible
factor-1α (HIF-1α) (29).
Experimentally, PARP-deficient mice have been shown to exhibit a
decreased level of HIF-1α. This transcription factor plays a major
role in stimulating tumor angiogenesis and is a subunit of the
heterodimer HIF-1 together with HIF-1β. HIF-1β is a nuclear
constitutively expressed protein that does not undergo regulation
by the oxygen level (30); by
contrast, HIF-1α is a cytoplasmic protein whose activation is
dependent on the oxygen concentration. In particular, in oxygenated
microenvironmental conditions, HIF-1α undergoes hydroxylation by
prolyl hydroxylases on its prolyl residues in the oxygen-dependent
degradation domain and this event leads to its binding to von
Hippel-Lindau protein, before being degraded in the
ubiquitin-proteasome pathway. Meanwhile, at low oxygen tension,
prolyl hydroxylase is inactive, resulting in HIF-1α stabilization,
which allows its migration to the nucleus, where it binds HIF-1β,
finally forming the HIF-1 complex (31). The HIF-1 complex targets a consensus
hypoxia response element in the promoter of several pro-angiogenic
genes, in particular VEGF, activating their transcription (32).
From a biological point of view, in vivo and
in vitro data have suggested that angiogenesis and
tumorigenesis in EOC is promoted by PARP-1 overexpression and due
to the increasing level of VEGF-A, PARP-1 can be considered a
potential therapeutic target (33).
Experimental data employing reverse transcription-
quantitative polymerase chain reaction demonstrated that SKOV3
human ovarian cancer cells transfected with PARP-1 small
interfering RNA express lower levels of VEGF-A mRNA compared with
SKOV3 cell cultures transfected with negative control-small
interfering-RNA (26). Moreover, the
knockdown of PARP-1 was shown to decrease VEGF-A levels in SKOV3
cells, as demonstrated by western blot analysis. These results were
confirmed by ELISA, revealing the presence of VEGF-A in the
supernatant of SKOV3 cell cultures transfected with negative
control-small interfering RNA.
Notably, in addition to these data, the PARP
inhibitor, N- (6-Oxo-5,6-dihydro-phenanthridin- 2-yl)-
N,N-dimethylacetamide (PJ-34), has been demonstrated to be endowed
with anti-angiogenic activity by the in vitro inhibition of
growth and migration of human umbilical vein endothelial cells
(34), probably due to the reduction
of nitric oxide, guanylylcyclase and the cGMP pathway, which
represent the drivers of the VEGF effect on endothelial cells.
Evidently, the effects of VEGF are also mediated by the binding to
VEGF receptors that, in turn, activates the intracellular pathways
of Akt, ERK1/2 and p38 MAP kinase. In vitro studies have
demonstrated that PJ-34 inhibits the phosphorylation of these
kinases, suggesting that in the VEGF response in endothelial cells,
PARP exerts a key role. Similarly, the PARP inhibitor, GPI 15427,
has been found to exert anti-angiogenic effects in PARP-1-knockout
mice (35).
Clinical application of PARP inhibitors in
BRCA mutations, mechanisms of activity and resistance
PARP inhibitors prevent the repair of persistent
SSBs and the reconstitution of DSBs, through the replication fork.
PARP inhibitors have been developed for targeting cancer related to
BRCA1 or BRCA2 gene mutations, these genes being
responsible for the synthesis of proteins involved in the HR repair
pathway. Individuals with the wild-type phenotype have both
functioning copies of the BRCA genes; by contrast, patients
carrying a BRCA mutation have only one functioning copy,
which allows the correct process of DNA repair and viability. When
a mutation occurs in the only functioning gene copy, cells lose
their mitotic control, become susceptible to tumor growing and are
unable to undertake HR (36).
When tumor cells carry both abnormal copies of the
gene, being homozygous for BRCA1 or BRCA2 mutations,
cells undergo inadequate DNA repair and become sensitive to PARP
inhibitors. In the presence of an oncogene mutation, targeted drugs
or gene therapy should theoretically induce synthetic lethality in
neoplastic cells; in other words, synthetic lethality occurs when
two cellular events occur independently, but permit cell survival,
whereas in combination, they result in cell death (37–39).
All PARP inhibitors can inhibit both PARP-1 and
PARP-2 by suppressing PARP catalytic activity, avoiding the
formation of PAR polymers and, consequently, blocking the binding
of NAD+ at the site of DNA damage. These effects
compromise the cellular ability to overcome DNA damage (20). Recent studies have investigated
possible biomarkers of the response to PARP inhibition by measuring
both the biosynthesis of RAD51 foci and PAR poly(ADP-ribose) and
53BP1 expression levels in cancer cells (37). However, efforts to identify an
efficient and more specific biomarker are imperative in order to
optimize clinical outcomes to PARP inhibitor treatment.
PARP inhibitors share similar toxic effects with
other chemotherapeutic agents, such as nausea, fatigue, vomiting,
anemia and abdominal pain, which are the most frequently reported
adverse effects (40).
