Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most
lethal gastrointestinal malignancy with close parallels between
incidence and mortality (1,2). In China, ~90,100 new cases of PDAC and
79,400 PDAC-associated mortality cases were estimated in 2015
(3). Although advancements have been
made in PDAC treatment, the prognosis of patients with PDAC remains
poor, primarily due to the frequency of distant metastasis.
Therefore, it is still urgent to investigate the molecular
mechanisms underlying PDAC progression and identify novel
therapeutic strategies to improve the outcome of patients with
PDAC.
The DEP domain containing 1B (DEPDC1B) gene is
located at the human chromosome 5q12.1, and encodes a protein
containing a Dishevelled, EGL-10 and pleckstrin (DEP) domain and a
Rho-GTPase-activating protein-like domain. The precise function of
DEPDC1B has not yet been clarified, but studies have shown its
association with a variety of cell events, including cell
proliferation, cell adhesion and cell cycle regulation (4–6). In
addition, previous studies have also claimed that DEPDC1B has an
oncogenic effect in breast carcinoma, non-small cell lung cancer
(NSCLC), prostate cancer, malignant melanoma and oral cancer
(7–11). However, the biological function of
DEPDC1B in PDAC has not yet been investigated.
Tumor metastasis is the leading cause of
cancer-associated mortality (12).
It is well known that epithelial-to-mesenchymal transition (EMT)
plays a crucial role in the initiation and promotion of cancer cell
invasion and dissemination (13–15).
Cancer cells that have undergone EMT lose their epithelial
characteristics and obtain the ability to invade other tissues
(16). Loss of E-cadherin is
regarded as a key event of EMT (17). At the same time, during EMT, the
expression levels of mesenchymal markers such as N-cadherin and
Vimentin are often upregulated (18). Previous studies have reported that
EMT is involved in the dissemination of cancer cells in PDAC
(18–20). It has also been reported that the
Akt/glycogen synthase kinase-3β (GSK3β)/Snail signaling pathway
regulates EMT and metastasis (21–23).
Activation of Akt can enhance Snail nucleus accumulation by
facilitating GSK3β degradation, subsequently inducing EMT and
cancer metastasis (24–26).
In the present study, the role of DEPDC1B in PDAC
and the association between DEPDC1B expression in PDAC tissues and
patient prognosis was analyzed. DEPDC1B silencing was found to
inhibit PDAC cell proliferation, migration and invasion, whereas
DEPDC1B overexpression exerted the opposite effect. Results also
suggested that DEPDC1B activated the Akt/GSK3β/Snail signaling
pathway and triggered EMT. Collectively, the present findings
suggested that DEPDC1B may be an important regulator of PDAC
progression and a potential target for future PDAC treatment.
Materials and methods
Gene Expression Omnibus (GEO) data
analysis
A total of three PDAC datasets [GSE15471 (27), GSE16515 (28) and GSE32676 (29)] were obtained from the GEO database
(https://www.ncbi.nlm.nih.gov/geo/).
The raw probe-level data (CEL files) and annotations for the probe
arrays downloaded from GEO were used for analysis conducted by R
software (version 3.5.0). The robust multi-array average algorithm
in the Affy package of R was used for data pre-processing including
background correction and quantile normalization. Then the probe ID
was transformed to gene symbols. For multiple probe sets
corresponding to the same gene, their average expression value was
taken as the gene expression value.
Patients and tissue samples
A total of 79 pairs of PDAC tissues and adjacent
non-cancerous tissues were collected from patients who underwent
surgical resection at Ruijin Hospital Affiliated with Shanghai Jiao
Tong University School of Medicine (Shanghai, China) between
January 2011 and December 2017. All patients were diagnosed with
PDAC via pathological diagnosis and had not received chemotherapy
or radiotherapy before resection. The present study was approved by
the Ethics Committee of Ruijin Hospital, and informed consent was
obtained from all patients included in the study.
Cell culture and reagents
The human pancreatic cancer cell lines BxPC-3, MIA
PaCa-2, PANC-1 and SW1990 were purchased from the Cell Bank of the
Chinese Academy of Sciences. Capan-1, Capan-2 and the normal human
pancreatic ductal cell line hTERT-HPNE were purchased from the
American Type Culture Collection. Cells were cultured in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.) containing 10% fetal bovine serum (FBS; Lonsera Science) and
1% penicillin/streptomycin at 37°C with 5% CO2. LY294002
was purchased from Absin (cat. no. abs810001).
Immunohistochemistry (IHC) and tissue
microarray (TMA)
A TMA including 79 pairs of matched PDAC tissues and
adjacent non-cancerous tissues was analyzed. The
immunohistochemical procedure was described in our previous study
(30). An antibody against DEPDC1B
(1:100; cat. no. PA5-72875; Invitrogen; Thermo Fisher Scientific,
Inc.) was used to detect the protein expression levels according to
the standard procedure (30). The
final IHC score was calculated by multiplying the percentage of
positive cells (0, ≤5%; 1, 5–25%; 2, 25–50%; 3, 50–75%; 4, >75%)
by the intensity scores (0, negatively stained; 1, weakly stained;
2, moderately stained; 3, strongly stained). Patients with a score
≥4 were regarded as having high expression of DEPDC1B. In contrast,
patients with an IHC score <4 were described as having low
expression.
RNA extraction and reverse
transcription quantitative PCR (RT-qPCR)
Total RNA of hTERT-HPNE and pancreatic cancer cells
was extracted using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). A total of 2 µg of total RNA was used to
synthesize cDNA with the First Strand cDNA Synthesis SuperMix for
RT-qPCR (Shanghai Yeasen Biotechnology Co., Ltd.), according to the
manufacturer's protocol. Then, qPCR reactions were performed using
SYBR Green Master Mix (Shanghai Yeasen Biotechnology Co., Ltd.) on
a StepOnePlus Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions. Relative mRNA expression was analyzed using the
2−ΔΔCq method and normalized to GAPDH (31). RT was performed at 42°C for 15 min,
followed by 2 min at 85°C. The qPCR program used included an
initial denaturation at 90°C for 5 min, followed by 40 cycles of
denaturation at 90°C for 10 sec and annealing/elongation at 60°C
for 30 sec. The primers used were as follows: DEPDC1B: Forward,
5′-CCACCAGGCACTTCACAAGAGAAC-3′; and reverse,
5′-CGGTGGACAAGACGGCAAGC-3′; GAPDH: Forward,
5′-CAGGAGGCATTGCTGATGAT-3′; and reverse,
5′-GAAGGCTGGGGCTCATTT-3′.
Western blot analysis
Total protein was isolated from hTERT-HPNE and
pancreatic cancer cells using RIPA containing 1% PMSF (both from
Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Protein concentrations were determined
using the BCA Protein Assay kit (Beyotime Institute of
Biotechnology). Protein samples (30–60 µg per lane) were separated
with 10% SDS-PAGE gels and transferred to PVDF membranes (EMD
Millipore). Then, the membranes were blocked in TBS-Tween-20
containing 5% fat-free milk for 2 h at room temperature and
incubated with the primary antibodies overnight at 4°C with gentle
shaking. The primary antibodies included DEPDC1B (1:1,000; cat. no.
PA5-72875; Invitrogen; Thermo Fisher Scientific, Inc.), E-cadherin
(1:1,000; cat. no. 3195T), N-cadherin (1:1,000; cat. no. 131116T),
Vimentin (1:1,000; cat. no. 3932S), Snail (1:1,000; cat. no.
