Introduction
Tumor immunotherapy has become an effective
treatment option following surgery, chemotherapy and radiotherapy
in some specific types of cancer, such as acute lymphoblastic
leukemia (1). Cancer immunotherapy
includes immune checkpoint inhibitors, adoptive cell transfer
therapy (ACT), tumor-specific vaccines and small molecule
inhibitors (1).
Tumor antigens can be divided into tumor-specific
antigens (TSAs), tumor-associated antigens (TAAs) and cancer-testis
antigens. TAAs are overexpressed in tumor cells; however, they are
typically present in low amounts in healthy cells. During thymic
development, T cells undergo positive and negative selection, in
which T cells with high affinity to autoantigens are eliminated
naturally, and only the T cells with low affinity to autoantigens
develop and mature (2). At present,
vaccines targeting TAAs are often affected by central or peripheral
immune tolerance or cause serious side effects (2).
New tumor antigens (neoantigens), also known as
TSAs, are novel antigens encoded by a mutation in tumor cells,
which are not expressed in healthy host cells. Major somatic
mutations, such as gene fusion, point and deletion mutations, can
produce abnormal proteins (3). These
proteins can be specifically recognized by T cells following
presentation, resulting in an immune response (3). Therefore, immunotherapy targeting
different tumor neoantigens has become a novel prospect in the
treatment of solid tumors.
Individualized immunotherapy and precision medicine
have emerged as the future of malignant tumor treatment.
Individualized tumor immunotherapy is a novel type of treatment
based on identifying specific tumor neoantigens expressed in each
individual patient. This type of therapy uses sequencing and
bioinformatic analyses on specific tumor tissues of patients
(4), and includes the use of
personalised vaccines or ACT to activate the immune response using
a combination of experimental screening and synthesis of peptides
(5).
In the present study, genes with a high frequency of
point mutations in colorectal, lung and liver cancer were screened
using The Cancer Genome Atlas (TCGA) database, and three groups of
antigen epitope peptides with a strong affinity for major
histocompatibility complex (MHC) molecules were selected using a
computer prediction algorithm, which identified mutant peptides of
KRAS proto-oncogene (KRAS) G12V, tumor protein p53 (TP53) R158L and
catenin β1 (CTNNB1) K335I. The ideal epitope peptides that could
specifically activate T-cell immune responses were screened using
immunogenicity tests. The present study aimed to perform a
preliminary screening of tumor neoantigens and immunogenic
epitopes, to provide a foundation for individualized immunotherapy
and precision medical treatment in the late stages of oncogenesis
(6).
Materials and methods
Reagents
The human HCT116 colorectal cancer, T2 cell lines,
human HepG2 liver cancer and human NCI-H292 lung cancer cell lines
were obtained from and authenticated using short-tandem repeat
analysis by The Cell Bank of Type Culture Collection of the Chinese
Academy of Sciences. Human peripheral blood was collected from
laboratory healthy volunteers [pre-determined human leukocyte
antigen (HLA)-A2 positive] and written informed consent was
provided by each volunteer.
The synthesis of wild-type and mutant peptides was
performed by Shanghai Biology Co., Ltd. (http://www.chinapeptides.com/). The upstream and
downstream primers reverse transcription-quantitative (RT-q)PCR
were synthesized by Beijing Qingke Biotechnology Co., Ltd..
Lipofectamine® 3000 and TRIzol® were
purchased from Invitrogen (Thermo Fisher Scientific, Inc.). The
SYBR Green qPCR mix was purchased from Promega Corporation.
BamH1 and Sal1 restriction endonucleases were
purchased from New England BioLabs, Inc. CD3 (cat. no. 05131-20)
and CD28 (cat. no. 10311-20) monoclonal antibodies were purchased
from PeproTech, Inc. Carboxyfluorescein succinimidyl ester
(CFSE)-fluorescein isothiocyanate (FITC) fluorescent dye,
phycoerythrin (PE) anti-human CD3 (cat. no. 300307), FITC
anti-human CD4 (cat. no. 357405), FITC anti-human CD8 (cat. no.
344703), PE-Cyanine 7 (Cy7) anti-human CD25 (cat. no. 302611), FITC
anti-human HLA-A2 (cat. no. 343303) and granzyme B antibodies (cat.
no. 515403) were purchased from BioLegend, Inc. X–VIVO culture
medium was purchased from Lonza Group, Ltd.. The plasmid extraction
kit was purchased from Omega Bio-Tek, Inc. The ELISA kit for IFN-γ
(cat. no. 1110002) was purchased from Neobioscience Technology Co.,
Ltd.. Calcein-acetoxymethyl (Cal-AM) and propidium iodide (PI)
fluorescent dyes were purchased from the Japan Research Institute,
Ltd.. Recombinant human (rh)interleukin (IL)-2, rhIL-7, rhIL-15,
rhIL-4, tumor necrosis factor (rhTNF)-α and granulocyte-macrophage
colony-stimulating factor (rhGM-CSF) all were recombinant human
cytokines, which were purchased from Sangon Biotech Co., Ltd.
TCGA database screening of gene
mutation sites
The ‘mutant gene’ column of colon, lung and liver
cancer was searched in TCGA database (https://portal.gdc.cancer.gov/), and the top 10 genes
with the highest mutation frequency in these three types of cancer
were identified. The mutation sites with a single base substitution
were screened out and used for further evaluation.
The immune presentation ability of
epitope peptides evaluated using the SYFPEITHI, BIMAS prediction
system
The epitope peptide score of colon, lung and liver
cancer containing a gene mutation site was predicted (9 aa,
HLAA201). According to the location of mutation in wild-type and
mutant peptides, Epitope peptides with higher scores (≥8 points)
before (wild-type) and after gene locus mutation (mutant) were
screened out for further evaluation. A total of 6 epitope peptides
(Table I) were screened and divided
into the following three groups: Wild-type and mutant peptides of
the KRAS group, TP53 group, and the CTNNB1 group, respectively,
which were synthesized by the aforementioned company. (SYFPEITHI
prediction system: The immune presentation ability of epitope
peptides could be evaluated by these prediction systems. The
prediction score of epitope peptide indicated the ability of immune
presentation (7,8).
 | Table I.WT and mutant epitope peptides. |
Table I.
WT and mutant epitope peptides.
| Gene | Mutation locus | WT epitope peptide
sequence (name) | Mutant epitope
peptide sequence (name) |
|---|
| KRAS | G12V | YKLVVVGAG (KRAS
WT) | YKLVVVGAV (KRAS
mutant) |
| TP53 | R158L | VRAMAIYKQ, (TP53
WT) | VLAMAIYKQ (TP53
mutant) |
| CTNNB1 | K335I | IMRTYTYEK, (CTNNB1
WT) | IMRTYTYEI (CTNNB1
mutant) |
Verifying the affinity of epitope
peptides to HLA-A2 molecules
T2 cells are B cells with the HLA-A2 gene. These
cells are deficient in the antigen polypeptide transporter, which
is required for endogenous antigen presentation, as they are unable
to present their own antigens on the cell surface. Therefore, MHC I
molecules on the T2 cell surface are often used to present foreign
antigens for immune cell recognition in immunological functional
testing experiments (9,10).
T2 cells were inoculated in a 6-well plate with
RPMI-1640 containing 10% FBS in 37°C and 5% CO2
incubators, at a density of 1×106 cells/ml. A total of 3
groups of the aforementioned wild-type and mutant epitope peptides
(20 ug/ml) were added to the 6-well plate with a well for each
peptide. Following a 4-h incubation at 37°C, the mean fluorescence
intensity (MFI) was detected using a Gallios flow cytometer
(Beckman Coulter, Inc.) using a labelled HLA-A2-FITC fluorescent
antibody (ready to use, incubation in 4°C for 20 min). The affinity
was measured using the flowing equation: Fluorescence index=(MFI of
epitope peptide-background MFI)/background MFI. Fluorescence index
>1.0 indicated the strong stability, fluorescence index <0.5
indicated the weakest stability.
Specific cytotoxic T lymphocytes
(CTLs) induced by epitope peptides in vitro
Dendritic cells (DCs) cultured in vitro
Human peripheral blood mononuclear cells obtained
from laboratory healthy volunteers (PBMCs; HLA-A2+, 300 × g
centrifugation in 37°C for 30 min) were cultured in 37°C and 5%
CO2 incubators for 4 h. The culture medium was removed
and the adherent cells were isolated. rhGM-CSF [1,000 international
units (IU)/ml] and rhIL-4 (500 IU/ml) were added to stimulate the
growth of monocytes. On the 3rd and 6th day, 10 µg/ml rhTNF-α was
added to promote the maturation of DCs and supplemented with
cytokines [rhGM-CSF (1,000 IU/ml), rhIL-4 (500 IU/ml), rhTNF-α (10
µg/ml)]. From the 6th day, rhTNF-α (10 µg/ml) was added every day
until the 9th day, and mature DCs were harvested by washing the
cells with PBS.
Specific CTLs induced by wild-type and mutant
peptides in each group
In the first round of stimulation, PBMCs
(2×106 cells/well) were inoculated in a 6-well plate and
the corresponding three groups of wild-type peptides (20 µg/ml),
mutant peptides (20 µg/ml) and β2-microglobulin (PeproTech, 3
µg/ml) were added to each well. IL-2 (100 IU/ml) and TNF-α (800
IU/ml) were added to each well every 2 days for 7 days. In the
second round of stimulation, mature DCs were added to the three
groups of wild-type and mutant peptides (20 µg/ml) and subsequently
co-cultured with PBMCs at a ratio of 1:10. The proliferation of T
lymphocytes was stimulated by adding the cytokines, IL-2, IL-7 and
IL-15 (10 ng/ml), every 3 days for 7 days. In the third round of
stimulation, CD3 and CD28 monoclonal antibodies (2 µg/ml) were
added to stimulate the proliferation of specific T lymphocytes.
Specific effective T cells were collected on the 21st day (300 × g
centrifugation in 37°C for 5 min) for functional detection.
Therefore, six corresponding specific T lymphocyte subsets
targeting three groups of wild-type and mutant peptides were
obtained (Table II).
| Table II.Specific CTLs induced by WT and
mutant peptides. |
Table II.
Specific CTLs induced by WT and
mutant peptides.
| Group | WT epitope peptide
sequence to induce CTLs (name) | Mutant epitope
peptide sequence to induce CTLs (name) |
|---|
| KRAS | YKLVVVGAG
(CTLKRAS WT) | YKLVVVGAV
(CTLKRAS mutant) |
| TP53 | VRAMAIYKQ
(CTLTP53 WT) | VLAMAIYKQ
(CTLTP53 mutant) |
| CTNNB1 | IMRTYTYEK
(CTLCTNNB1 WT) | IMRTYTYEI
(CTLCTNNB1 mutant) |
Detection of subsets of specific T
lymphocytes
The CTLs from culture on day 21 were labelled with
ready to use CD3-PE, CD8-FITC, CD4-FITC and CD25-PE-Cy7-flow
fluorescent antibodies and incubated at 4°C for 20 min in the dark.
Each tube was washed twice with PBS containing 2% FBS (Gibco;
Thermo Fisher Scientific, Inc.). The supernatant was discarded
following centrifugation at 400 × g in 37°C for 5 min. The cells
were resuspended with PBS in flow tube and analysed using flow
cytometry.
Detection of CTL cell proliferation
CTLs from culture on day 14 were placed into
Eppendorf® tubes and incubated with CFSE fluorescent dye
(final concentration, 1 µmol/l) for 10 min at 37°C. CTLs were
washed twice with X–VIVO medium and cultured into a 6-well plate at
a density of 1×106 cells/well in X–VIVO medium in an
incubator at 37°C with 5% CO2. A total of
1×105 cells were detected using flow cytometry for 4
consecutive days from the 16th to the 19th day. The cell
proliferation was analysed using the Modfit software (Verity
Software House, v5.0.9). The software would automatically fit cell
different generations, when the cells were in the state of
proliferation and division.
ELISA for the detection of IFN-γ secretion in T
lymphocytes stimulated by epitope peptides in vitro
T2 cells treated with 3 groups of epitope peptides
and the peptide-induced specific T-lymphocytes were mixed and
cultured at a density of 1×106 cells/well at 37°C for 24
h. The cell culture supernatant was extracted from each group (500
µl) following centrifugation at 400 × g at 37°C for 5 min.
Detection of IFN-γ secretion using ELISA was performed according to
the manufacturer's protocol. The experimental groups are shown in
Table III.
| Table III.T2 cells loaded with WT or mutant
peptides co-cultured with peptide-induced CTLs. |
Table III.