Recent evidence has shed light on an acquired
resistance to PARP inhibitors developed by neoplastic cells. A
number of mechanisms of pharmacological resistance have been
proposed; these include, in particular, the ability of tumor cells
to reverse the mutation in the BRCA gene, which restores HR
function (41). Other possible
mechanisms of resistance to PARP inhibitors can be either a
decrease in NHEJ, the reduction of PARP-1 enzymatic activity or the
increased activity of RAD51, an essential protein involved in HR
function (37). In the light of all
considerations and hypotheses, the resistance mechanisms to PARP
inhibitors are important aspects to ascertain knowledge for, in
order to forecast the efficacy of treatments.
Clinical trials with PARP inhibitors in
EOC
DNA repair processes induced by both BRCA and
PARP pathways are considered key elements for tumor aggressiveness.
If PARP is inhibited by a molecule that modifies its function,
cells genetically deficient in BRCA die, and this occurs in
EOC with BRCA mutations (42).
Olaparib
Olaparib was the first PARP inhibitor to be used in
clinical practice for the treatment of EOC; it acts as a single
therapeutic agent, with a 30–50% response rate when used in
second-line or subsequent line treatments in patients carrying
BRCA1-2 mutations (40,43–47). The
clinical activity of the drug is greater in the presence of
platinum-sensitive tumors, although platinum-resistant cancer also
responds to therapy. In addition, olaparib has been tested in
patients with the wild-type phenotype affected by high-grade serous
EOC, although the response rates have not been satisfactory in this
group of patients.
A double-blind randomized controlled phase II trial
enrolled patients with high-grade OC who were pre-treated with a
platinum-based second-line chemotherapeutic regimen, resulting in a
complete response (CR) or partial response (PR), to receive either
olaparib or a placebo as the maintenance therapy (48). The study demonstrated a better
outcome with regard to PFS time (median, 8.4 months vs. 4.8 months;
hazard ratio, 0.35; P<0.001) in patients treated with olaparib
compared with placebo. Furthermore, patients with a documented
germline BRCA mutation experienced a tripling of the time to
disease progression (median, 11.2 months vs. 4.3 months; hazard
ratio, 0.18; P<0.0001) (49).
The long-term follow-up of patients enrolled in the
aforementioned study (48)
demonstrated a favorable impact on OS time due to the maintenance
strategy. A similar outcome was recorded in a subsequent randomized
phase III trial testing olaparib vs. placebo in patients with
platinum-sensitive recurrent EOC, with a CR or PR after ≥2 lines of
platinum-based chemotherapy (50).
The overall median PFS time was 19.1 months for active maintenance
therapy vs. 5.5 months for placebo, respectively (hazard ratio,
0.30; P<0.0001).
Apart from its use in the maintenance approach,
olaparib has recently been approved by the FDA in monotherapy for
women who carry germline BRCA mutations with a diagnosis of
EOC and who have received a minimum of three prior lines of
cytotoxic chemotherapy (51).
Promising data from more recent studies might extend the clinical
indications of the drug within the near future.
Rucaparib
Rucaparib is a PARP inhibitor approved by the FDA as
a single-agent treatment in patients with EOC who carry either a
germline or a somatic BRCA mutation and who have received
pre-treatment with a minimum of two prior lines of chemotherapy. A
phase II trial demonstrated an objective response rate (ORR) of
~80% in patients with a BRCA mutation (52).
By contrast, in patients with the wild-type
phenotype with a high loss of heterozygosity, the ORR was reduced
to 44%, while in treated patients with a low loss of
heterozygosity, only 20% experienced a response (52). These data suggest that defects in DNA
repair mechanisms can be considered appropriate targets for therapy
with a PARP inhibitor.
A recent randomized phase III trial evaluating the
effects of maintenance therapy with rucaparib following second-line
chemotherapy demonstrated a significantly improved PFS time
(53), highlighting the candidacy of
this clinical approach to be once more a new standard of care for
patients with platinum-sensitive EOC.
Niraparib
Niraparib is the third PARP inhibitor available for
clinical use in the USA; it has been approved on the basis of the
results of a randomized phase III trial testing the drug compared
with a placebo, for maintenance therapy in patients who obtained a
CR or PR after second-line platinum-based chemotherapy (54). In women affected by EOC who carry a
BRCA mutation, the median PFS time of 21 months was higher
than the 5.5 months found for patients treated with placebo (hazard
ratio, 0.27). Favorable results have been observed even in the
overall non-germline BRCA patient population, with a median
PFS time of 9.3 months compared with 3.9 months (hazard ratio,
0.45) for placebo (54).
Moreover, within the same niraparib trial (54), an attempt was made to identify a
biomarker to recognize patients whose tumors could be particularly
susceptible to treatment, despite the absence of a germline
BRCA mutation. In patients with the wild-type phenotype with
tumors characterized by homologous recombinant deficiency (HRD),
maintenance therapy with niraparib exhibited a statistically
significant 12.9-month median PFS time compared with the 3.8 months
(hazard ratio, 0.38) found for placebo (54). From this objective evidence, the FDA
approved niraparib for clinical use as a second-line maintenance
strategy, following a response to platinum-based treatment without
requiring the use of a molecular biomarker, either BRCA
mutation or HRD-positive (Table
I).
 | Table I.Studies of PARP-inhibitors in
epithelial ovarian cancer. |
Table I.