3879S), Slug (1:1,000; cat. no. 9585T), Akt (1:1,000; cat. no.
4691S), and phosphorylated (p)-GSK-3β (1:1,000; cat. no. 5558S)
(all from Cell Signaling Technology, Inc.), Twist (1:1,000; cat.
no. ab49254; Abcam), p-Akt-S473 (1:1,000; cat. no. AP0140; ABclonal
Biotech Co., Ltd.), GSK-3β (1:1,000; cat. no. A16868; ABclonal
Biotech Co., Ltd.), and GAPDH (1:2,000, cat. no. GB12002,
Servicebio; Stratech). After washing with TBST three times, the
membranes were incubated with the corresponding horseradish
peroxidase-conjugated secondary antibodies (1:5,000; cat. no.
7074P2 and 7076S; Cell Signaling Technology, Inc.) at room
temperature for 1 h. Signals were detected using enhanced
chemiluminescence regent (GE Healthcare Bio-Sciences).
Lentivirus-mediated interference or
overexpression
Validated small interfering (si)RNA targeting human
DEPDC1B (5′-TGCTAGATTGGTAACGTTT-3′) and its corresponding negative
control (NC; 5′-TTCTCCGAACGTGTCACGT-3′) were synthesized and
inserted into the lentiviral vector GV248 by GeneChem, Inc. For
DEPDC1B overexpression, the coding sequence (CDS) of DEPDC1B was
amplified and cloned into pHBLV-CMV-MCS-3FLAG-EF1-ZsGreen-T2A-PURO
vector. The empty vector was used as the negative control for the
overexpression assay. Recombinant lentiviruses overexpressing
DEPDC1B and the negative control were provided by Hanbio
Biotechnology Co., Ltd.
For the inhibition and overexpression of DEPDC1B,
PDAC cells (Capan-1, SW1990, MIA PaCa-2 and PANC-1) were cultured
to 30–50% confluence and transfected with lentivirus and polybrene
overnight at the MOI (multiplicity of infection) of 10–30 according
to the manufacturer's protocol. Stable transfected cells were
selected in culture medium containing puromycin (2 µg/ml) for 10
days.
Cell Counting Kit-8 (CCK-8) assay
To measure cell viability, the transfected
pancreatic cancer cells were seeded in 96-well plates
(1×103 cells/well). A total of 100 µl complete culture
medium containing 10 µl CCK-8 reagent (Beyotime Institute of
Biotechnology) was added to each well at different time points of
interest. After incubation in the dark at 37°C for 2 h, the
absorption at 450 nm was detected. Each group had five duplicate
wells and the experiments were repeated in triplicate.
Colony formation assay
Different cells were plated in 6-well plates (500
cells/well) in triplicate. Cells were maintained in culture for 14
days, and were then fixed with 4% paraformaldehyde (Beyotime
Institute of Biotechnology) at room temperature for 20 min followed
by staining with crystal violet solution (Beyotime Institute of
Biotechnology) at room temperature for 30 min. Images were captured
with a camera and the colonies were counted manually to access cell
proliferation ability.
Wound healing assay
Capan-1, SW1990, MIA PaCa-2 and PANC-1 cells were
seeded into 6-well plates at a density of 5×105/well and
maintained in culture until 100% confluent. At that point, wounds
were made using a standard 200 µl pipette tip. Cells were washed
with PBS to remove debris and cultured in DMEM without FBS. Images
were captured at 0 and 48 h after scratching with a light
microscope (magnification, x200; Nikon Corporation). The
cell migratory ability was evaluated by measuring the percentage of
wound healing. The 48 h wound healing percentage=(0 h area of
wound-48 h area of wound)/(0 h area of wound).
Transwell assay
Transwell chambers (Corning, Inc.) precoated with or
without Matrigel (BD Biosciences) for 30 min at 37°C were used for
invasion and migration assays, respectively. A total of 600 µl of
DMEM containing 10% FBS was added to the lower chambers, and
5×104 cells suspended in 200 µl of serum-free DMEM were
placed in the upper chambers. After 24 h in culture, migrated and
invaded cells were fixed with 4% paraformaldehyde and stained with
crystal violet staining solution at room temperature for 10 min.
Cells in the lower chamber were removed using a cotton-tipped swab.
Finally, the numbers of migrated and invaded cells in five randomly
selected fields were counted under a light microscope
(magnification, x200; Nikon Corporation).
Immunofluorescence
Cells were seeded on glass coverslips and maintained
in culture for 24 h. Then cells were fixed with 4% paraformaldehyde
at room temperature for 15 min, permeabilized with 0.3% Triton
X-100 at room temperature for 10 min and blocked with 0.3% BSA
(Shanghai Yeasen Biotechnology Co., Ltd.) in PBS at room
temperature for 30 min. Then, primary antibodies against E-cadherin
(1:50; cat. no. 20874-1-AP), N-cadherin (1:100; cat. no.
66219-1-Ig) (both from Proteintech Group, Inc.) and Vimentin
(1:100; cat. no. 3932S; Cell Signaling Technology, Inc.) were added
and incubated overnight in a humidified chamber at 4°C. On the
following day, Alexa Fluor 555-labeled Donkey Anti-Rabbit IgG (H+L)
(1:200; cat. no. A0453; Beyotime Institute of Biotechnology) or
IFKine™ Red Donkey Anti-Mouse IgG (1:200; cat. no. A24411; Abbkine
Scientific Co., Ltd.) were applied and incubated for 1 h in the
dark at room temperature. Finally, the nuclei of cells were
counterstained with DAPI (Servicebio; Stratech) at room temperature
for 5 min. All washes were performed with PBS. Images were captured
with a fluorescence microscope (magnification, x400; Nikon
Corporation).
Xenograft tumor study
A total of 10 female BALB/c nude mice (age, 4–5
weeks; body weight, 12–16 g) were purchased from the Shanghai
Experimental Animal Center of the Chinese Academy of Sciences and
housed (5 mice per cage) under standard conditions (26°C, 40–60%
relative humidity, 12 h light/dark cycles) with unlimited access to
food and water. Animal health and behavior were monitored every
day. For tumor metastasis assays, DEPDC1B knockdown and negative
control Capan-1 cells (3×106 cells) were intravenously
injected into the tail vein of female 6-week-old nude mice (n=5 per
group). After 10 weeks, the mice were euthanized in a gradually
filled CO2 chamber with a flow rate of ≤30%
CO2 of the chamber volume/min. The death was verified by
the absence of respiratory movement and heartbeat. The lungs of the
mice were dissected for H&E staining to assess the lung
metastatic foci. All animal experiments were approved by the
Institutional Animal Care and Use Committee of Shanghai Jiaotong
University School of Medicine (IACUC approval no. B-2019-004).