T2 cells loaded with WT or mutant
peptides co-cultured with peptide-induced CTLs.
| Group | Peptide (WT) + CTL
(WT) | Peptide (mutant) +
CTL (mutant) |
|---|
| KRAS | T2peptide KRAS
WT + CTLKRAS WT | T2peptide KRAS
mutant + CTLKRAS mutant |
| TP53 | T2peptide TP53
WT + CTLTP53 WT | T2peptide TP53
mutant + CTLTP53 mutant |
| CTNNB1 | T2peptide
CTNNB1 WT + CTLCTNNB1 WT | T2peptide
CTNNB1 mutant + CTLCTNNB1 mutant |
Preliminary detection of cytotoxic activity of
specific CTLs using Cal-AM release assay
The greater release of Cal-AM from tumor cells, the
stronger the cytotoxic activity. T2 cells were stained with 2 µg/ml
Cal-AM at 37°C for 10 min in the dark. Following centrifugation in
300 × g at 37°C for 5 min, twice with sterile PBS, T2 cells were
added to the three groups of epitope peptides (20 µg/ml) and
incubated at 37°C for 4 h. Target cells were inoculated in 96-well
plates (1×105 cells/ml; 100 µl/well). Effective CTLs
were added according to the effective target ratio of 20:1 with a
volume of 100 µl/well. The maximum release group (2% Triton X-100
treated T2 cells for 24 h) and the self-releasing group (T2 cells
only) were prepared. The final volume of each well was 200 µl, and
each group was analyzed in triplicate. Following co-culture at 37°C
for 24 h (including the experimental group, the maximum release
group, the self-releasing group), 80 µl/well supernatant was
collected by centrifugation in 300 × g at 37°C for 5 min, and the
optical density (OD) values of each group were measured using an
automatic microplate reader (Thermo Fisher Scientific, Inc.) at an
excitation wavelength of 485 nm and an emission wavelength of 536
nm. The killing rate was calculated as follows: Killing rate
(%)=(ODexperimental group-ODself-release
group)/(ODmaximum release
group-ODself-release group) ×100. The experimental
groups are shown in Table III.
For the antibody blocking experiments, homotypic
control antibody (FITC Mouse lgG2b, Isotype Control, ready to use,
cat. no. 555057, eBioscience; Thermo Fisher Scientific, Inc.) or
HLA-A2 antibody were added to the target cells (T2 cells) and
incubated at 37°C with 5% CO2 for 60 min. Target cells
were added to 96-well plates (1×105 cells/ml; 100
µl/well) with the three groups of mutant peptide-induced CTLs
(1×105 cells/ml; 100 µl/well), which were tested
according to the aforementioned Cal-AM release assay.
Construction of recombinant eukaryotic expression
plasmids
TRIzol® was used to extract RNA from
HCT116 colon cancer cells, and the RNA was reverse transcribed into
cDNA. The reverse transcription conditions were as follows: 25°C
For 5 min, 42°C for 60 min, 70°C for 15 min, and 4°C for 10 min
(GoScript™ Reverse Transcription Mix, Promega Corportation).
Upstream and downstream primers for mutant genes were designed and
synthesized (Table IV). Using the
site-directed mutagenesis PCR method (11), the 12th amino acid translated from
the KRAS gene was transformed from glycine (G) to valine (V) by
changing the codon from GGT to GTT. The PCR amplification
conditions were as follows: 98°C For 10 sec, 58°C for 5 sec and
72°C for 90 sec, for 35 cycles (PrimeSTAR Max DNA Polymerase,
Takara Biotechnology Co., Ltd.). The high-fidelity enzyme
amplification product was identified using 1% agarose gel
electrophoresis with ethidium bromide. The mutant KRAS G12V gene
fragment was ligated with the pIRES2-EGFP plasmid (Guangzhou Aiji
Biotechnology Biological Co., Ltd.) using BamH1 and
Sal1 restriction endonucleases (New England BioLabs, Inc.)
at 16°C overnight. The recombinant plasmid, KRAS G12V-pIRES2-EGFP,
was obtained from the correctly sequenced (sequenced by Guangzhou
Qingke Biotechnology Co., Ltd.) genetically engineered bacteria
(DH5α) using a removing endotoxin plasmid extraction kit (Omega
Bio-Tek, Inc.).
| Table IV.Primers used in the present
study. |
Table IV.
Primers used in the present
study.
| Primer name | Sequence,
5′→3′ | Product, bp |
|---|
| KRAS-G12V-F |
ATGACTGAATATAAACTTGTGGTAGTTGGAGCTGTTGGCGTAGGCA | 46 |
| KRAS-G12V-R |
TTACATTATAATGCATTTTTTAATTTTCACACAGC | 35 |
| TP53-R158L-F1 |
ATGGAGGAGCCGCAGTCAGATCCTA | 25 |
| TP53-R158L-R1 |
TAGATGGCCATGGCGAGGACGC | 22 |
| TP53-R158L-F2 |
CGCGTCCTCGCCATGGCCATCT | 22 |
| TP53-R158L-R2 |
TCAGTCTGAGTCAGGCCCTTCTGT | 24 |
|
CTNNB1-K335I-F1 |
ATGGCTACTCAAGCTGATTTGATGGAGTT | 29 |
|
CTNNB1-K355I-R1 |
TGGTCCACAGTAGTATTTCGTAAGTATAGGTCCT | 34 |
|
CTNNB1-K335I-F2 |
TATACTTACGAAATACTACTGTGGACCACAAGCAGAGT | 38 |
|
CTNNB1-K355I-R2 |
TTACAGGTCAGTATCAAACCAGGCCAGCT | 29 |
| KRAS-qPCR-F |
ACTTGTGGTAGTTGGAGCTGGTGGCGTAGG | 30 |
| KRAS-qPCR-R |
GCACTGTACTCCTCTTGACCTGCTGTGTCG | 30 |
| CTNNB1-qPCR-F |
GGCTTGGAATGAGACTGCTGAT | 22 |
| CTNNB1-qPCR-R |
GCTGATTGCTGTCACCTGGAG | 21 |
| TP53-qPCR-F |
CCGTCTGGGCTTCTTGCATT | 20 |
| TP53-qPCR-R |
CGCCTCACAACCTCCGTCAT | 20 |
| GAPDH-F |
GGTGAAGGTCGGAGTCAACG | 20 |
| GAPDH-R |
CAAAGTTGTCATGGATGHACC | 20 |
The RNA of NCI-H292 lung cancer cells was extracted
and reverse transcribed into cDNA as aforementioned. Using the
site-directed mutagenesis PCR method, TP53 mutation gene was needed
to be amplified by two pairs PCR primers (TP53-F1,R1,F2,R2). The
158th amino acid translated from the TP53 gene was transformed from
arginine (R) to leucine (L) by changing the codon from CGC to CTC.
The steps of plasmid construction were the same as aforementioned,
obtaining the recombinant plasmid TP53-R158L-pIRES2-EGFP.
The RNA of HepG2 liver cancer cells was extracted
and reverse transcribed into cDNA as aforementioned. Using the
site-directed mutagenesis PCR method, CTNNB1 mutation gene was
needed to be amplified by two pairs PCR primers
(CTNNB1-F1,R1,F2,R2). The 335th amino acid translated from the
CTNNB1 gene was transformed from lysine (K) to isoleucine (I) by
changing the codon from AAA to ATA. The steps of plasmid
construction are the same as aforementioned, obtaining the
recombinant plasmid CTNNB1-K335I-pIRES2-EGFP. The PCR primers used
are shown in Table IV.
Sequencing comparison between the KRAS G12V, TP53
R158L, CTNNB1 K335I mutant gene clone and their respective
wild-type gene of KRAS, TP53, CTNNB1, was analyzed through the
tools of Sequence Alignment in the Vector builder analysis system
(https://en.vectorbuilder.com/tool/sequence-alignment.html).
Transfection of tumor cells with recombinant
plasmid
The extracted recombinant plasmids KRAS-G12V-
pIRES2-EGFP, TP53-R158L-pIRES2-EGFP and CTNNB1-K335I-pIRES2-EGFP
(800 ug/ul) were transfected into HCT116 colon cancer cells
(HLA-A2+; 1×106 cells/well), NCI-H292 lung cancer cells
(HLA-A2+; 1×106 cells/well) and HepG2 liver cancer cells
(HLA-A2+; 1×106 cells/well), respectively, using
Lipofectamine® 3000 according to the manufacturer's
protocol. The expression of green fluorescent protein (GFP) was
observed using an inverted fluorescence microscope at 200×
magnification (Olympus IX51; Olympus Corporation) 24 h following
transfection (maximum excitation wavelength, 490 nm), and the
transfection efficiency was detected using flow cytometry. The
positive cells expressing GFP were collected using a flow cell
sorter (MoFlo XDP; Beckman Coulter, Inc.).
Expression of mutated genes in tumor cells using
RT-qPCR
KRAS-G12V-pIRES2-EGFP, TP53-R158L-pIRES2-EGFP and
CTNNB1-K335I-pIRES2-EGFP recombinant plasmids were transfected into
HCT116, NCI-H292 and HepG2 cells, respectively. RNA was extracted
from wild-type and mutant tumor cells expressing recombinant
plasmids and reverse transcribed to cDNA, 24 h following
transfection, as aforementioned. The expression of the mutant genes
in transfected tumor cells was detected using RT-qPCR and the SYBR
Green qPCR mix (primers are shown in Table IV). The following thermocycling
conditions (Roche Diagnostics) were used: Initial denaturation at
95°C for 2 min, followed by 40 cycles of 95°C for 15 sec, 55°C for
30 sec and 72°C for 15 sec. GAPDH was used as the internal control.
The mRNA expression levels of each gene were calculated using the
2−ΔΔCq method (12).
Cytotoxic effect of specific CTLs on tumor cells
using the Cal-AM release assay
The 3 groups of mutant and wild-type tumor cells
were collected into Eppendorf® tubes and incubated with
2 µg/ml Cal-AM for 10 min at 37°C in the dark. The target cells
were seeded into a 96-well plate at a density of 1×105
cells/ml (100 µl/well). The corresponding peptide-induced CTLs were
added for co-culture for 24 h at 37°C (1×105 cells/ml;
100 µl/well). The remaining steps of the Cal-AM release assay used
was as aforementioned. The experimental groups are shown in
Table V.
| Table V.Co-culture of CTLs induced by WT and
mutant tumor cells and peptides. |
Table V.
Co-culture of CTLs induced by WT and
mutant tumor cells and peptides.
| Group | Cancer cells (type)
+ CTLWT | Cancer cells (type)
+ CT mutant |
|---|
| KRAS | HCT116 (WT) +
CTLKRAS WT | HCT116 (WT) +
CTLKRAS mutant |
|
| HCT116 (mutant) +
CTLKRAS WT | HCT116 (mutant) +
CTLKRAS mutant |
| TP53 | NCI-H292 (WT) +
CTLTP53 WT | NCI-H292 (WT) +
CTLTP53 mutant |
|
| NCI-H292 (mutant) +
CTLTP53 WT | NCI-H292 (mutant) +
CTLTP53 mutant |
| CTNNB1 | HepG2 (WT) +
CTLCTNNB1 WT | HepG2 (WT) +
CTLCTNNB1 mutant |
|
| HepG2 (mutant) +
CTLCTNNB1 WT | HepG2 (mutant) +
CTLCTNNB1 mutant |
Cytotoxic effect of specific CTLs on tumor cells
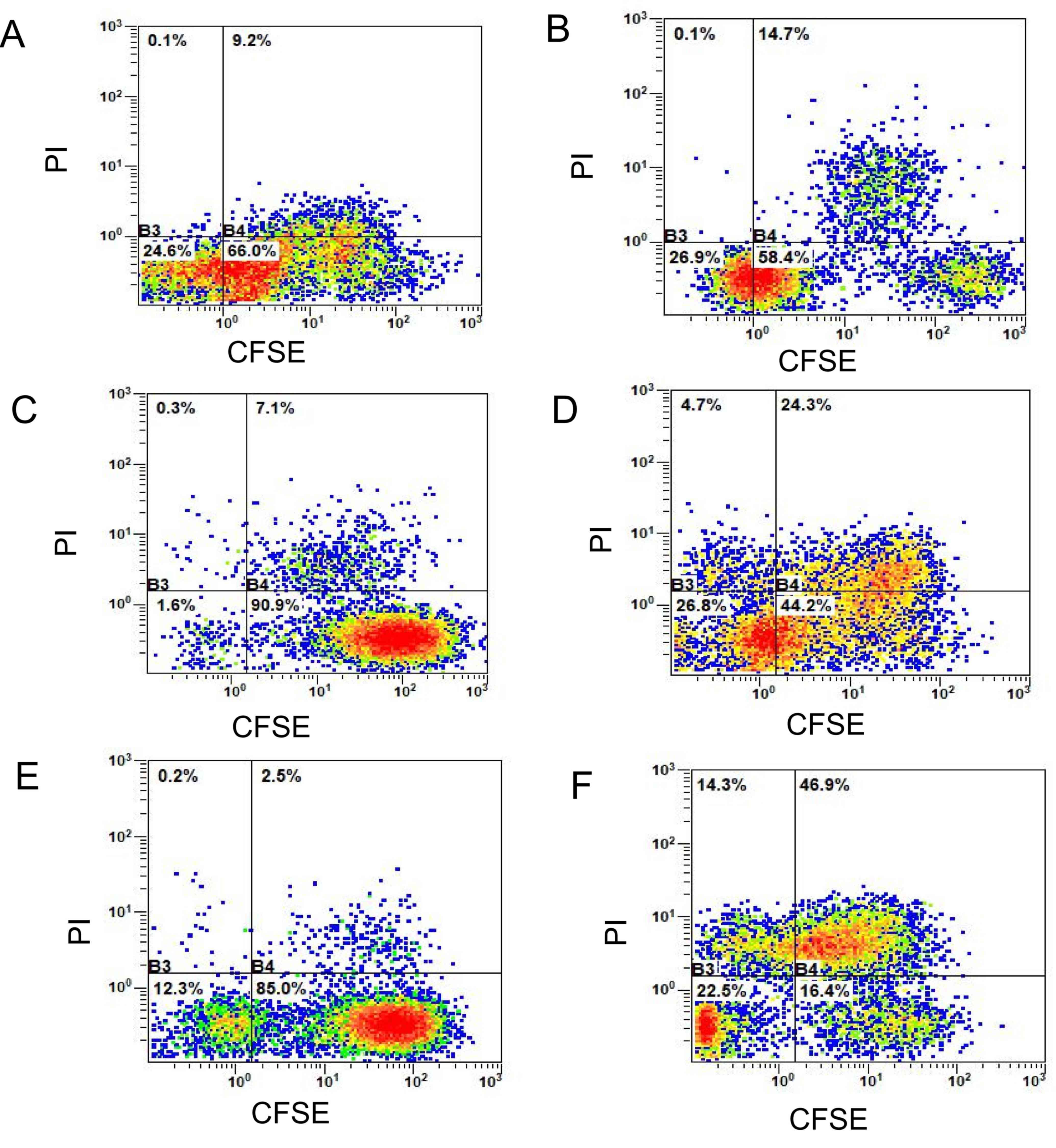
using the CFSE-PI assay
The 3 groups of wild-type and mutant tumor cells
were respectively collected into Eppendorf® tubes,
stained with CFSE fluorescent dye (final concentration, 1 µmol/l),
for 10 min at 37°C in the dark and subsequently washed twice with
PBS. The tumor cells of each group labelled with CFSE dye were
added to a 24-well plate at a density of 1×105 cells/ml
in X–VIVO medium. The mature specific T lymphocytes
(1×105 cells/ml) were co-cultured at 37°C and 5%
CO2. The cells were collected and stained with PI dye
(final concentration, 10 µg/ml, stained at 37°C for 10 min),
following 24 h of culture. The ratio of double-positive cells was
detected using flow cytometry. The experimental groups are shown in
Table VI.