Studies of PARP-inhibitors in
epithelial ovarian cancer.
| First author,
year | Patient
Population | Therapies and
doses | Outcomes | (Refs.) |
|---|
| Audeh et al,
2010 | Two cohorts of
women (aged ≥18 years) with confirmed genetic BRCA1 or
BRCA2 mutations and recurrent, measurable disease. | First cohort
(n=33): Continuous oral olaparib at the maximum tolerated dose of
400 mg twice daily. Second cohort (n=24): Continuous oral olaparib
at 100 mg twice daily. | ORR was 11 (33%)
out of 33 patients (95% CI, 20–51) in the cohort assigned 400 mg
olaparib twice daily and 3 (13%) out of 24 (4–31) in the
cohort assigned 100 mg twice daily. | (45) |
| Gelmon et
al, 2011 | Women with advanced
high-grade serous and/or undifferentiated ovarian carcinoma or
triple-negative breastancer were cstratified according to whether
they had a BRCA1 or BRCA2 mutation or not. A total of
91 patients were enrolled (65 with ovarian cancer and 26 breast
cancer). | Olaparib at 400 mg
twice daily. | In the ovarian
cancer cohorts, confirmed objective responses were seen in 7 (41%;
95% CI, 22–64) out of 17 patients with BRCA1 or BRCA2
mutations and 11 (24%; 95% CI, 14–38) out of 46 patients without
mutations. No confirmed objective responses were reported in
patients with breast cancer. | (46) |
| Swisher et
al, 2017 | A total of 204
patients with recurrent, platinum-sensitive, high-grade ovarian
carcinoma were classified into one of three predefined homologous
recombination deficiency subgroups on the basis of tumor mutational
analysis: BRCA mutant (deleterious germline or somatic),
BRCA wild-type and LOH high (LOH high group), or BRCA
wild-type and LOH low (LOH low group). | Rucaparib at 600 mg
twice daily for continuous 28 day cycles. | Progression-free
survival was significantly longer in the BRCA mutant (hazard
ratio, 0.27; 95% CI,0.16-0.44; P<0.0001) and LOH high (hazard
ratio, 0.62; 95% CI, 0.42-0.90; P=0·011) subgroups compared with
that in the LOH low subgroup. | (52) |
| Mirza et al,
2016 | A total of 553
patients were enrolled and categorized according to the presence or
absence of a germline BRCA mutation (gBRCA cohort
andnon-gBRCA cohort). Of these, 203 were in the gBRCA
cohort (with 138assigned niraparib and 65 placebo), and 350
patients were in the non-gBRCA cohort (with 234 assigned
niraparib and 116 placebo). | Niraparib at 300 mg
or placebo once daily. | Patients in the
niraparib group had significantly longer median PFS times than
those in the placebo group, namely 21.0 months vs. 5.5 in the
gBRCA cohort (hazard ratio, 0.27; 95% confidence interval
[CI], 0.17 to 0.41), as compared with 12.9 months vs. 3.8 months in
the non-gBRCA cohort for patients who had tumors with
homologous recombination deficiency (hazard ratio, 0.38; 95% CI,
0.24 to 0.59) and 9.3 months vs. 3.9 months in the overall
non-gBRCA cohort (hazard ratio, 0.45; 95% CI, 0.34 to 0.61;
P<0.001 for all three comparisons). | (54) |
Veliparib
Veliparib is a PARP inhibitor that is still under
investigation. A phase II study evaluated the effects of the use of
oral veliparib at 400 mg twice daily in 50 patients who underwent a
maximum of three prior chemotherapy regimens, with measurable
disease and who had never benefitted from another previous
treatment with a PARP inhibitor. The response rate was 26%, with a
median PFS time of 8.18 months (55). Another phase I/II study revealed a
65% overall response rate in platinum-resistant or partially
platinum-sensitive patients with a relapse of EOC carrying a
germline BRCA mutation treated with maintenance oral
veliparib 300 mg twice daily (56).
These preliminary data gave raise to other ongoing phase III
clinical trials testing veliparib not only in ovarian cancer, but
also in non-small cell lung and triple-negative breast cancer.
Talazoparib
Talazoparib is a new PARP inhibitor in clinical
development for patients with advanced or recurrent solid tumors.
In vitro studies have demonstrated that talazoparib exhibits
potent activity in tumor cells with BRCA or PTEN
mutations compared to other PARP inhibitors (57). In a multicenter phase I study, among
patients with BRCA-mutated ovarian cancer, talazoparib
exhibited a response in 5 out of 12 patients (42%), with a median
PFS time of 36.4 weeks (58).