Statistical analysis
All data were analyzed using SPSS software (version
20.0; IBM Corp.) and are presented as the mean ± standard error
(n≥3). Student's t-test was used to assess the statistical
significance between two groups. One-way ANOVA followed by the
Least Significant Difference post hoc test was used to compare
multiple groups. The χ2 test was used to analyze the
association between DEPDC1B expression and clinicopathological
parameters. Survival analysis was evaluated using the Kaplan-Meier
method and log-rank test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Upregulation of DEPDC1B in tumor
tissues is associated with poor prognosis in patients with
PDAC
The three datasets (GSE15471, GSE16515 and GSE32676)
were analyzed from the GEO database and the results showed that
DEPDC1B mRNA was significantly upregulated in PDAC tissues compared
with that in adjacent non-cancerous tissues (Fig. 1A). To further confirm this finding,
immunohistochemical analysis were performed in a TMA containing 79
samples of PDAC and adjacent non-cancerous tissues (Fig. 1B). According to the IHC score, the
expression levels of DEPDC1B were higher in PDAC tissues than those
in paired non-cancerous tissues (Fig. 1C
and D). No significant association between DEPDC1B expression
levels and the clinicopathological characteristics of the samples
was observed (Table I). However,
Kaplan-Meier survival analysis revealed that patients with PDAC
with higher expression levels of DEPDC1B could have a shorter
survival time (Fig. 1E). Meanwhile,
the data from The Cancer Genome Atlas also verified the association
between high DEPDC1B expression and poor prognosis (Fig. 1F).
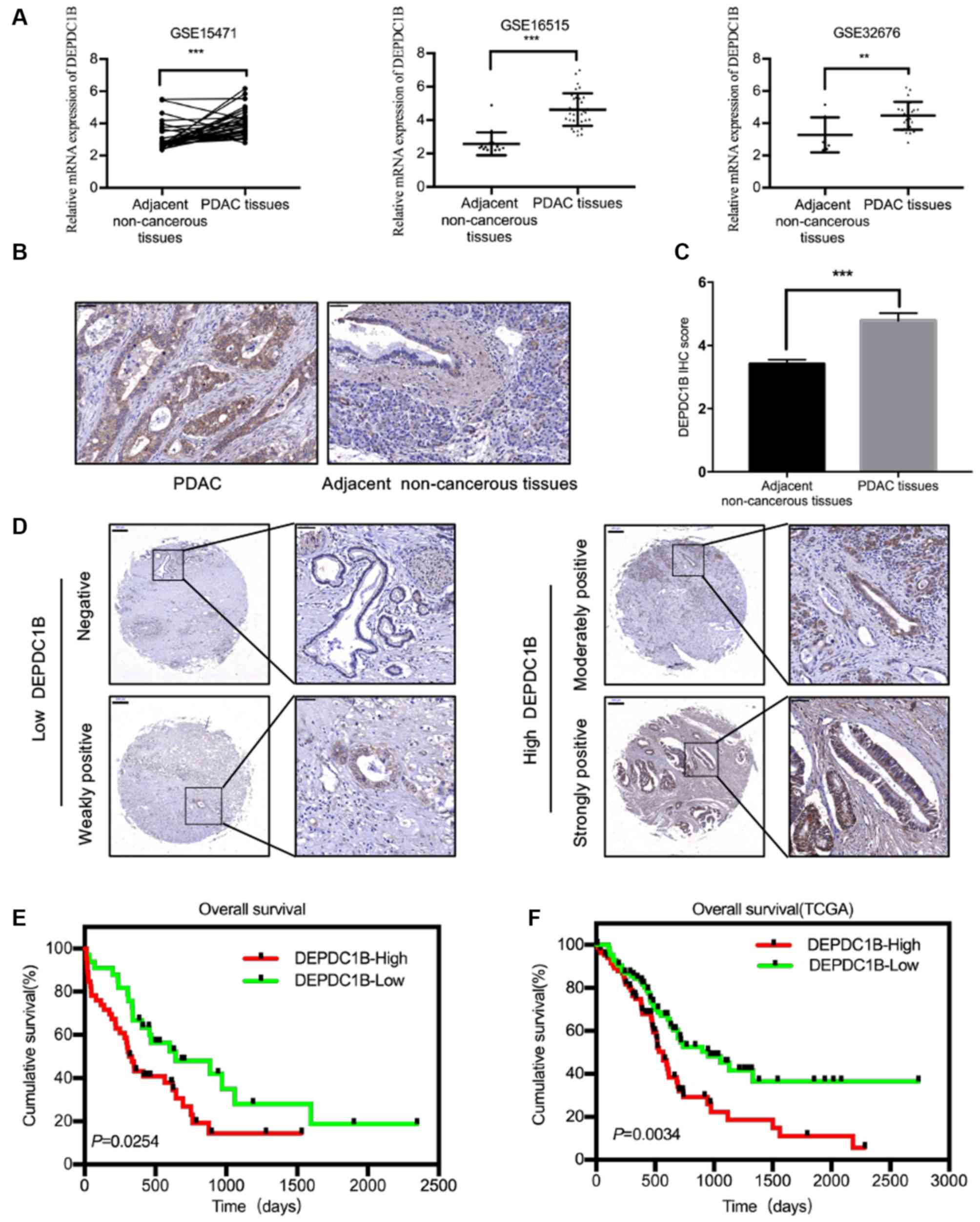 | Figure 1.DEPDC1B expression is increased in
PDAC tissues. (A) The GEO datasets (GSE15471, GSE16515 and
GSE32676) showed that the DEPDC1B mRNA expression levels were
upregulated in PDAC tissues. (B) Representative IHC staining images
for DEPDC1B in PDAC and adjacent non-cancerous tissues. Scale bar,
50 µm. (C) DEPDC1B IHC score in PDAC tissues and adjacent
non-cancerous tissues on TMAs. (D) Representative IHC staining
images for DEPDC1B expression levels in TMAs. Scale bar, 200 µm
(left), 50 µm (right). (E and F) Kaplan-Meier analysis showed the
association between DEPDC1B expression and patient prognosis.
**P<0.01; ***P<0.001. DEPDC1B, DEP domain containing 1B; GEO,
Gene Expression Omnibus; PDAC, pancreatic ductal adenocarcinoma;
IHC, immunohistochemistry; TCGA, The Cancer Genome Atlas; TMAs,
tissue microarrays. |
 | Table I.Associations between DEPDC1B low
expression (n=32) and DEPDC1B high expression (n=47) and
clinicopathologic characteristics of patients with pancreatic
ductal adenocarcinoma. |
Table I.
Associations between DEPDC1B low
expression (n=32) and DEPDC1B high expression (n=47) and
clinicopathologic characteristics of patients with pancreatic
ductal adenocarcinoma.