| Table VI.Co-culture of WT and mutant tumor
cells with peptide-induced CTLs. |
Table VI.
Co-culture of WT and mutant tumor
cells with peptide-induced CTLs.
| Group | Peptide (WT) + CTL
(WT) | Peptide (mutant) +
CTL (mutant) |
|---|
| KRAS | HCT116 (WT) +
CTLKRAS WT | HCT116 (mutant) +
CTLKRAS mutant |
| TP53 | NCI-H292 (WT) +
CTLTP53 WT | NCI-H292 (mutant) +
CTLTP53 mutant |
| CTNNB1 | HepG2 (WT) +
CTLCTNNB1 WT | HepG2 (mutant) +
CTLCTNNB1 mutant |
Statistical analysis
All data were expressed as the mean ± SD, which were
repeated in triplicate. Comparisons among multiple groups were
performed using two-way ANOVA and the Bonferroni's method (the post
hoc test) as indicated using the GraphPad Prism software v5.0
(GraphPad Software, Inc.). Student's unpaired t-test was used to
compare the means between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Screening epitope peptides using TCGA
database
The loci of single base substitutions in colon, lung
and liver cancer were screened using TCGA database (Table VII). Based on SYFPEITHI and BIMAS
prediction and evaluation system, three groups of epitope peptides
containing wild-type and mutant gene loci, with increased
immune-presentation capability, were selected and synthesized (9
aa, HLAA201). These included the epitope peptide, YKLVVVGAG,
expressed by the KRAS wild-type gene; YKLVVVGAV, expressed by the
KRAS mutant gene; the VRAMAIYKQ wild-type peptide, expressed by the
TP53 gene; the VLAMAIYKQ mutant peptide, expressed by the
TP53-mutant gene; the IMRTYTYEK wild-type peptide, expressed by the
CTNNB1 gene; and the IMRTYTYEI mutant peptide, expressed by the
CTNNB1-mutant gene (Table
VIII).
| Table VII.Cancer gene mutation sites in The
Cancer Genome Atlas database. |
Table VII.
Cancer gene mutation sites in The
Cancer Genome Atlas database.
| A, Mutations in
liver cancer |
|---|
|
|---|
| Gene | Mutant locus | Mutation frequency
(the number of patient cases) |
|---|
| TP53 | R249S | 11/364 |
| CTNNB1 | S45P | 11/364 |
| CTNNB1 | D32G | 7/364 |
| CTNNB1 | K335I | 6/364 |
| CTNNB1 | S33C | 6/364 |
|
| B, Mutation in
lung cancer |
|
| Gene | Mutant
locus | Mutation
frequency |
|
| KRAS | G12C | 62/1062 |
| KRAS | G12V | 39/1062 |
| EGFR | L858R | 23/1062 |
| PIK3CA | E545K | 21/1062 |
| TP53 | R158L | 20/1062 |
| KRAS | G12D | 20/1062 |
| PIK3CA | E542K | 18/1062 |
|
| C, Mutations in
colon cancer |
|
| Gene | Mutant
locus | Mutation
frequency |
|
| KRAS | G12D | 60/537 |
| KRAS | G12V | 50/537 |
| BRAF | V600E | 50/537 |
| KRAS | G13D | 41/537 |
| TP53 | R175H | 39/537 |
| PIK3CA | E545K | 35/537 |
| Table VIII.Wild-type and mutant epitope
peptides. |
Table VIII.
Wild-type and mutant epitope
peptides.
|
|
|
| Mutant locus | Wild-type epitope
peptide |
|---|
|
|
|
|
|
|
|---|
| Type of cancer | Gene | Mutant epitope
peptide | Sequence | Score | Sequence | Score |
|---|
| Colon | KRAS | G12V |
YKLVVVGAG | 9 |
YKLVVVGAV | 19 |
| Lung | TP53 | R158L | VRAMAIYKQ | 10 | VLAMAIYKQ | 20 |
| Liver | CTNNB1 | K335I |
IMRTYTYEK | 16 |
IMRTYTYEI | 24 |
Affinity of epitope peptides with
HLA-A2 molecule
As shown in Fig. 1,
the HLA-A2 concentration, as expressed by T2 cells treated with the
KRAS mutant peptide (Fig. 1B), was
increased by 5.6±4% compared with that in the cells with wild-type
peptide (Fig. 1A). The expression of
HLA-A2 was 9.6±3% higher in T2 cells with the TP53 mutant peptides
(Fig. 1D) compared with that in the
cells with the wild-type peptides (Fig.
1C). The expression of HLA-A2 was significantly increased by
30.4±2% in T2 cells loaded with CTNNB1 mutant peptides (Fig. 1F) compared with that with wild-type
peptides (Fig. 1E). Notably, the
fluorescence index of the CTNNB1 group increased the most
(P<0.001; Fig. 2). The present
results indicated that the mutant peptides promoted the higher
expression of the HLA-A2 molecule in T2 cells, which formed a
compound with high affinity and strong stability with MHC I
molecules. The affinity observed in the TP53 mutant epitope
peptides was weakest (fluorescence index <0.5), with the weakest
occurring in the KRAS group (Fig.
2).
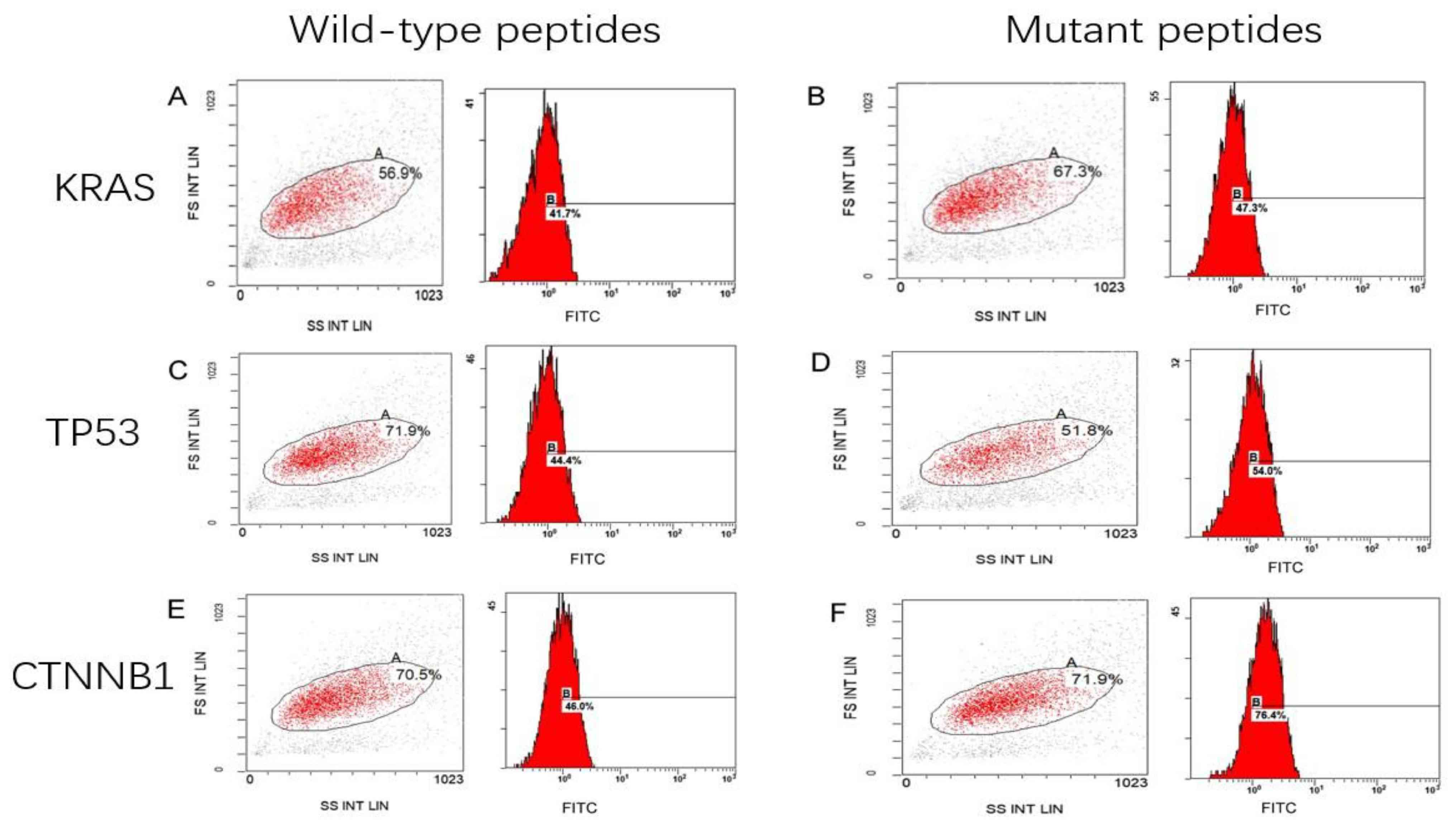 | Figure 1.Mean fluorescence intensity of
peptide-treated T2 cells. T2 cells treated with (A) KRAS wild-type
peptide (41.7%), (B) KRAS mutant peptide (47.3%), (C) TP53
wild-type peptide (44.4%), (D) TP53 mutant peptide (54.0%), (E)
CTNNB1 wild peptide (46.0%) and (F) CTNNB1 mutant peptide (76.4%).