Selection of PARP inhibitors in EOC
To date, to the best of our knowledge, no direct
trial comparing the three commercially available PARP inhibitors
has been performed; therefore, a summary report about the relative
efficacy or toxicity of each drug is not immediately being
attempted. Thanks to the results of the discussed clinical trials
and despite the different adverse events (AEs) they induce, all
agents have obtained regulatory approval for use in different
clinical settings. The large majority of patients enrolled in
non-randomized and randomized trials for all drugs have continued
treatment (if permitted by the protocol), despite recognized
toxicity, often with appropriate dose modifications and treatment
interruptions to permit recovery from the AEs (40,43–54).
All currently available PARP inhibitors have some
common side effects, in particular low-grade nausea, fatigue and
myelosuppression, which can compromise the quality of life of
patients, despite no evidenced cancer-related symptoms (59).
The selection of the right PARP inhibitor remains a
challenge. At the current time, olaparib is the only PARP inhibitor
approved for first-line therapy owing to the results of the SOLO1
clinical trial. By contrast, for second-line treatment the matter
is still under debate and the selection could be based on certain
specific differences in toxicity. Niraparib exerts a more potent
effect on platelet counts (54),
while rucaparib induces an increase in creatinine and transaminases
(52,53), both of which are essentially
false-positives, as they are not associated with real kidney
toxicity or liver toxicity. The cause of this peculiar effect
remains under investigation, as it seems to be associated with the
interaction of the PARP inhibitor with certain transport proteins,
which is responsible for the difficulty in monitoring the clinical
effect of the drug in patients with kidney and liver comorbidities
(60–62).
More frequent toxicities of PARP inhibitors can be
grouped as follows: i) Hematological: Anemia, thrombocytopenia and
neutropenia (55,63,64).
These outcomes provoke different degrees of bone marrow
suppression, depending upon the dose, which must be prescribed only
after blood counts have been taken. It is considered best practice
to follow-up new patients with lower blood counts at a weekly
frequency, particularly in the case of bone marrow suppression due
to previous chemotherapeutic regimens. ii) Gastrointestinal:
Nausea, vomiting, diarrhea, constipation, difficulty in eating and
anorexia (65), which tend to reduce
in severity with time. iii) Other: Fatigue.
In the SOLO-1/2 trial, 3 patients in the olaparib
group developed acute myeloid leukemia (AML) or myelodysplastic
syndrome (MDS). Therefore, patients who begin therapy with a PARP
inhibitor must be advised about this 3% risk of developing AML or
MDS (50).
Niraparib can cause hypertension, tachycardia and
headaches due to its interaction with the norepinephrine dopamine
carriers; therefore, patients affected by hypertension have to
monitor their blood pressure regularly (66).
Rucaparib can cause a benign elevation both in liver
function tests and in serum cholesterol during the first few weeks
of treatment (60).
Future development of PARP inhibitors in
EOC
Ongoing trials are questioning the new possible
clinical applications of PARP inhibitors in EOC, as first-line
maintenance therapy or in combination with chemotherapy, but also
the potential cross-resistance among all PARP inhibitors. In fact,
the pharmacological strategy based on PARP-after-PARP could be
considered in women who have failed to respond to one PARP agent or
who have progressed after the initial response.
Maintenance therapy plays a central role in the
clinical use of PARP agents, both as a first-line and a second-line
response to platinum-based chemotherapy and as third-line
maintenance.
Other clinically relevant questions to elaborate on
in the treatment of EOC with PARP inhibitors are the possible
combination therapies with standard platinum-based cytotoxic
therapy, bevacizumab, a checkpoint inhibitor and a topoisomerase I
inhibitor, such as topotecan.
The last cited strategy is based on the activity of
topoisomerase I to bind DNA during its replication or repair,
tempering SSBs and diminishing the associated distortional tension
(67). Topotecan is still approved
for EOC therapy due to its ability to induce de-stabilization of
the replication forks promoting DNA lesions (68,69).
PARP-1 is activated by topotecan and induces DNA lesion-reducing
DNA breakage (69). In the light of
these considerations, topoisomerase I inhibition with topotecan in
combination with PARP inhibition could lead to a magnification and
strengthening of the anti-EOC response. This therapeutic strategy
has been explored in pre-clinical studies and is now under clinical
investigation, as in vitro anti-tumor effects have been
shown to be highly potentiated (70–73).
Another clinical approach involving PARP inhibitors
is represented by their potential ability to radiosensitize EOC
(74). In a recently published
study, BRCA1-deficient high-grade ovarian cancer cells were
shown to be more sensitive to radiotherapy alone after
olaparib-mediated radiosensitization, compared with
BRCA1-proficient cells. Furthermore, when used in
association with radiotherapy, olaparib inhibited DNA damage repair
and PARP-1 activity, increased apoptosis and increased OS.
All these notable and potentially revolutionary
clinical applications of PARP inhibitors must be considered
alongside the potential associated toxicities, first of which is
the onset of MDS and AML; this toxicity seems to be due to prior
DNA damage caused by cytotoxic chemotherapy. The overall risk of
MDS and AML after PARP inhibitors is <3%, with a number of
patients having received >5 years of continuous PARP inhibitor
therapy without onset. In the future, longer treatments in a larger
population of patients will be required; therefore, the incidence
of these serious events must be carefully evaluated.
