Introduction
Gliomas are the most common type of neuroepithelial
tumor of the central nervous system. Glioblastoma multiforme (GBM),
referring to grade-IV glioma, is one of the deadliest of human
cancer types. Based on the clinical course and molecular
characteristics, GBMs may be classified into two subtypes (1–3). Primary
GBMs refers to the vast majority of GBMs, which are thought to form
de novo in the elderly, with a median overall survival of 15
months after maximal surgical resection, chemotherapy and
radiotherapy (RT) (4). Secondary
GBMs (sGBMs) typically progress from lower-grade tumors and affect
younger patients. Since the Philadelphia chromosome was discovered
in chronic myeloid leukemia in 1960, studies performed over the
past six decades have identified fusion genes and proteins in a
multitude of other types of cancer and through several different
approaches (5). Fusion transcripts
between fibroblast growth factor receptor 3 and transforming acidic
coiled-coil containing protein 3 (6), and between the MYB proto-oncogene and
the quaking homolog KH domain RNA binding protein were initially
reported as the recurrent fusion transcripts in GBMs and pediatric
gliomas, respectively (7). A
previous study by our group identified a novel, recurrent fusion
rearrangement involving the protein tyrosine phosphatase receptor
type Z1 (PTPRZ1) and MET proto-oncogene receptor tyrosine kinase
(MET) genes (ZM) in 15% of sGBMs (8). However, the mechanisms by which these
genes contribute to gliomagenesis and progression in ZM-negative
sGBM have remained to be fully elucidated.
In the present study, RNA sequencing was performed
on 42 sGBM samples with or without ZM fusion. mRNAs with
differentially expressed genes between patients with and without ZM
fusion were identified and data were analyzed using a univariate
Cox regression model in R language. A total of six mRNAs were
selected to develop the risk score with random repeated sampling.
ZM-negative patients with high risk scores had a relatively shorter
overall survival (OS) time compared with those with low risk
scores. The risk score was independent of clinical observations,
including sex, age, isocitrate dehydrogenase (IDH) mutation status,
O-6-methylguanine-DNA methyltransferase (MGMT) methylation status
and therapeutic strategies. The prognostic value of the risk score
on patients' OS was higher compared with the aforementioned
clinical information. The immune cell response was enhanced in
ZM-negative patients with high risk scores. Increased macrophages
rather than endothelial score and NF-κB enrichment were identified
in patients with high risk scores. In summary, the risk score
signature may be robust in predicting the survival of patients with
ZM-negative sGBM and may help identify novel therapeutic targets
for further treatment for patients with ZM-negative sGBM.
Materials and methods
Samples
In the present study, 42 tumor samples of confirmed
sGBM were selected for the analysis. The samples were collected
from January 2005 through to December 2018. The methods of the RNA
sequencing were provided in a previous study (9). Our data used in the study were original
samples to be used to generate the dataset. The samples were
collected at Beijing Tiantan Hospital (Beijing, China) by our group
and the RNA sequencing data were uploaded to the Chinese Glioma
Genome Atlas (CGGA) RNA sequencing dataset (http://www.cgga.org.cn). For each patient, the
following clinical information was collected: Sex, age, IDH status,
MGMT promoter methylation status, RT, temozolomide (TMZ)
chemotherapy and OS. Among the 42 cases, 35 were ZM
fusion-negative, whereas the other 7 cases were ZM fusion-positive.
Among the patients, 24 were male and 11 were female. The average
age was 38.7 years (range, 8–58 years). The sample IDs of ZM
fusion-negative samples are provided in supplementary Table SI. The clinical and molecular
information of the 42 patients was obtained from the CGGA database
and held in the medical records. The tumor tissues included in the
CGGA database were obtained during surgery and informed consent was
obtained from all patients.
Gene selection
Student's t-test was used to identify the
differentially expressed genes between ZM-positive and -negative
cases with P<0.05 used as a selection threshold. Univariate Cox
regression was used to determine the genes associated with
survival. The differentially expressed genes associated with
survival were selected for further analysis. The hazard ratio (HR)
of univariate Cox regression was used to develop the gene
signature. The risk score model was developed using the following
formula: Risk Score = ∑ni=1
βi xi, where βi indicates the HR
for each gene in the CGGA database of ZM fusion-negative cases and
xi indicates the expression value of each gene (10).