KRAS, KRAS proto-oncogene; TP53, tumor protein 53; CTNNB1, catenin
β1; FS, forward scatter; SS, side scatter; INT, intensity; LIN,
linear; FITC, fluorescein isothiocyanate. |
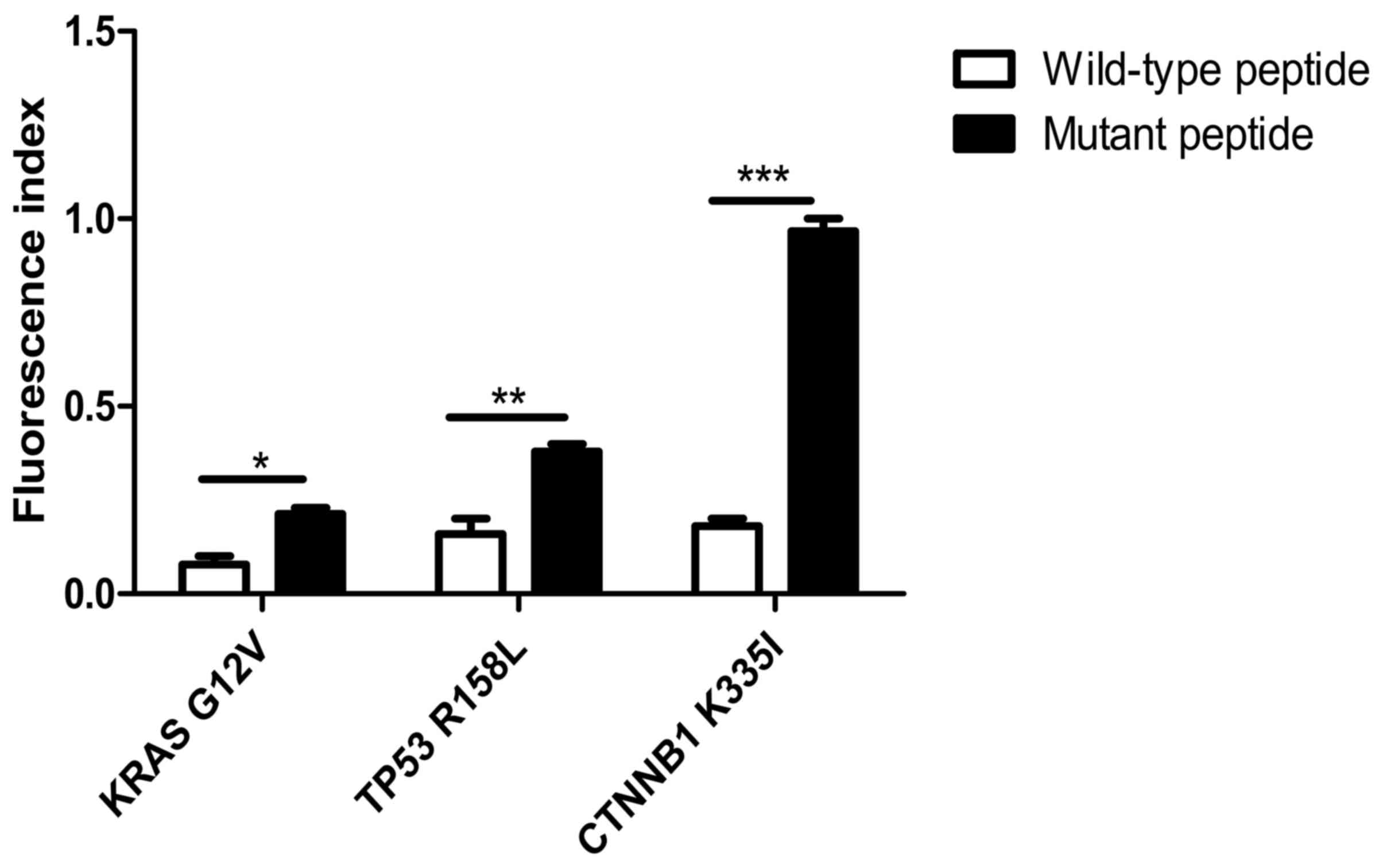 | Figure 2.FI in peptide-treated T2 cells. The
FI in T2 cells treated with the KRAS mutant peptide increased by
5.6±4% compared with that in cells treated with the wild-type
peptide. The FI in T2 cells loaded with TP53 mutant peptides was
9.6±3% higher compared with that in cells treated with the
wild-type peptides. The FI in T2 cells treated with CTNNB1 mutant
peptides was significantly increased by 30.4±2% compared with that
in cells loaded with the wild-type peptides. *P<0.05;
**P<0.01; ***P<0.001. FI, fluorescence index; KRAS, KRAS
proto-oncogene; TP53, tumor protein 53; CTNNB1, catenin β1; G,
glycine; V, valine; R, arginine; L, leucine K, lysine; I,
isoleucine. |
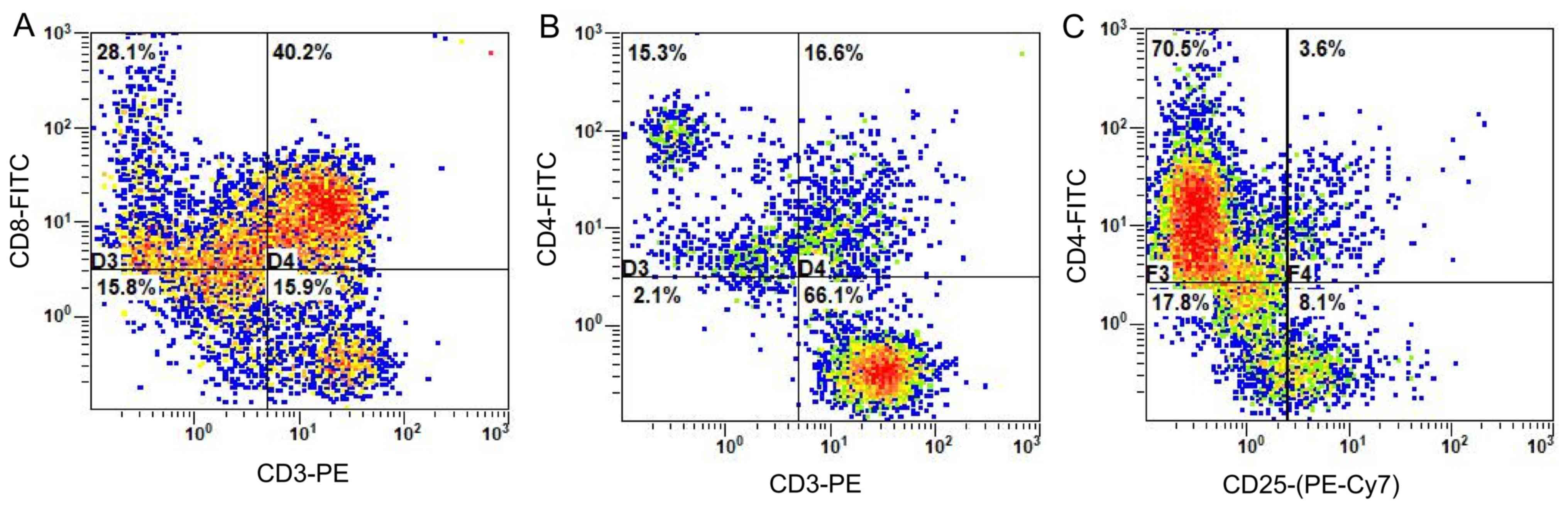
Flow cytometry for phenotypic
detection of CTLs
CTLs were cultured for 21 days before being
collected and the surface antigens were detected using flow
cytometry. As shown in Fig. 3, the
proportion of CD8+ T cells (CD3+ CD8+T) among
the lymphocytes was 40.2% (Fig. 3A),
whereas T helper cells (CD3+ CD4+T) accounted for 16.6%
of lymphocytes (Fig. 3B), while 3.6%
were comprised of regulatory T cells
(CD4+CD25+T) (Fig.
3C).
Proliferation of specific CTLs
Following CFSE staining and flow detection of CTLs
cultured in vitro for 14 days, the Modfit software was used
to analyze the results and determine the proliferation index of T
lymphocytes. Between the 16 and the 19th day, the proliferation
index of T cells increased gradually from 1.45 to 15.13, 18.55 and
18.86, respectively (Fig. 4). This
indicated that T lymphocytes were in a state of proliferation and
division. However, as the cells were in the third stage of
differentiation (Fig. 4C and D for
days 16–19), induced by antigenic peptides, the growth and
proliferation rate of the cells tended to be flat (Fig. 4).
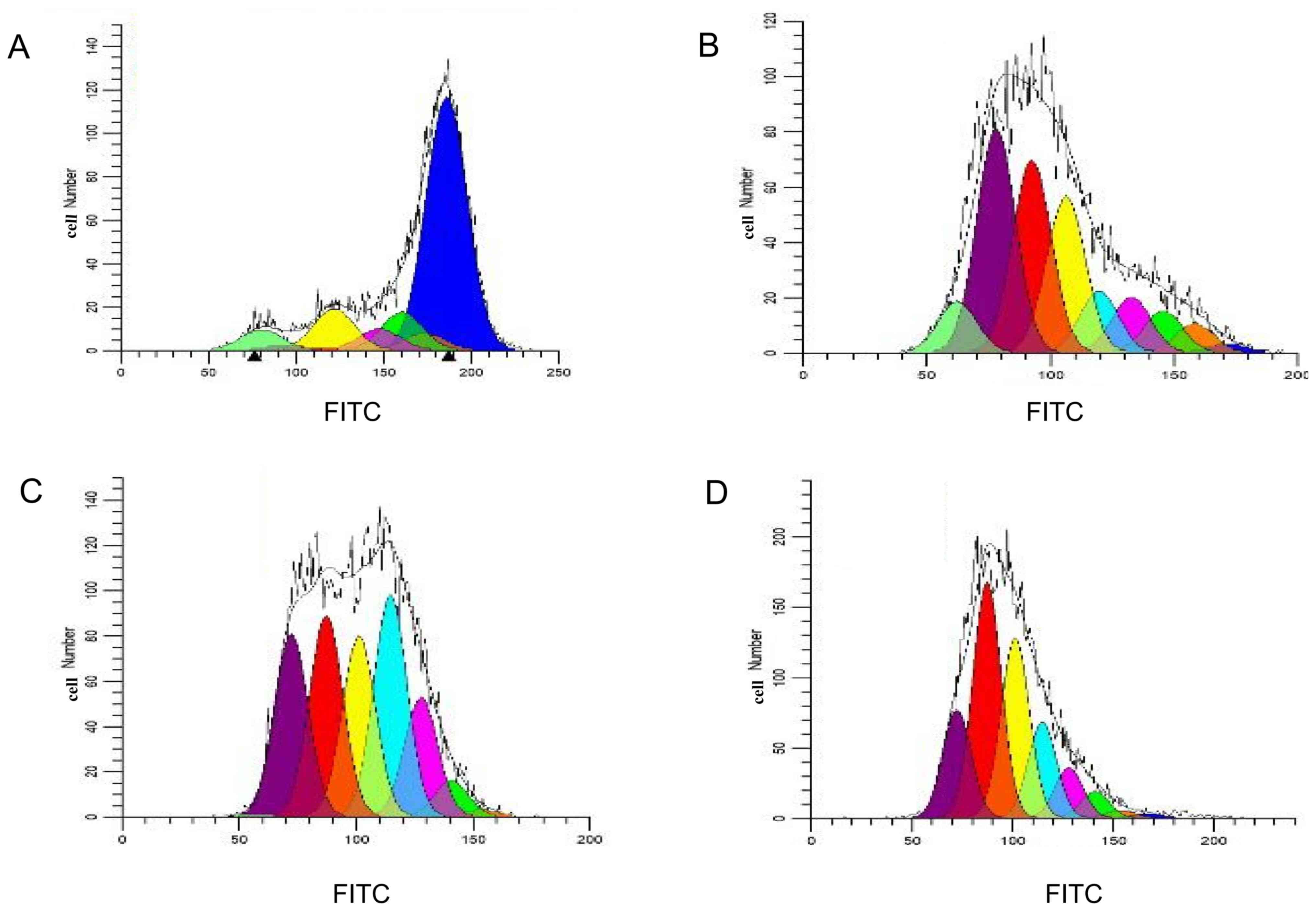 | Figure 4.Modfit analysis of cell proliferation
and division. Cell proliferation and division on (A) the 1st, (B)
the 2nd, (C) the 3rd and (D) the 4th day following
carboxyfluorescein succinimidyl ester staining. Blue, the parental
cell; orange, generation 2; green, generation 3; pink, generation
4; light blue, generation 5; yellow, generation 6; red, generation
7; purple, generation 8; and laurel-green, generation 9. FITC,
fluorescein isothiocyanate. |
IFN-γ secretion of specific T cells
stimulated by epitope peptides in vitro
As shown in Fig. 5,
the secretion of IFN-γ from CTLs, induced by mutant peptides of the
KRAS, TP53 and CTNNB1 groups, was 160±10, 174±5 and 180±6 ng/ml,
respectively. The IFN-γ secretion of specific CTLs induced by
mutant peptides in the three groups was higher compared with that
in the CTLs induced by the wild-type peptides, and the difference
was determined statistically significant using the Bonferroni's
test (the post hoc test following an ANOVA, P<0.05). Notably,
the results indirectly indicated that the mutant antigenic peptides
of the three groups improved the cellular immune function of
specific CTLs, especially in the CTNNB1 group.