|
|
| DEPDC1B level |
|
|---|
|
|
|
|
|
|---|
| Clinicopathological
characteristics | Total (n=79) | Low, n | High, n | P-value |
|---|
| Sex |
|
|
| 0.156 |
|
Female | 32 | 16 | 16 |
|
|
Male | 47 | 16 | 31 |
|
| Age, years |
|
|
| 0.107 |
|
<60 | 17 | 4 | 13 |
|
|
≥60 | 62 | 28 | 34 |
|
| Tumor size, cm |
|
|
| 0.094 |
|
<5 | 56 | 26 | 30 |
|
| ≥5 | 23 | 6 | 17 |
|
| Primary
tumor(T) |
|
|
| 0.352 |
|
T1+T2 | 52 | 23 | 29 |
|
|
T3+T4 | 27 | 9 | 18 |
|
| Lymph node
metastasis |
|
|
| 0.957 |
|
Absent | 59 | 24 | 35 |
|
|
Present | 20 | 8 | 12 |
|
| Metastasis |
|
|
| 0.391 |
| No | 76 | 32 | 44 |
|
|
Yes | 3 | 0 | 3 |
|
| TNM [AJCC
(44)] |
|
|
| 0.464 |
|
I+II | 38 | 17 | 21 |
|
|
III+IV | 41 | 15 | 26 |
|
| Nerve invasion |
|
|
| 0.143 |
|
Absent | 35 | 11 | 24 |
|
|
Present | 44 | 21 | 23 |
|
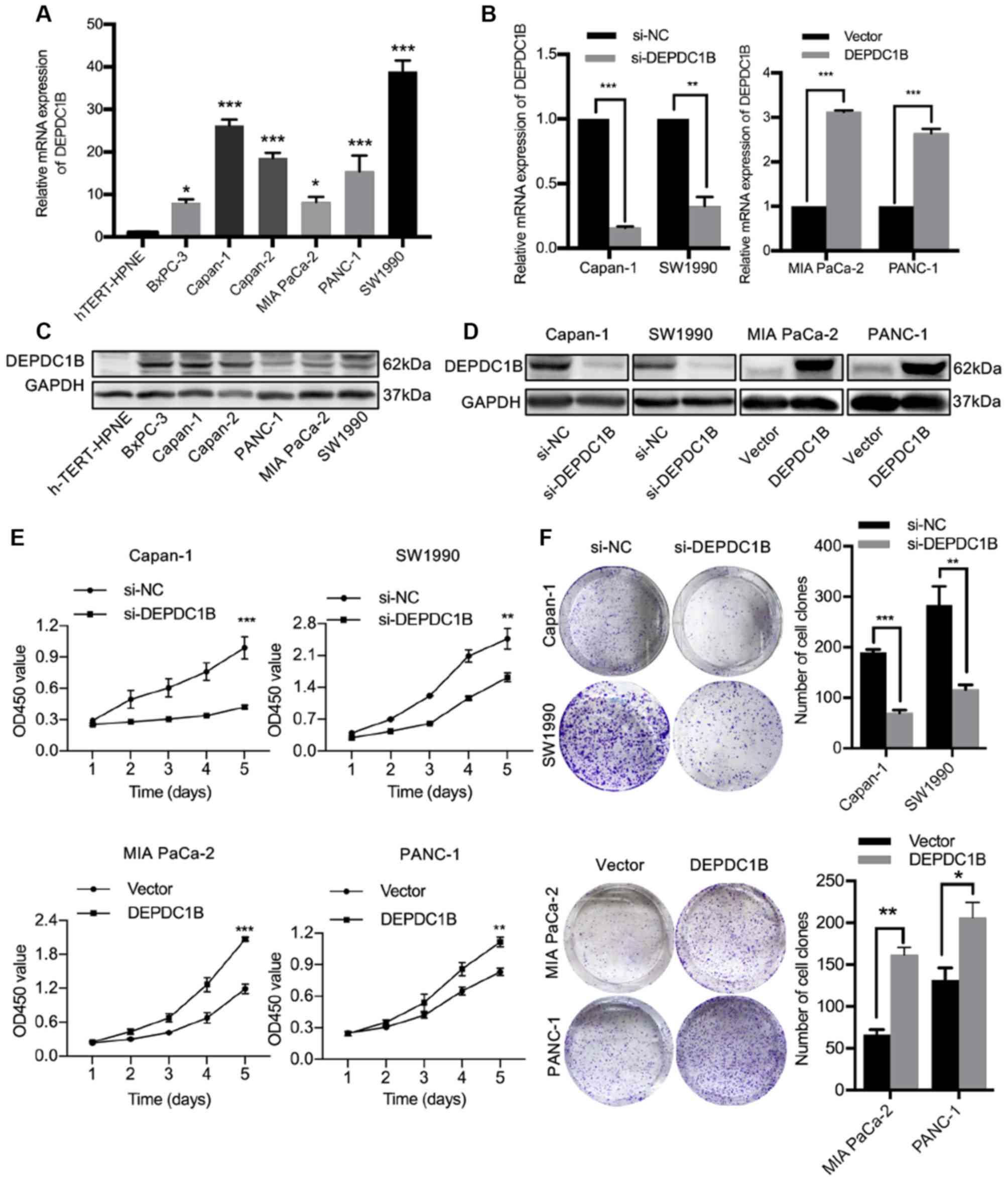
DEPDC1B expression in PDAC cell lines
and construction of a stable cell line
DEPDC1B expression was detected in six PDAC cell
lines and the normal pancreatic cells hTERT-HPNE by RT-qPCR and
western blotting. Higher expression of DEPDC1B was observed in PDAC
cell lines compared with hTERT-HPNE (Fig. 2A and C). Capan-1 and SW1990 cells
with high DEPDC1B expression, MIA PaCa-2 and PANC-1 cells with low
DEPDC1B expression were selected for following experiments. Through
a lentiviral delivery system, DEPDC1B was stably knocked down in
Capan-1 and SW1990 cells and overexpressed in MIA PaCa-2 and PANC-1
cells. The transfection efficiency was confirmed by RT-qPCR and
western blotting (Fig. 2B and
D).
DEPDC1B promotes the proliferation,
migration and invasion of PDAC cells
Considering the association between high DEPDC1B
expression and poor prognosis, the role of DEPDC1B in cell
proliferation, migration and invasion was further investigated.
CCK-8 assay and colony formation assays revealed that cell
proliferation was inhibited by DEPDC1B knockdown and promoted by
DEPDC1B overexpression (Fig. 2E and
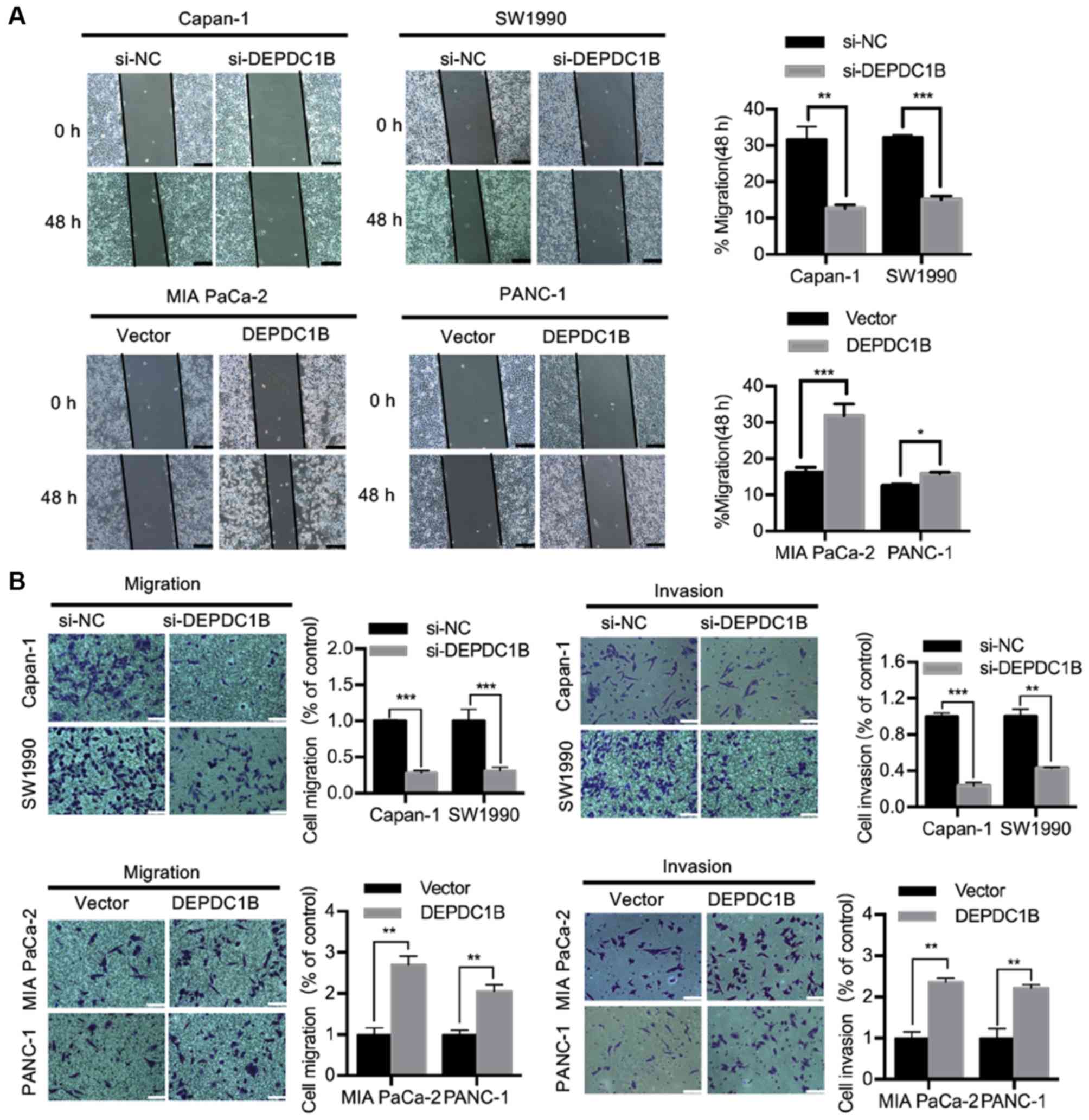
F). Wound healing and Transwell assays were performed to
investigate the role of DEPDC1B in cell migration and invasion. The
wound healing assay showed that silencing DEPDC1B inhibited the
migratory ability of Capan-1 and SW1990 cell lines, whereas DEPDC1B
overexpression increased the wound healing rate of MIA PaCa-2 and
PANC-1 cells (Fig. 3A). Transwell
assays indicated that knockdown of DEPDC1B suppressed the migration
and invasion of PDAC cells, while DEPDC1B overexpression promoted
PDAC cell migration and invasion (Fig.