Finally, PARP inhibitors represent a new important
weapon against EOC, which is known to be associated with a poor
prognosis, with a few therapeutic options. The potential clinical
efficacy of PARP inhibitors lies not only in their peculiar
mechanisms of action, but also in the number of clinical approaches
they are involved with. The near future may provide the answers to
all questions related to PARP inhibitors in this context.
Acknowledgements
The authors would like to thank Mrs Daniela Simone
(IRCCS Istituto di Ricovero e Cura a Carattere Scientifico
‘Giovanni Paolo II’, Bari, Italy) for her assistance in the
purchase of the manuscripts useful for the elaboration of this
review.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
GR and GC conceived and designed the study of
comparison among PARP inhibitors. VL, AK, MDL, VDV and EN performed
the literature review, selecting information and clinical trials.
VL wrote the original draft of the manuscript and ML revised
English language and syntaxes. EN, ML, CDG, GG, EC and GR
critically revised the manuscript for important intellectual
content in terms of clinical trial results, adverse reactions and
future perspectives in therapy. GC, GG, EN, CDG and GR supervised
the study. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ozols RF, Bundy BN, Greer BE, Fowler JM,
Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM
and Baergen R; Gynecologic Oncology Group, : Phase III trial of
carboplatin and paclitaxel compared with cisplatin and paclitaxel
in patients with optimally resected stage III ovarian cancer: A
gynecologic oncology group study. J Clin Oncol. 21:3194–3200. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Perren TJ, Swart AM, Pfisterer J,
Ledermann JA, Pujade- Lauraine E, Kristensen G, Carey MS, Beale P,
Cervantes A, Kurzeder C, et al: A phase 3 trial of bevacizumab in
ovarian cancer. N Engl J Med. 365:2484–2496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burger RA, Brady MF, Bookman MA, Fleming
GF, Monk BJ, Huang H, Mannel RS, Homesley HD, Fowler J, Greer BE,
et al: Incorporation of bevacizumab in the primary treatment of
ovarian cancer. N Engl J Med. 365:2473–2483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ranieri G, Ferrari C, Di Palo A, Marech I,
Porcelli M, Falagario G, Ritrovato F, Ramunni L, Fanelli M, Rubini
G and Gadaleta CD: Bevacizumab-based chemotherapy combined with
regional deep capacitive hyperthermia in metastatic cancer
patients: A pilot study. Int J Mol Sci. 18:14582017. View Article : Google Scholar
|
|
5
|
Ranieri G, Patruno R, Ruggieri E,
Montemurro S, Valerio P and Ribatti D: Vascular endothelial growth
factor (VEGF) as a target of bevacizumab in cancer: From the
biology to the clinic. Curr Med Chem. 13:1845–1857. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gadducci A and Guerrieri ME: PARP
inhibitors alone and in combination with other biological agents in
homologous recombination deficient epithelial ovarian cancer: From
the basic research to the clinic. Crit Rev Oncol Hematol.
114:153–165. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sunada S, Nakanishi A and Miki Y:
Crosstalk of DNA double-strand break repair pathways in
poly(ADP-ribose) polymerase inhibitor treatment of breast cancer
susceptibility gene 1/2-mutated cancer. Cancer Sci. 109:893–899.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bitler BG, Watson ZL, Wheeler LJ and
Behbakht K: Behbakht K: PARP inhibitors: Clinical utility and
possibilities of overcoming resistance. Gynecol Oncol. 147:695–704.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alsop K, Fereday S, Meldrum C, deFazio A,
Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, et
al: BRCA mutation frequency and patterns of treatment response in
BRCA mutation-positive women with ovarian cancer: A report from the
Australian ovarian cancer study group. J Clin Oncol. 30:2654–2663.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hennessy BT, Timms KM, Carey MS, Gutin A,
Meyer LA, Flake DD II, Abkevich V, Potter J, Pruss D, Glenn P, et
al: Somatic mutations in BRCA1 and BRCA2 could expand the number of
patients that benefit from poly (ADP ribose) polymerase inhibitors
in ovarian cancer. J Clin Oncol. 28:3570–3576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Richter S, Haroun I, Graham TC, Eisen A,
Kiss A and Warner E: Variants of unknown significance in BRCA
testing: Impact on risk perception, worry, prevention and
counseling. Ann Oncol. 24 (Suppl 8):viii69–viii74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frank TS, Deffenbaugh AM, Reid JE, Hulick
M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV,
Pruss DR and Critchfield GC: Clinical characteristics of
individuals with germline mutations in BRCA1 and BRCA2: Analysis of
10,000 individuals. J Clin Oncol. 20:1480–1490. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Foulkes WD, Metcalfe K, Sun P, Hanna WM,
Lynch HT, Ghadirian P, Tung N, Olopade OI, Weber BL, McLennan J, et
al: Estrogen receptor status in BRCA1- and BRCA2-related breast
cancer: The influence of age, grade, and histological type. Clin
Cancer Res. 10:2029–2034. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bhattacharjee S and Nandi S: Rare genetic
diseases with defects in DNA repair: Opportunities and challenges
in orphan drug development for targeted cancer therapy. Cancers
(Basel). 10:2982018. View Article : Google Scholar
|
|
15
|
Andor N, Maley CC and Ji HP: Genomic
instability in cancer: Teetering on the limit of tolerance. Cancer
Res. 77:2179–2185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
O'Connor MJ: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Langelier MF, Eisemann T, Riccio AA and
Pascal JM: PARP family enzymes: Regulation and catalysis of the
poly(ADP-ribose) posttranslational modification. Curr Opin Struct
Biol. 53:187–198. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grimaldi G and Corda D: ADP-ribosylation
and intracellular traffic: An emerging role for PARP enzymes.