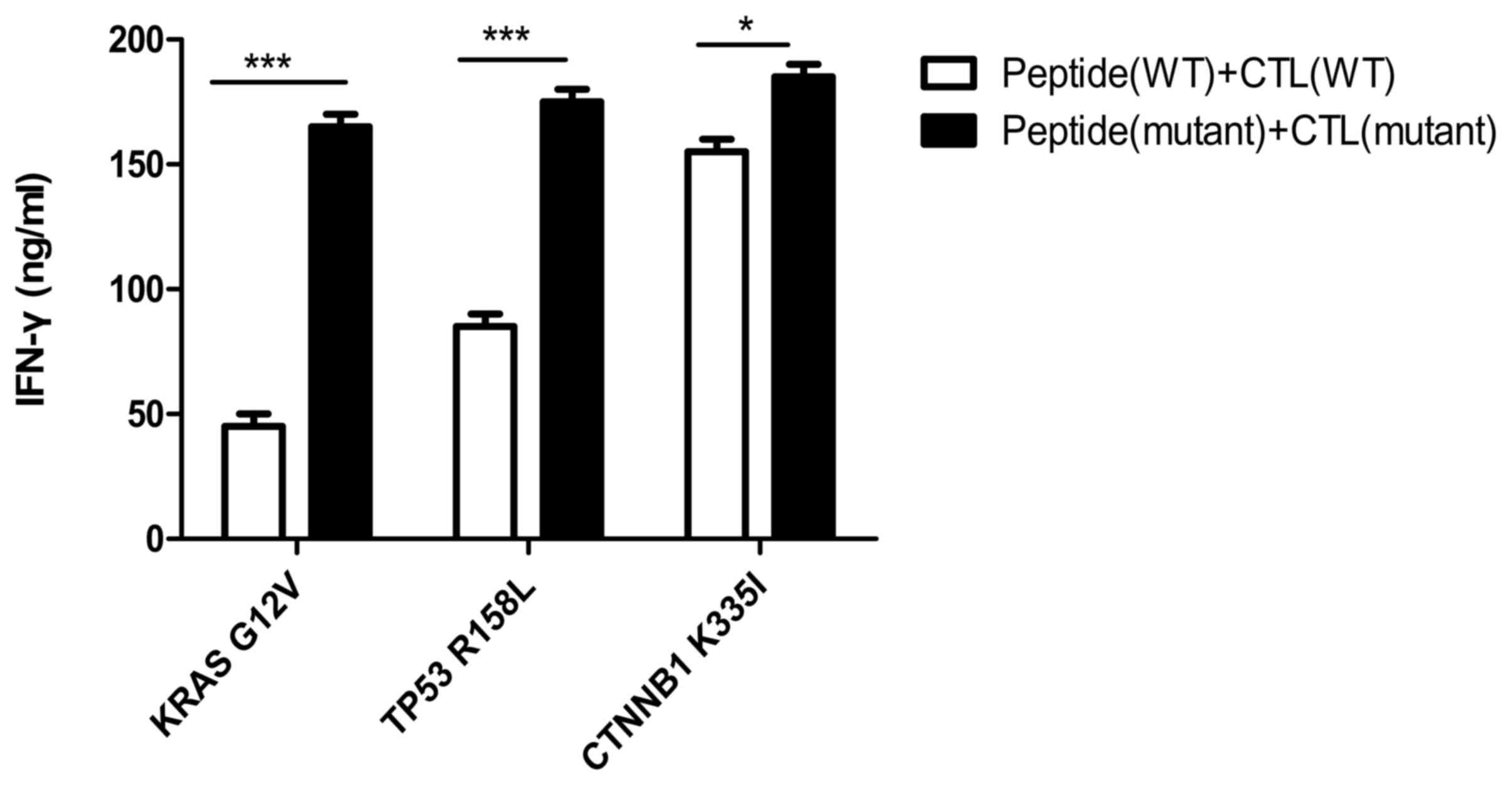 | Figure 5.IFN-γ secretion of specific T cells
stimulated by epitope peptides. IFN-γ secretion of CTLs induced by
mutant peptides in the KRAS, TP53 and CTNNB1 groups, was 160±10,
174±5 and 180±6 ng/ml, respectively. Experiments were repeated
three times. Date are presented as the mean ± SD. *P<0.05;
***P<0.001. IFN, interferon; CTL, cytotoxic T lymphocyte; KRAS,
KRAS proto-oncogene; TP53, tumor protein 53; CTNNB1, catenin β1;
WT, wild-type; G, glycine; V, valine; R, arginine; L, leucine K,
lysine; I, isoleucine. |
Preliminary detection of cytotoxic
activity in specific CTLs against T2 cells with added peptide
As shown in Fig. 6A,
the killing rates of CTLs induced by mutant peptides in the KRAS,
TP53 and CTNNB1 groups were 35±3, 48±2 and 50±2%, respectively. The
target cell killing rate of mutant peptide-specific CTLs in the
CTNNB1 group was higher compared with that in the other two groups.
However, compared with wild-type peptides, the increased killing
rate of CTLs induced by the mutant peptides in vitro was the
highest in the TP53 group, and the difference was determined to be
statistically significant using Bonferroni's post hoc test
following an ANOVA (P<0.01). It was initially determined that,
compared with wild-type peptides, the induction of specific CTLs by
the three groups of mutant peptides improved their affinity for
target cells.
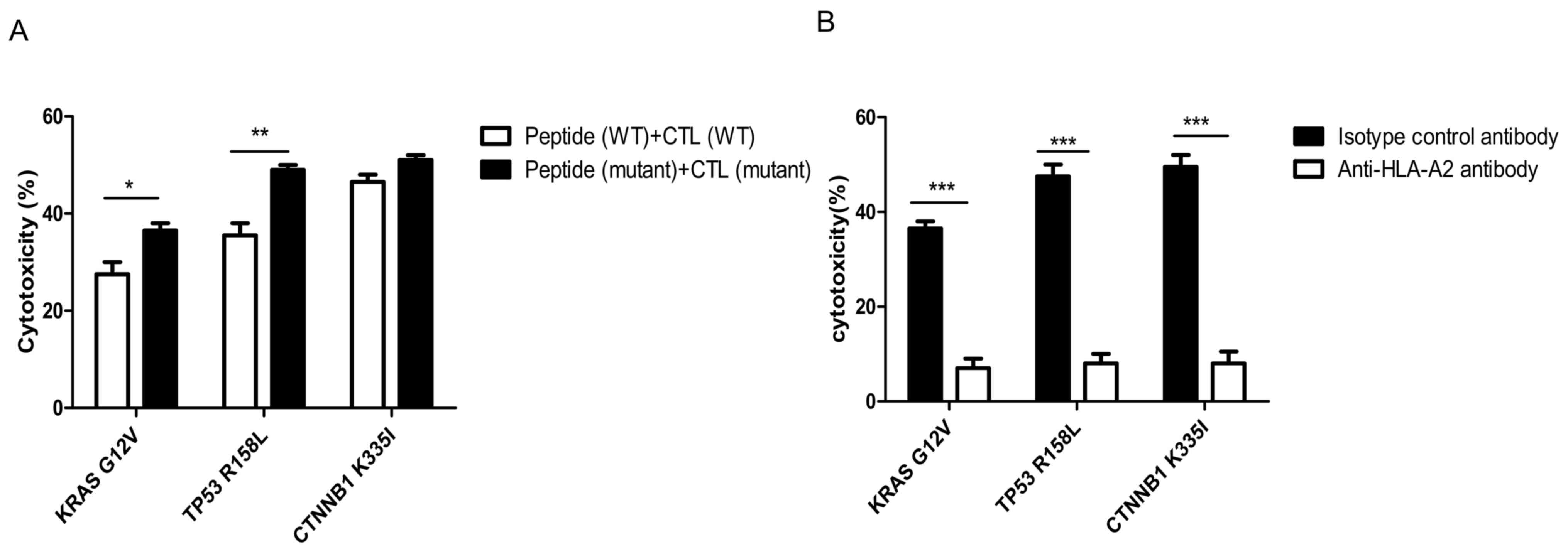 | Figure 6.Cytotoxicity of specific CTLs
simulated by antigenic peptides. (A) Cytotoxicity of specific CTLs
to T2 cells treated with peptides detected using the
calcein-acetoxymethyl release assay. The killing rates of CTLs
induced by mutated peptides in the KRAS, TP53 and CTNNB1 groups
were 35±3, 48±2 and 50±2%, respectively. The amplified cytotoxic
function of CTLs induced by mutant peptides in vitro was the
largest in the TP53 group. (B) HLA-A2 antibody blocking assay. The
difference between mutant peptide-loaded T2 cells treated with
isotype control antibody and anti-HLA-A2 antibody in each group was
statistically significant. Experiments were repeated three times.
Date are presented as the mean ± SD. *P<0.05; **P<0.01;
***P<0.001. CTL, cytotoxic T lymphocyte; KRAS, KRAS
proto-oncogene; TP53, tumor protein 53; CTNNB1, catenin β1; WT,
wild-type; HLA, human leukocyte antigen; G, glycine; V, valine; R,
arginine; L, leucine K, lysine; I, isoleucine. |
The specificity of CTLs towards the mutant
peptide-treated T2 cells in each group was significantly decreased
in the cells treated with anti-HLA-A2 antibody compared with that
in the isotype control antibody (P<0.001; Fig. 6B).
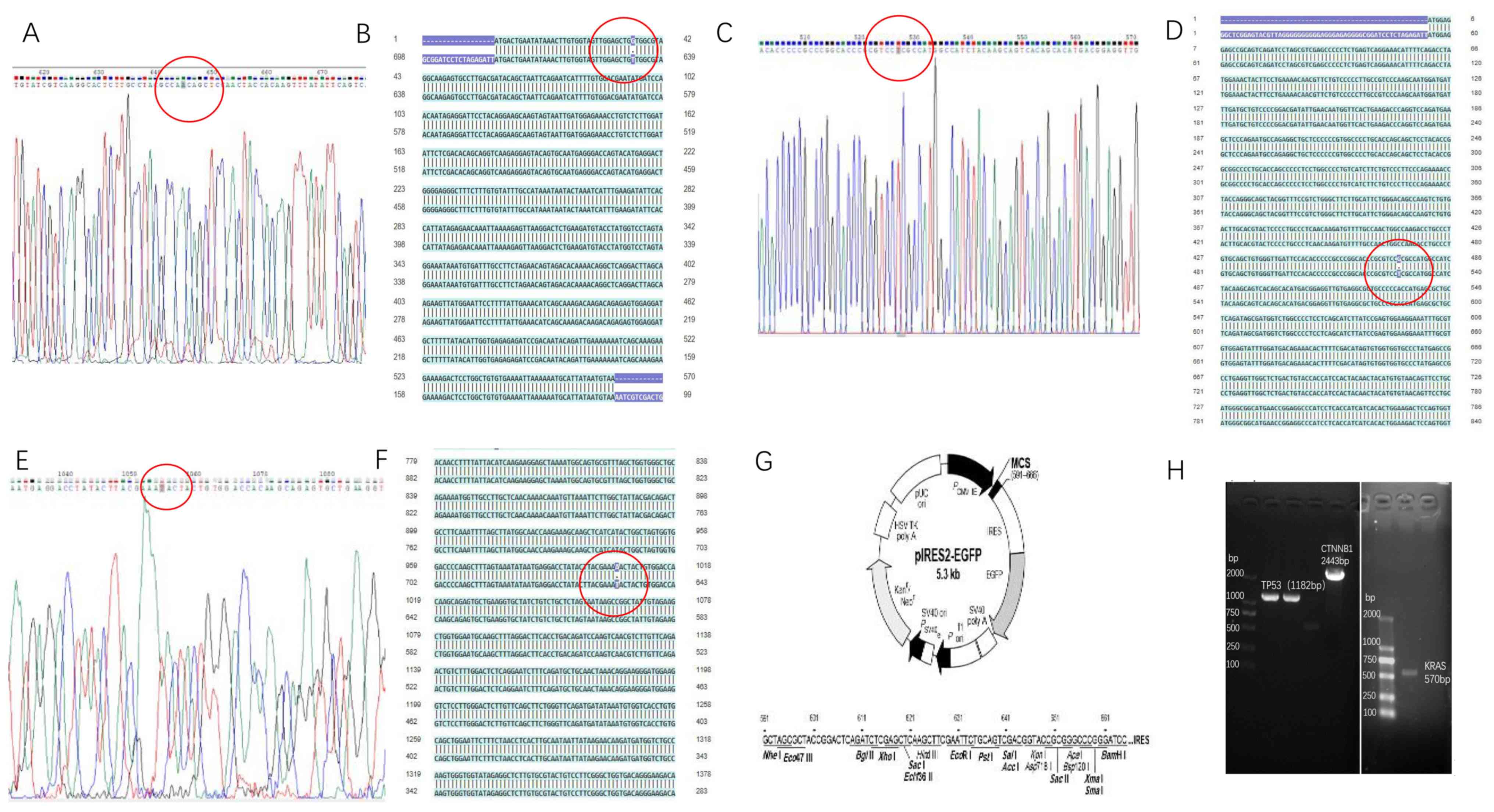
Construction of recombinant eukaryotic
expression plasmids
Using the site-directed mutagenesis PCR method, PCR
products were determined to contain the KRAS G12V, TP53 R158L and
CTNNB1 K335I mutated genes using agarose gel electrophoresis,
yielding sizes of 570, 1,182 and 2,443 bp, respectively. The
electrophoresis bands were consistent with the expected sizes of
the target fragments (Fig. 7H).
Following sequencing of the mutant genetically
engineered bacteria (about 5 clones were used for the sequencing of
the mutant genes), the recombinant plasmid of the site-directed
mutant KRAS G12V-pIRES2-EGFP was compared with that for the
wild-type in the HCT116 cell line (Fig.
7A). The 12th amino acid encoding the KRAS protein was changed
from G to V by substituting the codon from GGT to GTT, while none
of the other bases were mutated in the KRAS gene (Fig. 7B). The TP53 R158L (Fig. 7C and D) and CTNNB1 K335I (Fig. 7E and F) mutant gene clones were also
successfully constructed in the pIRES2-EGFP plasmid (Fig. 7G).
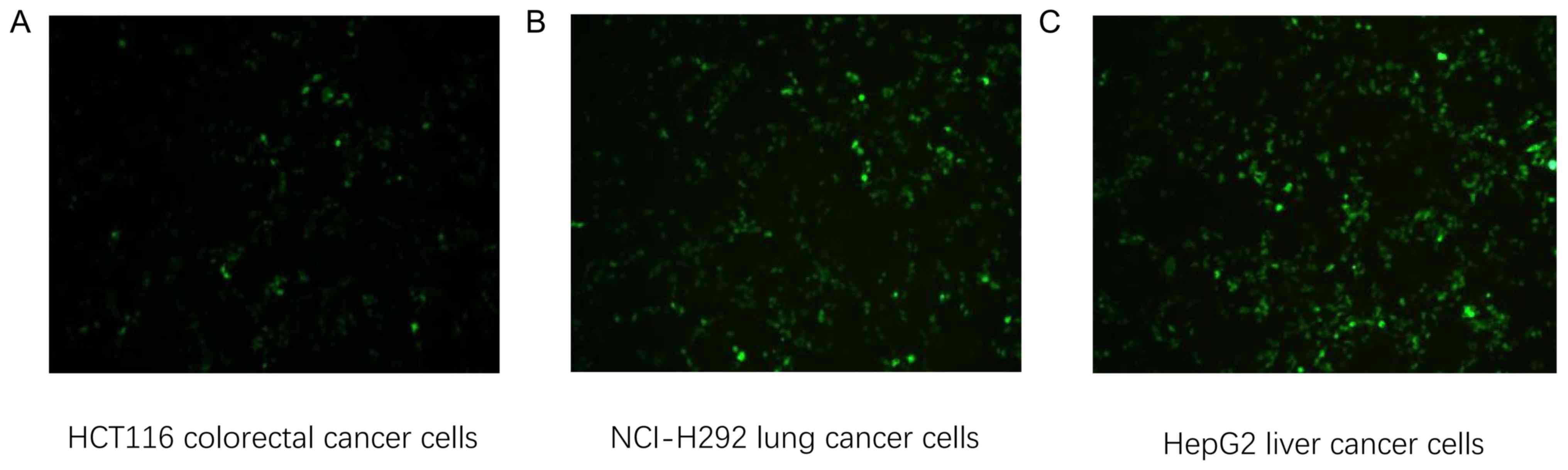
Transfection of tumor cells with
recombinant plasmid and detection of transfer efficiency
HCT116 colorectal (HLA-A2+), NCI-H292 lung (HLA-A2+)
and HepG2 liver cancer cells (HLA-A2+) were transfected with KRAS
G12V-EGFP-pIRES2, TP53 R158L-pIRES2-EGFP and CTNNB1
K335I-pIRES2-EGFP recombinant plasmids, respectively. After 24 h,
GFP expression was confirmed in all three cell lines using an
inverted fluorescence microscope, 24 h following transfection
(Fig. 8).
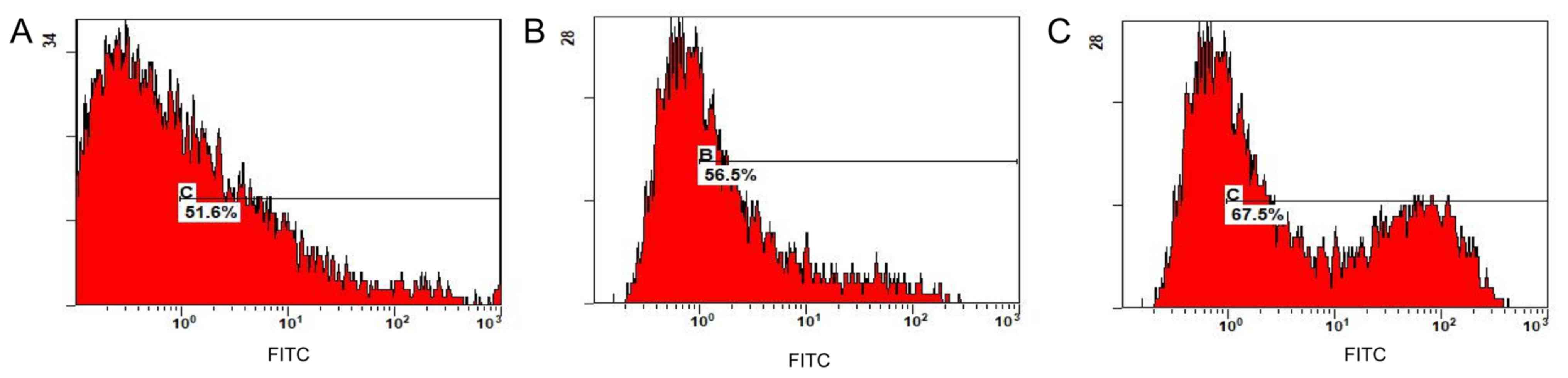
Flow cytometry revealed that the transfer efficiency
of the KRAS G12V-pIRES2-EGFP, TP53 R158L-pIRES2-EGFP and CTNNB1
K335I-pIRES2-EGFP recombinant plasmids was 51.6, 56.5 and 67.5%,
respectively (Fig. 9). The positive
cells expressing GFP were separated and collected using a flow cell
sorter for follow-up experiments.
Expression of mutant genes in tumor
cells
Following transfection of the recombinant plasmids,
KRAS G12V-pIRES2-EGFP, TP53 R158L-pIRES2-EGFP and CTNNB1
K335I-pIRES2-EGFP into HCT116, NCI-H292 and HepG2 cells,
respectively, RT-qPCR results indicated that the mutant gene was
overexpressed in the transfected tumor cells compared with that in
the wild-type cells (P<0.001; Fig.
10).
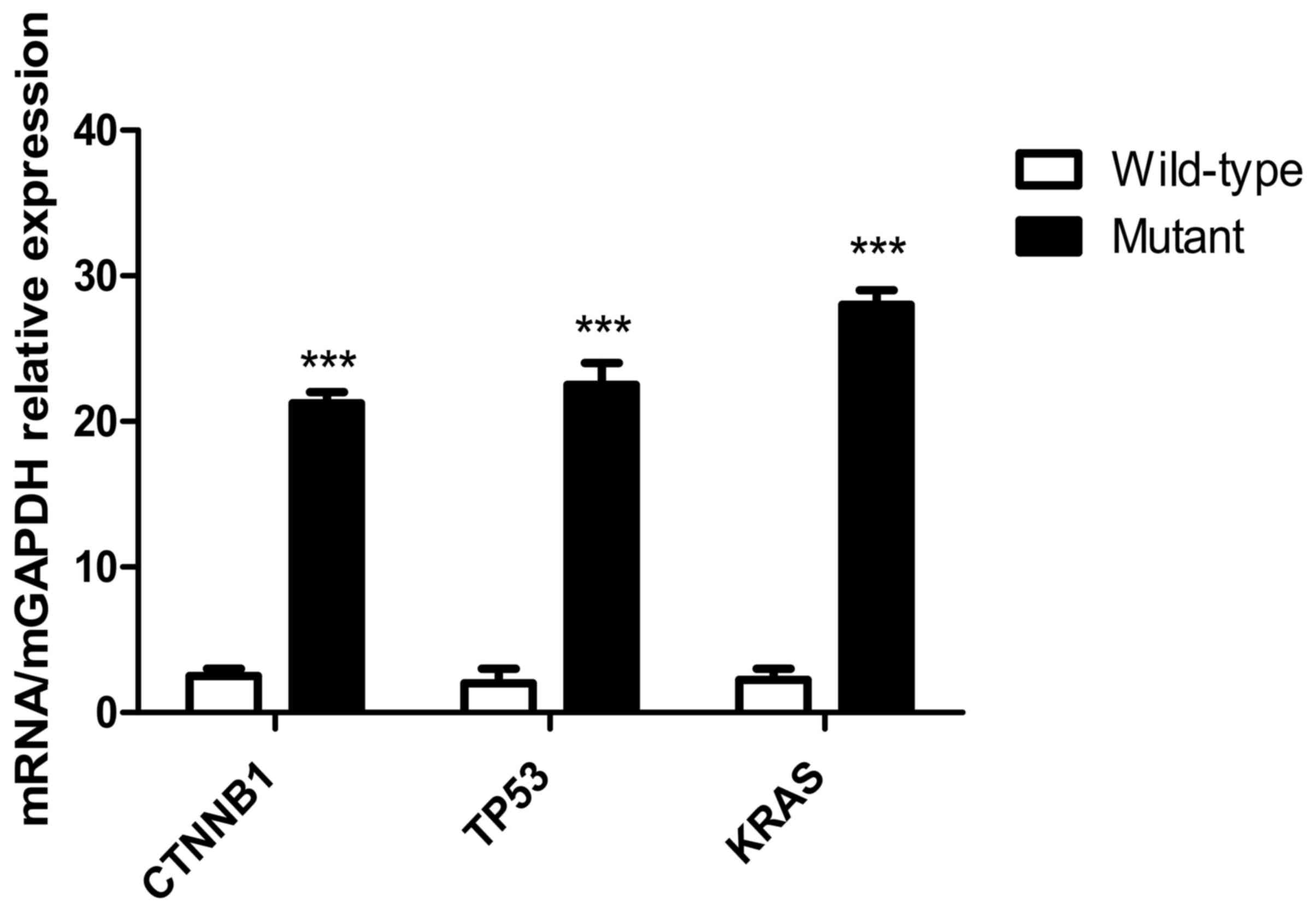 | Figure 10.Expression levels of mutant genes in
tumor cells detected using RT-qPCR. Expression levels of mutant
genes in the CTNNB1, TP53 and KRAS groups. The results of the
RT-qPCR indicated that following transfection of KRAS
G12V-pIRES2-EGFP, TP53 R158L-pIRES2-EGFP and CTNNB1
K335I-pIRES2-EGFP recombinant plasmids into HCT116, NCI-H292 and
HepG2 tumor cells, respectively. The KRAS, TP53, CTNNB1 mutant gene
was overexpressed in the transfected tumor cells compared with that
in the wild-type cells. ***P<0.001. RT-qPCR, reverse
transcription-quantitative PCR; KRAS, KRAS proto-oncogene; TP53,
tumor protein 53; CTNNB1, catenin β1. |
Cytotoxicity of peptide-induced
specific CTLs on wild-type and mutant tumor cells
In the KRAS, TP53 and CTNNB1 groups, the killing
rates of mutant peptide-induced specific CTLs towards mutant tumor
cells [cancer cells (mutant) + CTLs (mutant)] were 30±3, 32±4 and
42±3%, respectively (Fig. 11). The
killing rates of CTLs induced by mutant peptides in the three
groups on wild-type and mutant tumor cells were similar. However,
the cytotoxic specificity of wild-type peptide-induced CTLs to both
wild-type and mutant tumor cells was low. In addition, the killing
rates of specific CTLs induced by mutant peptides in the KRAS, TP53
and CTNNB1 groups were significantly higher compared with those
induced by wild-type peptides. The difference was statistically
significant between the cancer cell (WT)+CTL (WT) and cancer cell
(mutant)+CTL (mutant) groups (P<0.0001; Fig. 11). The target cell killing rate of
CTLs induced by CTNNB1 mutant peptides was higher compared with
that in the KRAS and TP53 groups, and the results were consistent
with the cytotoxicity of CTLs against T2 cells treated with
peptides (Fig. 6A). Furthermore, the
specificity and cytotoxicity of CTLs induced by mutant peptides in
the CTNNB1 group were enhanced, when compared with those induced by
wild-type peptides.
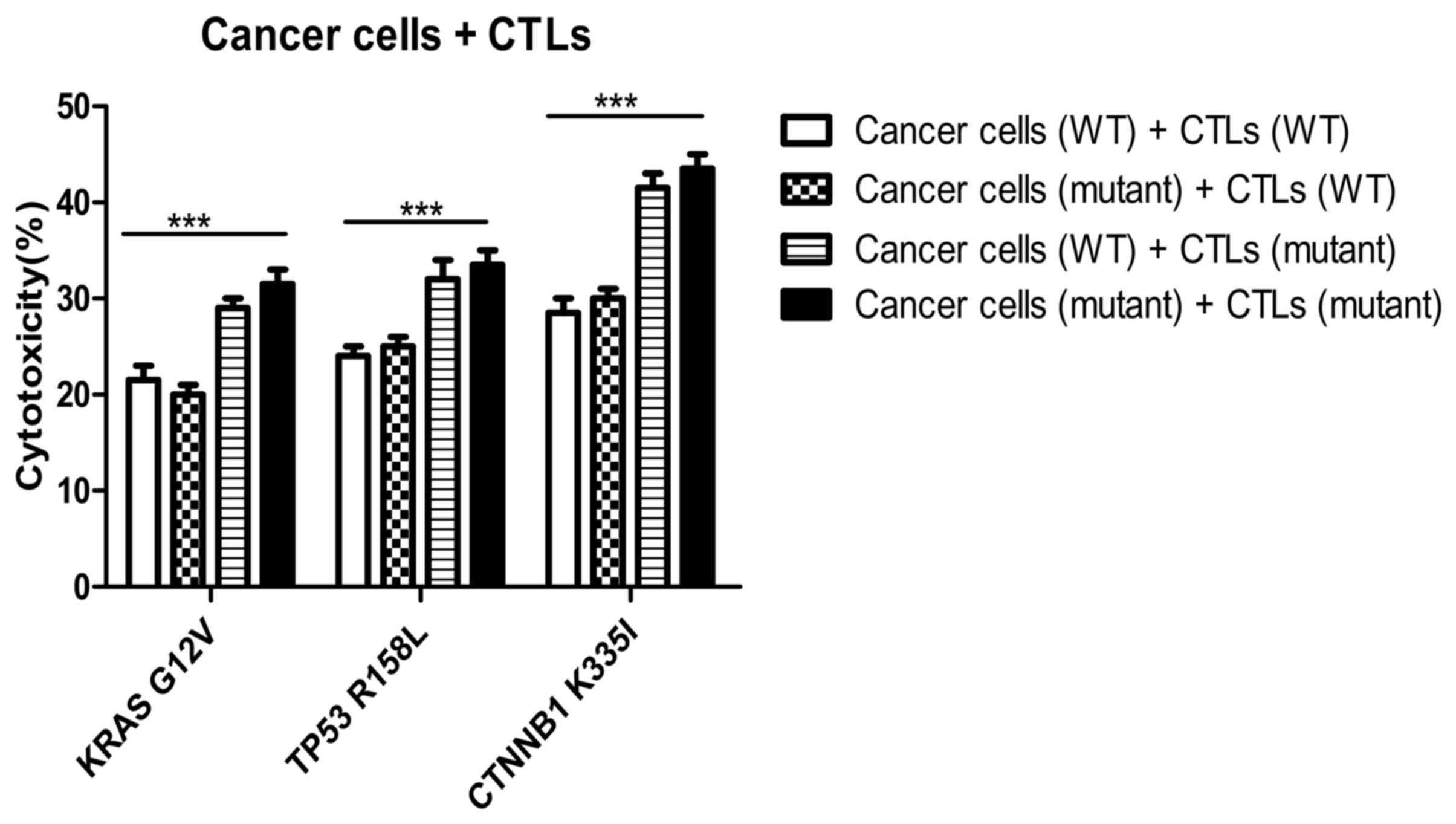 | Figure 11.Cytotoxicity of specific CTLs on
wild-type and mutant tumor cells. The killing rates of mutant
peptide-induced specific CTLs to mutant tumor cells were 30±3, 32±4
and 42±3% in the KRAS, TP53 and CTNNB1 groups, respectively. The
killing rates of specific CTLs induced by mutant peptides in the
KRAS, TP53, and CTNNB1 groups were higher compared with those
induced by WT peptides. ***P<0.0001. KRAS, KRAS proto-oncogene;
TP53, tumor protein 53; CTNNB1, catenin β1; CTL, cytotoxic T
lymphocyte; WT, wild-type; G, glycine; V, valine; R, arginine; L,
leucine K, lysine; I, isoleucine. |
Detection of cytotoxicity of specific
CTLs on tumor cells using CFSE-PI staining
The double-positive rate
(CFSE+PI+, the first quadrant) of mutant
peptide-induced CTLs in the three groups (Fig. 12) was significantly higher compared
with that in the wild-type peptide group, and this difference was
statistically significant according to the Bonferroni post hoc test
following an ANOVA. (P<0.0001; Fig.
13).
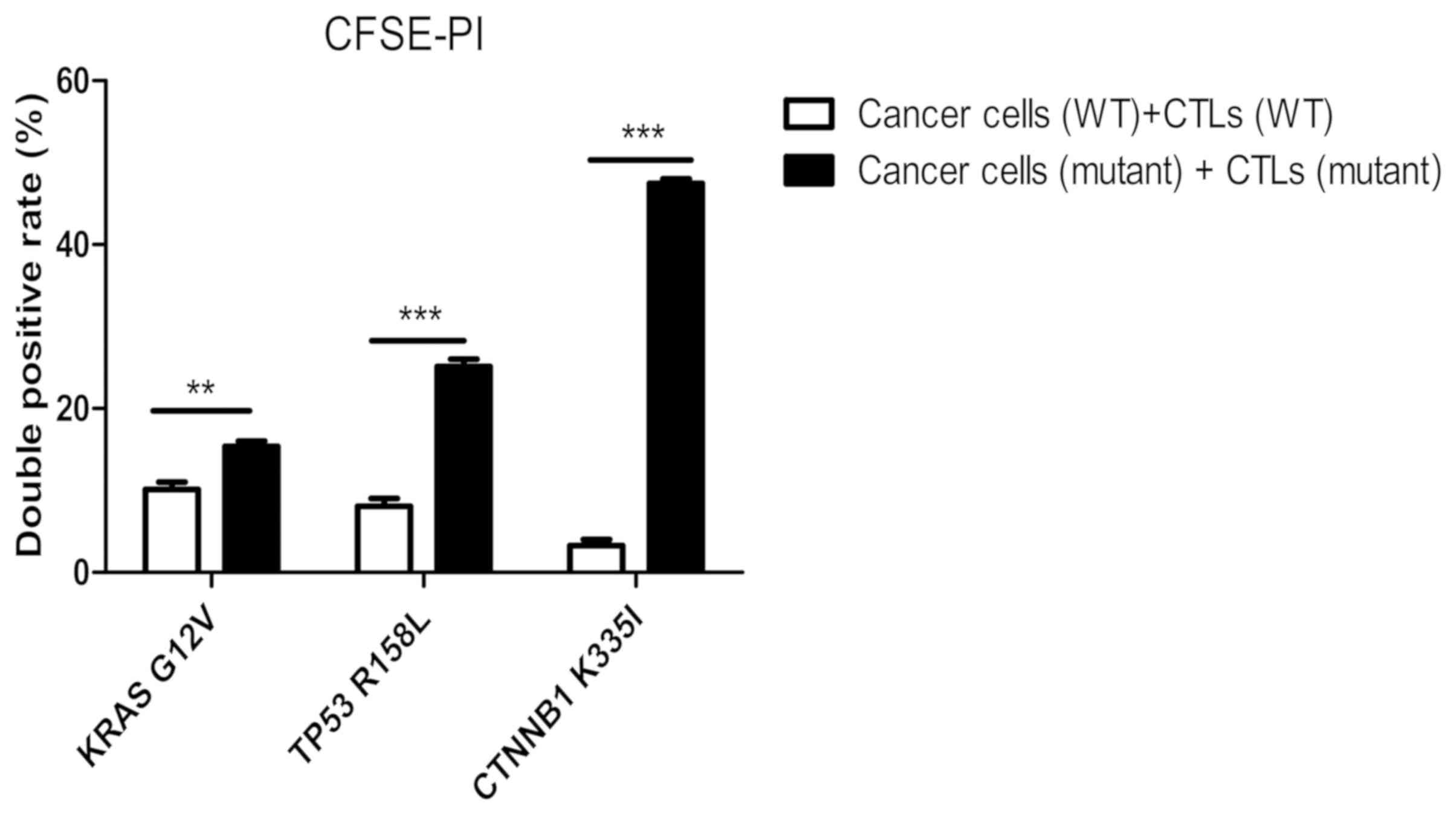 | Figure 13.Detection of the killing rate of
specific CTLs on wild-type and mutant tumor cells using CFSE-PI
staining. The killing rate of the mutant peptide-induced CTLs on
mutant target tumor cells in the KRAS, TP53 and the CTNNB1 groups
increased by 5.5±3, 17.2±4 and 44.4±2.5%, respectively compared
with that in their respective WT peptide-induced specific CTLs in
the wild-type tumor cells. The differences were statistically
significant according to the Bonferroni's post hoc test following
an ANOVA. **P<0.01; ***P<0.001. CFSE, carboxyfluorescein
succinimidyl ester; PI, propidium iodide; KRAS, KRAS
proto-oncogene; TP53, tumor protein 53; CTNNB1, catenin β1; CTL,
cytotoxic T lymphocyte; WT, wild-type; G, glycine; V, valine; R,
arginine; L, leucine K, lysine; I, isoleucine. |
When compared with the wild-type peptide-induced
specific CTLs, the killing rate of mutant peptide-induced target
cells in the KRAS group increased by 5.5±3.0% (Fig. 12A and B), whereas in the TP53 group
it increased by 17.2±4.0% (Fig. 12C and
D) and in the CTNNB1 group by 44.4±2.5% (Fig. 12E and F). The killing rates of
target cells induced by mutant peptides in the CTNNB1 group were
the highest, followed by those in the TP53 and KRAS groups
(Figs. 12 and 13). The results were consistent with those
of Cal-AM detection (Fig. 11),
indicating that the CTLs induced by mutant peptides of the CTNNB1
group had a stronger cytotoxic effect on tumor cells compared with
those induced by mutant peptides of the TP53 and KRAS groups.
Discussion
Tumorigenesis has been associated with gene
mutations, such as gene fusion, point and deletion mutations
(9). When genes encoding the normal
proteins are mutated, it may lead to abnormal functions within the
cell, such as exponential growth, proliferation and metastasis, as
well as evading the immune system (10). TAAs are primarily derived from
autoantigens, which are highly expressed by tumor cells, however
these are expressed in low amounts in healthy somatic cells
(13). Therefore, it is difficult to
activate the low affinity, naive T cells involved in immune
tolerance. During T-cell development in thymus and positive and
negative selection, progenitor T cells will eliminate immature T
cells with high affinity to autoantigens, and the remaining cells
will become naive T cells with a low affinity to autoantigens to
develop and mature (14). Therefore,
it is difficult to activate naive T cells with low affinity
involved in immune tolerance, and immunotherapy targeting TAAs
cannot achieve the ideal curative effect, such as tumor regression.
However, new tumor antigens, also known as neoantigens or TSAs, are
epitope-specific antigens produced on the cell surface due to
mutations within cancer cells (15).
These antigenic peptides are tumor-specific and can therefore be
recognized by T lymphocytes, directly inducing an immune response
(15). Furthermore, TSAs are only
expressed on the tumor cells and not on healthy cells. The binding
affinity of the T-cell receptor (TCR) to T cells is much higher
compared with that of TAA (16). T
cell activation and cytotoxicity have been associated with the
affinity of TCR-MHC antigenic peptide complexes, therefore TSAs
have the potential to be effective targets for tumor immunotherapy
(17).
During the cellular immune response, mutant peptides
of tumor cells can be presented on the surface of cells by MHC I
molecules, which are then recognized by the TCR of T cells
(18). However, the majority of
tumor cells evade recognition by the immune system, as they can
downregulate or delete the expression of MHC I molecules (18). Therefore, at present, polypeptide
synthesis has been used to produce mutated peptides of tumor cells
in vitro, which are then introduced into the human body
through individualized vaccines or ACT to stimulate the
proliferation of specific T cells that recognize these neoantigens
in vivo, thus specifically killing tumor cells (19).
TCGA database contains gene expression profiling,
somatic mutations, copy number variation and DNA methylation data
which has been produced by sequencing tumors and paracancerous
tissues for >30 types of cancer (20). In the present study, point mutation
gene loci of high-frequency mutations of colorectal, lung and liver
cancer were identified from TCGA. A total of 3 short epitope
peptides with high affinity to HLA-A201 molecules were screened
using a peptide prediction algorithm and directed at these new
tumor antigens. The tumor antigens of cancer cells were recognized
by the immune system through the epitope peptides. The activation
effect of the epitope peptide on T cells was analyzed using
immunogenicity tests of specific CTLs in vitro, to identify
the epitope peptides which could activate specific T cells to
recognize tumor antigens, which may be used in future immunotherapy
studies (21).
In the present study, KRAS G12V, TP53 R158L and
CTNNB1 K335I gene point mutation loci were identified in
colorectal, lung and liver cancer to design antigenic peptides,
which were used to induce CTL proliferation. CD8+ T
cells accounted for about 40% of all lymphocytes after 21 days.
From the immunogenicity experiments of antigen epitope peptides
containing point mutations, it was demonstrated that specific T
cells induced by mutant antigen peptides in the CTNNB1 group had
the strongest cytotoxic affinity towards mutant HepG2 liver cancer
cells. The cytotoxic effects of the TP53 mutant antigen
peptide-induced CTLs were weaker compared with those in the CTNNB1
group, and with the TP53 wild-type antigen peptide; however, the
immunogenicity was markedly increased, and TP53 mutant
peptideinduced specific T cells, which could target the tumor cells
with this point mutation more effectively. The immunogenicity of
the mutant antigenic peptide in the KRAS group was lower compared
with that in the TP53 and CTNNB1 groups; however, the
cytotoxiceffect of specific CTLs on HCT116 colorectal cancer cells
was slightly improved. The present results suggest that point
mutations in the antigenic peptides targeting tumor neoantigens may
improve the stability of binding to the MHC molecules, enhancing
immunogenicity and activating CD8+ T cells effectively.
The cytotoxiceffect of CTLs induced by mutant peptides in the three
groups was similar in both wild-type and mutant tumor cells, which
may be associated with the cross-recognition effect of T cells. As
there was only one amino acid difference between the mutant and
wild-type peptides, there were two types of antigenic peptide-MHC
complexes that were recognized by specific, effective
CD8+ T cells (22). The
present results indicate that specific CTLs, induced by mutant
peptides, can kill both mutant and wild-type tumor cells. When the
anti-HLA-A2 antibody was added, the cytotoxic effect of specific
CTLs in the three groups was significantly reduced, indicating that
the cytotoxic effect was performed in an HLA-A2-dependent manner.