3B).
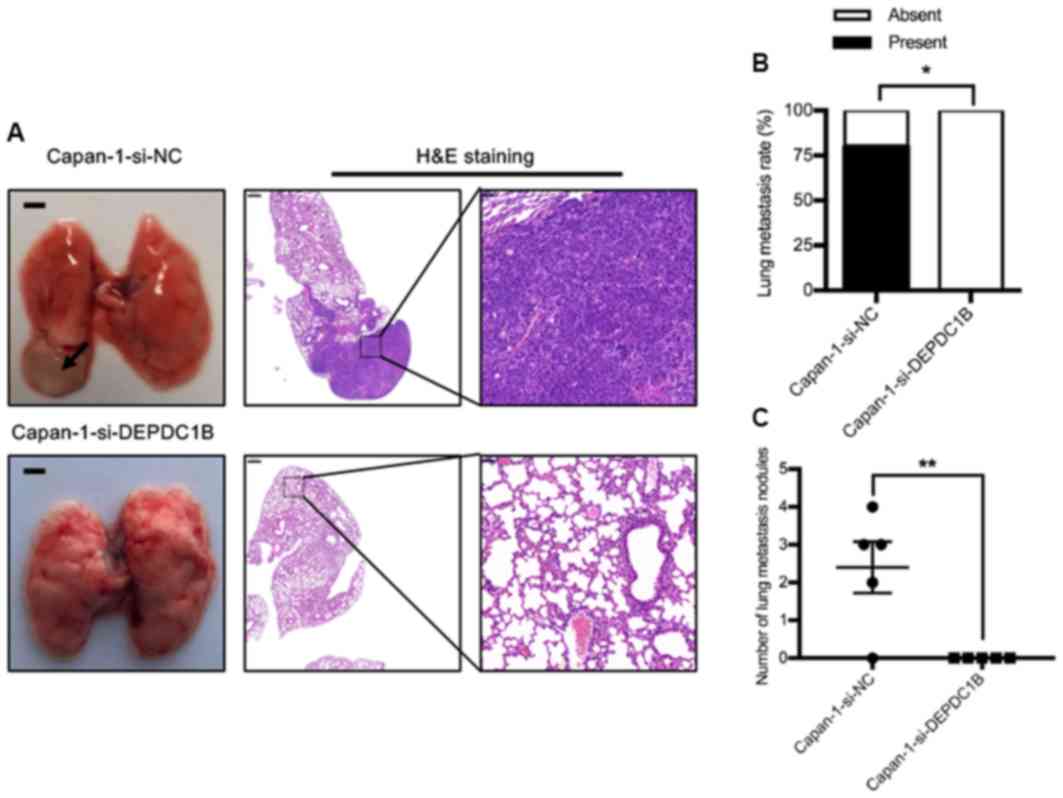
To further investigate the effect of DEPDC1B on
tumor metastasis in vivo, a lung metastatic model was
established in nude mice. The results demonstrated that the
incidence of lung metastasis in the si-DEPDC1B group was lower than
that in the control group (Fig. 4).
The mean tumor diameter in the control group was 1.0 mm and the
largest tumor measured 6.0 mm. No metastatic nodules were observed
in the si-DEPDC1B group.
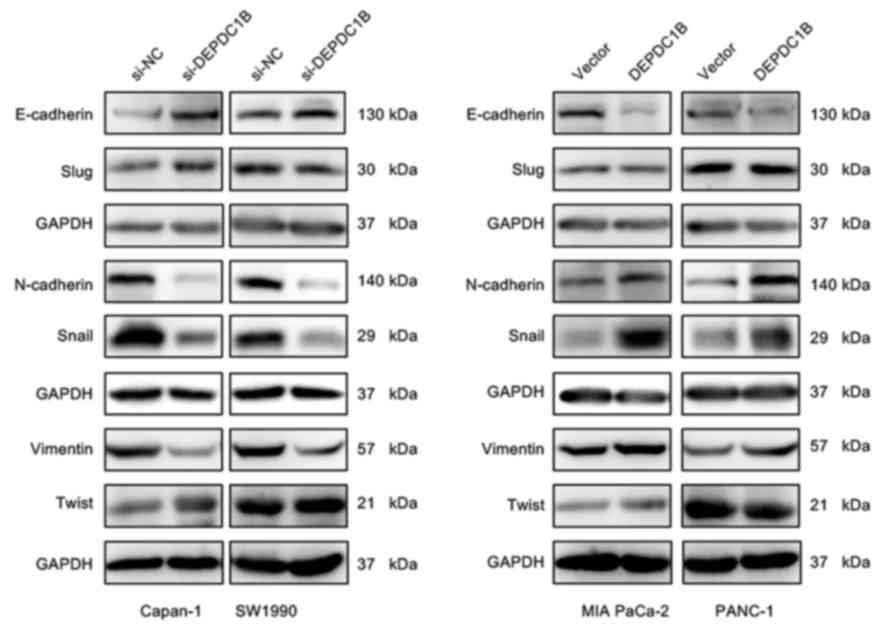
DEPDC1B regulates EMT in PDAC
cells
EMT is an important event in tumor metastasis, which
allows cancer cells to acquire invasive properties and to develop
metastatic growth characteristics (16). Therefore, the present study
hypothesized that DEPDC1B may be involved in the EMT process. To
investigate this effect, the expression of EMT markers and
EMT-inducing transcription factors (EMT-TFs) was analyzed in the
present study. Western blotting assays revealed that the expression
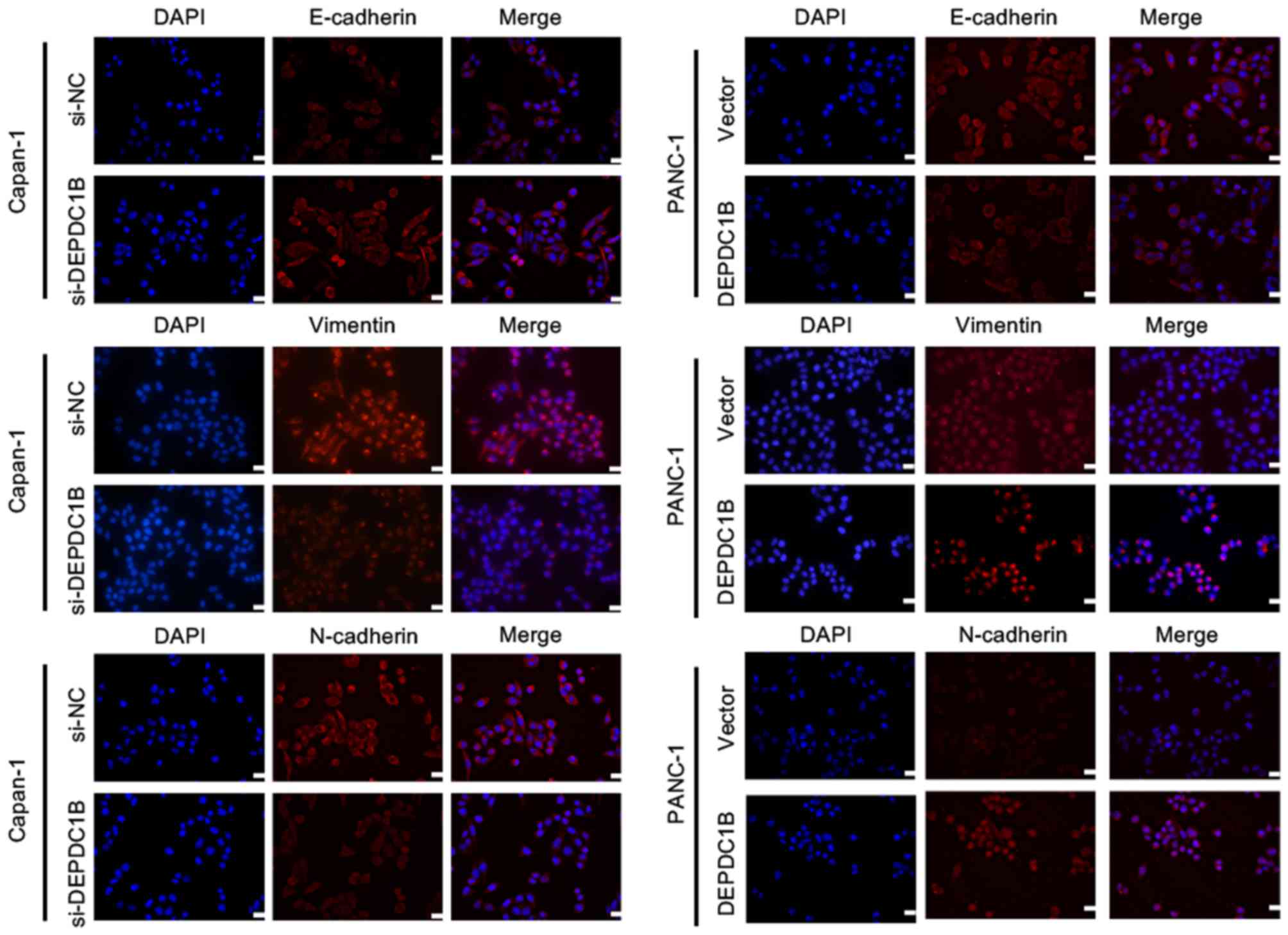
of the epithelial marker E-cadherin was increased, whereas the
mesenchymal markers Vimentin and N-cadherin, and the EMT-TF Snail
were markedly decreased in DEPDC1B knockdown cells (Fig. 5). Conversely, downregulation of
E-cadherin and upregulation of Vimentin, N-cadherin and Snail were
detected in the cell lines overexpressing DEPDC1B (Fig. 5). There was no significant difference
in the expression of Slug and Twist between the NC and treatment
group. In addition, immunofluorescent staining was performed to
analyze the protein expression of E-cadherin, Vimentin and
N-cadherin. The results of the immunofluorescence assay further
verified the alteration of EMT makers in DEPDC1B knockdown and
overexpression cells (Fig. 6). These
results indicated that DEPDC1B may be involved in the EMT process
in PDAC cells.
DEPDC1B modulates the Akt/GSK3β/Snail
signaling pathway in PDAC cells
As the main member of EMT-TFs, Snail can bind to the
E-box area of E-cadherin and suppress its expression, thereby
inducing EMT (32–34). Numerous studies have revealed that
Akt can inhibit GSK3β activation and subsequently lead to the
stabilization of endogenous Snail (24,25,35). The
present study hypothesized that DEPDC1B may induce EMT via the
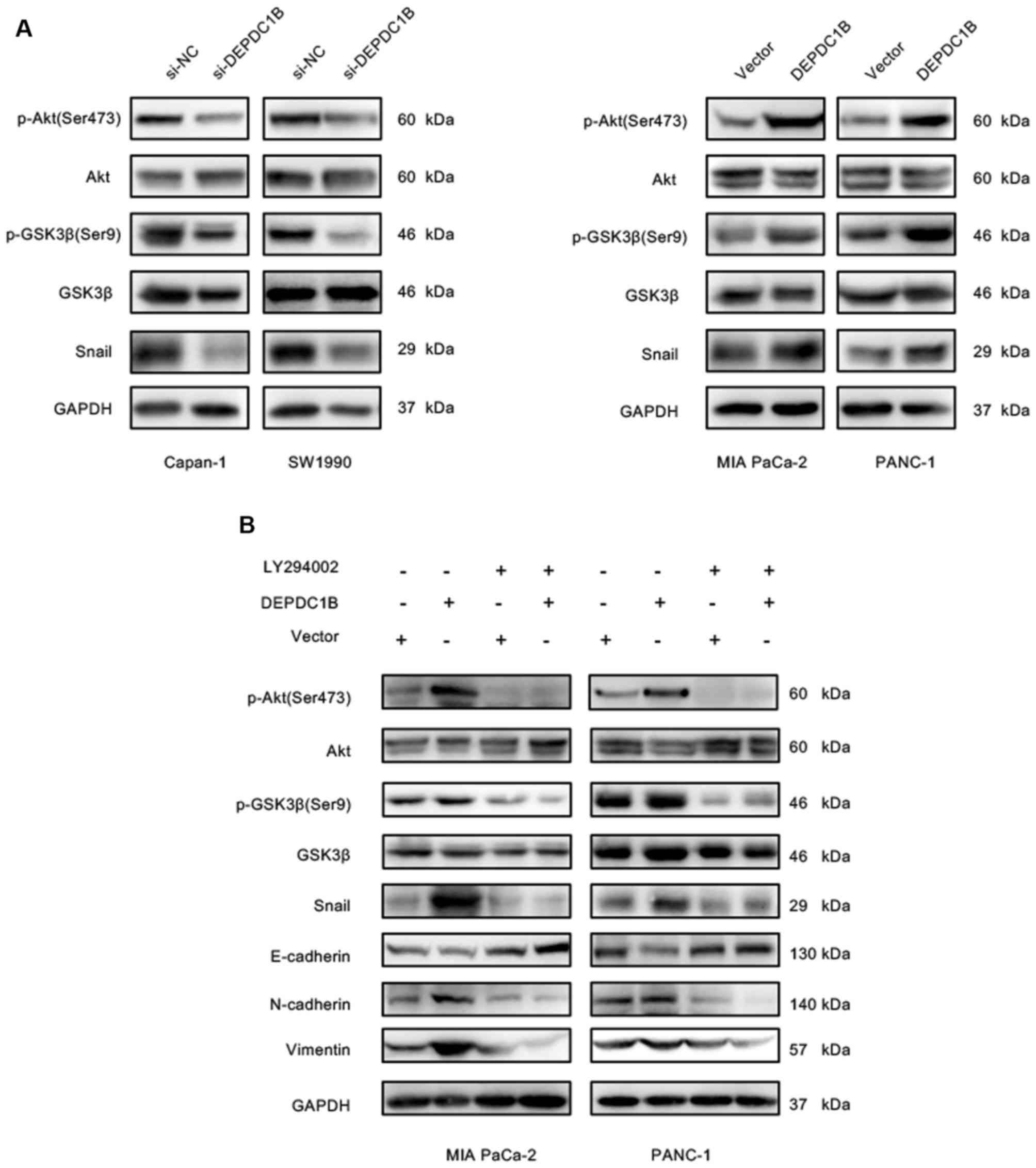
Akt/GSK3β/Snail pathway. As presented in Fig. 7A, DEPDC1B overexpression
significantly increased the levels of p-Akt at S473, p-GSK3β and
Snail, whereas these protein levels were decreased in DEPDC1B
knockdown cells. These findings suggested that DEPDC1B may enhance
Snail expression via the Akt/GSK3β pathway. To further verify
whether Snail upregulation was mediated by Akt, DEPDC1B
overexpressing cells were treated with the Akt inhibitor LY294002.
The results showed that LY294002 treatment reversed the
upregulation of p-Akt, p-GSK3β and Snail induced by the
overexpression of DEPDC1B in both PANC-1 and MIA PaCa-2 cells
(Fig. 7B). In addition, the
inhibition of Akt increased the protein expression levels of
E-cadherin and decreased Vimentin and N-cadherin expression
(Fig. 7B). These results suggested
that DEPDC1B promoted EMT via the Akt/GSK3β/Snail signaling
pathway.
Discussion
PDAC remains to be one of the most devastating
diseases, and metastasis is a major challenge in clinical
treatment. Conventional treatments including surgery, chemotherapy
and radiation have little effect on the course of the disease
(36). In addition, clinical trials
of various biological agents targeting specific signaling pathways
or transcription factors have proved disappointing (37). For example, a randomized,
double-blind, placebo-controlled trial (NCT01231581) of gemcitabine
plus trametinib (an oral MEK inhibitor) did not improve OS, PFS,
ORR or DOR in chemotherapy-naive patients with metastatic PDAC
(38). Therefore, a deeper
understanding of the molecular mechanisms underlying PDAC and
finding new molecules to establish novel therapeutic targets remain
an urgent requirement. Previous research investigating DEPDC1B has
demonstrated its upregulation in several types of cancer (7–11). These
studies have shown that DEPDC1B promotes the development of oral
cancer via Rac-ERK1/2 pathway and enhances migration and invasion
of NSCLC through Wnt/β-catenin signaling.
In the present study, the expression of DEPDC1B was
found to be significantly increased in PDAC tissues compared with
adjacent non-cancerous tissues. Furthermore, clinical data showed
that upregulation of DEPDC1B is associated with poor prognosis of
patients with PDAC. These results indicated that DEPDC1B may be
involved in PDAC progression. In addition, the present study
confirmed the important role of DEPDC1B in promoting PDAC migration
and invasion. Likewise, DEPDC1B overexpression was found to induce
EMT process and increase the expression of Snail. Further analysis
of metastasis-associated pathways revealed that p-Akt and p-GSK3β
were involved in the observed effects driven by DEPDC1B on PDAC
cells. Previous studies have revealed that the Akt/GSK3β/Snail
signaling pathway regulates EMT (21–23), and
in the present study, western blot results showed that
overexpression of DEPDC1B enhanced the expression of p-Akt, p-GSK3β
and Snail. In contrast, decreased expression of p-Akt, p-GSK3β and
Snail was observed when DEPDC1B was knocked down. In addition,
treatment with the Akt inhibitor LY294002 decreased the expression
of p-Akt, p-GSK3β and Snail, which subsequently affected
E-cadherin, Vimentin and N-cadherin expression. Taken together,
these findings indicate that DEPDC1B may mediate EMT through the
Akt/GSK3β/Snail signaling pathway.
The present study reported the role of DEPDC1B in
PDAC and the association between DEPDC1B and the Akt/GSK3β/Snail
pathway. The detailed interaction between DEPDC1B and Akt pathway
may be attributed to the DEP domain that can regulate G
protein-coupled receptor (GPCR)-initiated pathways via docking onto
GPCRs (39,40), which can consequently activate the
PI3K/Akt pathway (41–43). Therefore, further investigation on
whether DEPDC1B activates the Akt pathway through the interaction
between its DEP domain and GPCRs would be helpful to improve the
current understanding of the mechanism underlying PDAC metastasis.
In addition, there are a number of limitations to the present
study, such as the lack of healthy controls and the lack of
experiments performed on the same cell line.
In summary, the current study demonstrated that
DEPDC1B is upregulated in PDAC tissues and cell lines, and it may
work as a prognosis predictive marker for patients with PDAC.
Furthermore, DEPDC1B promotes tumor migration and invasion and
induces EMT via the regulation of Akt/GSK3β/Snail signaling
pathway, which may represent a new therapeutic target for PDAC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. NSFC 81870385,
NSFC81672719, NSFC81702740 and NSFC81800491).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL, TL, QW and LFW conceived and designed the study.
XL wrote the manuscript. XL and TL performed the majority of the
experiments. XH and HX assisted with the collection of clinical
samples and data. WW, JL, YQ and LW analyzed the experimental data.
QW and LFW reviewed the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Ruijin Hospital. Written informed content was obtained
from all patients. All animal experiments were approved by the
institutional animal care and use committee of Shanghai Jiaotong
University School of Medicine (IACUC approval no. B-2019-004).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marchesi S, Montani F, Deflorian G,
D'Antuono R, Cuomo A, Bologna S, Mazzoccoli C, Bonaldi T, Di Fiore
PP and Nicassio F: DEPDC1B coordinates de-adhesion events and
cell-cycle progression at mitosis. Dev Cell. 31:420–433. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garcia-Mata R: Arrested detachment: A
DEPDC1B-mediated de-adhesion mitotic checkpoint. Dev Cell.
31:387–389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahuja P and Singh K: In silico approach
for SAR analysis of the predicted model of DEPDC1B: A novel target
for oral cancer. Adv Bioinformatics. 2016:31360242016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boudreau HE, Broustas CG, Gokhale PC,
Kumar D, Mewani RR, Rone JD, Haddad BR and Kasid U: Expression of
BRCC3, a novel cell cycle regulated molecule, is associated with
increased phospho-ERK and cell proliferation. Int J Mol Med.
19:29–39. 2007.PubMed/NCBI
|
|
8
|
Yang Y, Liu L, Cai J, Wu J, Guan H, Zhu X,
Yuan J and Li M: DEPDC1B enhances migration and invasion of
non-small cell lung cancer cells via activating Wnt/β-catenin
signaling. Biochem Biophys Res Commun. 450:899–905. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bai S, Chen T, Du T, Chen X, Lai Y, Ma X,
Wu W, Lin C, Liu L and Huang H: High levels of DEPDC1B predict
shorter biochemical recurrence-free survival of patients with
prostate cancer. Oncol Lett. 14:6801–6808. 2017.PubMed/NCBI
|
|
10
|
Su YF, Liang CY, Huang CY, Peng CY, Chen
CC, Lin MC, Lin RK, Lin WW, Chou MY, Liao PH and Yang JJ: A
putative novel protein, DEPDC1B, is overexpressed in oral cancer
patients, and enhanced anchorage-independent growth in oral cancer
cells that is mediated by Rac1 and ERK. J Biomed Sci. 21:672014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Y, Sun W, Zheng B, Liu X, Luo Z, Kong
Y, Xu M and Chen Y: DEPDC1B knockdown inhibits the development of
malignant melanoma through suppressing cell proliferation and
inducing cell apoptosis. Exp Cell Res. 379:48–54. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Valastyan S and Weinberg Robert A: Tumor
metastasis: Molecular insights and evolving paradigms. Cell.
147:275–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial- mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chaffer CL, San Juan BP, Lim E and
Weinberg RA: EMT, cell plasticity and metastasis. Cancer Metastasis
Rev. 35:645–654. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen T, You Y, Jiang H and Wang ZZ:
Epithelial-mesenchymal transition (EMT): A biological process in
the development, stem cell differentiation, and tumorigenesis. J
Cell Physiol. 232:3261–3272. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mihaljevic AL, Michalski CW, Friess H and
Kleeff J: Molecular mechanism of pancreatic cancer-understanding
proliferation, invasion, and metastasis. Langenbecks Arch Surg.
395:295–308. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lan Y, Han J, Wang Y, Wang J, Yang G, Li
K, Song R, Zheng T, Liang Y, Pan S, et al: STK17B promotes
carcinogenesis and metastasis via AKT/GSK-3β/Snail signaling in
hepatocellular carcinoma. Cell Death Dis. 9:2362018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu L, Dai Y, Chen J, Zeng T, Li Y, Chen
L, Zhu YH, Li J, Li Y, Ma S, et al: Maelstrom promotes
hepatocellular carcinoma metastasis by inducing
epithelial-mesenchymal transition by way of Akt/GSK-3β/Snail
signaling. Hepatology. 59:531–543. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang H, Zhou Z, Jin S, Xu K, Zhang H and
Xu J, Sun Q, Wang J and Xu J: PRMT9 promotes hepatocellular
carcinoma invasion and metastasis via activating
PI3K/Akt/GSK-3β/Snail signaling. Cancer Sci. 109:1414–1427. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bachelder RE, Yoon SO, Franci C, de
Herreros AG and Mercurio AM: Glycogen synthase kinase-3 is an
endogenous inhibitor of Snail transcription: Implications for the
epithelial-mesenchymal transition. J Cell Biol. 168:29–33. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou BP and Hung MC: Wnt, hedgehog, and
snail: Sister pathways that control by GSK-3beta and beta-Trcp in
the regulation of metastasis. Cell Cycle. 4:772–776. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Badea L, Herlea V, Dima SO, Dumitrascu T
and Popescu I: Combined gene expression analysis of whole-tissue
and microdissected pancreatic ductal adenocarcinoma identifies
genes specifically overexpressed in tumor epithelia.
Hepatogastroenterology. 55:2016–2027. 2008.PubMed/NCBI
|
|
28
|
Pei H, Li L, Fridley BL, Jenkins GD,
Kalari KR, Lingle W, Petersen G, Lou Z and Wang L: FKBP51 affects
cancer cell response to chemotherapy by negatively regulating Akt.
Cancer Cell. 16:259–266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Donahue TR, Tran LM, Hill R, Li Y,
Kovochich A, Calvopina JH, Patel SG, Wu N, Hindoyan A, Farrell JJ,
et al: Integrative survival-based molecular profiling of human
pancreatic cancer. Clin Cancer Res. 18:1352–1363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shen R, Wang Q, Cheng S, Liu T, Jiang H,
Zhu J, Wu Y and Wang L: The biological features of PanIN initiated
from oncogenic Kras mutation in genetically engineered mouse
models. Cancer Lett. 339:135–143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu W, Yang Z and Lu N: A new role for the
PI3K/Akt signaling pathway in the epithelial-mesenchymal
transition. Cell Adh Migr. 9:317–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Shi J, Chai K, Ying X and Zhou BP:
The role of snail in EMT and tumorigenesis. Curr Cancer Drug
Targets. 13:963–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nieto MA: The snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qiao M, Sheng S and Pardee AB: Metastasis
and AKT activation. Cell Cycle. 7:2991–2996. 2014. View Article : Google Scholar
|
|
36
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:2140–2141. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Garrido-Laguna I and Hidalgo M: Pancreatic
cancer: From state-of-the-art treatments to promising novel
therapies. Nat Rev Clin Oncol. 12:319–334. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Infante JR, Somer BG, Park JO, Li CP,
Scheulen ME, Kasubhai SM, Oh DY, Liu Y, Redhu S, Steplewski K and
Le N: A randomised, double-blind, placebo-controlled trial of
trametinib, an oral MEK inhibitor, in combination with gemcitabine
for patients with untreated metastatic adenocarcinoma of the
pancreas. Eur J Cancer. 50:2072–2081. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ballon DR, Flanary PL, Gladue DP, Konopka
JB, Dohlman HG and Thorner J: DEP-domain-mediated regulation of
GPCR signaling responses. Cell. 126:1079–1093. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Consonni SV, Maurice MM and Bos JL: DEP
domains: Structurally similar but functionally different. Nat Rev
Mol Cell Biol. 15:357–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Khalil BD, Hsueh C, Cao Y, Abi Saab WF,
Wang Y, Condeelis JS, Bresnick AR and Backer JM: GPCR signaling
mediates tumor metastasis via PI3Kβ. Cancer Res. 76:2944–2953.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Murga C, Fukuhara S and Gutkind JS: A
novel role for phosphatidylinositol 3-kinase beta in signaling from
G protein-coupled receptors to Akt. J Biol Chem. 275:12069–12073.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dbouk HA, Vadas O, Shymanets A, Burke JE,
Salamon RS, Khalil BD, Barrett MO, Waldo GL, Surve C, Hsueh C, et
al: G protein-coupled receptor-mediated activation of p110beta by
Gbetagamma is required for cellular transformation and
invasiveness. Sci Signal. 5:ra892012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Allen PJ, Kuk D, Castillo CF, Basturk O,
Wolfgang CL, Cameron JL, Lillemoe KD, Ferrone CR, Morales-Oyarvide
V, He J, et al: Multi-institutional validation study of the
American joint commission on cancer (8th Edition) changes for T and
N staging in patients with pancreatic adenocarcinoma. Ann Surg.
265:185–191. 2017. View Article : Google Scholar : PubMed/NCBI
|