Gene ontology (GO) analysis
The correlation between the risk score and other
genes was analyzed by Pearson correlation analysis using R
programming language. The positively correlated genes (r>0.4;
P<0.05) and the negatively correlated genes (r<-0.4,
P<0.05) were selected for analysis in the Database for
Annotation, Visualization and Integrated Discovery (DAVID;
http://david.abcc.ncifcrf.gov/home.jsp) to detect
which functional terms were associated with the risk score
(11).
Gene set enrichment analysis
(GSEA)
GSEA was used to analyze the association between the
risk score and the hallmarks. The data were divided into two groups
based on the risk score (low and high) and subjected to GSEA as
previously described (12). The
analysis was performed using the GSEA java software v4.0.3
(12).
Statistical analysis
In the present study, SPSS 16.0 (SPSS, Inc.), R
software 3.2.5 (11) and GraphPad
Prism 7.0 statistical software (GraphPad Software, Inc.) were used
for data analysis and plotting. The R package ‘survival’
(http://www.bioconductor.org/) was used
to analyze the most significant survival-associated mRNAs with the
highest area under the curve (AUC) value in the dataset using the
receiver operating characteristic curve analysis. The percentage of
macrophages and endothelial cells was calculated by EPIC in R
(13). Differentially expressed
genes between ZM fusion-positive and -negative cases were
determined by Student's t-test, which was also used to compare the
risk scores between two groups. Survival analysis was performed
using Kaplan-Meier survival analysis and univariate and
multivariate Cox regression analysis. The analyzed factors included
risk score, sex, age, IDH status, MGMT promoter methylation status,
RT and TMZ chemotherapy. P<0.05 was considered to indicate
statistical significance.
Results
Identification of survival-associated
mRNAs
To characterize the transcriptomic RNA expression in
sGBMs, RNA was extracted from 42 sGBM specimens with or without ZM
fusion to perform whole-transcriptome sequencing. Student's t-test
was performed to identify differentially expressed genes between
samples with and without ZM fusion (Fig.
1A). Univariate Cox regression was performed in the ZM-negative
cohort and the genes associated with survival (P<0.05) were
selected. The overlapping genes in the two analyses were selected
for further analysis. Using the R package ‘survival’, the most
significant survival-associated mRNAs with the highest AUC value in
the dataset were identified (Fig.
1B), which yielded six genes: Signal regulatory protein δ
(SIRPD), kinesin family member 11 (KIF11), zinc finger protein 117
(ZNF117), base methyltransferase of 25S rRNA 2 homolog (C7orf60),
FH2 domain containing 1 (FHDC1) and dimethylarginine
dimethylaminohydrolase 2 (DDAH2). Each of these genes was
differentially expressed between ZM-positive and -negative tumor
tissues and demonstrated lower expression levels in patients
without ZM fusion (Fig. 1C). The HR
of the survival analysis of each gene was also determined; the HRs
of SIRPD, KIF11, ZNF117 and DDAH2 were positive, which indicated
oncogenic characteristics in cell biological processes, whereas the
HRs of C7orf60 and FHDC1 were negative, which indicated that these
genes may suppress tumor occurrence (Fig. 1D).