In summary, all three groups of the epitope peptides with higher
affinity to MHC molecules may induce specific CTLs against the
point mutant antigens and enhance the immune response. Among these
peptides, the point mutant antigenic peptide in the CTNNB1 group
demonstrated a potent ability to induce T-cell proliferation,
activation, specific recognition and apoptosis of tumor cells,
which may explain why the affinity of mutant peptides for MHC
molecules in the CTNNB1 group was higher compared with that in the
other two groups. In addition, the TCR gene family should be
further analyzed in future studies. Currently, an ideal and
effective TCR molecule is a key focus of research into TCR-T
immunotherapy. Therefore, the present study provides preliminary
screening data of effective TCR molecules, which may be beneficial
to develop TCR-T immunotherapy in the future.
Among solid tumors, melanoma has the highest
frequency of point mutations (such as BRAF V600E), followed by lung
(KRAS G12C), colorectal (KRAS G12D), gastric (BRAF V600E) and liver
cancer (TP53 R249S) (18). However,
there are relatively few mutations in blood tumors, such as acute
lymphocytic leukaemia and acute myelogenous leukaemia (23). Chimeric antigen receptor T-cell
(CAR-T) therapy has been effective in the treatment of some blood
tumors (such as acute lymphoblastic leukemia), but remains poorly
effective in solid tumors (such as liver cancer) (24). CAR-T therapy recognizes membrane
surface antigens (such as CD19 and B-cell maturation antigen);
however, as solid tumors lack cell-surface specific targets, TCR
T-cell (TCR-T) therapy may be more effective compared with CAR-T in
the treatment of solid tumors, since it recognizes TSAs that are
produced due to genetic mutations in cancer cells (24). Therefore, TSAs have become the key
targets for screening TCR molecules in TCR-T immunotherapy. At
present, the key to TCR-T immunotherapy is to identify an ideal TCR
molecule (24). Therefore, the
present study synthesized epitope peptides based on tumor
neoantigens and identified specific T-cell clones with significant
cytotoxic effects, which is conducive to screening effective TCR
molecules.
Recent preclinical studies have demonstrated that
individualized cancer vaccines based on new antigens have been used
effectively in melanoma and glioblastoma. Ott et al
(25) revealed that tumor regression
without recurrence was observed in patients with melanoma injected
with a long peptide (15 aa) vaccine targeting individualized new
antigens. Keskin et al (26)
observed that patients with glioblastoma inoculated with
multi-epitope new antigen vaccines exhibited an increase in the
number of new antigen-specific CD4 and CD8 tumor infiltrating
lymphocytes (TILs). With regards to ACT and based on new antigens,
the study by Cafri et al (27) used TILs to identify KRAS G12D mutants
to treat patients with colorectal and breast cancer. The results
revealed that patients with colorectal cancer treated high activity
CD8+ TILs, could identify KRAS G12D, and a complete
regression of the tumor was observed. Therefore, tumor
neoantigen-specific T cells serve an important role in tumor
immunotherapy (28). Previous
studies also suggest that transfecting the TCR gene, which
recognizes tumor neoantigens, into naive CD8+ T cells
may provide tumor neoantigen-specific TILs therapy and TCR-T
immunotherapy for patients which have the same antigen (29,30). At
present, vaccines and ACT based on neoantigens have achieved some
success (31), while immune
checkpoint inhibitors and combination therapies towards solid
tumors remain in the development stage (32,33).
With the development of next-generation sequencing
technology and peptide prediction algorithms (34), it is now possible to sequence
individual tumor tissues and identify non-synonymous mutations
which occur in tumor cells but not in healthy somatic cells
(35,36). To improve the accuracy of T-cell
immunotherapy for cancer treatment, and to fully achieve the
benefits of individual tumor immunotherapy, it is necessary to
design antigen epitope peptides with high affinity for MHC
molecules and high specificity to tumor neoantigens (37).
In conclusion, the point mutations in tumor
neoantigens identified in the three groups may improve the
cytotoxicity of specific T cells. The present study revealed that
the mutant peptides in the CTNNB1 group were effective at
activating the cellular immune response. Therefore, immunotherapies
using this tumor neoantigen epitope peptide should be investigated
in the future, to improve the accuracy of T-cell immunotherapy in
cancer treatment and to fully achieve personalized immunotherapy
and precision medical treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81703053, 21771042
and 31300737). The present study was also supported by the Natural
Science Foundation of Guangdong Province (grant nos.
2018A030313114, 2018A030313860 and 2016A030310298), and the
Innovation and University Promotion Project of Guangdong
Pharmaceutical University (grant nos. 2017KCXTD020 and
2017KZDXM049).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WW prepared and wrote the manuscript. YC performed
the experiments. LH and WL performed experimental data analysis. CT
critically revised the manuscript. HS designed the experimental
scheme, reviewed and edited the manuscript. WW, YC, LH, WL, CT and
HS contributed to data analysis, drafting or revising of the
manuscript, gave final approval of the version to be published and
agree to be accountable for all aspects of the work.
Ethics approval and consent to
participate
The present study follows international and national
regulations in accordance with the Declaration of Helsinki. The
present study was approved by the Ethics Committee of The First
Affiliated Hospital of Guangdong Pharmaceutical University
(Guangzhou, China; ref. no. 2017-003). Human peripheral blood was
collected from laboratory healthy volunteers and written informed
consent was provided by each volunteer.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TCGA
|
The Cancer Genome Atlas
|
|
CTL
|
cytotoxic T lymphocyte
|
|
MHC
|
major histocompatibility complex
|
|
HLA
|
human leukocyte antigen
|
|
ACT
|
adoptive cell transfer therapy
|
|
TAA
|
tumor-associated antigen
|
|
TSA
|
tumor-specific antigen
|
|
CFSE
|
carboxyfluorescein succinimidyl
ester
|
|
PI
|
propidium iodide
|
|
MFI
|
mean fluorescence intensity
|
|
DC
|
dendritic cell
|
|
PBMC
|
peripheral blood mononuclear cell
|
|
Cal-AM
|
calcein-acetoxymethyl
|
References
|
1
|
Efremova M, Finotello F, Rieder D and
Trajanoski Z: Neoantigens generated by individual mutations and
their role in cancer immunity and immunotherapy. Front Immunol.
8:16792017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei
J, Ren S and Zhou C: Tumor neoantigens: From basic research to
clinical applications. J Hematol Oncol. 12:932019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong
F, Guo C, Wu X, Li Y, Li X, et al: Neoantigen vaccine: An emerging
tumor immunotherapy. Mol Cancer. 18:1282019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou C, Zhu C and Liu Q: Toward in silico
identification of tumor neoantigens in immunotherapy. Trends Mol
Med. 25:980–992. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garcia-Garijo A, Fajardo CA and Gros A:
Determinants for neoantigen identification. Front Immunol.
10:13922019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wirth TC and Kühnel F: Neoantigen
targeting-dawn of a new era in cancer immunotherapy? Front Immunol.
8:18482017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rammensee H, Bachmann J, Emmerich NP,
Bachor OA and Stevanović S: SYFPEITHI: Database for MHC ligands and
peptide motifs. Immunogenetics. 50:213–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parker KC, Bednarek MA and Coligan JE:
Scheme for ranking potential HLA-A2 binding peptides based on
independent binding of individual peptide side-chains. J Immunol.
152:163–175. 1994.PubMed/NCBI
|
|
9
|
Richters MM, Xia H, Campbell KM,
Gillanders WE, Griffith OL and Griffith M: Best practices for
bioinformatic characterization of neoantigens for clinical utility.
Genome Med. 11:562019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tran E, Ahmadzadeh M, Lu YC, Gros A,
Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al:
Immunogenicity of somatic mutations in human gastrointestinal
cancers. Science. 350:1387–1390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carey MF, Peterson CL and Smale ST:
PCR-mediated site-directed mutagenesis. Cold Spring Harb Protoc.
2013:738–742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mazzei M, Vascellari M, Zanardello C,
Melchiotti E, Vannini S, Forzan M, Marchetti V, Albanese F and
Abramo F: Quantitative real time polymerase chain reaction
(qRT-PCR) and RNAscope in situ hybridization (RNA-ISH) as effective
tools to diagnose feline herpesvirus-1-associated dermatitis. Vet
Dermatol. 30:e491–e147. 2019. View Article : Google Scholar
|
|
13
|
Thaxton JE and Li Z: To affinity and
beyond: Harnessing the T cell receptor for cancer immunotherapy.
Hum Vaccin Immunother. 10:3313–3321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kisielow P: How does the immune system
learn to distinguish between good and evil? The first definitive
studies of T cell central tolerance and positive selection.
Immunogenetics. 71:513–518. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schumacher TN and Schreiber RD:
Neoantigens in cancer immunotherapy. Science. 348:69–74. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kanaseki T: Proteogenomics HLA Ligandome
analysis for cancer antigen research. Gan To Kagaku Ryoho.
46:1377–1381. 2019.(In Japanese). PubMed/NCBI
|
|
17
|
Matsushita H, Demachi-Okamura A and
Takahashi Y: Neoantigens are critical targets in naturally and
therapeutically induced immune responses to cancer. Gan To Kagaku
Ryoho. 46:1372–1376. 2019.(In Japanese). PubMed/NCBI
|
|
18
|
Garrido F and Aptsiauri N: Cancer immune
escape: MHC expression in primary tumors versus metastases.
Immunology. 158:255–266. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mandal R and Chan TA: Personalized
oncology meets immunology: The path toward precision immunotherapy.
Cancer Discov. 6:703–713. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Z, Jensen MA and Zenklusen JC: A
practical guide to the cancer genome atlas (TCGA). Methods Mol
Biol. 1418:111–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu YC and Robbins PF: Cancer immunotherapy
targeting neoantigens. Semin Immunol. 28:22–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ren L, Leisegang M, Deng B, Matsuda T,
Kiyotani K, Kato T, Harada M, Park JH, Saloura V, Seiwert T, et al:
Identification of neoantigen-specific T cells and their targets:
Implications for immunotherapy of head and neck squamous cell
carcinoma. Oncoimmunology. 8:e15688132019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen YP, Zhang Y, Lv JW, Li YQ, Wang YQ,
He QM, Yang XJ, Sun Y, Mao YP, Yun JP, et al: Genomic analysis of
tumor microenvironment immune types across 14 solid cancer types:
Immunotherapeutic implications. Theranostics. 7:3585–3594. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vormehr M, Diken M, Türeci Ö, Sahin U and
Kreiter S: Personalized neo-epitope vaccines for cancer treatment.
Recent Results Cancer Res. 214:153–167. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J,
Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al: An
immunogenic personal neoantigen vaccine for patients with melanoma.
Nature. 547:217–221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Keskin DB, Anandappa AJ, Sun J, Tirosh I,
Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E,
et al: Neoantigen vaccine generates intratumoral T cell responses
in phase Ib glioblastoma trial. Nature. 565:234–239. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cafri G, Yossef R, Pasetto A, Deniger DC,
Lu YC, Parkhurst M, Gartner JJ, Jia L, Ray S, Ngo LT, et al: Memory
T cells targeting oncogenic mutations detected in peripheral blood
of epithelial cancer patients. Nat Commun. 10:4492019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tran E, Turcotte S, Gros A, Robbins PF, Lu
YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS,
et al: Cancer immunotherapy based on mutation-specific CD4+ T cells
in a patient with epithelial cancer. Science. 344:641–645. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tran E, Robbins PF, Lu YC, Prickett TD,
Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al:
T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J
Med. 375:2255–2262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zacharakis N, Chinnasamy H, Black M, Xu H,
Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al:
Immune recognition of somatic mutations leading to complete durable
regression in metastatic breast cancer. Nat Med. 24:724–730. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shindo Y, Hazama S and Nagano H: Cancer
vaccine focused on neoantigens. Gan To Kagaku Ryoho. 46:1367–1371.
2019.(In Japanese). PubMed/NCBI
|
|
32
|
Chu Y, Liu Q, Wei J and Liu B:
Personalized cancer neoantigen vaccines come of age. Theranostics.
8:4238–4246. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou J, Zhao W, Wu J, Lu J, Ding Y, Wu S,
Wang H, Ding D, Mo F, Zhou Z, et al: Neoantigens derived from
recurrently mutated genes as potential immunotherapy targets for
gastric cancer. Biomed Res Int. 2019:81031422019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vilimas T: Measuring tumor mutational
burden using whole-exome sequencing. Methods Mol Biol. 2055:63–91.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smith CC, Chai S, Washington AR, Lee SJ,
Landoni E, Field K, Garness J, Bixby LM, Selitsky SR, Parker JS, et
al: Machine-learning prediction of tumor antigen immunogenicity in
the selection of therapeutic epitopes. Cancer Immunol Res.
7:1591–1604. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Riley TP, Keller GLJ, Smith AR, Davancaze
LM, Arbuiso AG, Devlin JR and Baker BM: Structure based prediction
of neoantigen immunogenicity. Front Immunol. 10:20472019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brennick CA, George MM, Corwin WL,
Srivastava PK and Ebrahimi-Nik H: Neoepitopes as cancer
immunotherapy targets: Key challenges and opportunities.
Immunotherapy. 9:361–371. 2017. View Article : Google Scholar : PubMed/NCBI
|