Introduction
Esophageal cancer (EC) was the sixth leading cause
of cancer-associated mortality worldwide in 2014 (1). Adenocarcinoma and squamous cell
carcinoma are the primary types of EC (2). Squamous cell carcinoma is more common
in Asia and developing countries (3), and its prognosis is poor (4).
Chemotherapy is a routine method for EC treatment.
Commonly used chemotherapeutic agents include platinum drugs,
taxanes and epirubicin (EPI) (5).
However, low solubility in water and poor selective capability have
greatly limited the clinical application of chemotherapeutic agents
(6). Therefore, it is necessary to
develop a novel drug delivery system (DDS) to enhance drug
solubility, promote drug accumulation in tumor cells and achieve
‘on-demand’ drug release, to result in increased efficacy of EC
treatment.
Recently, DDSs based on nanoparticles have been
developed to deliver antitumor drugs. For example, Fan et al
(5) developed EPI-loaded near
infrared fluorescent peptide nanoparticles for esophageal cancer
therapy; the results revealed that these nanoparticles could
significantly enhance the efficiency of EPI and decrease its system
toxicity. These ‘nano-DDSs’ can improve the water solubility,
biocompatibility, and tumor-tissue accumulation of a drug via the
enhanced permeability and retention effect, and reduce the side
effects of a drug (7–9). To achieve on-demand release of a drug,
various stimuli-responsive DDSs have been developed. Various
endogenous signals, such as pH and glutathione, have been employed
as stimuli to trigger drug release (10). For example, Zhang et al
(11) developed a redox-responsive
polymeric micelle co-loaded paclitaxel/apatinib for effectively
reversing cancer multidrug resistance; the results revealed that in
the presence of glutathione, both drugs could rapidly be released
to kill cancer cells.
ATP is considered the ‘molecular unit of currency’
of intracellular energy transfer. ATP exhibits a high concentration
in the cytosol of tumor cells (1–10 mM) compared with the
extracellular concentration of ATP (<0.4 mM) (12). Therefore, ATP can serve as a stimulus
to trigger the release of chemotherapeutic agents.
Aptamers are oligonucleotide/peptide molecules that
bind to a specific target molecule (13). Binding of aptamers to ATP has been
reported to promote release of preloaded therapeutics directly
through a ‘conformational switch’ that is recognized and activated
specifically by ATP (14–16).
Anthracyclines are traditional anticancer drugs.
They can destroy cancer cells efficaciously because they interact
with the GC pairs of DNA, and inhibit the growth of tumor cells by
interfering with DNA transcription (17). Therefore, anthracyclines can be
loaded into double-stranded DNA (‘DNA duplex’)-containing GC
pairs.
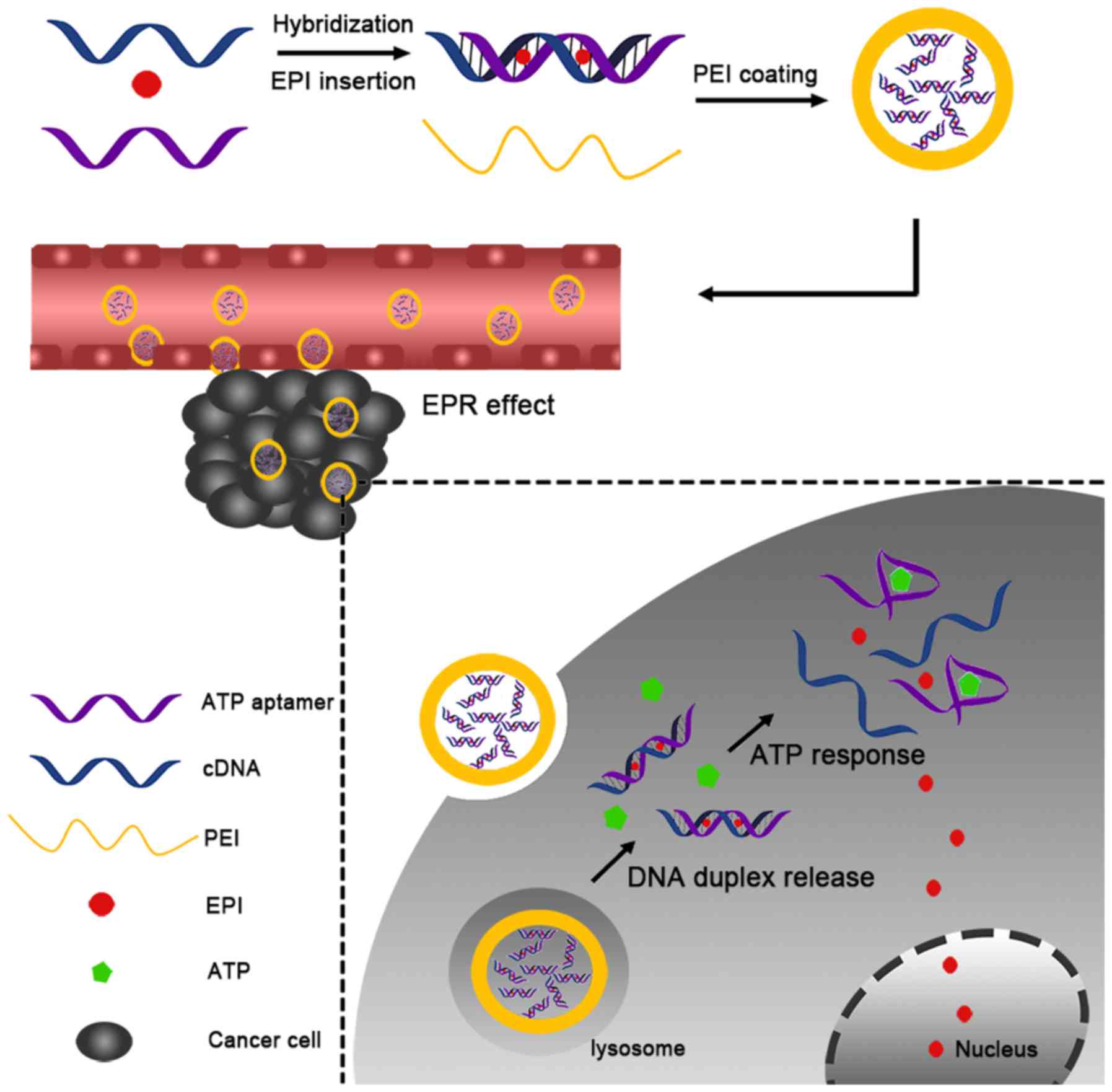
In the present study, a nano-DDS composed of an ATP
aptamer (Ap) and its complementary single-stranded DNA (cDNA), EPI
and polyethyleneimine (PEI) was developed. First, the Ap interacted
with cDNA to form a duplex by complementation. Subsequently, EPI
was loaded into the duplex DNA through interaction with the GC
pairs in the duplex (Ap-EPI). Finally, PEI (which has a positive
charge) underwent electronic interaction with the DNA duplex to
condense the DNA duplex into nanoparticles (Fig. 1). It was hypothesized that PEI-Ap-EPI
nanoparticles could increase accumulation in tumor cells and
release EPI rapidly in the presence of a high level of ATP, thereby
improving treatment efficacy considerably.
Materials and methods
Materials
The Ap (5′-ACCTGGGGGAGTATTGCGGAGGAAGGT-3′), cDNA of
the Ap (5′-ACCTTCCTCCGCAATACTCCCCCAGGT-3′), control aptamer
(5′-ACCTGGTTTAGGCGGCTCGGGAAT-3′) and cDNA of the control aptamer
(5′-ATTCCCGAGCCGCCTAAACCAGGT-3′) were purchased from Sangon Biotech
Co., Ltd. Trypan Blue dye was obtained from Generay Biotech Co.,
Ltd. RPMI-1640 medium and FBS were purchased from Invitrogen;
Thermo Fisher Scientific, Inc. Penicillin was supplied by CSPC
Pharmaceutical Group, Ltd. Streptomycin was obtained from Merro
Pharmaceutical Co., Ltd. MTT was purchased from Sigma-Aldrich;
Merck KGaA. PBS was obtained from Beyotime Institute of
Biotechnology. Ethylenediaminetetraacetic acid and MgCl2
were obtained from Sinopharm Chemical Reagent Co., Ltd. Water was
purified and deionized using a Milli-Q™ system from EMD
Millipore.
Cell culture
The EC KYSE-70 and EC109 cell lines were obtained
from the American Type Culture Collection, and incubated in
RPMI-1640 medium supplemented with 10% (v/v) FBS, 100 U/ml
penicillin and 100 mg/ml streptomycin in an atmosphere of 5%
CO2 at 37°C. Cells were counted using a hemocytometer
(Sigma-Aldrich; Merck KGaA). Cell viability was assessed by
exclusion of Trypan Blue dye (0.4%). In brief, 10 µl Trypan Blue
dye solution was added to 100 µl cell suspension, and maintained at
room temperature for 3–5 min. Subsequently, 10 µl cells suspension
was added onto the hemocytometer and observed using a Nikon TS100
light microscope (Nikon Corporation).
DDS preparation
The Ap and its cDNA were dissolved in PBS
supplemented with MgCl2 (5 mM), mixed and agitated for
24 h at room temperature. Subsequently, EPI was added to the DNA
duplex and incubated for 2 h at room temperature to form Ap-EPI.
The amount of intercalated EPI was determined based on the
fluorescence intensity, which was recorded using a microplate
reader (Tecan Group, Ltd.).
Next, PEI-Ap-EPI nanoparticles were created.
Briefly, Ap-EPI (2.5 µM) was reacted with branched PEI (25 k) at a
ratio of 1:2 in aqueous solution for 1 h at room temperature.
Excess PEI was removed by centrifugation at 2,000 × g for 5 min at
room temperature. As a result, PEI-Ap-EPI was obtained. The final
nanoparticles were prepared by resuspending PEI-Ap-EPI in ultrapure
water.
The morphology of PEI-Ap-EPI nanoparticles was
determined by transmission electron microscopy (TEM) using a Tecnai
G2 system (FEI; Thermo Fisher Scientific, Inc.) and their size
distribution was determined by differential light scattering (DLS)
using a Zs90 setup (Malvern Instruments, Ltd.).
Characterization of Ap-EPI
Ap-EPI (1.5 µM) was incubated with ATP (0, 0.2, 0.4,
1, 2, 4 and 8 mM) for 15 min at 37°C, and the fluorescence
intensity was measured to evaluate the ATP response of Ap-EPI. To
evaluate the specificity of the ATP response, Ap-EPI (2 µM) was
added to PBS supplemented with MgCl2 (5 mM), and
incubated with ATP, GTP, uridine triphosphate (UTP) or cytidine
triphosphate (CTP), respectively, for 15 min at 37°C. All compounds
were used at 0, 0.5, 1, 2, 4 and 8 mM, respectively. Finally,
fluorescence spectroscopy of EPI was performed at an excitation
wavelength of 480 nm to examine the specificity of Ap-EPI.
ATP-triggered EPI release in
vitro
A total of 2 mg of PEI-Ap-EPI (containing 224 µg
EPI) was dispersed into PBS containing 5 mM MgCl2.
Subsequently, ATP (0.4 and 4 mM) was added, and the mixture was
incubated at 37°C for 0, 0.5, 1, 2, 4, 8, 12 or 24 h. The amount of
EPI released was determined via measurement of fluorescence
intensity at the aforementioned time points. In addition,
PEI-control (Ctrl) Ap-EPI mixed with ATP (4 mM) was used as a
negative control.
Stability of PEI-Ap-EPI
nanoparticles
To determine the stability of PEI-Ap-EPI
nanoparticles, they were dispersed in PBS with 10% FBS and
incubated at 37°C. The size distribution and zeta potential of
PEI-Ap-EPI nanoparticles were measured using DLS at 0, 2, 4, 6, 8,
12 and 24 h.
EPI accumulation in cells
EPI accumulation in KYSE-70 and EC109 cells was
measured by fluorescence microscopy. Cells (~105
cells/well) were seeded in a six-well plate. After 24 h, EPI (0.5
µg/ml), Ap-EPI and PEI-Ap-EPI dispersed in serum-free medium were
added to the six-well plate, and incubated at 37°C for another 24
h. Subsequently, the drugs were removed and cells were washed three
times with PBS. Subsequently, cells were incubated with 4%
paraformaldehyde for 15 min at room temperature. Finally, confocal
laser scanning microscopy (CLSM) was performed using an LSM 710
setup (Zeiss AG) after cells had been treated with an
anti-fluorescence quencher.
Cell viability assay
KYSE-70 and EC109 cells were seeded into 96-well
plates at a density of 5×103 cells per well and cultured
in medium for 24 h. Then, these two cell lines were treated with
increasing concentrations (0.1, 0.5, 1, 2, 4, 8 and 16 µg/ml EPI
for KYSE 70 cells; 0, 0.06, 0.16, 0.32, 0.64, 1.28 and 2.56 µg/ml
EPI for EC109 cells; different concentrations were used due to the
different sensitivity of the cells to EPI) of free EPI, Ap-EPI or
PEI-Ap-EPI, and incubated at 37°C for 48 h. Afterwards, these drugs
were removed and 20 µl MTT (5 mg/ml) was added to each well and
incubated for another 4 h. Finally, the medium in each well was
replaced with 150 µl dimethyl sulfoxide. The absorbance was scanned
at 490 nm by FlexStation™ 3 (Molecular Devices, LLC). Each
experiment was repeated at least three times. The IC50
value was calculated using GraphPad Prism 6 (GraphPad Software,
Inc.).
Apoptosis detection
Apoptosis of KYSE-70 and EC109 cells was detected
using the APO-BrdU TUNEL Assay kit (Life Technologies; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
These two cell types were seeded in six-well plates at a density of
105 cells per well. Following culture for 12 h, KYSE-70
cells were incubated with 0.5 µg/ml EPI, Ap-EPI or PEI-Ap-EPI for
another 12 h. EC109 cells were treated with the three formulations
of EPI (0.1 µg/ml) for 12 h. Subsequent steps were carried out in
accordance with the manufacturer's protocol. In brief, cells were
stained with BrdU solution (10 µM) at 37°C for 2 h, washed with PBS
for three times, fixed using 4% formaldehyde at room temperature
for 15 min, washed with PBS for three times, incubated with Triton
X-100 permeabilization buffer (1 ml/well) for 20 min at room
temperature, cultured with 2N HCl (1 ml) for 10 min at room
temperature, incubated with phosphate/citric acid buffer (pH 7.4, 1
ml/well) for 10 min at room temperature, washed with Triton X-100
permeabilization buffer, stained with anti-BrdU primary antibody at
room temperature overnight, washed with Triton X-100
permeabilization buffer, and finally stained with FITC-labeled
secondary antibody at room temperature for 1 h. Cells were observed
under a fluorescence microscope (magnification, ×40; Leica
Microsystems GmbH). Green fluorescence indicated apoptotic cells.
Nuclei were stained with DAPI (10 µg/ml) at room temperature for 15
min and emitted blue fluorescence. Apoptosis of KYSE70 and EC109
cells following treatment with the three drugs was assessed by
counting the percentage of apoptotic cells (green) among the total
cells (blue) according to the method of Takeda et al
(18).
Determination of cell cycle
phases
For analyses of cell cycle arrest, KYSE70 and EC109
cells were seeded in a six-well plate at a density of
105 cells per well and incubated with EPI, Ap-EPI or
PEI-Ap-EPI for 48 h. Then, cells were collected and fixed with 70%
ethanol overnight at 4°C. Cells were washed with PBS, exposed to
PI/RNase staining buffer (BD Biosciences) and incubated in the dark
for 30 min. Cells were counted using a flow cytometer
(FACScalibur™; BD Biosciences) and analyzed using FlowJo 7.6
(FlowJo LLC) (19).
Statistical analysis
Each experiment was repeated three times. Numerical
data are presented as the mean ± SD. One-way single factorial ANOVA
followed by Tukey's post hoc test was performed to determine
statistical significance of the data using SPSS software (version
19.0; IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
EPI loading and ATP-response
release
First, an Ap and its cDNA were hybridized to
construct a carrier to load free EPI. The Ap has 27 base pairs rich
in GC that can accommodate EPI (12). To evaluate the number of EPI
molecules loaded into the ATP-Duplex, changes in the fluorescence
intensity of EPI were measured when it was mixed with various
concentrations of the ATP-Duplex. Different concentrations of the
ATP-Duplex were added to a fixed concentration of EPI. It was
demonstrated that the fluorescence intensity of EPI decreased with
an increasing concentration of the ATP-Duplex. Fluorescence nearly
disappeared when the molar ratio of duplex: EPI was 1:2 (Fig. 2A).
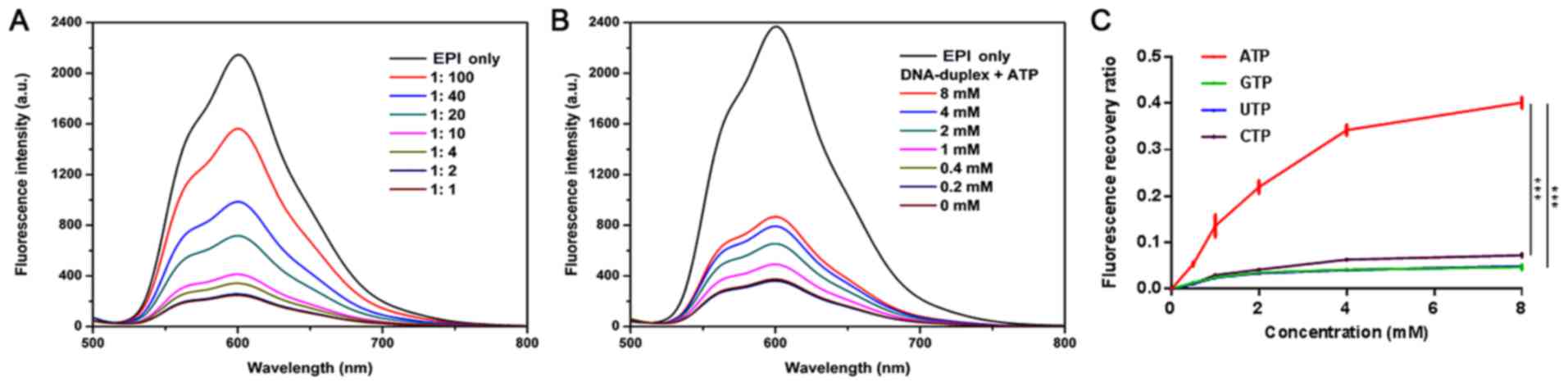 | Figure 2.Characterization of Ap-EPI. (A)
Fluorescence spectra of EPI solution (1.5 µM) with an increasing
concentration of the hybridized DNA duplex of the ATP aptamer and
its complementary single-stranded DNA following incubation for 15
min in PBS with MgCl2 (5 mM). (B) Fluorescence spectra
of Ap-EPI (equivalent to 1.5 µM EPI) in the presence of ATP (0,
0.2, 0.4, 1, 2, 4 and 8 mM) following incubation for 15 min. (C)
Fluorescence recovery of Ap-EPI following addition of ATP, GTP, CTP
and UTP (0, 0.5, 1, 2, 4 and 8 mM). Error bars indicate the SD
(n=3). ***P<0.001. Ap, ATP aptamer; EPI, epirubicin; CTP,
cytidine triphosphate; UTP, uridine triphosphate. |
In the present design, the duplex dissociated once
the Ap combined with ATP [which has a different extracellular
concentration (<0.4 mM) and cytosol concentration (1–10 mM)],
and then EPI was released from the ATP-Duplex. Therefore, to assess
the ATP-Duplex response to ATP, the fluorescence intensity of
released EPI was recorded after Ap-EPI (1.5 µM) had been incubated
with ATP (0, 0.2, 0.4, 1, 2, 4 or 8 mM). In the ATP-Duplex group,
the fluorescence intensity strengthened upon a gradual increase in
the ATP concentration. When the ATP concentration reached 4 mM, the
fluorescence intensity of EPI was 2-fold higher than that when the
ATP concentration was 0.4 mM (Fig.
2B). These results suggested that the ATP-Duplex was sensitive
to ATP, and that EPI release was dependent on the ATP
concentration.
To demonstrate the specificity of ATP-triggered EPI
release, the release of EPI from Ap-EPI was investigated under GTP,
UTP and CTP conditions. Ap-EPI (2 µM) was treated with GTP, UTP,
CTP or ATP at different concentrations for 12 h, and EPI release
was measured using a microplate reader. EPI release was low from
Ap-EPI following incubation with GTP, UTP or CTP (Fig. 2C). Furthermore, when the
concentration of GTP, UTP and CTP increased to 8 mM, ~5% of
fluorescence was recovered. Conversely, fluorescence recovery was
~40% when 8 mM ATP was added, which was 8-fold higher than that
when GTP, UTP or CTP was added. This result illustrated that the
ATP-Duplex response to ATP was specific.
Preparation and characterization of
PEI-Ap-EPI
Dizaj et al (20) demonstrated that the DNA formed in
nanoparticles can protect them from enzymatic digestion. To avoid
the EPI-loaded DNA duplex being digested by enzymes during
administration, PEI (a polymer molecule with a positive charge) was
employed to condense Ap-EPI into nanoparticles through charge
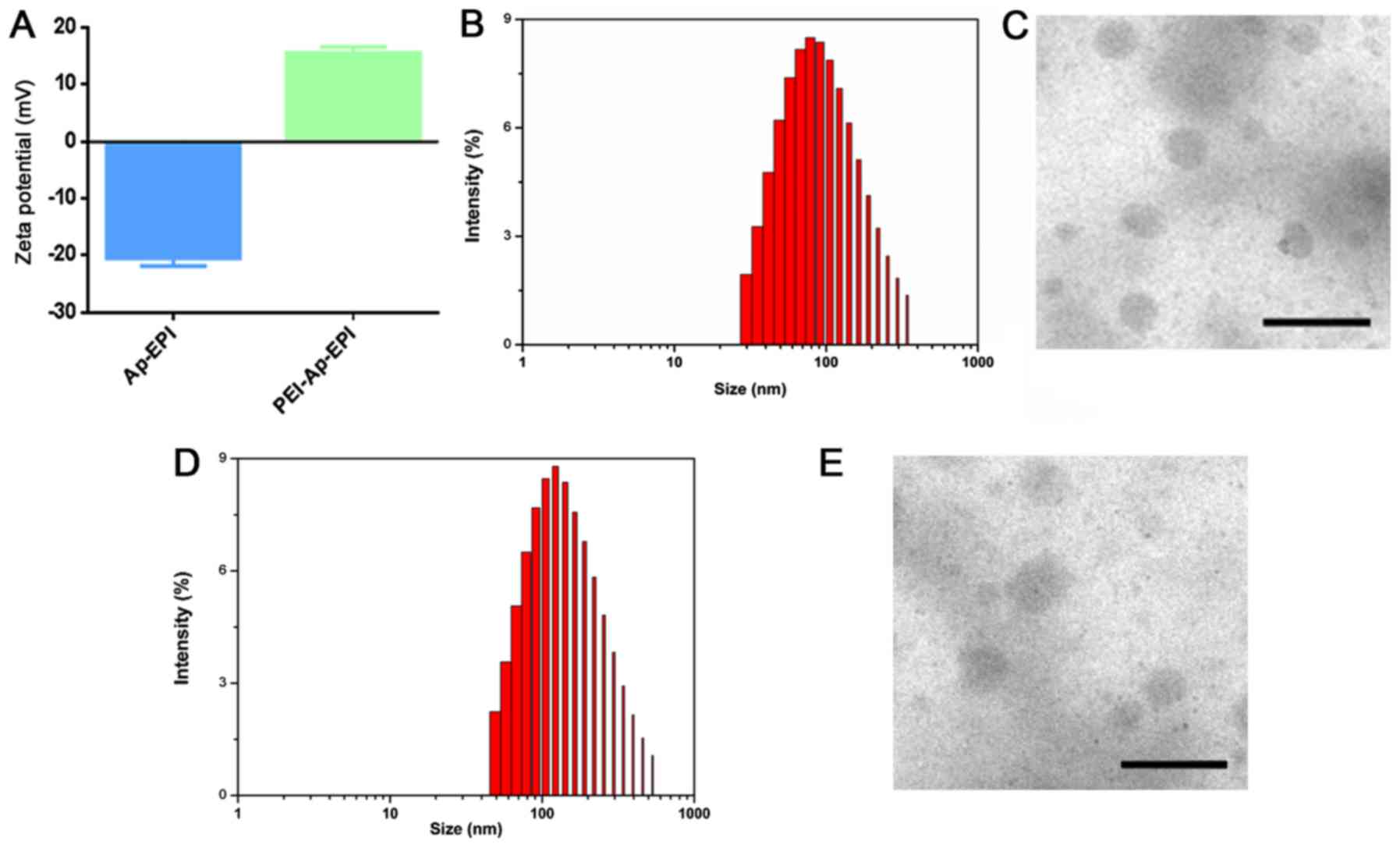
interaction. To confirm this phenomenon, the zeta-potential changes
on the surface of Ap-EPI were measured. The surface charge of
Ap-EPI was −20.4 mV, whereas its zeta potential changed to positive
(~15.5 mV) after it interacted with PEI (Fig. 3A). This result demonstrated that
Ap-EPI interacted completely with PEI. In addition, the size
distribution and morphology of PEI-Ap-EPI were examined further by
DLS and TEM, respectively. The size of PEI-Ap-EPI measured by DLS
was ~100 nm and the polydispersity index was 0.2 (Fig. 3B), demonstrating that PEI-Ap-EPI was
small and of narrow dispersion. In accordance with the results of
DLS, TEM also revealed a uniform spheroid structure of size ~100 nm
(Fig. 3C). Additionally,
PEI-Ctrl-EPI exhibited a narrow dispersion with uniform spheroid
structure (Fig. 3D and E).
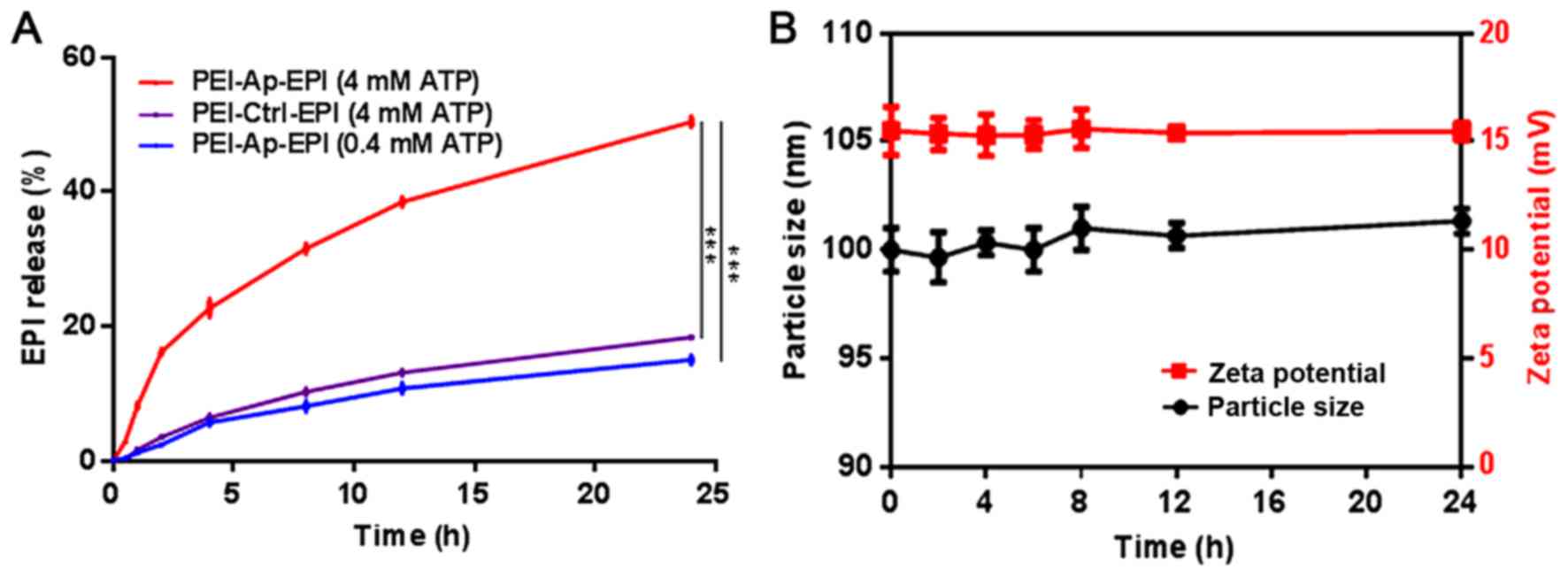
Subsequently, to evaluate the ATP-responsivity of
PEI-Ap-EPI, the EPI released from PEI-Ap-EPI nanoparticles
triggered by ATP was investigated. Typically, ATP of various
concentrations was added to PEI-Ap-EPI solutions and incubated for
24 h at 37°C. The fluorescence of PEI-Ap-EPI solutions at different
time points was recorded to depict the profile of EPI release
(Fig. 4A). After 24 h, >50% EPI
was released from PEI-Ap-EPI in the presence of 4 mM ATP, which was
significantly higher compared with the control groups, exhibiting a
time-dependent release. In the 0.4 mM ATP group, ~15% EPI was
released. Notably, in the PEI-Ctrl-EPI group, only ~18% EPI was
released after 24 h in the presence of 4 mM ATP, indicating that
PEI-Ctrl-EPI exhibited little response to ATP.
To investigate the stability of PEI-Ap-EPI, the
particle size and zeta potential were monitored for 24 h when it
was incubated in PBS with 10% FBS at pH 7.4. There was little
change in particle size and zeta potential, which demonstrated that
PEI-Ap-EPI was quite stable in the circulation (Fig. 4B).
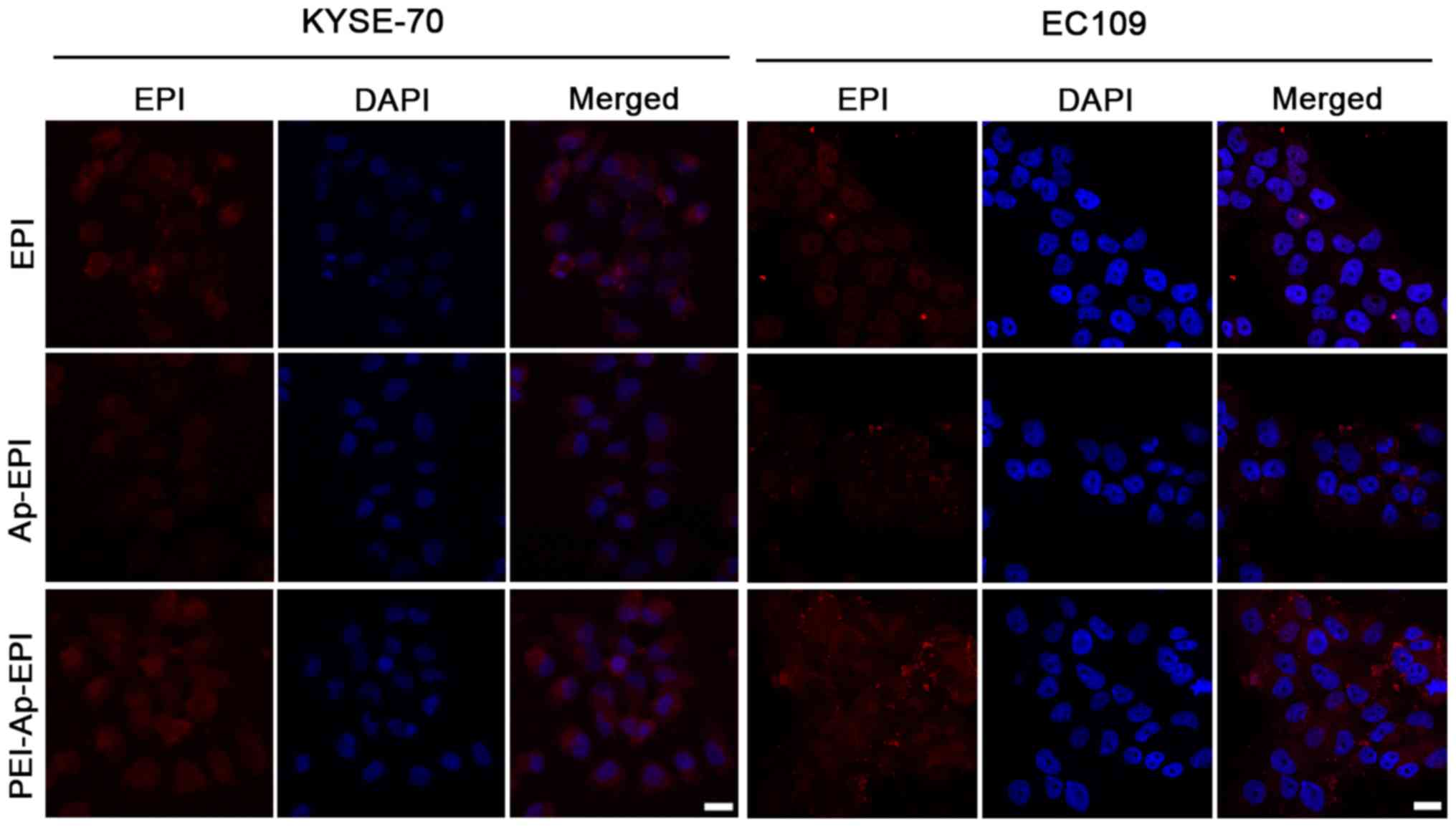
Intracellular accumulation
Sufficient accumulation of a drug in cancer cells is
indispensable for efficacious treatment (21). In the present study, intracellular
accumulation of EPI, Ap-EPI and PEI-Ap-EPI was measured via
fluorescence imaging using CLSM after KYSE-70 and EC109 cells had
been treated with these three formulations (Fig. 5). The fluorescence intensity was
strongest following treatment with PEI-Ap-EPI, showing the greatest
intracellular accumulation. This result could have been due to
PEI-Ap-EPI having greater drug loading and being internalized more
readily by cancer cells due to the reversal of surface charge after
Ap-EPI coating with PEI. Ap-EPI showed low accumulation. The
negative surface charge of Ap-EPI likely impeded its
internalization.
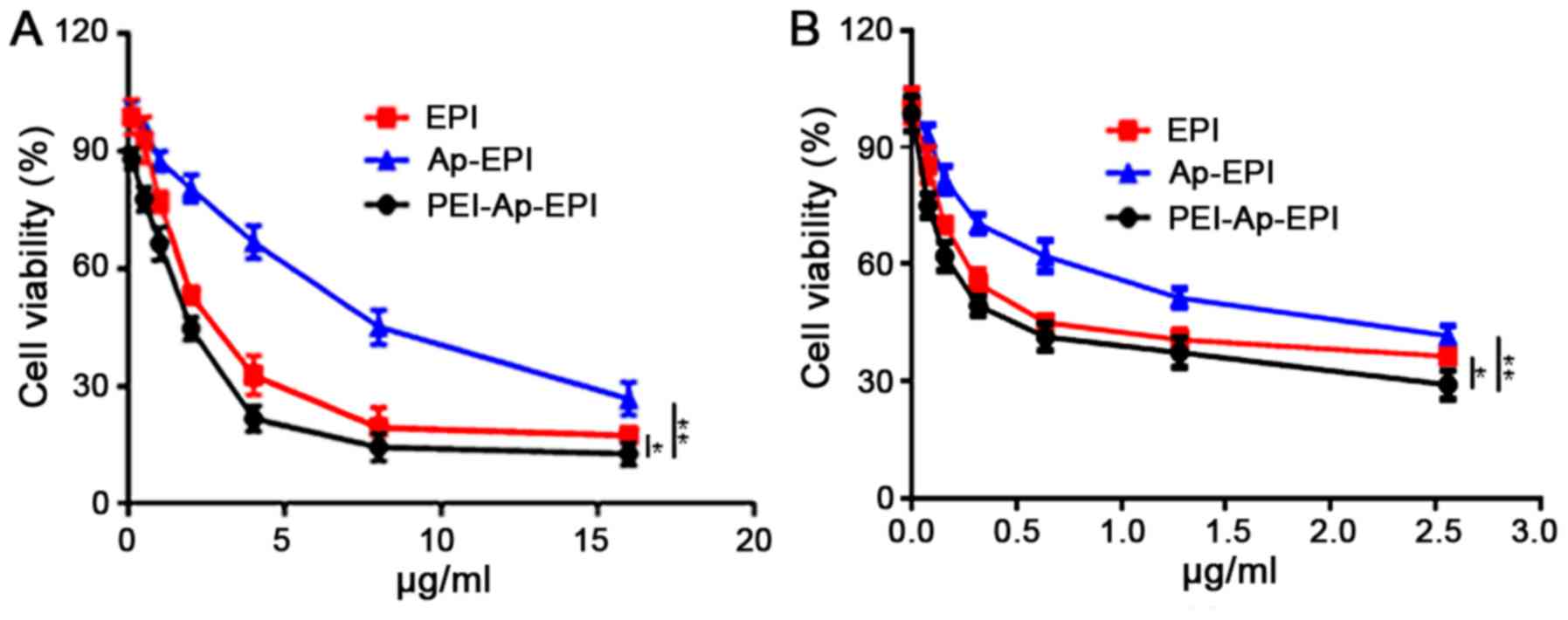
Cytotoxicity in vitro
The toxicity of EPI, Ap-EPI and PEI-Ap-EPI in
KYSE-70 and EC109 cells was evaluated using an MTT assay. The
cytotoxicity of the drugs increased with increasing drug dose
(Fig. 6). In KYSE-70 cells, the
IC50 of PEI-Ap-EPI was 2 µg/ml, which was lower than
that of EPI (2.5 µg/ml), but the IC50 of Ap-EPI (7.5
µg/ml) was ~3- and 3.7-fold higher than that of EPI and PEI-Ap-EPI,
respectively. In EC109 cells, the IC50 of PEI-Ap-EPI was
0.41 µg/ml, which was markedly lower than that of EPI and Ap-EPI,
and this was consistent with the result in KYSE-70 cells. The
reason may be that, without a PEI coating, the surface negative
charge of Ap-EPI limits its internalization, impeding its response
to extracellular ATP (which is at a low concentration). This result
demonstrated that PEI-Ap-EPI had the highest toxicity in KYSE-70
and EC109 cells.
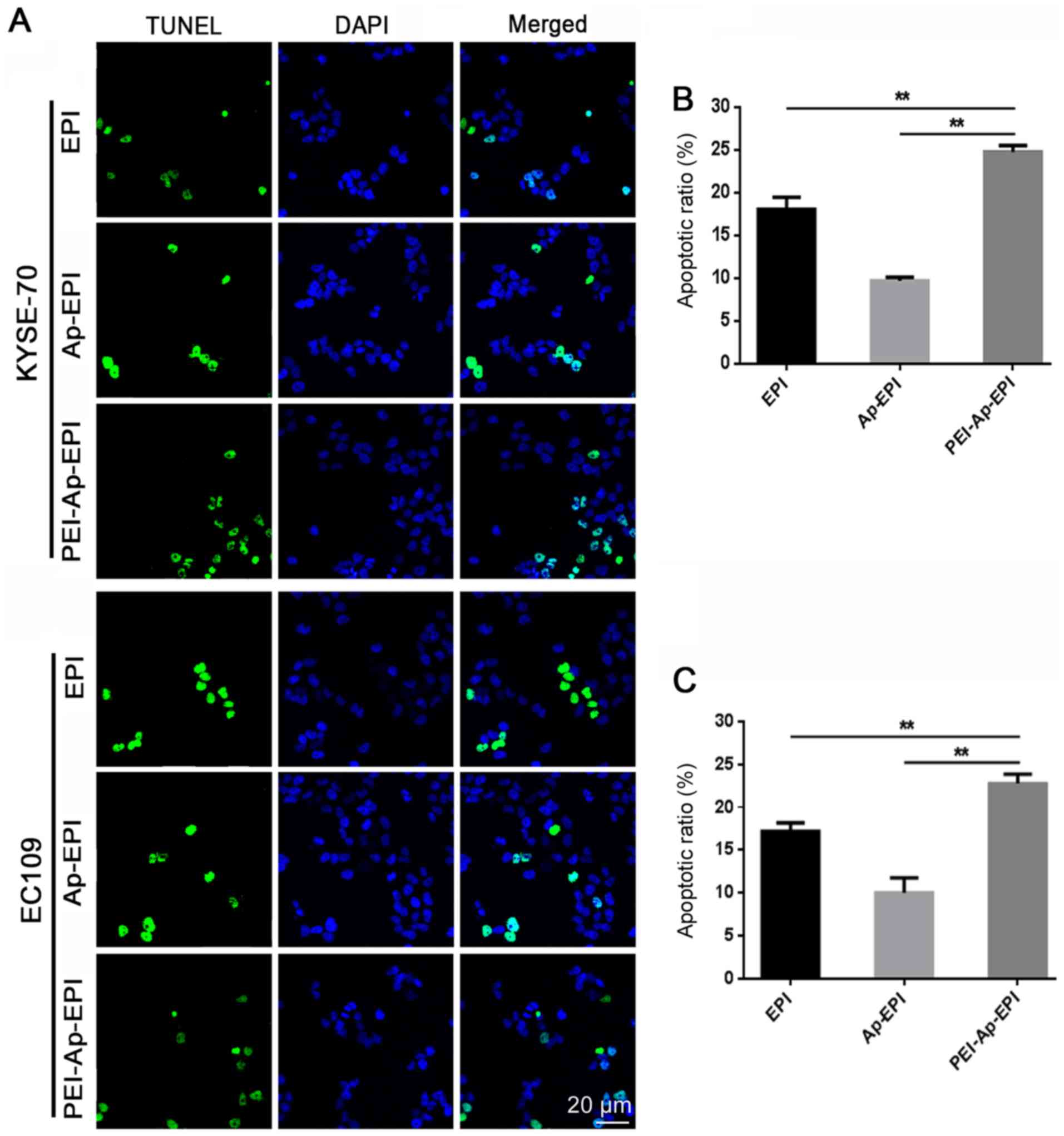
TUNEL assay
The TUNEL assay was utilized to demonstrate the DNA
damage that PEI-Ap-EPI can elicit. After KYSE-70 and EC109 cells
were incubated with EPI, Ap-EPI or PEI-Ap-EPI, a TUNEL assay was
performed and fluorescence microscopy was used to capture images.
Blue fluorescence indicated nuclei whereas green fluorescence
indicated apoptotic cells (Fig. 7A).
In the PEI-Ap-EPI group, more green fluorescence was observed,
indicating that it caused more severe damage than the other two
groups. After counting the number of apoptotic cells in the three
groups, it was demonstrated that PEI-Ap-EPI had the strongest
ability to cause DNA damage. The apoptotic index (AI) in KYSE-70
cells was ~25%, which was higher than that of EPI (18%; P=0.008)
and Ap-EPI (10%; P=0.005), respectively (Fig. 7B). The AI of PEI-Ap-PEI in EC109
cells was similar to that in KYSE cells (~23%) and the AI of EPI
and Ap-EPI was 17% (P=0.007) and 10% (P=0.004), respectively
(Fig. 7C).
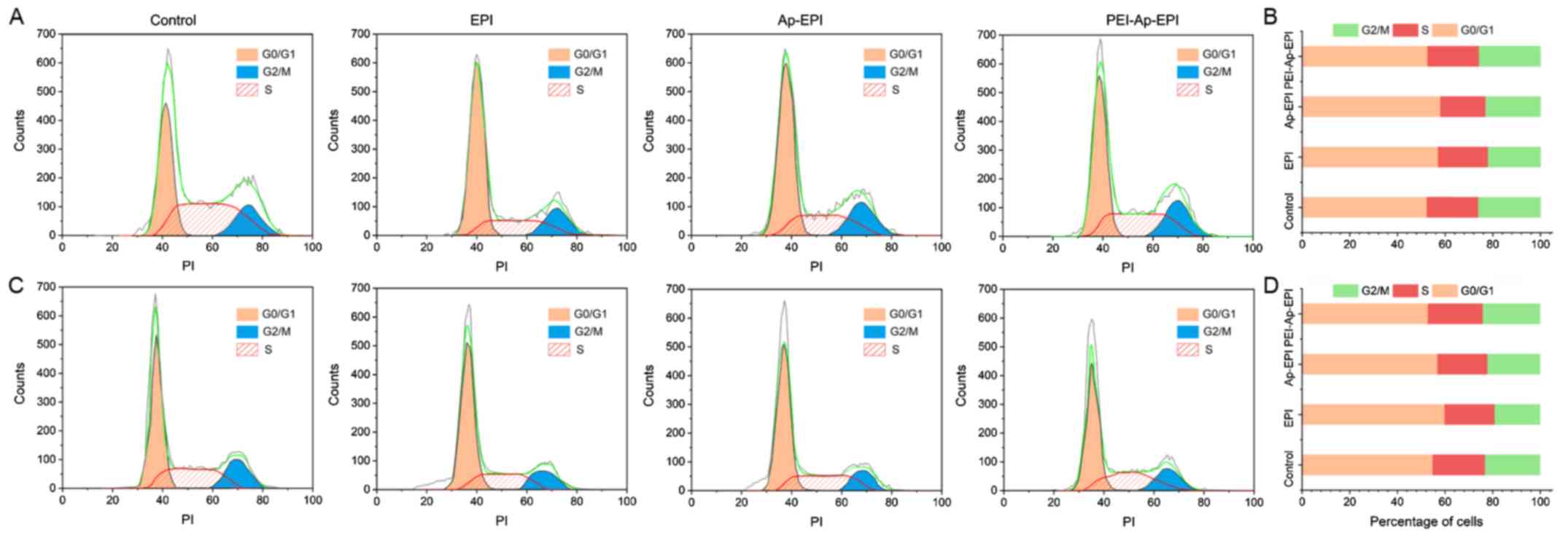
Cell cycle arrest
To investigate the influence of different
formulations of EPI on cell-cycle arrest, cells at each phase were
counted by flow cytometry (Fig. 8).
In KYSE70 and EC109 cells, after they had been treated with the
three formulations of EPI, the proportion of cells in the
G0/G1 phase was the greatest, and the cell
cycle distribution did not differ among the three formulations.
Discussion
Chemotherapy is a conventional way to inhibit tumor
growth. However, some disadvantages, such as poor selectivity,
weaken the efficacy of chemotherapy. Therefore, overcoming the
shortcomings of chemotherapeutics to improve the efficacy of EC
therapy would be helpful, and using nanoparticles for drug delivery
may be an effective method.
ATP is considered to be the ‘molecular unit of
currency’ of intracellular energy transfer. ATP is present at high
concentrations in the cytosol of tumor cells (1–10 mM) compared
with the extracellular concentration of ATP (<0.4 mM) (12). The distinct difference in the ATP
levels between the extracellular and intracellular milieu is the
biological principle for the design of ATP-responsive carriers. In
the present study, an ATP-responsive nanoplatform for EPI delivery
for EC treatment was developed. It was demonstrated that an Ap
composed of 27 base pairs rich in GC accommodated EPI following
interaction with its cDNA. The in vitro drug release
experiments demonstrated that EPI can be rapidly released in the
presence of intracellular ATP (4 mM), but only a small amount of
EPI is released in the presence of extracellular ATP (0.4 mM).
These actions contributed to high drug loading and release of the
active drug in cells, resulting in toxicity to tumor cells.
PEI was selected to condense the DNA duplex due to
four main reasons. First, through the charge interaction between
PEI and DNA duplex, nanoparticles can be formed, which contribute
to drug accumulation in tumor tissues (22). Second, condensation with PEI can
overcome DNA instability in vivo and reduce premature
leakage of EPI (12). Third, PEI can
become protonated in the acid environment of lysosomes, resulting
in disassembly of PEI-Ap-EPI nanoparticles and promotion of
DNA-duplex escape from lysosomes (23), which, ultimately, helps to enhance
therapy efficacy. Fourth, the DNA duplex can have a negative
charge, thereby impeding its internalization by cancer cells
(24). Following interaction with
PEI, the charge of the obtained nanoparticles changed to positive,
which was conducive to cell internalization.
KYSE-70 and EC109 cells were used to investigate the
efficacy of PEI-Ap-EPI. It was demonstrated that more EPI
accumulated in cells after they had been treated with PEI-Ap-EPI. A
possible reason is that PEI-Ap-EPI has more drug-loading sites and
is internalized more readily by cancer cells due to surface-charge
reversal after Ap-EPI coating with PEI. The group treated with
Ap-EPI exhibited little accumulation of EPI. The negative surface
charge of Ap-EPI likely impeded its internalization.
In in vitro cytotoxicity experiments, the low
IC50 of PEI-Ap-EPI could be explained by relatively high
drug loading and effective internalization. In accordance with this
hypothesis, absence of PEI coating resulted in poor
internalization, which contributed to the high IC50. The
TUNEL assay result was consistent with the data from experiments on
intracellular accumulation and the MTT assay. These results
suggested that PEI-Ap-EPI had improved efficacy for EC cells. In
addition, the present study investigated the influence of drug
formulations on cell cycle arrest and but did not find a
significant difference among them.
The main limitation of the present study was the
lack of in vivo experiments. Clinical application of the
nano-DDS can occur only after rigorous in vivo experiments
have been completed.
In conclusion, a novel DDS (PEI-Ap-EPI
nanoparticles) for EC treatment was constructed. PEI-Ap-EPI
nanoparticles of size ~100 nm were responsive specifically to a
high concentration of ATP in EC cells, and were stable in the
presence of FBS. In vitro experiments demonstrated that
PEI-Ap-EPI could increase EPI accumulation in tumor cells.
PEI-Ap-EPI showed higher cytotoxicity, and caused more severe DNA
damage than Ap-EPI and EPI. These results illustrated that the
novel nano-DDS may be efficacious in EC treatment, and has higher
efficacy than EPI alone.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Qingdao
University Development and Innovation Found (grant no.
201825QUDIF124).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW and LX conceived the study, and analyzed and
interpreted the data. XL performed the experiments, analyzed data
and wrote the manuscript. RY and DW performed the experiments and
analyzed data. JW and DW designed experiments, analyzed the data,
polished the manuscript and guided the reply to the comments. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pakzad R, Mohammadian-Hafshejani A,
Khosravi B, Soltani S, Pakzad I, Mohammadian M, Salehiniya H and
Momenimovahed Z: The incidence and mortality of esophageal cancer
and their relationship to development in Asia. Ann Transl Med.
4:292016.PubMed/NCBI
|
|
2
|
Bandla S, Pennathur A, Luketich JD, Beer
DG, Lin L, Bass AJ, Godfrey TE and Litle VR: Comparative genomics
of esophageal adenocarcinoma and squamous cell carcinoma. Ann of
Thorac Surg. 93:1101–1106. 2012. View Article : Google Scholar
|
|
3
|
Zhang HZ, Jin GF and Shen HB:
Epidemiologic differences in esophageal cancer between Asian and
Western populations. Chin J Cancer. 31:281–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Crane SJ, Locke GR III, Harmsen WS,
Zinsmeister AR, Romero Y and Talley NJ: Survival trends in patients
with gastric and esophageal adenocarcinomas: A population-based
study. Mayo Clin Proc. 83:1087–1094. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan Z, Chang Y, Cui C, Sun L, Wang DH, Pan
Z and Zhang M: Near infrared fluorescent peptide nanoparticles for
enhancing esophageal cancer therapeutic efficacy. Nat Commun.
9:26052018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu Y, Zhang B and Yan B: Enabling
anticancer therapeutics by nanoparticle carriers: The delivery of
Paclitaxel. Int J Mol Sci. 12:4395–4413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wen H, Jung H and Li X: Drug delivery
approaches in addressing clinical pharmacology-related issues:
Opportunities and challenges. Aaps J. 17:1327–1340. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Greish K: Enhanced permeability and
retention (EPR) effect for anticancer nanomedicine drug targeting.
Methods Mol Biol. 624:25–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patra JK, Das G, Fraceto LF, Campos EVR,
Rodriguez-Torres MDP, Acosta-Torres LS, Diaz-Torres LA, Grillo R,
Swamy MK, Sharma S, et al: Nano based drug delivery systems: Recent
developments and future prospects. J Nanobiotechnology. 16:712018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mo R and Gu Z: Tumor microenvironment and
intracellular signal-activated nanomaterials for anticancer drug
delivery. Mater Today. 19:274–283. 2016. View Article : Google Scholar
|
|
11
|
Zhang X, Ren X, Tang J, Wang J, Zhang X,
He P, Yao C, Bian W and Sun L: Hyaluronic acid reduction-sensitive
polymeric micelles achieving co-delivery of tumor-targeting
paclitaxel/apatinib effectively reverse cancer multidrug
resistance. Drug Deliv. 27:825–835. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mo R, Jiang T, DiSanto R, Tai W and Gu Z:
ATP-triggered anticancer drug delivery. Nat Commun. 5:33642014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan Y, Shi YS, Wu XD, Liang HY, Gao YB, Li
SJ, Zhang XM, Wang F and Gao TM: DNA aptamers that target human
glioblastoma multiforme cells overexpressing epidermal growth
factor receptor variant III in vitro. Acta Pharmacol Sin.
34:1491–1498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huizenga DE and Szostak JW: A DNA aptamer
that binds adenosine and ATP. Biochemistry. 34:656–665. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Modh H, Witt M, Urmann K, Lavrentieva A,
Segal E, Scheper T and Walter JG: Aptamer-based detection of
adenosine triphosphate via qPCR. Talanta. 172:199–205. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heilkenbrinker A, Reinemann C, Stoltenburg
R, Walte JG, Jochums A, Stahl F, Zimmermann S, Strehlitz B and
Scheper T: Identification of the target binding site of
ethanolamine-binding aptamers and its exploitation for ethanolamine
detection. Anal Chem. 87:677–685. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Canzoneri JC and Oyelere AK: Interaction
of anthracyclines with iron responsive element mRNAs. Nucleic Acids
Res. 36:6825–6834. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takeda K, Uchiyama K, Kinukawa M, Tagami
T, Kaneda M and Watanabe S: Evaluation of sperm DNA damage in bulls
by TUNEL assay as a parameter of semen quality. J Reprod Dev.
61:185–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo JR, Chen QQ, Lam CW and Zhang W:
Effects of karanjin on cell cycle arrest and apoptosis in human
A549, HepG2 and HL-60 cancer cells. Biol Res. 48:402015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dizaj SM, Jafari S and Khosroushahi AY: A
sight on the current nanoparticle-based gene delivery vectors.
Nanoscale Res Lett. 9:2522014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang L, Lin X, Wang J, Hu Z, Ji Y, Hou S,
Zhao Y, Wu X and Chen C: Novel insights into combating cancer
chemotherapy resistance using a plasmonic nanocarrier: Enhancing
drug sensitiveness and accumulation simultaneously with localized
mild photothermal stimulus of femtosecond pulsed laser. Adv Funct
Mater. 24:4229–4239. 2014. View Article : Google Scholar
|
|
22
|
Blanco E, Shen H and Ferrari M: Principles
of nanoparticle design for overcoming biological barriers to drug
delivery. Nat Biotechnol. 33:941–951. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Akinc A, Thomas M, Klibanov AM and Langer
R: Exploring polyethylenimine-mediated DNA transfection and the
proton sponge hypothesis. J Gene Med. 7:657–663. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Y, Xu CF, Iqbal S, Yang XZ and Wang J:
Responsive nanocarriers as an emerging platform for cascaded
delivery of Nucleic acids to cancer. Adv Drug Deliv Rev.
115:98–114. 2017. View Article : Google Scholar : PubMed/NCBI
|