Biochem Soc Trans. 47:357–370. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ricks TK, Chiu HJ, Ison G, Kim G, McKee
AE, Kluetz P and Pazdur R: Successes and challenges of PAPR
inhibitors in cancer therapy. Front Oncol. 5:2222015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dziadkowiec KN, Gasiorowska E,
Nowak-Markwitz E and Jankowska A: PARP inhibitors: Review of
mechanisms of action and BRCA1/2 mutation targeting. Prz
Menopauzalny. 15:215–219. 2016.PubMed/NCBI
|
|
21
|
Davar D, Beumer JH, Hamieh L and TawbiH:
Role of PARP inhibitors in cancer biology and therapy. Curr Med
Chem. 19:3907–3921. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bhattacharjee S and Nandi S: Choices have
consequences: The nexus between DNA repair pathways and genomic
instability in cancer. Clin Transl Med. 5:452016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bhattacharjee S and Nandi S: DNA damage
response and cancer therapeutics through the lens of the fanconi
anemia DNA repair pathway. Cell Commun Signal. 15:412017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghosh S, Lu Y, Katz A, Hu Y and Li R:
Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter
of the aromatase gene (CYP19) in human adipose stromal cells. Am J
Physiol Endocrinol Metab. 292:E246–E252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bhattacharjee S and Nandi S: Synthetic
lethality in DNA repair network: A novel avenue in targeted cancer
therapy and combination therapeutics. IUBMB Life. 69:929–937. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen A: PARP inhibitors: Its role in
treatment of cancer. Chin J Cancer. 30:463–471. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ranieri G: Biological basis of tumor
angiogenesis and therapeutic intervention: Past, present, and
future. Int J Mol Sci. 19:16552018. View Article : Google Scholar
|
|
28
|
Prat J; FIGO Committee on Gynecologic
Oncology, : FIGO's staging classification for cancer of the ovary,
fallopian tube, and peritoneum: Abridged republication. J Gynecol
Oncol. 26:87–89. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martin-Oliva D, Aguilar-Quesada R, O'valle
F, Muñoz-Gámez JA, Martínez-Romero R, García Del Moral R, Ruiz de
Almodóvar JM, Villuendas R, Piris MA and Oliver FJ: Inhibition of
poly(ADP-ribose) polymerase modulates tumor-related gene
expression, including hypoxia-inducible factor-1 activation, during
skin carcinogenesis. Cancer Res. 66:5744–5756. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizukami Y, Kohgo Y and Chung DC: Hypoxia
inducible factor-1 independent pathways in tumor angiogenesis. Clin
Cancer Res. 13:5670–5674. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zimna A and Kurpisz M: Hypoxia-inducible
factor-1 in physiological and pathophysiological angiogenesis:
Applications and therapies. Biomed Res Int. 2015:5494122015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Balamurugan K: HIF-1 at the crossroads of
hypoxia, inflammation, and cancer. Int J Cancer. 138:1058–1066.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Del Rivero J and Kohn EC: PARP inhibitors:
The cornerstone of DNA repair-targeted therapies. Oncology
(Williston Park). 31:265–273. 2017.PubMed/NCBI
|
|
34
|
Wei W, Li Y, Lv S, Zhang C and Tian Y:
PARP-1 may be involved in angiogenesis in epithelial ovarian
cancer. Oncol Lett. 12:4561–4567. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tentori L, Lacal PM, Muzi A, Dorio AS,
Leonetti C, Scarsella M, Ruffini F, Xu W, Min W, Stoppacciaro A, et
al: Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene
deletion reduces angiogenesis. Eur J Cancer. 43:2124–2133. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Benafif S and Hall M: An update on PARP
inhibitors for the treatment of cancer. Onco Targets Ther.
8:519–528. 2015.PubMed/NCBI
|
|
37
|
Oplustilova L, Wolanin K, Mistrik M,
Korinkova G, Simkova D, Bouchal J, Lenobel R, Bartkova J, Lau A,
O'Connor MJ, et al: Evaluation of candidate biomarkers to predict
cancer cell sensitivity or resistance to PARP-1 inhibitor
treatment. Cell Cycle. 11:3837–3850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arnaudeau C, Lundin C and Helleday T: DNA
double-strand breaks associated with replication forks are
predominantly repaired by homologous recombination involving an
exchange mechanism in mammalian cells. J Mol Biol. 307:1235–1245.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tutt AN, Lord CJ, McCabe N, Farmer H,
Turner N, Martin NM, Jackson SP, Smith GC and Ashworth A:
Exploiting the DNA repair defect in BRCA mutant cells in the design
of new therapeutic strategies for cancer. Cold Spring Harb Symp
Quant Biol. 70:139–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Domchek SM, Aghajanian C, Shapira-Frommer
R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G,
Fried G, Stemmer SM, et al: Efficacy and safety of olaparib
monotherapy in germline BRCA1/2 mutation carriers with advanced
ovarian cancer and three or more lines of prior therapy. Gynecol
Oncol. 140:199–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sakai W, Swisher EM, Karlan BY, Agarwal
MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ,
Couch FJ, et al: Secondary mutations as a mechanism of cisplatin
resistance in BRCA2-mutated cancers. Nature. 451:1116–1120. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lord CJ and Ashworth A: PARP inhibitors:
Synthetic lethality in the clinic. Science. 355:1152–1158. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fong PC, Yap TA, Boss DS, Carden CP,
Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S,
Messiou C, et al: Poly(ADP)-ribose polymerase inhibition: Frequent
durable responses in BRCA carrier ovarian cancer correlating with
platinum-free interval. J Clin Oncol. 28:2512–2519. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Audeh MW, Carmichael J, Penson RT,
Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN,
Oaknin A, Loman N, et al: Oral poly(ADP-ribose) polymerase
inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and
recurrent ovarian cancer: A proof-of-concept trial. Lancet.
376:245–251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gelmon KA, Tischkowitz M, Mackay H,
Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M,
Gilks B, et al: Olaparib in patients with recurrent high-grade
serous or poorly differentiated ovarian carcinoma or
triple-negative breast cancer: A phase 2, multicentre, open-label,
nonrandomised study. Lancet Oncol. 12:852–861. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kaufman B, Shapira-Frommer R, Schmutzler
RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G,
Stemmer SM, Hubert A, et al: Olapari bmonotherapy in patients with
advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol.
33:244–250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ledermann J, Harter P, Gourley C,
Friedlander M, Vergote I, Rustin G, Scott C, Meier W,
Shapira-Frommer R, Safra T, et al: Olaparib maintenance therapy in
platinum-sensitive relapsed ovarian cancer. N Engl J Med.
366:1382–1392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ledermann J, Harter P, Gourley C,
Friedlander M, Vergote I, Rustin G, Scott CL, Meier W,
Shapira-Frommer R, Safra T, et al: Olaparib maintenance therapy in
patients with platinum-sensitive relapsed serous ovarian cancer: a
preplanned retrospective analysis of outcomes by BRCA status in a
randomized phase 2 trial. Lancet Oncol. 15:852–861. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pujade-Lauraine E, Ledermann JA, Selle F,
Gebski V, Penson RT, Oza AM, Korach J, Huzarski T, Poveda A,
Pignata S, et al: Olaparib tablets as maintenance therapy in
patients with platinum-sensitive, relapsed ovarian cancer and a
BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised,
placebo-controlled, phase 3 trial. Lancet Oncol. 18:1274–1284.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim G, Ison G, McKee AE, Zhang H, Tang S,
Gwise T, Sridhara R, Lee E, Tzou A, Philip R, et al: FDA approval
summary: Olaparib monotherapy in patients with deleterious germline
BRCA-mutated advanced ovarian cancer treated with three or more
lines of chemotherapy. Clin Cancer Res. 21:4257–4261. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Swisher EM, Lin KK, Oza AM, Scott CL,
Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O'Malley DM,
et al: Rucaparib in relapsed, platinum-sensitive high-grade ovarian
carcinoma (ARIEL2 Part 1): An international, multicentre,
open-label, phase 2 trial. Lancet Oncol. 18:75–87. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Coleman RL, Oza AM, Lorusso D, Aghajanian
C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G,
et al: Rucaparib maintenance treatment for recurrent ovarian
carcinoma after response to platinum therapy (ARIEL3): A
randomised, double-blind, placebo-controlled, phase 3 trial.
Lancet. 390:1949–1961. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mirza MR, Monk BJ, Herrstedt J, Oza AM,
Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I,
et al: Niraparib maintenance therapy in platinum-sensitive,
recurrent ovarian cancer. N Engl J Med. 375:2154–2164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Coleman RL, Sill MW, Bell-McGuinn K,
Aghajanian C, Gray HJ, Tewari KS, Rubin SC, Rutherford TJ, Chan JK,
Chen A and Swisher EM: A phase II evaluation of the potent, highly
selective PARP inhibitor veliparib in the treatment of persistent
or recurrent epithelial ovarian, fallopian tube, or primary
peritoneal cancer in patients who carry a germline BRCA1 or BRCA2
mutation-an NRG oncology/gynecologic oncology group study. Gynecol
Oncol. 137:386–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Steffensen KD, Adimi P and Jakobsen A:
Veliparib monotherapy to patients with BRCA germ line mutation and
platinum-resistant or partially platinum-sensitive relapse of
epithelial ovarian cancer: A phase I/II study. Int J Gynecol
Cancer. 27:1842–1849. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shen Y, Rehman FL, Feng Y, Boshuizen J,
Bajrami I, Elliott R, Wang B, Lord CJ, Post LE and Ashworth A: BMN
673, a novel and highly potent PARP1/2 inhibitor for the treatment
of human cancers with DNA repair deficiency. Clin Cancer Res.
19:5003–5015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
de Bono J, Ramanathan RK, Mina L, Chugh R,
Glaspy J, Rafii S, Kaye S, Sachdev J, Heymach J, Smith DC, et al:
Phase I, dose-escalation, two-part trial of the PARP inhibitor
talazoparib in patients with advanced germline BRCA1/2 mutations
and selected sporadic cancers. Cancer Discov. 7:620–629. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
LaFargue CJ, Dal Molin GZ, Sood AK and
Coleman RL: Exploring and comparing adverse events between PARP
inhibitors. Lancet Oncol. 20:e15–e28. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rucaparib, . LiverTox: Clinical and
research information on drug-induced liver injury [Internet].
(Bethesda (MD)). National Institute of Diabetes and Digestive and
Kidney Diseases. Jun 1–2012-2017.
|
|
61
|
Olaparib, . LiverTox: Clinical and
research information on drug-induced liver injury [Internet].
(Bethesda (MD)). National Institute of Diabetes and Digestive and
Kidney Diseases. Jun 1–2012-2017.
|
|
62
|
Niraparib, . LiverTox: Clinical and
research information on drug-induced liver injury [Internet].
(Bethesda (MD)). National Institute of Diabetes and Digestive and
Kidney Diseases. Jun 20–2012-2017.
|
|
63
|
Zhou J, Feng L and Zhang X: Risk of severe
hematologic toxicities in cancer patients treated with PARP
inhibitors: A meta-analysis of randomized controlled trials. Drug
Des Devel Ther. 11:3009–3017. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Alecu I, Milenkova T and Turner SR: Risk
of severe hematologic toxicities in cancer patients treated with
PARP inhibitors: Results of monotherapy and combination therapy
trials. Drug Des Devel Ther. 12:347–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu Y, Meng J and Wang G: Risk of selected
gastrointestinal toxicities associated with poly (ADP-ribose)
polymerase (PARP) inhibitors in the treatment of ovarian cancer: A
meta-analysis of published trials. Drug Des Devel Ther.
12:3013–3019. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Moore KN, Mirza MR and Matulonis UA: The
poly (ADP ribose) polymerase inhibitor niraparib: Management of
toxicities. Gynecol Oncol. 149:214–220. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hsiang YH and Liu LF: Identification of
mammalian DNA topoisomerase I as an intracellular target of the
anticancer drug camptothecin. Cancer Res. 48:1722–1726.
1988.PubMed/NCBI
|
|
68
|
Staker BL, Hjerrild K, Feese MD, Behnke
CA, Burgin AB Jr and Stewart L: The mechanism of topoisomerase I
poisoning by a camptothecin analog. Proc Natl Acad Sci USA.
99:15387–15392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
ten Bokkel Huinink W, Gore M, Carmichael
J, Gordon A, Malfetano J, Hudson I, Broom C, Scarabelli C, Davidson
N, Spanczynski M, et al: Topotecan versus paclitaxel for the
treatment of recurrent epithelial ovarian cancer. J Clin Oncol.
15:2183–2193. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
D'Onofrio G, Tramontano F, Dorio AS, Muzi
A, Maselli V, Fulgione D, Graziani G, Malanga M and Quesada P:
Poly(ADP-ribose) polymerase signaling of topoisomerase 1-dependent
DNA damage in carcinoma cells. Biochem Pharmacol. 81:194–202. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Patel AG, Flatten KS, Schneider PA, Dai
NT, McDonald JS, Poirier GG and Kaufmann SH: Enhanced killing of
cancer cells by poly(ADP-ribose) polymerase inhibitors and
topoisomerase I inhibitors reflects poisoning of both enzymes. J
Biol Chem. 287:4198–4210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wahner Hendrickson AE, Menefee ME,
Hartmann LC, Long HJ, Northfelt DW, Reid JM, Boakye-Agyeman F,
Kayode O, Flatten KS, Harrell MI, et al: A phase I clinical trial
of the Poly(ADP-ribose) polymerase inhibitor veliparib and weekly
topotecan in patients with solid tumors. Clin Cancer Res.
24:744–752. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hjortkjær M, Kanstrup H, Jakobsen A and
Steffensen KD: Veliparib and topotecan for patients with
platinum-resistant or partially platinum-sensitive relapse of
epithelial ovarian cancer with BRCA negative or unknown BRCA
status. Cancer Treat Res Commun. 14:7–12. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bi Y, Verginadis II, Dey S, Lin L, Guo L,
Zheng Y and Koumenis C: Radiosensitization by the PARP inhibitor
olaparib in BRCA1-proficient and deficient high-grade serous
ovarian carcinomas. Gynecol Oncol. 150:534–544. 2018. View Article : Google Scholar : PubMed/NCBI
|