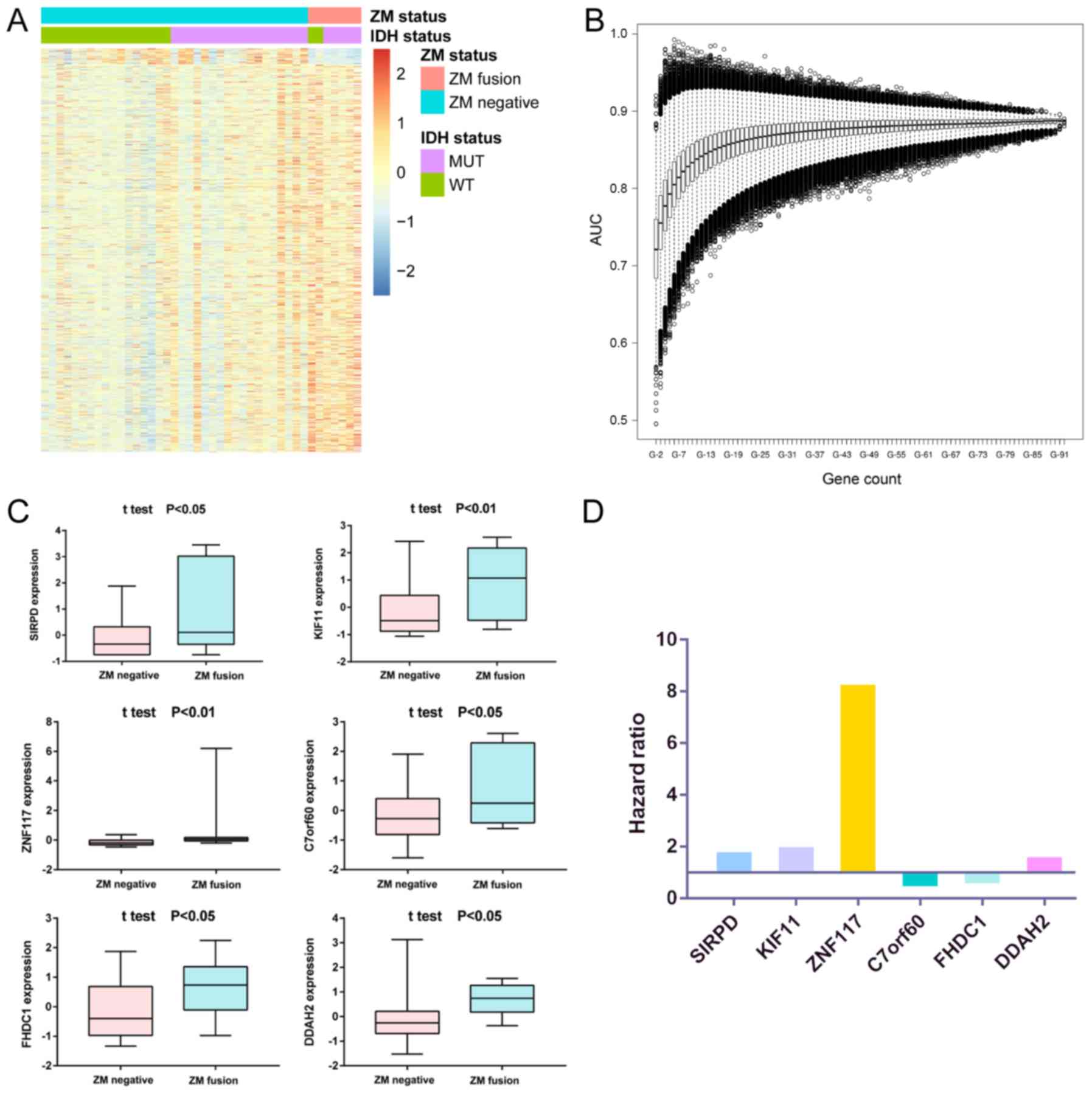 | Figure 1.(A) Differentially expressed genes
between ZM-positive and ZM-negative samples. (B) ROC curve analysis
of different numbers of genes to select to institute the risk score
which predict the 1-year survival with the best prediction. The
horizontal lines indicate the median AUC. The bars indicate the max
and min AUC. The boxes indicated the 25–75% AUC. The circles
represent the extreme value. (C) Expression of the six genes
between ZM-positive and ZM-negative samples. The horizontal lines
indicate the median expression. The bars indicate the maximum and
minimum expression. The boxes indicated the 25–75% expression. (D)
Hazard ratio of the six genes by univariate Cox regression
analysis. ZM, protein tyrosine phosphatase receptor type Z1-MET
proto-oncogene receptor tyrosine kinase fusion; AUC, area under the
curve; IDH, isocitrate dehydrogenase; MUT, mutated; WT, wild-type;
SIRPD, signal regulatory protein δ; KIF11, kinesin family member
11; ZNF117, zinc finger protein 117; C7orf60, base
methyltransferase of 25S rRNA 2 homolog; FHDC1, FH2 domain
containing 1; DDAH2, dimethylarginine dimethylaminohydrolase 2. |
Clinical and genomic characteristics
of risk score in patients with ZM-negative sGBM
To investigate the clinical and genomic
characteristics of the six selected genes, different weights were
assigned to each gene for an independent risk score. The risk score
of each patient was determined as follows: Risk score=(0.5368×SIRPD
expression) + (0.6481 × KIF11 expression) + (2.1026 × ZNF117
expression) + (−0.6095 × C7orf60 expression) + (−0.4264 × FHDC1
expression) + (0.4152 × DDAH2 expression). The corresponding
coefficients and P-values are presented in Table I. The high-risk score group was
distinguished from the low-risk score group by the median score
value as the cut-off value. Patients with ZM-negative sGBM with
high risk scores exhibited poor prognosis, with significantly
shorter OS times compared with those of patients with sGBM with low
risk scores (median OS: High risk score, 233 days; low risk score,
572 days; P<0.0001, log-rank test; Fig. 2A). In addition, the progression-free
survival (PFS) time in the high- and low-risk groups was evaluated
and patients with ZM-negative sGBM in the high-risk group
demonstrated a significantly shorter PFS time compared with
patients in the low-risk group, consistent with the trends of the
OS time (Fig. 2B). The correlation
between the risk score and mRNA expression was analyzed; the
results indicated that the risk score may be a potential prognostic
indicator (Fig. 2C).
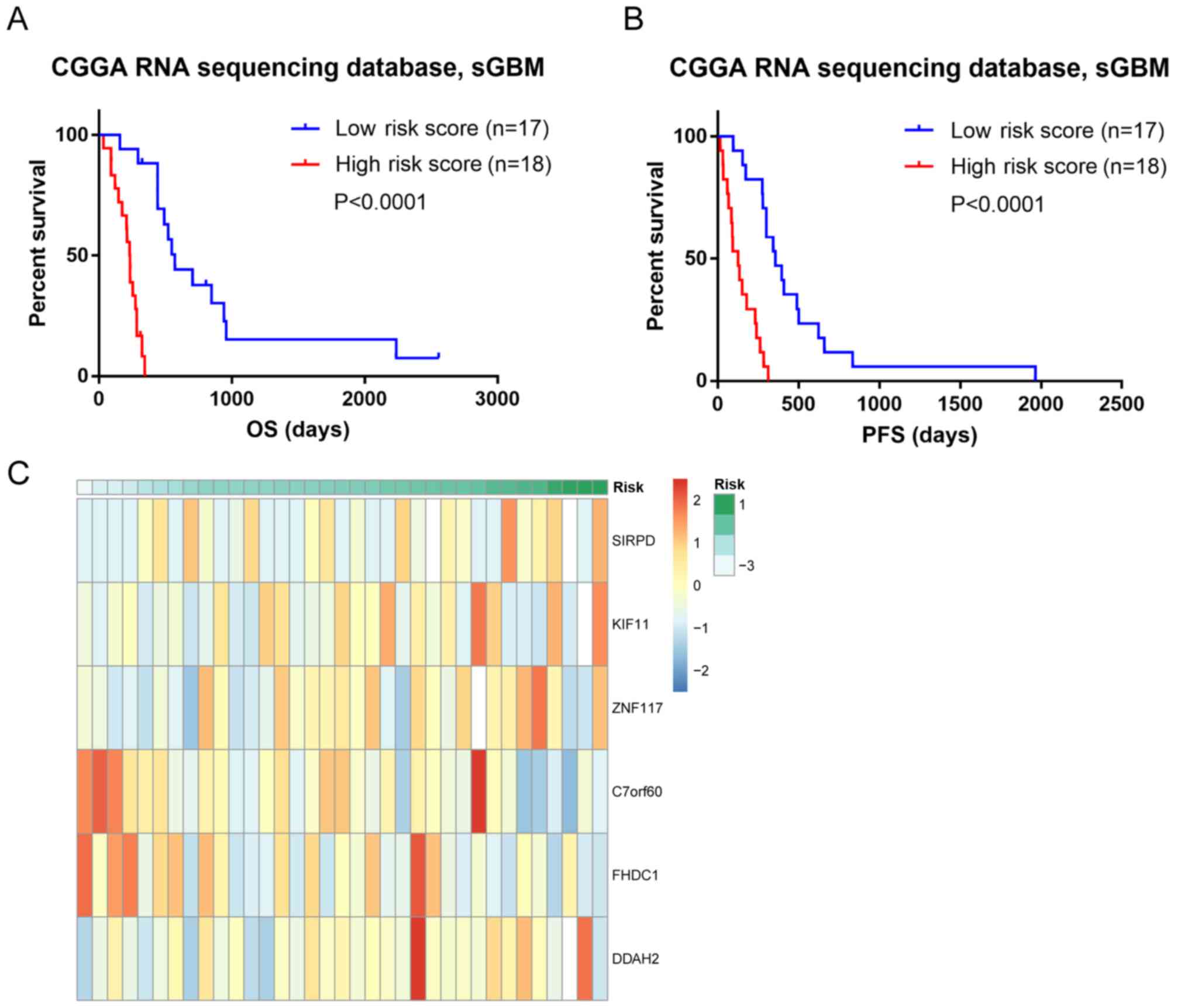 | Figure 2.(A) Survival analysis of patients
with ZM-negative sGBM in CGGA dataset. (B) PFS analysis in patients
with ZM-negative sGBM in CGGA dataset. (C) The correlation between
the risk score and the six gene expression. ZM, protein tyrosine
phosphatase receptor type Z1-MET proto-oncogene receptor tyrosine
kinase fusion; sGBM, secondary glioblastoma multiforme; CGGA,
Chinese Glioma Genome Atlas; OS, overall survival; PFS,
progression-free survival; SIRPD, signal regulatory protein δ;
KIF11, kinesin family member 11; ZNF117, zinc finger protein 117;
C7orf60, base methyltransferase of 25S rRNA 2 homolog; FHDC1, FH2
domain containing 1; DDAH2, dimethylarginine dimethylaminohydrolase
2. |
 | Table I.HR (95% CI) and P-values generated
from the univariate Cox regression analysis in the dataset for each
gene. |
Table I.
HR (95% CI) and P-values generated
from the univariate Cox regression analysis in the dataset for each
gene.
| Gene | HR | 95% CI | P-value |
|---|
| SIRPD | 1.711 | 1.025–2.855 | 0.040 |
| KIF11 | 1.912 | 1.125–3.248 | 0.017 |
| ZNF117 | 8.187 | 1.306–51.334 | 0.025 |
| C7orf60 | 0.544 | 0.339–0.871 | 0.011 |
| FHDC1 | 0.653 | 0.441–0.966 | 0.033 |
| DDAH2 | 1.515 | 1.079–2.126 | 0.016 |
Clinical indications of risk score as
an independent marker for prognosis
The association between the risk score and clinical
indicators, including sex, age, IDH status, MGMT promoter
methylation status, RT and TMZ chemotherapy, was analyzed. The
results demonstrated that the risk score was independent of sex,
age, IDH mutation status, RT and TMZ chemotherapy, but associated
with MGMT promoter methylation status (Fig. 3A). Uni- and multivariate Cox
regression analysis was performed on these clinical factors. IDH
status, risk score and TMZ chemotherapy were identified as
potential prognostic markers by univariate Cox analysis (Fig. 3B). In the multivariate analysis, the
risk score was the only factor identified as an independent
prognostic marker in ZM-negative sGBMs (Table II). Therefore, the risk score was
the most significant marker to indicate survival in patients with
ZM-negative sGBM.
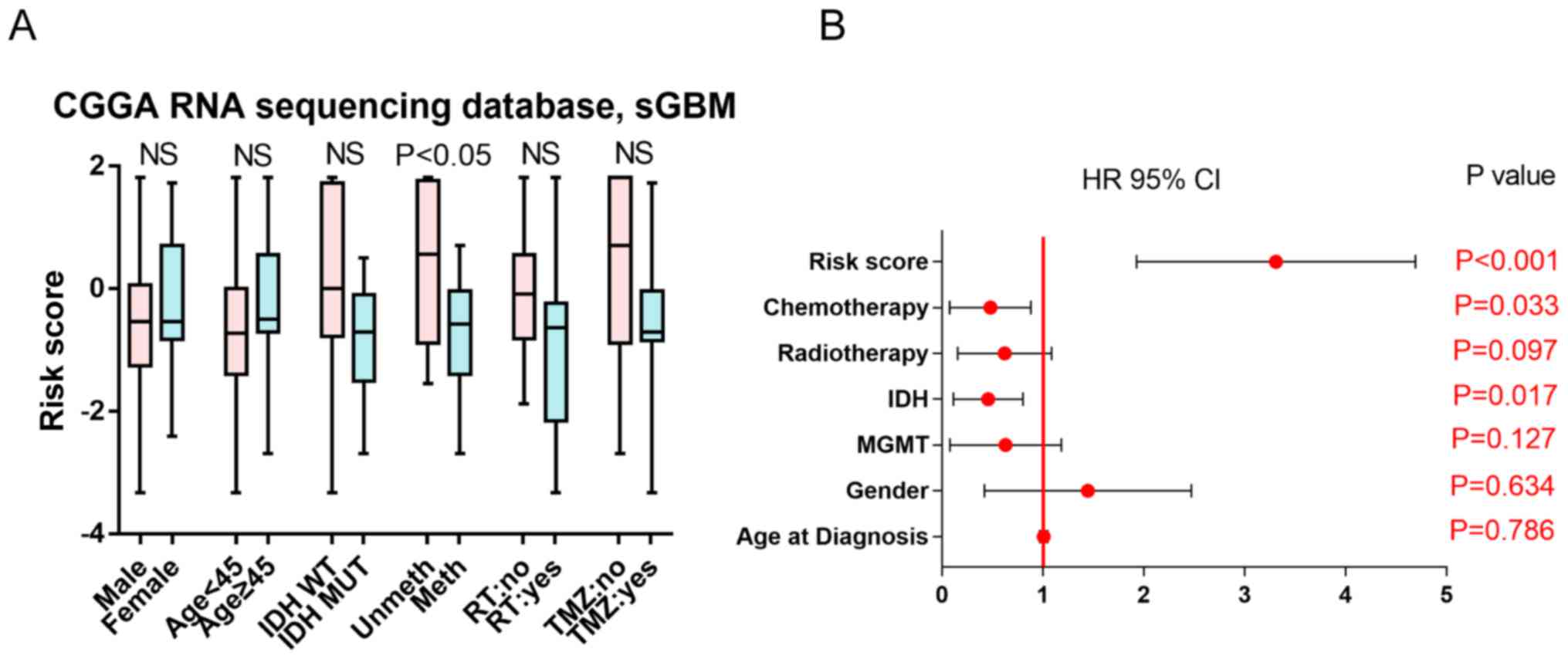 | Figure 3.(A) Risk score associated with
different clinical characteristics of patients. The horizontal
lines indicate the median risk score. The bars indicate the maximum
and minimum risk score. The boxes indicated the 25–75% risk score.
(B) Forest plot of univariate Cox analysis. HR, hazard ratio; IDH,
isocitrate dehydrogenase; MGMT, O-6-methylguanine-DNA
methyltransferase; NS, no significance; MUT, mutated; WT,
wild-type; unmeth, unmethylated MGMT promoter; CGGA, Chinese Glioma
Genome Atlas; sGBM, secondary glioblastoma multiforme; RT,
radiotherapy; TMZ, temozolomide. |
| