Introduction
Gastric cancer (GC) is a leading cause of
cancer-related deaths worldwide and is known to be an age-related
disease (1). The mean age of
patients with GC at diagnosis is ~60 years and <3% of GC cases
are reported in patients <30 years of age (2,3). Several
reports have found that younger patients (~30 years) are often
diagnosed with advanced stages of GC and have worse prognosis
compared with that in elderly patients (~60 years) (4,5). In
younger patients, the cancer was found to spread rapidly and was
biologically more aggressive (6).
Integrative genomic approaches, which associate genomic and
transcriptomic analysis have been found to increase the
identification of numerous events and processes associated with
tumor aggressiveness and may assist in selecting candidate genes
from normal and pathological samples for targeted therapy (7). A previous study aimed to investigate
the biological and genomic mechanisms in the progression of GC has
led to the development of target-oriented therapy for advanced GC
to be used in a clinical setting, with base substitution mutations
in genes such as tumor protein p53 (TP53), AT-rich
interaction domain 1A (ARID1A), KRAS proto-oncogene, GTPase
(KRAS), phosphatidylinositol-4,5-bisphosphate 3-kinase
catalytic subunit alpha (PIK3CA), ring finger protein 43
(RNF43), APC regulator of WNT signaling pathway
(APC), RAS p21 protein activator 1 (RASA1) and erb-b2
receptor tyrosine kinase 2 (ERBB2) identified as important
therapeutic targets in anti-cancer treatments (8). Developing personalized therapy for
optimal individual cancer patient outcome has become more feasible
due to the rapid advancement in next-generation sequencing
techniques and technologies that enable fast and comprehensive
characterizations of tumors at the molecular level (9–12). As
every patient harbors a unique combination of variants that
influence the risk, onset, and progression of the disease, an
effective personalized therapy is dependent on a well-profiled
genome from the individual patient with cancer and understanding
the oncogenic mechanisms that regulate the progression of the tumor
(13). With a view to identifying
potential clinically actionable therapeutic targets that may inform
individualized treatment strategies, the whole genome was sequenced
in a 26-year-old patient with advanced stage GC, by integrating the
whole genome and whole transcriptome sequencing data.
Materials and methods
Sample collection and
preservation
A 26-year-old female patient was admitted to the
First Affiliated Anhui Medical Hospital in December 2016 having
presented with GC-related symptoms. The patient felt uncomfortable
in the upper left quadrant of her abdomen and had repeated black
melena for one month. This symptom recurred and became more serious
for 2 weeks. The patient felt full and uncomfortable in the upper
left quadrant of her abdomen repeatedly following a meal, one month
prior to the development of black melena. She also had nausea,
although there was no hematemesis or hematochezia. The abdominal
discomfort worsened and the patient received treatment in Luan
Jinkai Hospital. Thereafter, the patient visited the First
Affiliated Hospital of Anhui Medical University where the present
study was conducted. The gastroscopy of the patient indicated GC
with obstruction, while pathology indicated antrum considered as
carcinoma mucocellulare. Pathological examination of the samples
was performed using hematoxylin and eosin staining. Briefly,
tissues were sliced with a dimension of 1.5×1.5×0.2-0.3 cm and
soaked in 40°C warm water. The sections were deparaffinized in
xylene and were gradually hydrated through graded ethanol (100, 95,
80 and 70%) each for 3 min at 25°C. The sections were stained in
hematoxylin solution for 10 min at 25°C and differentiated in 1%
hydrochloric alcohol. Then the slides were rinsed with tap water
and distilled water until the nuclei became blue, and dehydrated in
95% ethanol. The sections were counterstained in 1% eosin solution
for 5 min at 25°C, washed with 70% ethanol twice and absolute
ethanol, and were cleared in two washes of xylene. The sections
were mounted with neutral balsam and observed under a light
microscope magnification, ×400 (×40 objective and ×10 ocular
magnification; BX-42; Olympus). An abdominal computer tomography
scan + enhancement showed that both armpits had multiple small
lymph nodes and the gastric antrum had thickened walls along with
multiple swollen lymph nodes in the surrounding area. The patient
was diagnosed with a T3N3bM0 (advanced stage IIIC) GC using AJCC
TNM staging system 7th edition (14).
The patient underwent a gastrostomy for tumor
removal. Tumor tissue and adjacent normal tissue ~5-cm away from
the tumor were resected and preserved in liquid nitrogen until
use.
DNA and RNA extraction and library
preparation
Genomic DNA was isolated from the resected tissues
using a genomic DNA isolation kit (Bioo Scientific; http://www.biooscientific.com) according to the
manufacturer's recommendations. Extracted DNA was quantified using
a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies; Thermo
Fisher Scientific, Inc.) and the integrity was assessed using 1%
agarose gel electrophoresis and visualized in gel imager (Tanon;
http://www.biotanon.com). RNA was extracted from
the tumor and adjacent normal tissue using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.), and eluted in
RNAse-free water. RNA quantity and quality were assessed using an
Agilent 2100 Bioanalyzer (Agilent Technologies Inc.). DNA and RNA
libraries were prepared according to standard protocols according
to Bioo Scientific.
Whole transcriptome sequencing and
pre-analysis
For whole transcriptome sequencing, raw sequencing
reads were produced using an Illumina X10 sequencer (Illumina Inc.)
with 150-bp paired-end reads according to the manufacturer's
instructions. Raw RNA-Sequencing (RNA-Seq) data was trimmed by
removing the adaptors and low quality reads using trim_galore
v0.4.4 software (15). The data was
subsequently aligned to the human reference genome GRCh37.p13 using
STAR v2.6.1a software (16) in a
2-pass mapping. Read count in the tumor and adjacent normal tissues
was calculated using htseq-count v0.10.0 software (17), and differentially expressed genes
were detected using edgeR v3.24.3 software (18). Genes with P≤0.001 and log2
fold change ≥1 were considered as differentially expressed.
Whole genome sequencing and
pre-analysis
Whole genome sequencing of the prepared libraries
was performed using the Illumina X10 sequencer (Illumina Inc.) with
150-bp paired-end reads according to the manufacturer's
instructions. The raw fastq data was mapped to the human reference
genome (b37) provided by the Broad Institute with the
Burrows-Wheeler Aligner v.7.0.12 (19). Read duplications were marked using
the Picard tool v.2.10.10; local realignment and base quality score
recalibration was performed using Genome Analysis ToolKit (GATK) v.
3.8-0 (20) and the final bam files
were used for variant calling.
Somatic and germline variants
detection
Germline variants were called using the GATK
HaplotyperCaller joint v3.8 software (21) and then filtered using GATK variant
quality score recalibration (VQSR). Variants that passed using the
VQSR module with coverage ≥6X and supporting reads ≥0 were
retained, while the variants showing genotype ‘0/0’ or ‘./.’ in the
tumor sample were filtered out. For somatic variant calling, three
different types of software were used: GATK HaplotypeCaller joint
v3.8, Mutect v1.1.4 and MuTect2 v3.8 (22). In Mutect and MuTect2, all variants
that were flagged as ‘PASS’ were kept. Other variants were retained
with a coverage ≥5X and supporting ≥3 reads, if they were flagged
among a subset of ‘alternative (23)
allele in normal’, ‘triallelic site’, ‘possible contamination’,
‘clustered read position’ in Mutect, or ‘germline risk’, ‘alt
allele in normal’, ‘t lod fstar’ (tumor does not meet likelihood
threshold), ‘str contraction’ (site filtered due to contraction of
short tandem repeat region), ‘triallelic site’ (site filtered
because > two alternate alleles pass tumor likelihood threshold)
in MuTect2. Germline variants were removed from both the Mutect and
MuTect2 output data. In all three softwares, variant allele
frequency in the tumor sample had to be 3 times higher compared
with that in the normal sample, and >5% of the variant reads in
the tumor sample was required. Functional annotations for both
germline and somatic variants were added to each mutation using the
ANNOVAR software v.2018Apr16 (17)
and several publicly available databases, such as 1,000 Genome
Project (24), the Exome Aggregation
Consortium (25), the Genome
Aggregation Database (26) and the
compiled scores prediction system dbnsfp33a (27), dbSNP (28) and COSMIC (29).
Transition to transversion ratio
calculation
The transition to transversion ratio for germline
variants was calculated by dividing total transitions by total
transversions. Transitions refer to variations that involve a
change from purine to purine, while transversions refer to
variations that involve a change from purine to pyrimidine or vice
versa (30). The transition to
transversion ratio between homologous strands of DNA is generally
~2, and for human, the ratio is ~2.1 (31).
Copy number alternation and structural
variation analysis
Copy number variants (CNV), tumor purity and ploidy
was analyzed using cnv_facets software (https://github.com/dariober/cnv_facets) based on
FACETS v0.5.14 (32), and run with
the ‘-cval 25 400’ command. Duplicated and deleted segments were
retained and annotated using an ENCODE gene symbol in R v3.5.1
(33). iCallSV (https://github.com/rhshah/iCallSV), which applied
Delly v0.7.5 prediction method (34)
was used to detect structural variations and fusions using default
parameters. Inter- and intra-chromosomal fusions >50 kb were
retained and only in-frame deletions, duplications and fusions were
selected for subsequent analysis.
Mutation significance analysis
The 1,000 Genome Project (24), the Exome Aggregation Consortium
(25) and the Genome Aggregation
Database (26) provided alternative
allele frequency data in different populations that ANNOVAR
software applied to the present variations. Annotations using a set
of algorithm scoring system from dbNSFP v.33a, including FATHMM,
LRT, Mutation Assessor, Mutation Taster, M-CAP, Polyphen2, PROVEAN
and SIFT (35), were also applied to
the variations. Then the common variants were filtered out, which
are sites with a minor allele frequency ≥1%, to get the most
informative ones. Mutations predicted to be deleterious by at least
two algorithms of dbNSFP were considered to be potential pathogenic
sites of these informative variants.
Identification of mutations in cancer
associated genes
To identify genes with a high possibility of
involvement in cancer development, the Cancer Gene Census (CGC)
(https://cancer.sanger.ac.uk/census)
(36) and Cancer Predisposition
Genes (CPG) (37) databases were
used. In total, 850 candidate genes from cancer-associated
databases (723 genes from CGC and 114 genes from CPG with 87
overlapping genes), were investigated. The gene lists were
downloaded directly from the two databases with no specific
selection criteria.
Pathway analysis
Gene enrichment was performed using the R package
clusterProfiler v3.10.1 software (38), which implements a hypergeometric
model to test for gene set overrepresentation relative to a
background gene set. Enrichment was analyzed using the functions:
‘Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG)’,
‘gseKEGG’ (Gene Set Enrichment Analysis (GSEA) (39) of KEGG, and a function of
clusterProfiler), P<0.05, OrgDb=org.Hs.eg.db (a database for Genome Wide
Annotation for Human, referenced by clusterProfiler).
Integrated analysis of whole genome
and transcriptome sequencing data
The differentially expressed genes were compared
with mutated genes identified by whole genome sequencing. To
achieve this, genes that were mutated and with differentially
expressed transcripts were merged in R v3.5.1 (33).
Analysis of public GC RNA-Seq
data
The RNA-Seq data from The Cancer Genome Atlas
stomach adenocarcinoma (TCGA STAD) was downloaded from TCGAbiolinks
online tool (https://github.com/BioinformaticsFMRP/TCGAbiolinks).
To identify the most significantly differentially expressed genes
in GC, the results from three RNA-Seq differential expression
analysis tools, including limma v3.42.2 (40), DEseq2 v1.26.0 (41) and edgeR v3.28.1 (18), were combined and the overlapping
genes were separated for further analysis. For all three tools,
P<0.05 and the average log2 fold change ± standard
deviation were used as the cut-off values. Gene Ontology (42) enrichment analysis was performed using
the Database for Annotation, Visualization and Integrated Discovery
(DAVID) v6.7 functional annotation clustering tool to determine the
potential functions of the differentially expressed genes. The KEGG
pathway analysis was performed to determine the involvement of
differentially expressed genes in different biological pathways.
The-log10 (P) denotes the enrichment score, indicating the
significance of the pathway associations.
Kaplan-Meier plotter (KM-Plotter)
analysis
KM-plotter (https://kmplot.com/analysis/) is an online database
and tool which can be used for the discovery and validation of
survival biomarkers (43). It
contains cancer gene expression data and survival information from
Gene Expression Omnibus (44),
European Genome-phenome Archive (EGA) (https://www.ebi.ac.uk/ega/home), and TCGA (https://www.cancer.gov/tcga), and uses each percentile
of mRNA expression between the lower (25%) and upper quartiles
(75%) of expression as a cut-off point to divide patients into high
and low expression groups. The KM-plotter software, which included
available transcriptome and survival data (both overall and
disease-free survival times), was used to analyze a total of 1,440
patients with GC, based on default parameters.
Results
Tumor purity and coverage
statistics
The proportion and the ploidy of tumor cells in the
sample were ~22% and 1.74, respectively. Using whole genome
sequencing, the total number of paired reads were 8.13 and 8.34
billion, the mean read coverage was 37.16 and 35.48, while the mean
library insert size was 319 and 323 bp for normal and tumor tissue
samples, respectively. For the consensus coding sequence region,
~90% of the bases had over 30X coverage. Using whole transcriptome
sequencing, 1.04 and 1.11 billion clean reads were obtained with an
average input read length of 253 and 261 bp for normal and tumor
tissue, respectively.
Gene expression analysis
The RNA-Seq pipeline was used (Fig. S1A) for the whole transcriptome
sequencing. Genes with P≤0.001 and absolute log2 fold
change value ≥1 were considered as significantly differentially
expressed genes. In total, 766 down- and 765 upregulated genes were
obtained (Fig. S1B). The RNA-Seq
result was subsequently matched to the whole genome sequencing data
for further analysis.
Germline mutation analysis
Germline mutation analysis revealed a total of
4,228,339 single nucleotide polymorphisms (SNPs) and Indels with
95.03% of the sites previously reported in the dbSNP database. The
transition to transversion ratio was 2.05, indicating that the
germline analysis had high confidence, as the empirical value for
whole genome analysis is ~2.1 (31).
Among the germline variants, 21,405 SNPs/Indels were located in the
exonic regions, of which 10,136 were predicted to alter the
protein, including 9,504 non-synonymous single nucleotide
variations (SNVs), 112 frameshift deletions, 90 frameshift
insertions, 173 non-frameshift deletions, 169 non-frameshift
insertions, 78 stop-gain, and 10 stop-loss. More than 1% minor
allele frequency variants from publicly available databases were
removed, as those sites are common in the population. Of the
candidate genes from the CGC and CPG databases, 13 SNPs and two
Indels were identified in 15 genes (Table I). A deleterious score evaluation of
the 15 variants revealed that mutations in DOCK8 (p.D63N),
HLF (p.V59A), NKX2-1 (p.G322S), NRG1 (p.S17C),
PRDM2 (p.S823T), WNK2 (p.C652R) were deleterious,
while mutations in KMT2D (p.V401M), RAD51D (p.A181V)
and ROS1 (p.E1605Q) were possibly damaging.
 | Table I.Functional assessment of the germline
mutations and prediction of their effect. |
Table I.
Functional assessment of the germline
mutations and prediction of their effect.
| Gene | Databse | Chromosome | Position | Reference
allele | Alteration | Type | AA. Change | Cosmic82 | Final
prediction |
|---|
| AKAP9 | CGC | 7 | 91739445 | C | T | Non-SNV | p.T3899I | N | U |
| DOCK8 | CPG | 9 | 286491 | G | A | Non-SNV | p.D63N | Y | D |
| HIF1A | CGC | 14 | 62207747 | C | A | Non-SNV | p.T669N | N | U |
| HLF | CGC | 17 | 53345172 | T | C | Non-SNV | p.V59A | N | D |
| KMT2D | CGC | 12 | 49446404 | C | T | Non-SNV | p.V401M | N | P |
| MLLT3 | CGC | 9 | 20414344 | CTG | – | Non-FS DEL | p.163_164del | Y | U |
| MN1 | CGC | 22 | 28194933 | – | TGCTGC | Non-FS | p.Q533delins | N | U |
|
|
|
|
|
| TGCTGC | INS | QQQQQ |
|
|
| NKX2-1 | CGC | 14 | 36986635 | C | T | Non-SNV | p.G322S | N | D |
| NRG1 | CGC | 8 | 32505286 | C | G | Non-SNV | p.S17C | N | D |
| PRDM2 | CGC | 1 | 14107360 | T | A | Non-SNV | p.S823T | Y | D |
| PTPRC | CGC | 1 | 198675962 | A | G | Non-SNV | p.N101S | N | U |
| RAD51D | CPG | 17 | 33428245 | G | A | Non-SNV | p.A181V | N | P |
| RNF213 | CGC | 17 | 78346531 | C | A | Non-SNV | p.P4250T | N | U |
| ROS1 | CGC | 6 | 117662652 | C | G | Non-SNV | p.E1605Q | N | P |
| WNK2 | CGC | 9 | 96015284 | T | C | Non-SNV | p.C652R | N | D |
Somatic mutation analysis
A total of 3 softwares from the Broad Institute were
used to identify the somatic variants and 114 variants in the
exonic regions were found. This included 45 non-synonymous SNVs, 6
frameshift deletions, 11 frameshift insertions, 17 non-frameshift
deletions, 3 non-frameshift insertions, 2 stop-gain and 30
synonymous SNVs. Similar to the germline mutation analysis, common
variants were filtered out with publicly available datasets to
select the most informative somatic variants. In total, 29 variants
were obtained, including 13 mutations which were reported in the
Catalogue of Somatic Mutations in Cancer database (Table SI). Of these, 3 protein-truncating
variants (BPNT1, p.S144I; FRG1, p.N153D; and
TAS2R31, p.L59F) were predicted to be deleterious and one
variant (KRTAP5-3, p.C185S) was predicted to be possibly
damaging. However, for transcriptional regulation, 3 genes
(MUC2, MUC4 and SLC8A2) were found to be
differentially expressed.
Large-scale variation analysis
To identify large-scale variations in the patient,
the copy number alterations and complex structural events in the
tumor sample were investigated. The structural variant discovery
tool, DELLY, reported 701 structural events; however, only in-frame
events or fusions over 50 kb between two genes were retained.
Ultimately, 20 significant structural variations were reported
(Table SII). Another sensitive
tool, FACETS, detected 58 large segments, of which 62 were
deletions and 112 were duplications found in the coding regions
(Table SIII). Combining the results
from the two tools, nine genes (CDKN1B, FBXW7, MUC4, CCND3,
ETV6, FGFR2, FGFR3, TFEB and KMT2C) were reported in
either the CGC or CPG databases. CDKN1B, FBXW7 and
MUC4 showed duplications, and there were deletions in
CCND3, ETV6, FGFR2, FGFR3 and TFEB. Although
KMT2C rearranged with BAGE2, the latter was not
reported in the 2 databases. The mRNA expression levels of the
genes with structural variations was also investigated and 21 were
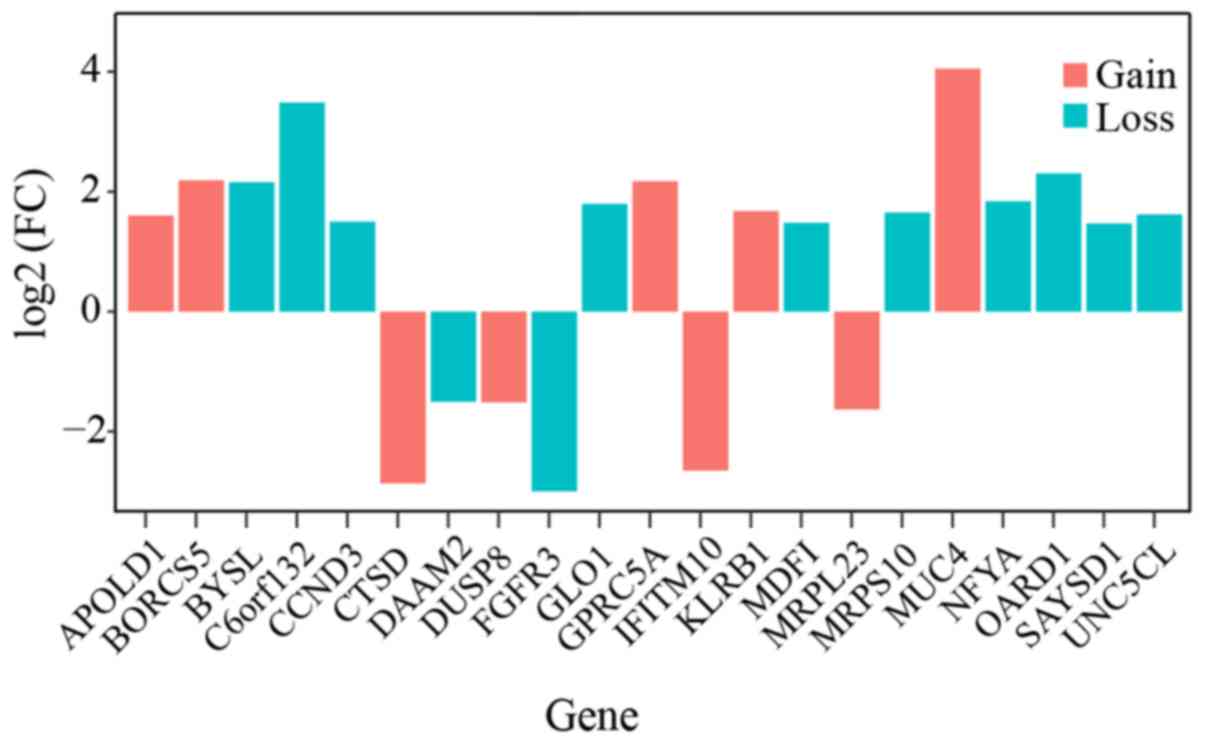
found to be differentially expressed in the tumor sample (Fig. 1).
Integrated pathway analysis
results
RNA-Seq and mutation data were analyzed to
characterize the genomic alterations in known signaling pathways.
The GO enrichment for somatic mutated genes revealed terms, which
included 3 mucin family genes, MUC2, MUC4 and MUC6,
and were enriched in ‘maintenance of gastrointestinal epithelium’,
‘O-glycan processing’, and digestive system process, while
KRTAP5-3, TCHH, RPTN, POU3F1 were enriched in epidermal cell
differentiation pathway (Table
SIV). The KEGG enrichment analysis revealed SLC8A2 and
SLC25A5 were enriched in calcium signaling and cGMP-PKG
signaling pathways (Table SIV).
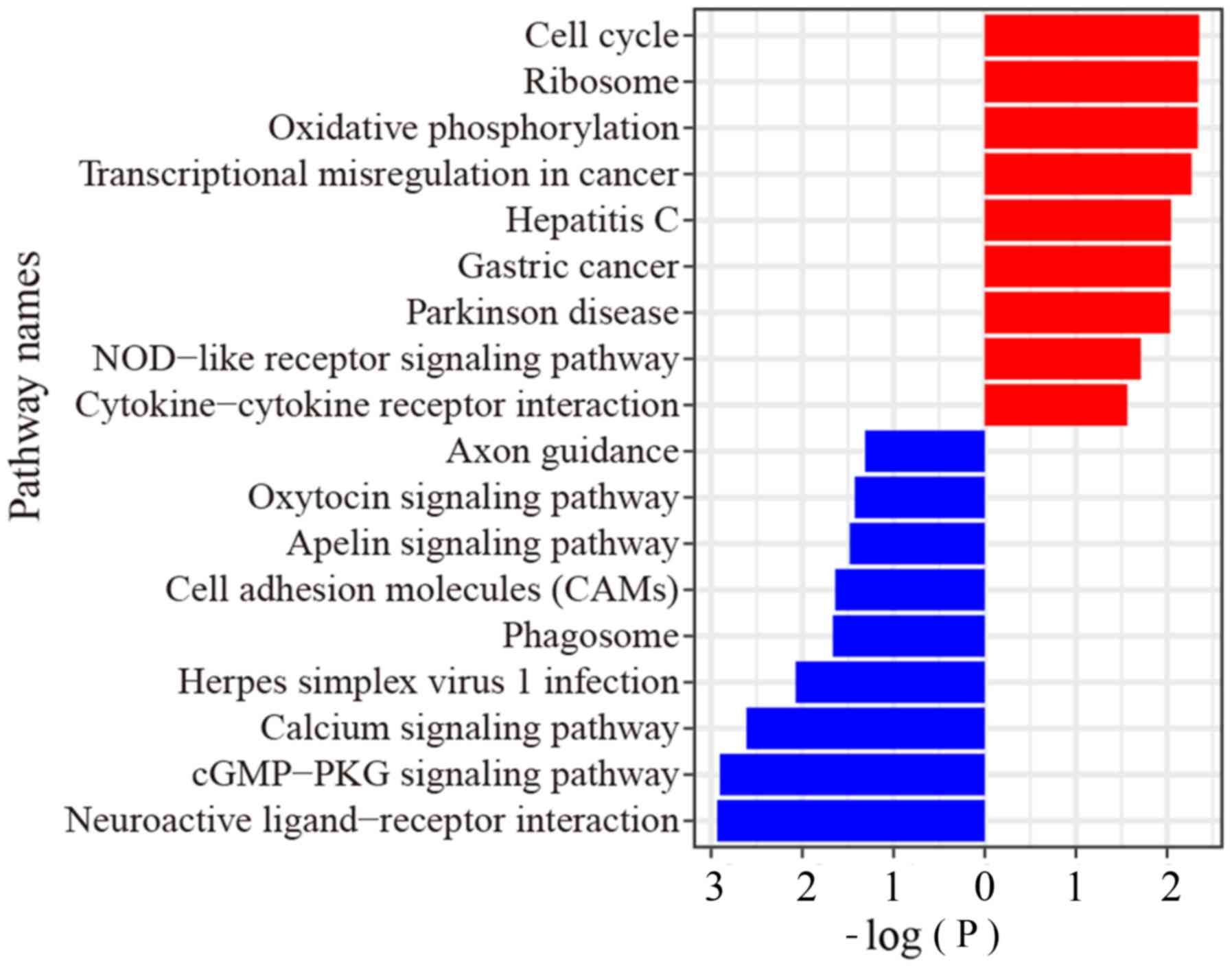
From the RNA-Seq data, GSEA showed that the upregulated genes were
enriched in ‘cGMP-PKG signaling’, ‘calcium signaling’, and
‘oxytocin signaling’ pathways, while the downregulated genes were
enriched in ‘cell cycle’ pathway, ‘oxidative phosphorylation’
pathway, ‘transcriptional mis-regulation in cancer’ pathway and ‘GC
pathway’ (Fig. 2).
Comparison with TCGA STAD GC
features
To ensure the present study was representative and
valuable, the genomic features of the patient was compared with
data from TCGA STAD dataset, which was downloaded from
TCGAbiolinks, and 329 (136 up- and 193 downregulated genes)
differentially expressed genes were identified, which were also
found in the RNA-Seq analysis. A total of three tools were used to
identify the most significantly differentially expressed genes, and
136 up- and 193 downregulated genes were identified (Fig. S2A), and were subsequently
investigated in the tumor tissue sample obtained from the patient
with GC and compared with that in the adjacent normal tissue. In
total, 27 genes were identified, including five that were up- and
22 that were downregulated (Fig.
3A). GO and KEGG pathway enrichment analysis revealed one
(GO:MF) and three (KEGG) significant terms, respectively, including
digestion and gastric acid secretion (Fig. 3B).
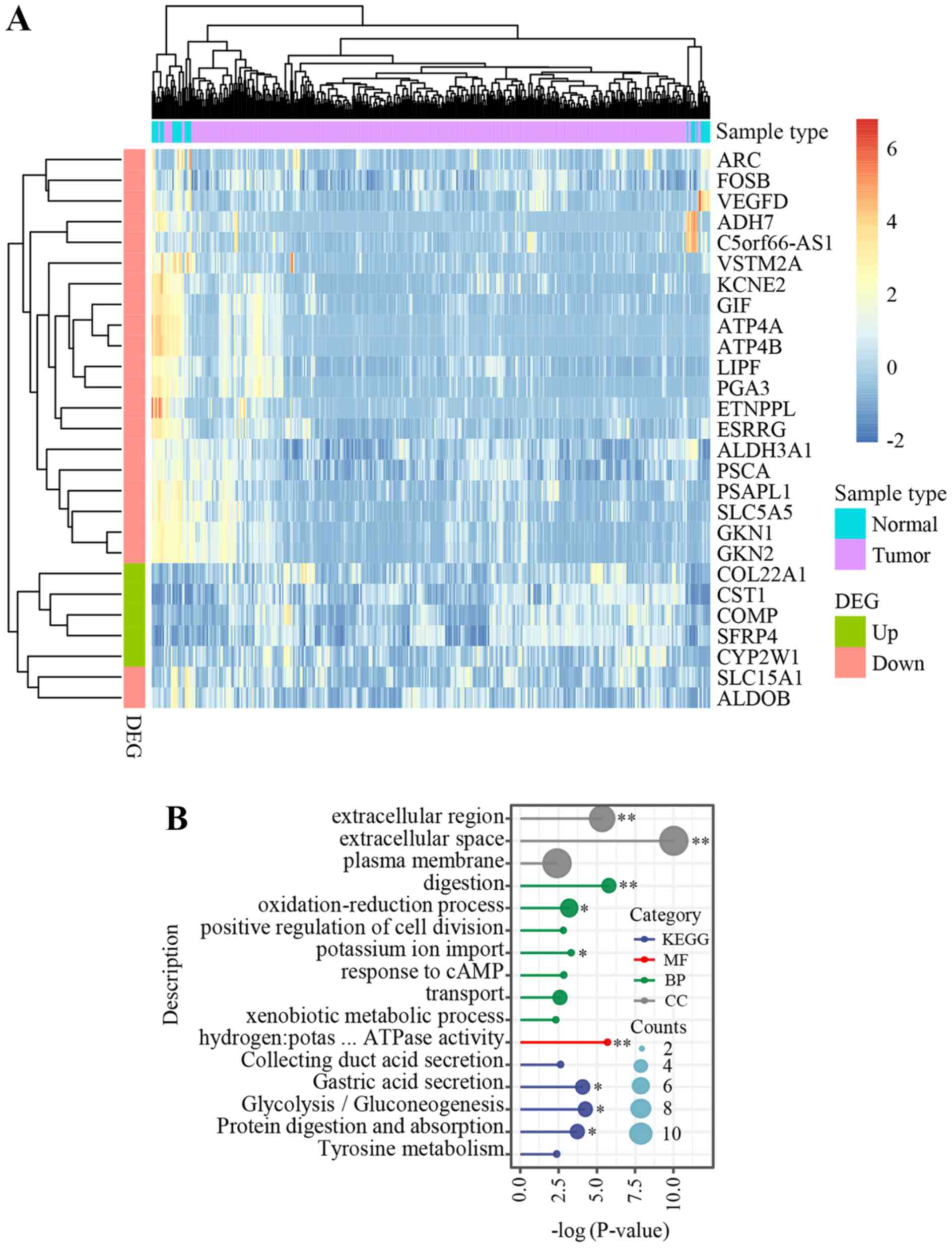 | Figure 3.Genomic features in the young patient
in the present study compared with that in patients from TCGA STAD
dataset. (A) Heatmap of the 27 genes, which were dysregulated in
both TCGA STAD and in the present study. Counts per million (CPM)
indicates the expression level of each gene and log (CPM+1), which
ranged from −2 (blue) to 6 (red), was used for sample clustering
and visualization. (B). Gene Ontology enrichment analysis and KEGG
pathway analysis identified several significant pathways.
*P<0.05 and **P<0.01. DEG, differentially expressed gene;
KEGG, Kyoto Encyclopedia of Genes and Genomes; MF, molecular
functions; CC, cellular components; BP, biological processes; TCGA,
The Cancer Genome Atlas; STAD, stomach adenocarcinoma;
hydrogen:potas … ATPase activity, hydrogen:potassium-exchanging
ATPase activity. |
To determine the associated functional genomic
alterations of these 27 genes, the previous studies that reported
them were analyzed. In the present study, there were no somatic
mutations in the 27 genes, however, 12 of the genes, including
gastrokine 2 (GKN2), prosaposin-like 1 (PSAPL1),
ethanolamine-phosphate phospho-lyase (ETNPPL), cytochrome
P450 family 2 subfamily W member 1 (CYP2W1), SFRP4,
collagen type XXII α 1 chain, prostate stem cell antigen, lipase F,
gastric type, solute carrier family 15 member 1 (SLC15A1),
ATPase H+/K+ transporting subunit β (ATP4B), aldehyde
dehydrogenase 3 family member A1 (ALDH3A1) and cystatin SN
(CST1) were mutated in germline. Notably, two (SFRP4
and SLC15A1) of the 12 germline mutated genes have been
reported in previous studies. Germline mutations in SFRP4
(P320T and R340K) were reported to be strongly associated with
human cancer types (45) while
mutations in SLC15A1 were linked with bowel diseases
(46).
The KM plotter database was subsequently used with
the log-rank test to investigate the association between the
expression level of the 27 genes and clinical outcomes (both
disease-free and overall survival times). In the database, patients
with GC are divided into high and low expression groups, with each
percentile of mRNA expression between the lower (25%) and upper
quartiles (75%) used as a cut-off point. A total of 10 genes
(ALDH3A1, ETNPPL, activity regulated cytoskeleton associated
protein (ARC), ATP4B, CST1, CYP2W1, GKN2, PSAPL1,
SFRP4 and SLC15A1) with differential expression were
significantly associated (P<0.05) with survival times. In all
the 10 genes, those with high expression (red curve) were
correlated with a poorer survival while genes with low expression
(black curve) correlated with higher survival in patients with GC
(Fig. S2B).
Discussion
In an attempt to improve the understanding of the
pathobiology of GC in young patients and provide insights for
personalized treatment strategies, the genome of a 26-year old
patient with GC was performed using both whole genome and whole
transcriptome sequencing. Germline and somatic mutations analyses
of the whole genome sequencing data revealed 15 cancer related
genes with germline mutations, 27 genes with 29 informative somatic
mutations and nine genes with structural events that could underlie
the progression of GC in the patient. Of the 27 genes containing
somatic mutations, 26 have not been previously reported as driver
genes in TCGA (42). Only
MUC6, a immunohistochemical marker used to support the
diagnosis of gastric-type EA (47),
has been reported as a driver gene. In the mutation significance
analysis, a set of scoring system was used to predict deleterious
amino acid substitutions. This analysis revealed nine germline
variants and four somatic mutations with potentially pathogenic
sites. Compared with somatic mutations, more deleterious germline
sites were detected and genes containing these sites were all found
to be cancer-associated, suggesting that inheritance disorders
might be a key risk factor for the development of GC in the
patient.
To improve the understanding of the
inter-relationships between genomic alterations and the
transcriptome, the mutated genes at the transcriptional level were
also investigated. It was found that genes with somatic mutations,
such as MUC2, MUC4, SLC8A2, and with somatic structural
mutations, such as CCND3, FGFR2 and FGFR3, were
differentially expressed, suggesting that they could be promising
precision therapy targets.
Mucin genes are known to maintain the
gastrointestinal epithelium, and persistent mucosal inflammation
increase the risk of developing GC (48). In addition, the fibroblast growth
factor receptor (FGFR) pathway plays a key role in GC pathogenesis,
and detection of FGFR2 copy number in the plasma circulating tumor
DNA are potential predictive biomarkers to FGFR inhibition
(49).
With respect to heterogeneity of GC, it was
hypothesized that mutations in different genes that are involved in
the same pathway could result in similar clinical phenotypes.
Therefore, different pathways were investigated that could be
dysregulated in the patient with GC. The calcium signaling pathway,
cGMP-PKG signaling pathway and transcriptional mis-regulation were
enriched at both the genomic and transcriptomic levels, suggesting
that these pathways might play crucial roles in young patients with
GC. Notably, these pathways were not found from the analysis of
TCGA STAD dataset (42), suggesting
that the pathogenesis of GC in young individuals, particularly in
the patient in the present study may be different. Nevertheless,
the genomic features of the young patient with GC was compared with
TCGA STAD data and 27 differential expression genes were
identified, particularly germline mutations in SFRP4 (P320T
and R340K), which are strongly associated with human cancers
(45). SFRP4 is a member of
the SFRP family (50). SFRP4s
contain a cysteine-rich domain homologous to the putative
Wnt-binding site of Frizzled proteins (50) and act as soluble modulators of Wnt
signaling, which is a well-known pathway that is involved in
tumorigenesis of GC (51). This
indicates that such germline mutations could be informative in
designing patient specific therapies. Several germline mutations
were found in the 27 genes; however, no somatic alterations or
structural events were identified, suggesting that genetic factors
may be important in the development of early GC.
Notably, the youngest patient in TCGA STAD dataset
was 30-years-old and >99% (439/443) of the patients were >40
years old, thus, TCGA STAD data could have originated from elderly
patients with GC. This supports the requirement to investigate the
genome in younger patients with GC to identify possible therapeutic
targets. The patient in the present study was diagnosed at age 26,
which was much younger compared with patients in TCGA STAD dataset.
TCGA-STAD RNA-seq data was reanalyzed and 331 dysregulated genes
were found (138 were up- and 193 were downregulated), however, only
27 of these were found in the patient in the present study,
suggesting that there was a considerable difference between GC in
young and elderly patients. There may be indications that the
pathogenesis of the patient was substantially different from
typical patients with GC, as cancer-related pathways, which were
significantly enriched at both genomic and transcriptomic levels in
the patient were also not found in TCGA STAD dataset.
In conclusion, genes with germline (SFRP4),
and somatic mutations (MUC2, MUC4, SLC8A2), and those with
structural variations (CCND3, FGFR2 and FGFR3), which
were differentially expressed in the patient could be promising
precision therapy targets.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported in part by the Hefei
National Laboratory for Physical Sciences at Microscale (grant no.
HFNLM20180012).
Availability of data and materials
We have deposited the data to National Genomics Data
Center with the project number PRJCA002949. The datasets are not
publicly accessible due to local data protection laws. However,
access to the data can be granted upon a reasonable request to the
corresponding author.
Authors' contributions
QW and KH conceived the study and designed the
experiments. LL and ZH provided the tissue samples and supplied the
clinical and pathological information of the patient. QR, OEA and
SW performed sample preparation, DNA isolation for sequencing and
library construction. KH and WY performed the bioinformatics
analysis. OEA, WY, KH and QW drafted and revised the manuscript and
supplementary information. All authors approved the final version
of the manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
the First Affiliated Hospital of Anhui Medical University. The
patient provided written informed consent for the use of the
resected tissue, and the sample was obtained under Institutional
Review Board approval.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GC
|
gastric cancer
|
|
CGC
|
Cancer Gene Census
|
|
CPG
|
Cancer Predisposition Genes
|
|
Indels
|
insertions and deletions
|
|
SNP
|
single nucleotide polymorphism
|
|
SNV
|
single nucleotide variation
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
TCGA STAD
|
The Cancer Genome Atlas stomach
adenocarcinoma
|
References
|
1
|
De Vita F, Di Martino N, Fabozzi A,
Laterza MM, Ventriglia J, Savastano B, Petrillo A, Gambardella V,
Sforza V, Marano L, et al: Clinical management of advanced gastric
cancer: The role of new molecular drugs. World J Gastroentero.
20:14537–14558. 2014. View Article : Google Scholar
|
|
2
|
Mori M, Sugimachi K, Ohiwa T, Okamura T,
Tamura S and Inokuchi K: Early gastric carcinoma in Japanese
patients under 30 years of age. Br J Surg. 72:289–291. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lim S, Lee HS, Kim HS, Kim YI and Kim WH:
Alteration of E-cadherin-mediated adhesion protein is common, but
microsatellite instability is uncommon in young age gastric
cancers. Histopathology. 42:128–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smith BR and Stabile BE: Extreme
aggressiveness and lethality of gastric adenocarcinoma in the very
young. Arch Surg. 144:506–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Quijano Orvananos F, Moreno Paquentin E,
Alvarez JJ, Martinez Munive A and Butron Perez L: Gastric carcinoma
in patients under 35 years. Rev Gastroenterol Mex. 64:75–77.
1999.(In Spanish). PubMed/NCBI
|
|
6
|
Theuer CP, Kurosaki T, Taylor TH and
Anton-Culver H: Unique features of gastric carcinoma in the young:
A population-based analysis. Cancer. 83:25–33. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wierinckx A, Roche M, Raverot G,
Legras-Lachuer C, Croze S, Nazaret N, Rey C, Auger C, Jouanneau E,
Chanson P, et al: Integrated genomic profiling identifies loss of
chromosome 11p impacting transcriptomic activity in aggressive
pituitary PRL Tumors. Brain Pathol. 21:533–543. 2011.PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Research Network, .
Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard
B, Hinoue T, Laird PW, Curtis C, et al: Comprehensive molecular
characterization of gastric adenocarcinoma. Nature. 513:202–209.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chmielecki J and Meyerson M: DNA
sequencing of cancer: What Have We Learned? Annu Rev Med. 65:63–79.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garraway LA and Lander ES: Lessons from
the Cancer Genome. Cell. 153:17–37. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou SB, Diaz LA and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garraway LA: Genomics-driven oncology:
Framework for an emerging paradigm. J Clin Oncol. 31:1806–1814.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen R: Abstract 4495: Development and
clinical application of an integrative genomic approach to
personalized cancer therapy. Cancer Res. 762016.doi:
10.1158/1538-7445.AM2016-4495.
|
|
14
|
Edge SB and Compton CC: The American Joint
Committee on Cancer: The 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krueger F: Trim Galore: A wrapper tool
around Cutadapt and FastQC to consistently apply quality and
adapter trimming to FastQ files, with some extra functionality for
MspI-digested RRBS-type (Reduced Representation Bisufite-Seq)
libraries. 2012.
|
|
16
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Anders S, Pyl PT and Huber W: HTSeq-a
Python framework to work with high-throughput sequencing data.
Bioinformatics. 31:166–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H: Aligning sequence reads, clone
sequences and assembly contigs with BWA-MEM. (Submitted on 16 Mar
2013 (v1), last revised 26 May, 2013 (this version, v2)).
|
|
20
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, et
al: A framework for variation discovery and genotyping using
next-generation DNA sequencing data. Nat Genet. 43:491–498. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Poplin R, Ruano-Rubio V, DePristo MA,
Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD,
Levy-Moonshine A, Roazen D, et al: Scaling accurate genetic variant
discovery to tens of thousands of samples. BioRxiv. July
24–2017.doi: https://doi.org/10.1101/201178.
|
|
22
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Colaprico A, Silva TC, Olsen C, Garofano
L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM,
Castiglioni I, et al: TCGAbiolinks: An R/Bioconductor package for
integrative analysis of TCGA data. Nucleic Acids Res. 44:e712016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
1000 Genomes Project Consortium, . Auton
A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini
JL, McCarthy S, McVean GA and Abecasis GR: A global reference for
human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karczewski KJ, Weisburd B, Thomas B,
Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha
KE, Cummings BB, et al: The ExAC browser: Displaying reference data
information from over 60 000 exomes. Nucleic Acids Res.
45:D840–D845. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karczewski KJ and Karczewski LF: The
genome aggregation database (gnomAD). 2017.
|
|
27
|
Liu X, Wu C, Li C and Boerwinkle E: dbNSFP
v3.0: A One-stop database of functional predictions and annotations
for human nonsynonymous and splice-site SNVs. Hum Mutat.
37:235–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tate JG, Bamford S, Jubb HC, Sondka Z,
Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E,
et al: COSMIC: The catalogue of somatic mutations in cancer.
Nucleic Acids Res. 47:D941–D947. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kimura M: A simple method for estimating
evolutionary rates of base substitutions through comparative
studies of nucleotide-sequences. J Mol Evol. 16:111–120. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mwenifumbo JC and Marra MA: Cancer
genome-sequencing study design. Nat Rev Genet. 14:321–332. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen R and Seshan VE: FACETS:
Allele-specific copy number and clonal heterogeneity analysis tool
for high-throughput DNA sequencing. Nucleic Acids Res. 44:e1312016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Team RC: R: A language and environment for
statistical computing. 2013.
|
|
34
|
Rausch T, Zichner T, Schlattl A, Stutz AM,
Benes V and Korbel JO: DELLY: Structural variant discovery by
integrated paired-end and split-read analysis. Bioinformatics.
28:i333–i339. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle
E, Wang K and Liu X: Comparison and integration of deleteriousness
prediction methods for nonsynonymous SNVs in whole exome sequencing
studies. Hum Mol Genet. 24:2125–2137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Futreal PA, Coin L, Marshall M, Down T,
Hubbard T, Wooster R, Rahman N and Stratton MR: A census of human
cancer genes. Nat Rev Cancer. 4:177–183. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rahman N: Realizing the promise of cancer
predisposition genes. Nature. 505:302–308. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu GC, Wang LG, Han YY and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bailey MH, Tokheim C, Porta-Pardo E,
Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim
J, Reardon B, et al: Comprehensive characterization of cancer
driver genes and mutations. Cell. 174:1034–1035. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Szász AM, Lánczky A, Nagy Á, Förster S,
Hark K, Green JE, Boussioutas A, Busuttil R, Szabó A and Győrffy B:
Cross-validation of survival associated biomarkers in gastric
cancer using transcriptomic data of 1,065 patients. Oncotarget.
7:49322–49333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Taiaroa G, Rawlinson D, Featherstone L, et
al: Direct RNA sequencing and early evolution of SARS-CoV-2.
bioRxiv. 2020.(Epub ahead of print).
|
|
45
|
He ML, Chen Y, Chen Q, He Y, Zhao J, Wang
J, Yang H and Kung HF: Multiple gene dysfunctions lead to high
cancer-susceptibility: Evidences from a whole-exome sequencing
study. Am J Cancer Res. 1:562–573. 2011.PubMed/NCBI
|
|
46
|
Zucchelli M, Torkvist L, Bresso F,
Halfvarson J, Hellquist A, Anedda F, Assadi G, Lindgren GB,
Svanfeldt M, Janson M, et al: PepT1 oligopeptide transporter
(SLC15A1) gene polymorphism in inflammatory bowel disease. Inflamm
Bowel Dis. 15:1562–1569. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hodgson A, Parra-Herran C and Mirkovic J:
Immunohistochemical expression of HIK1083 and MUC6 in endometrial
carcinomas. Histopathology. 75:552–558. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li M, Huang L, Qiu H, Fu Q, Li W, Yu Q,
Sun L, Zhang L, Hu G, Hu J and Yuan X: Helicobacter pylori
infection synergizes with three inflammation-related genetic
variants in the GWASs to increase risk of gastric cancer in a
Chinese population. PLoS One. 8:e749762013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hierro C, Alsina M, Sanchez M, Serra V,
Rodon J and Tabernero J: Targeting the fibroblast growth factor
receptor 2 in gastric cancer: Promise or pitfall? Ann Oncol.
28:1207–1216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cruciat CM and Niehrs C: Secreted and
transmembrane wnt inhibitors and activators. Cold Spring Harb
Perspect Biol. 5:a0150812013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Herr P, Hausmann G and Basler K: WNT
secretion and signalling in human disease. Trends Mol Med.
18:483–493. 2012. View Article : Google Scholar : PubMed/NCBI
|