Introduction
Melanoma is a common solid malignant tumor that has
a high frequency of metastasis and relapse; in 2018, 287,723 new
cases of melanoma and 60,712 deaths were registered worldwide,
ranking as the 21st most common type of cancer worldwide
(1). High incidence of melanoma has
become a serious threat to human health worldwide. Traditional
therapeutic interventions including chemotherapy, radiotherapy and
surgery cannot effectively increase the overall survival time of
patients with melanoma, particularly those in advanced stages
(2). Thus, it is crucial to find
novel strategies to further improve the outcome of melanoma
therapy.
Evodiamine (EVO), a quinolone alkaloid derived from
the fruit of Evodia rutaecarpa (Chinese name, Wu-Zhu-Yu)
that has been used in traditional Chinese medicine to treat
headache and gastrointestinal disorders (3), has been demonstrated to exert various
biological effects including testosterone secretion (4), catecholamine secretion (5), antinociceptive (6), anti-inflammatory (7), antiobesity (8), vasodilatory (9), thermoregulatory (10) and uterotonic (11) effects. Numerous studies have revealed
that EVO exhibited antitumor activity by inhibiting the
proliferation, inducing cell cycle arrest and apoptosis, and
decreasing the invasion and migration of a variety of tumor cells
(12–18). In addition, the cytotoxicity of EVO
is tumor-specific, as demonstrated by EVO inducing limited toxicity
in normal human peripheral blood cells (14).
Reactive oxygen species (ROS), which have been aptly
described as a heterogeneous group of diatomic oxygen from free and
non-free radical species, have been implicated as mediators of
various biological processes, such cell proliferation, inflammation
and aging (19). Depending on the
levels, ROS is referred to as a ‘double-edged sword’ in tumor
cells: Persistent high levels of ROS compared with normal cells
often leads to increased cell proliferation and adaptive responses
that may contribute to tumorigenesis, metastasis and treatment
resistance; however, further exposure to exogenous ROS results in
tumor cell death (20,21). Redox biology, especially ROS, serves
a central role in all aspects of melanoma pathophysiology,
including initiation, progression and metastasis (22). EVO induces oxidative stress by
increasing ROS generation, as demonstrated by previous studies
(23,24); however, the association between ROS
and EVO-induced cell death remains unclear.
The present study aimed to investigate the effects
of EVO in human melanoma A-375 cells and the roles of ROS in
EVO-induced cell death.
Materials and methods
Reagents
EVO was supplied by MedChemExpress and was dissolved
(50 mM) in dimethyl sulfoxide (DMSO) and stored at −80°C. The
concentration of DMSO used for control treatments was based on the
highest concentration of EVO in each experiment. All other reagents
were purchased from Sigma-Aldrich; Merck KGaA, with the exception
of Z-VAD-fmk and Nec-1, which were also purchased from
MedChemExpress.
Cell culture
A-375 cells was obtained from the National
Infrastructure of Cell Line Resource and maintained under standard
culture conditions (37°C, 5% CO2) in RPMI-1640 medium
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.).
CCK-8 cell viability assay
A-375 cells were seeded into a 96-well plate at
1×104 cells per well with 100 µl culture medium and
cultured at 37°C with 5% CO2. A-375 cells were treated
with DMSO or 5–15 µM EVO for 24, 48 and 72 h, or pretreated with
100 µM Z-VAD-fmk, 50 µM Nec-1 or 2,000 U/ml catalase for 1 h prior
to treatment with 10 µM EVO for 24 h. After treatment, the medium
was replaced with 10% Cell Counting Kit-8 (Dojindo
Laboratories)-containing medium and incubated for 2 h at 37°C with
5% CO2. Following incubation, the plates were scanned
using a Power Wave XS microplate reader (BioTek Instruments, Inc.),
and the absorbance at 450 nm was recorded.
Determination of intracellular
ROS
Intracellular ROS levels were determined using a
DCFH-DA kit (Beyotime Institute of Biotechnology). In brief, A-375
cells were seeded in the 6-well plates (2×105
cells/well) and treated with DMSO, 10 µM EVO or 2,000 U/ml catalase
for 24 h, or pretreated with catalase (2,000 U/ml, 2 h
pre-treatment) and incubated in the medium containing 10 µM EVO and
2,000 U/ml catalase for 24 h. After treatment, the cells were
harvested by trypsinization and centrifugation (120 × g for 5 min
at 4°C), incubated with 50 µM DCFH-DA solution at 37°C in the dark
for 30 min and subjected to analysis using a BD Accuri C6 flow
cytometer with BD Accuri C6 software v1.0.264.21 (BD
Biosciences).
Cell cycle and apoptosis assays
A-375 cells were seeded in 6-well plates
(2×105 cells/well) and treated with DMSO or 10 µM EVO
for 24 h. After treatment, the cells were harvested by
trypsinization and centrifugation (120 × g for 5 min at 4°C), and
fixed with 70% ethanol at 4°C for ≥12 h. After rinsing twice with
phosphate buffer solution (PBS), the cells were re-suspended in a
DNA staining solution containing 40 mg/ml propidium iodide (PI) and
0.1 mg/ml RNase (Beyotime Institute of Biotechnology) at room
temperature in the dark for 30 min. The cells were analyzed with a
BD Accuri C6 Flow Cytometer (BD Biosciences) equipped with the
FlowJo software (FlowJo vX; FlowJo LLC). Then, the cell cycle
distribution was determined.
For the apoptosis assay, A-375 cells were treated
with DMSO (24 h), or EVO (10 µM) for 12 and 24 h, then the cells
were harvested by trypsinization and centrifugation (120 × g for 5
min at 4°C). After rinsing twice with PBS, the cells were
re-suspended in a solution containing Annexin-V and PI (EpiZyme
Biotech), and the cells were analyzed with a BD Accuri C6 Flow
Cytometer and BD Accuri C6 software.
Western blot analysis
A-375 cells were seeded in the 10-cm dishes
(1.2×106 cells/dish). The cells were treated with DMSO
for 24 h or with 10 µM EVO for 3, 6, 9, 12 and 24 h. After
treatment, the cells were rinsed with PBS (4°C). The cells were
lysed with RIPA lysis buffer (Beyotime Institute of Biotechnology)
at 4°C for 30 min and centrifuged (12,000 × g) at 4°C for 15 min.
For each treatment group, 40 µg protein determined using BCA assay
(Beyotime Institute of Biotechnology) was separated by SDS-PAGE
(10-12% gel) and transferred to a PVDF membrane. After blocking in
5% non-fat milk for 1 h, the membrane was incubated with the
primary antibodies at 4°C overnight, with the exception of β-actin,
which was used for an incubation of 1 h at room temperature. The
antibodies against phosphorylated (p-) cell division cycle (cdc) 2
(cat. no. 9114), cdc2 (cat. no. 77055), cyclin B1 (cat. no. 12231),
cdc25C (cat. no. 4688), caspase-3 (cat. no. 14220), caspase-9 (cat.
no. 9502), poly (ADP-ribose) polymerase 1 (PARP-1; cat. no. 9532),
BCL2 (cat. no. 4223), BAX (cat. no. 5023), p-receptor-interacting
serine/threonine kinase (RIP; cat. no. 65746), RIP (cat. no. 3493),
p-RIP3 (cat. no. 93654) and RIP3 (cat. no. 13526) were purchased
from Cell Signaling Technology, Inc., and the antibody against
β-actin (cat. no. AC028) was purchased from ABclonal Biotech Co.,
Ltd. The primary antibodies were diluted (1:1,000) with TBS-0.5%
Tween-20 (TBS-T) buffer with 5% BSA (Beyotime Institute of
Biotechnology), with the exception of horseradish peroxidase
(HRP)-conjugated β-actin, which was diluted (1:5,000) with TBS-T
buffer with 5% non-fat milk. The secondary antibody, HRP Goat
Anti-Rabbit IgG (H+L) (cat. no. AS014), was purchased from ABclonal
Biotech Co., Ltd., and diluted (1:5,000) with TBS-T buffer with 5%
non-fat milk. The signals were detected using an enhanced
chemiluminescence system (Beijing Kechuang Ruixin Biotechnology
Co., Ltd.) and quantified by ImageJ software (v1.8.0; National
Institutes of Health). Representative blots and quantification from
three independent experiments are presented in the figures.
Mitochondrial membrane potential (Δψm)
assay
Similar to the cell cycle assay, A-375 cells were
harvested, and a Mitochondrial Membrane Potential Assay kit with
JC-1 (Beyotime Institute of Biotechnology) was used for the
analysis of Δψm according to the manufacturer's instructions.
J-aggregates emitted red fluorescence in cells with a high Δψm,
whereas the JC-1 monomer emitted green fluorescence in cells with a
low Δψm. The value of Δψm was expressed as the ratio of red to
green fluorescence intensity.
Statistical analysis
Data are presented as the mean ± standard error.
Statistical analysis was performed by GraphPad Prism 7 software
(GraphPad Software, Inc.) using Student's t-test for two-group
comparisons or one-way ANOVA followed by Dunnett's post hoc test
for comparisons between treatment and control groups or by Tukey's
test for comparisons among multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
EVO inhibits A-375 cell
proliferation
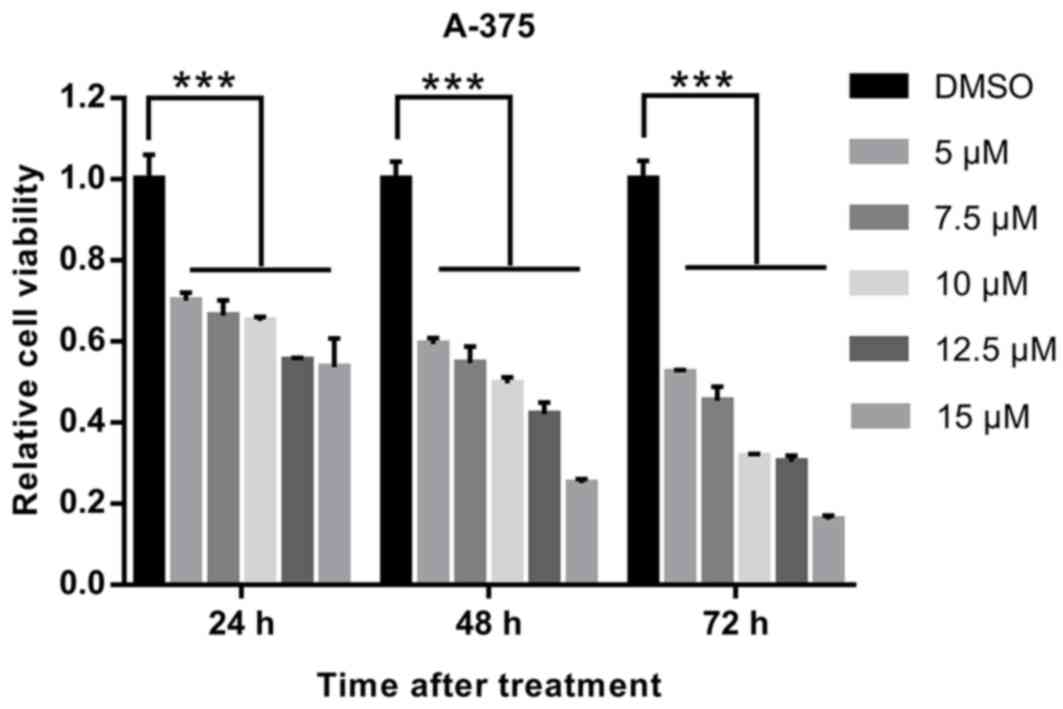
A CCK-8 assay was used to evaluate the effects of
EVO on the proliferation of human melanoma A-375 cells. As
presented in Fig. 1 and Table SI, EVO inhibited cell proliferation
in a dose- and time-dependent manner, and 10 µM was selected for
further experiments.
EVO induces G2/M cell cycle
arrest in A-375 cells
Since cell cycle arrest serves an important role in
cell proliferation inhibition, the cell cycle was assessed using
flow cytometry in the present study. As presented in Fig. 2A, the cell cycle of A-375 cells
exposed to EVO (10 µM, 24 h) was selectively arrested in the
G2/M phase. Further results demonstrated that EVO
treatment appeared to increase the phosphorylation levels of cdc2
at 3 and 24 h compared with those in the DMSO control, indicating
that EVO may activate the cdc2/cyclin B1 complex. Additionally,
following EVO exposure, the protein level of unphosphorylated
cdc25C (interphase cdc25C, 60 kDa) was attenuated, and a slower
migration form of cdc25C (mitotic cdc25C, 75 kDa) appeared to
increase over time (Fig. 2B), which
suggested that EVO selectively induced G2/M phase cell
cycle arrest by activating the cdc2/cyclin B1 complex.
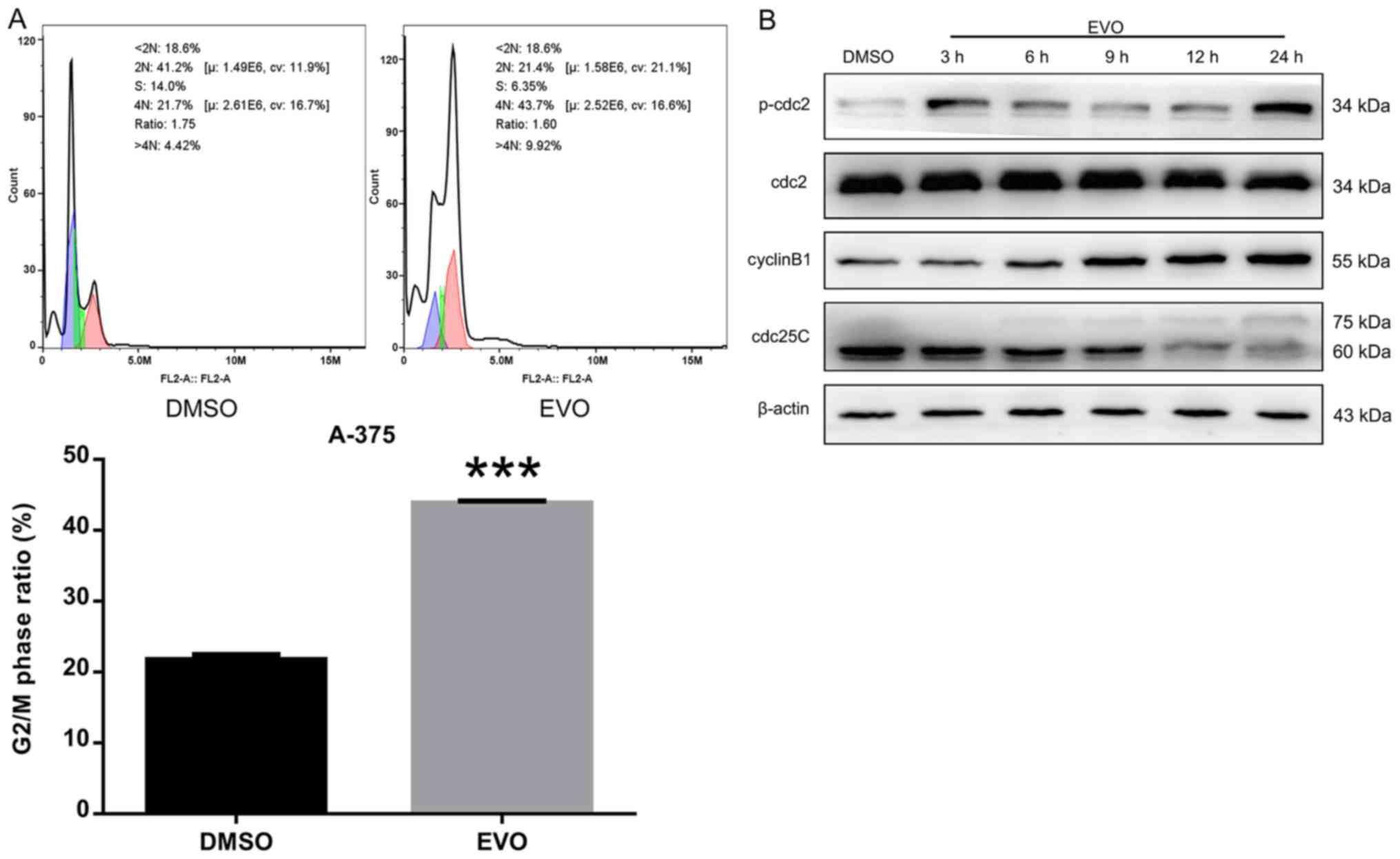 | Figure 2.EVO induces G2/M cell
cycle arrest in A-375 cells. (A) PI staining assay of A-375 cells
treated with DMSO or 10 µM EVO for 24 h examined by flow cytometry.
(B) A-375 cells were treated with DMSO or 10 µM EVO for 3, 6, 9, 12
or 24 h, and the levels of p-cdc2 (Thr161), cdc2, cyclin B1 and
cdc25C were examined by western blotting. ***P<0.001 vs. DMSO.
EVO, evodiamine; PI, propidium iodide; cdc, cell division cycle;
p-, phosphorylated. |
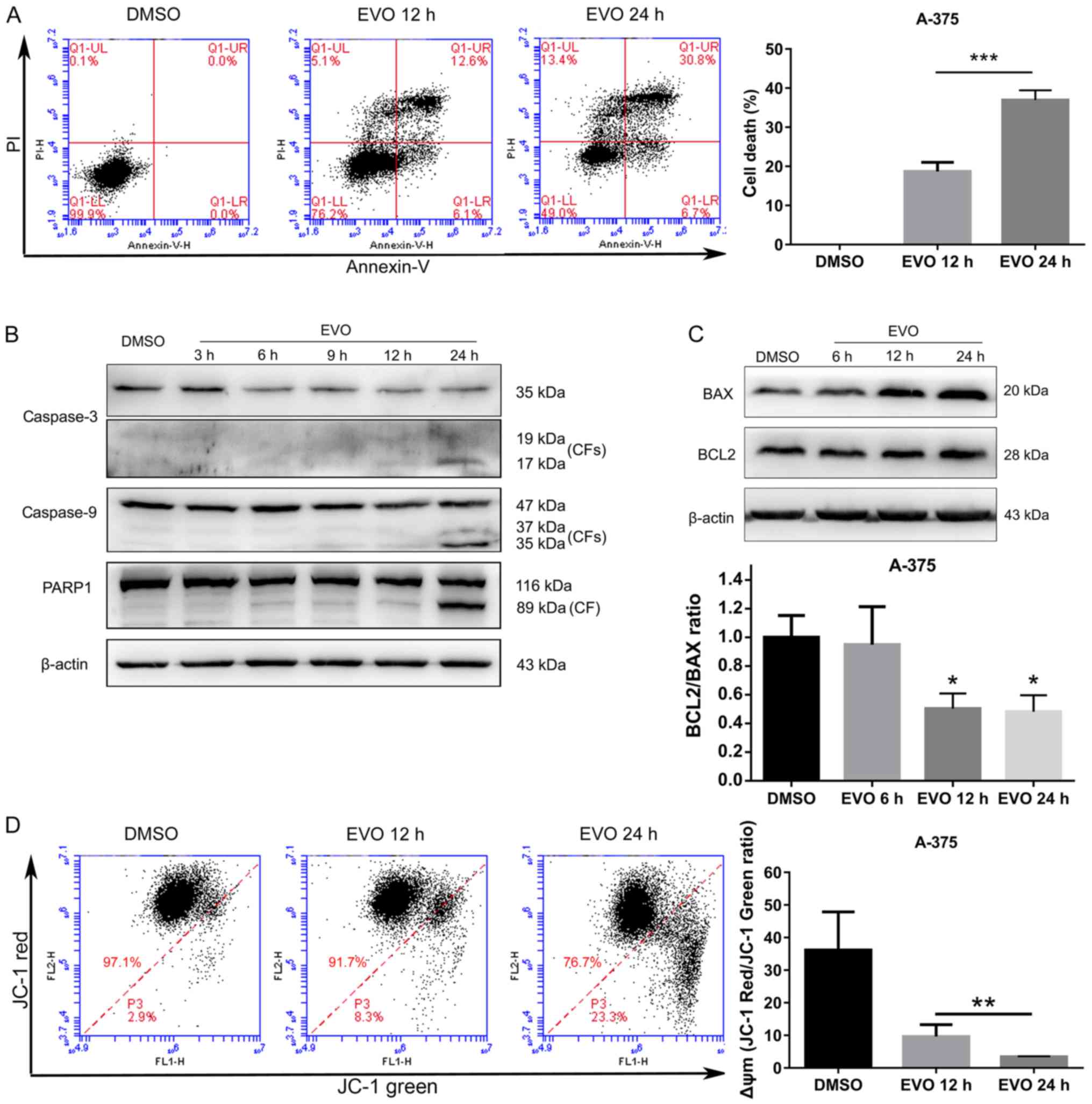
EVO induces apoptosis in A-375
cells
Apoptosis was assessed by flow cytometry with
Annexin-V/PI staining. As presented in Fig. 3A, the percentage of
Annexin-V-FITC-positive cells in A-375 cells exposed to EVO was
higher compared with that in DMSO-treated cells, indicating that
EVO induced apoptosis in A-375 cells. In addition, compared with
those in the DMSO group, activated (cleaved) caspase-3, caspase-9
and PARP1 appeared to increase over time in cells exposed to EVO
(Fig. 3B), confirming that EVO
induced apoptosis. The protein level of BAX, which is involved in
intrinsic apoptosis, was upregulated following EVO treatment
(Fig. 3C), and a significant
decrease in the BCL2/BAX ratio was observed in cells exposed to EVO
compared with that in DMSO-treated cells (Fig. 3C), indicating that intrinsic
apoptosis may be involved in EVO-induced cell death. EVO exposure
also induced significant Δψm dissipation compared with that in the
DMSO group, indicating that mitochondrial outer membrane
permeabilization (MOMP) occurred in A-375 cells exposed to EVO
(Fig. 3D).
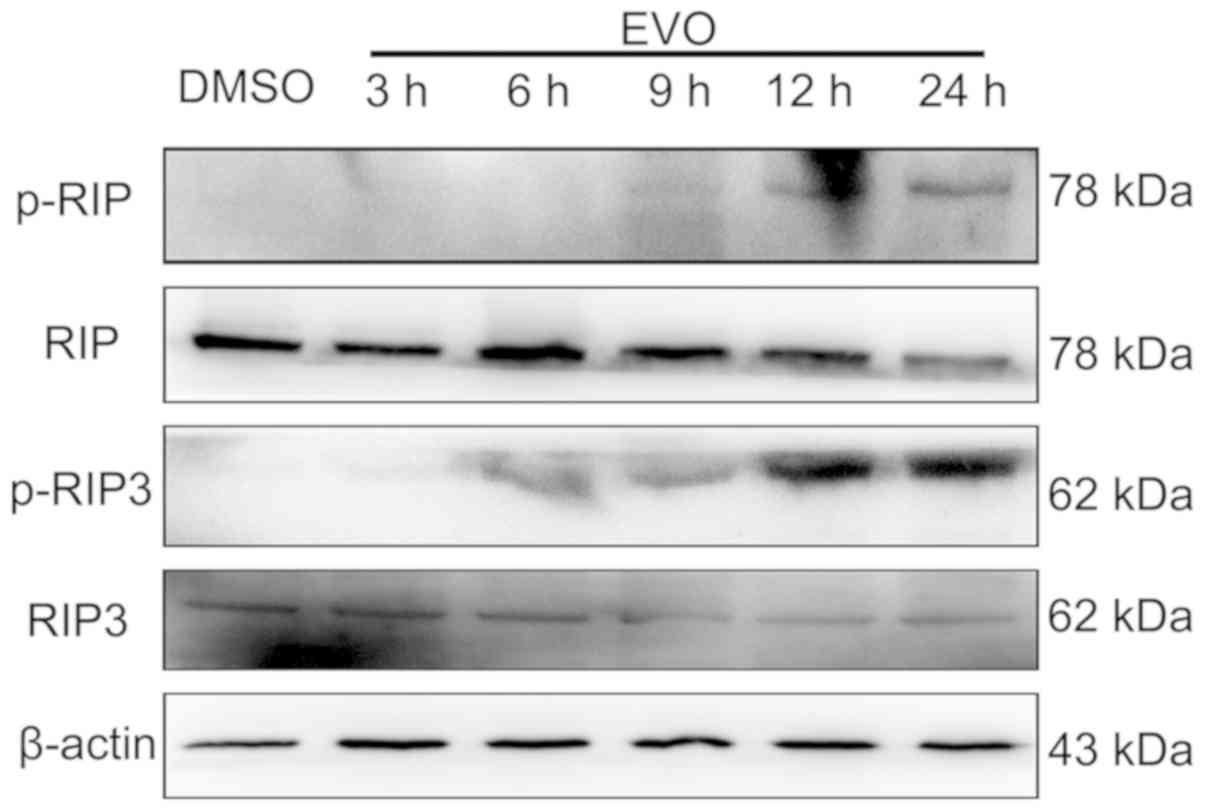
EVO induces necroptosis in A-375
cells
In addition to apoptosis, the present study examined
another type of regulated cell death, necroptosis, which generally
manifests with a necrotic morphological type and depends on the
sequential activation of RIP, RIP3 and mixed lineage kinase
domain-like pseudo kinase (MLKL) (25,26). As
presented in Fig. 4, the
phosphorylation levels of RIP and RIP3 were increased following
evodiamine treatment, indicating that RIP and RIP3 were activated
to precipitate necroptosis in cells exposed to EVO.
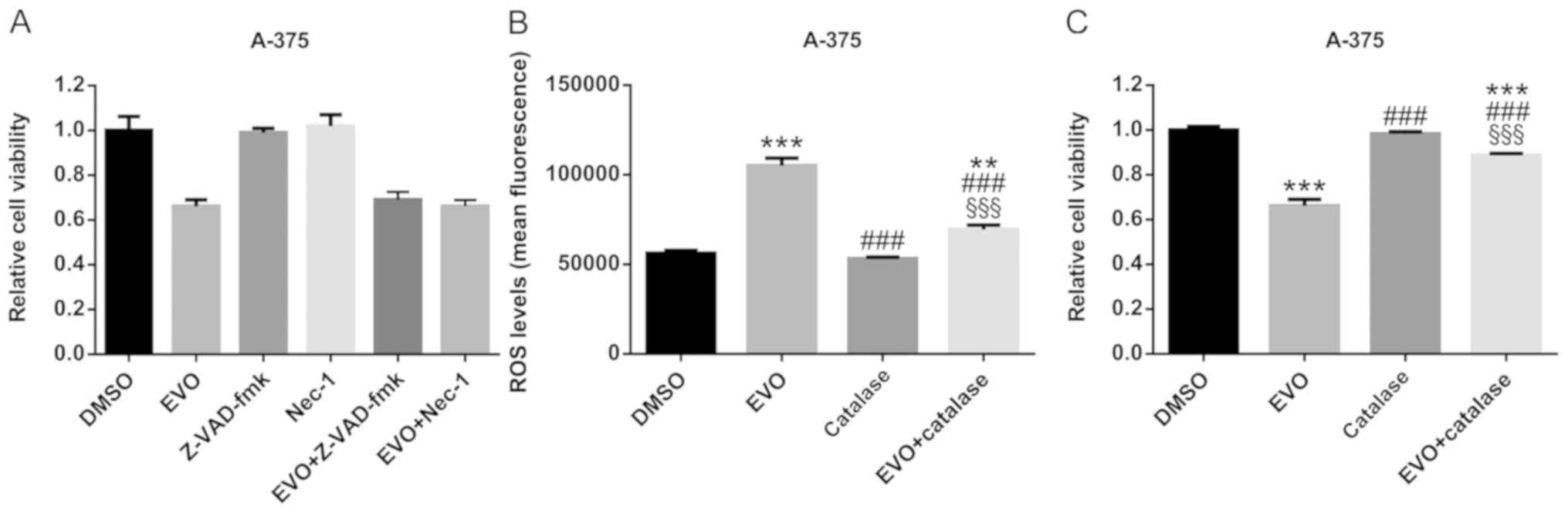
EVO induces cell death via ROS
generation in A-375 cells
In order to confirm that the apoptosis and
necroptosis involved in EVO-induced cell death, the pan-caspase
inhibitor Z-VAD-fmk and the RIP1 inhibitor Nec-1 were used, and no
significant differences were observed between the EVO and
EVO+Z-VAD-fmk or EVO+Nec-1 groups, which revealed that neither
Z-VAD-fmk nor Nec-1 attenuated the cytotoxicity of EVO (Fig. 5A). However, this did not suggest that
apoptosis or necroptosis was not involved in EVO-induced cell
death, but indicated that EVO cytotoxicity was a complicated
process.
In order to elucidate the mechanism of cell death
induced by EVO in A-375 cells, ROS levels were assessed in the
present study. The results revealed that EVO treatment (10 µM, 24
h) increased ROS generation compared with that in DMSO-treated
cells (Fig. 5B). In addition,
catalase, which quenches ROS (27),
significantly attenuated the EVO-induced ROS generation and
consequent cell death (Fig. 5B and
C), indicating that ROS may serve an important role in
EVO-induced cell death.
Discussion
EVO, a major quinazoline carboline alkaloid in
Evodia rutaecarpa, exerts cytotoxic effects on different
types of human cancer cells, including leukemic T-lymphocytes
(12), melanoma (13), breast (14), prostate (15), cervical (16), colon (17) and lung (18) cancer cells. However, the underlying
mechanism of EVO-induced cell death has not been fully elucidated.
The present study investigated the biological effects of EVO in
human melanoma A-375 cells.
The effect of EVO on A-375 cell proliferation was
assessed using a CCK-8 assay in the present study, and the results
revealed that EVO inhibited cell proliferation in a dose- and
time-dependent manner. Several previous studies have proposed that
EVO induces cell cycle arrest at the G2/M phase
(15,24,28),
which was in agreement with the results of the present study. Cell
cycle progression is controlled by various classes of cyclins,
cyclin-dependent kinases and other regulatory proteins (29). Among them, the activated cdc2/cyclin
B complex regulates the progression of the cell cycle from the
G2 to the M phase (30).
In the present study, EVO treatment upregulated the phosphorylation
levels of cdc2 at Thr161 compared with those in DMSO-treated cells,
but had no effects on the total levels of cdc2 protein, indicating
that EVO may activate cdc2. In addition, EVO exposure upregulated
cyclin B1 protein levels, which suggested that EVO may induce
G2/M arrest by activating the cdc2/cyclin B1 complex.
Activated cdc25C dephosphorylates cdc2 on Thr14 and Tyr15 and
triggers the activation of the cdc2/cyclin B1 complex (31). In the present study, EVO exposure
upregulated the levels of p-cdc25C and downregulated the levels of
unphosphorylated cdc25C compared with those in the DMSO group,
indicating that cdc25C may be activated by EVO to activate cdc2.
These results suggested that EVO may induce G2/M phase
arrest in A-375 cells by activating the cdc2/cyclin B1 complex.
Apoptosis, which is a type I form of cell death
classified by macroscopic morphological alterations, serves an
important role in EVO-induced cell death in colorectal cancer
(32) and hepatocellular carcinoma
(33) cells; consistently, in the
present study, EVO exposure significantly upregulated the apoptotic
rate of human melanoma A-375 cells compared with that in
DMSO-treated cells. There are two forms of apoptosis, extrinsic and
intrinsic, which may be involved in EVO-induced cell death as
demonstrated by the results of the present study. Intrinsic
apoptosis is also termed ‘mitochondrial apoptosis’, as irreversible
MOMP is the crucial step for intrinsic apoptosis (34). MOMP is controlled by members of the
BCL2 apoptosis regulator protein family (35). MOMP is mediated by BAX and/or BCL2
antagonist/killer 1 in response to apoptotic stimuli (36). By contrast, MOMP is antagonized by
antiapoptotic members of the BCL2 family, such as BCL2 (35). In the present study, EVO treatment
significantly upregulated the ratio of BCL2/BAX protein levels
compared with those in the DMSO-treated control group, indicating
that EVO may induce MOMP in A-375 cells. In addition, MOMP directly
promotes the cytosolic release of apoptogenic factors normally
located in the mitochondrial intermembrane space (36). The released mitochondrial protein
somatic cytochrome c binds to apoptotic peptidase-activating factor
1 and pro-caspase-9 to form the supramolecular complex referred to
as apoptosome, which is responsible for caspase-9 activation
(37). Activated caspase-9 can
catalyze the proteolytic activation of executioner caspases,
caspase-3 and −7, which are the enzymes responsible for cell
demolition during intrinsic and extrinsic apoptosis in mammalian
cells (38). In the present study,
EVO treatment induced a significant Δψm dissipation and activation
of caspase-9 and caspase-3 compared with that in DMSO-treated
cells, indicating that EVO may induce MOMP and consequent intrinsic
apoptosis in A-375 cells.
Activation of PARP1 was also observed in A-375 cells
exposed to EVO in the present study. PARP1 is a specific component
of the DNA damage response machinery, and its activation induces
the accumulation of poly (ADP-ribose) polymers and poly
(ADP-ribosylated) proteins in the mitochondria, resulting in Δψm
dissipation and MOMP (39). The
binding of the poly (ADP-ribose) polymers to apoptosis-inducing
factor mitochondria associated 1 (AIF) promotes the release of AIF
into the cytosol and its translocation into the nucleus, where it
mediates DNA fragmentation and chromatin condensation (39). This form of cell death induced by the
consequent activation of PARP1 and AIF is known as
caspase-independent apoptosis, currently termed ‘parthanatos’ by
the Nomenclature Committee on Cell Death (40).
The present study also revealed that in human
melanoma A-375 cells, EVO exposure induced the upregulation of RIP
and RIP3 phosphorylation compared with that in DMSO-treated cells,
indicating that necroptosis may also be involved in EVO-induced
cell death. The inhibitors of caspase (Z-VAD-fmk) and RIP (Nec-1)
were selected to confirm that apoptosis and necroptosis were
involved in EVO-induced cell death in the present study. However,
Z-VAD-fmk and Nec-1 both failed to rescue A-375 cells from the cell
death induced by EVO. Of note, increasing evidence has indicated
that the pharmacologic inhibition of the processes which are
commonly considered necessary for cell death execution often does
not avoid cellular demise, but alters its biochemical and
morphologic manifestations (41).
Thus, the inhibitors of caspase (Z-VAD-fmk and broad-spectrum
caspase inhibitors), PARP1 and RIP (Nec-1) may fail to reverse cell
death induced by pharmacologic interventions despite significantly
blocking the activation of caspases, PARP1 and RIP (42–44).
However, the proportion of cells eventually succumbing to regulated
cell death (RCD) does not change (41). Thus, the results of the present study
suggested that Z-VAD-fmk or Nec-1 may block the corresponding cell
death phenotype and cause the switch towards another type in
EVO-induced cell death.
Early-stage biochemical processes, declining ATP
levels and redox alterations in RCD can be reversed to restore
cellular homeostasis (41). In the
present study, ROS generation was observed in A-375 cells exposed
to EVO, and catalase significantly attenuated EVO-induced ROS
generation and cell death, which indicated that ROS may serve an
important role in EVO-induced cell death. In conclusion, the
present study demonstrated that EVO induced apoptosis and
necroptosis in human melanoma A-375 cell via a ROS-dependent
pathway.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Yinlin Ge
(Institute of Biochemistry, Medical College, Qingdao University,
Qingdao, China) for the help in revising the manuscript.
Funding
The present study was supported by the Shandong Key
R&D Program (grant no. 2017GSF218084) and the Qingdao Applied
Basic Research Program (grant no. 19-6-2-31-cg).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
NL performed the majority of the experiments and
analyzed the data. YL assisted with the experiments. GC
participated in the analysis, data interpretation and experiment
design. KG designed the experiments and wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leonardi GC, Falzone L, Salemi R, Zanghì
A, Spandidos DA, Mccubrey JA, Candido S and Libra M: Cutaneous
melanoma: From pathogenesis to therapy (Review). Int J Oncol.
52:1071–1080. 2018.PubMed/NCBI
|
|
3
|
Lee SH, Son JK, Jeong BS, Jeong TC, Chang
HW, Lee ES and Jahng Y: Progress in the studies on rutaecarpine.
Molecules. 13:272–300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin H, Tsai SC, Chen JJ, Chiao YC, Wang
SW, Wang GJ, Chen CF and Wang PS: Effects of evodiamine on the
secretion of testosterone in rat testicular interstitial cells.
Metabolism. 48:1532–1535. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoshizumi M, Houchi H, Ishimura Y, Hirose
M, Kitagawa T, Tsuchiya K, Minakuchi K and Tamaki T: Effect of
evodiamine on catecholamine secretion from bovine adrenal medulla.
J Med Invest. 44:79–82. 1997.PubMed/NCBI
|
|
6
|
Kobayashi Y: The nociceptive and
anti-nociceptive effects of evodiamine from fruits of Evodia
rutaecarpa in mice. Planta Med. 69:425–428. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chiou WF, Sung YJ, Liao JF, Shum AY and
Chen CF: Inhibitory effect of dehydroevodiamine and evodiamine on
nitric oxide production in cultured murine macrophages. J Nat Prod.
60:708–711. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kobayashi Y, Nakano Y, Kizaki M, Hoshikuma
K, Yokoo Y and Kamiya T: Capsaicin-like anti-obese activities of
evodiamine from fruits of Evodia rutaecarpa, a vanilloid
receptor agonist. Planta Med. 67:628–633. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chiou WF, Chou CJ, Shum AY and Chen CF:
The vasorelaxant effect of evodiamine in rat isolated mesenteric
arteries: Mode of action. Eur J Pharmacol. 215:277–283. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsai TH, Lee TF, Chen CF and Wang LC:
Thermoregulatory effects of alkaloids isolated from Wu-chu-yu in
afebrile and febrile rats. Pharmacol Biochem Behav. 50:293–298.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
King CL, Kong YC, Wong NS, Yeung HW, Fong
HH and Sankawa U: Uterotonic effect of Evodia rutaecarpa
alkaloids. J Nat Prod. 43:577–582. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee TJ, Kim EJ, Kim S, Jung EM, Park JW,
Jeong SH, Park SE, Yoo YH and Kwon TK: Caspase-dependent and
caspase-independent apoptosis induced by evodiamine in human
leukemic U937 cells. Mol Cancer Ther. 5:2398–2407. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang C, Wang MW, Tashiro SI, Onodera S and
Ikejima T: Evodiamine induced human melanoma A375-S2 cell death
partially through interleukin 1 mediated pathway. Biol Pharm Bull.
28:984–989. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liao CH, Pan SL, Guh JH, Chang YL, Pai HC,
Lin CH and Teng CM: Antitumor mechanism of evodiamine, a
constituent from Chinese herb Evodiae fructus, in human
multiple-drug resistant breast cancer NCI/ADR-RES cells in vitro
and in vivo. Carcinogenesis. 26:968–975. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kan SF, Yu CH, Pu HF, Hsu JM, Chen MJ and
Wang PS: Anti-proliferative effects of evodiamine on human prostate
cancer cell lines DU145 and PC3. J Cell Biochem. 101:44–56. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fei XF, Wang BX, Li TJ, Tashiro SI, Minami
M, Xing DJ and Ikejima T: Evodiamine, a constituent of Evodiae
fructus, induces anti-proliferating effects in tumor cells. Cancer
Sci. 94:92–98. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ogasawara M, Matsubara T and Suzuki H:
Inhibitory effects of evodiamine on in vitro invasion and
experimental lung metastasis of murine colon cancer cells. Biol
Pharm Bull. 24:917–920. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ogasawara M, Matsunaga T, Takahashi S,
Saiki I and Suzuki H: Anti-invasive and metastatic activities of
evodiamine. Biol Pharm Bull. 25:1491–1493. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dickinson BC and Chang CJ: Chemistry and
biology of reactive oxygen species in signaling or stress
responses. Nat Chem Biol. 7:504–511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kong Q, Beel JA and Lillehei KO: A
threshold concept for cancer therapy. Med Hypotheses. 55:29–35.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schafer FQ and Buettner GR: Redox
environment of the cell as viewed through the redox state of the
glutathione disulfide/glutathione couple. Free Radic Biol Med.
30:1191–1212. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Obrador E, Liu-Smith F, Dellinger RW,
Salvador R, Meyskens FL and Estrela JM: Oxidative stress and
antioxidants in the pathophysiology of malignant melanoma. Biol
Chem. 400:589–612. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang R, Deng D, Shao N, Xu Y, Xue L, Peng
Y, Liu Y and Zhi F: Evodiamine activates cellular apoptosis through
suppressing PI3K/AKT and activating MAPK in glioma. OncoTargets
Ther. 11:1183–1192. 2018. View Article : Google Scholar
|
|
24
|
Yang J, Wu LJ, Tashino S, Onodera S and
Ikejima T: Protein tyrosine kinase pathway-derived ROS/NO
productions contribute to G2/M cell cycle arrest in
evodiamine-treated human cervix carcinoma HeLa cells. Free Radic
Res. 44:792–802. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Linkermann A and Green DR: Necroptosis. N
Engl J Med. 370:455–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Murphy JM, Czabotar PE, Hildebrand JM,
Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D,
Webb AI, et al: The pseudokinase MLKL mediates necroptosis via a
molecular switch mechanism. Immunity. 39:443–453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Glorieux C and Calderon PB: Catalase, a
remarkable enzyme: Targeting the oldest antioxidant enzyme to find
a new cancer treatment approach. Biol Chem. 398:1095–1108. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang DM, Guh JH, Huang YT, Chueh SC,
Chiang PC and Teng CM: Induction of mitotic arrest and apoptosis in
human prostate cancer pc-3 cells by evodiamine. J Urol.
173:256–261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Taylor WR and Stark GR: Regulation of the
G2/M transition by p53. Oncogene. 20:1803–1815. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Booher RN, Holman PS and Fattaey A: Human
Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not
Cdk2 activity. J Biol Chem. 272:22300–22306. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sui H, Zhou LH, Zhang YL, Huang JP, Liu X,
Ji Q, Fu XL, Wen HT, Chen ZS, Deng WL, et al: Evodiamine suppresses
ABCG2 mediated drug resistance by inhibiting p50/p65 NF-kB pathway
in colorectal cancer. J Cell Biochem. 117:1471–1481. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang J, Cai X, Lu W, Hu C, Xu X, Yu Q and
Cao P: Evodiamine inhibits STAT3 signaling by inducing phosphatase
shatterproof 1 in hepatocellular carcinoma cells. Cancer Lett.
328:243–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Delbridge AR, Grabow S, Strasser A and
Vaux DL: Thirty years of BCL-2: Translating cell death discoveries
into novel cancer therapies. Nat Rev Cancer. 16:99–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Riedl SJ and Salvesen GS: The apoptosome:
Signalling platform of cell death. Nat Rev Mol Cell Biol.
8:405–413. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fatokun AA, Dawson VL and Dawson TM:
Parthanatos: Mitochondrial-linked mechanisms and therapeutic
opportunities. Br J Pharmacol. 171:2000–2016. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Galluzzi L, Bravo-San Pedro JM, Vitale I,
Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D,
Annicchiarico-Petruzzelli M, et al: Essential versus accessory
aspects of cell death: Recommendations of the NCCD 2015. Cell Death
Differ. 22:58–73. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Prabhakaran K, Li L, Borowitz JL and Isom
GE: Caspase inhibition switches the mode of cell death induced by
cyanide by enhancing reactive oxygen species generation and PARP-1
activation. Toxicol Appl Pharmacol. 195:194–202. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Steinhart L, Belz K and Fulda S: Smac
mimetic and demethylating agents synergistically trigger cell death
in acute myeloid leukemia cells and overcome apoptosis resistance
by inducing necroptosis. Cell Death Dis. 4:e8022013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dunai ZA, Imre G, Barna G, Korcsmaros T,
Petak I, Bauer PI and Mihalik R: Staurosporine induces necroptotic
cell death under caspase-compromised conditions in U937 cells. PLoS
One. 7:e419452012. View Article : Google Scholar : PubMed/NCBI
|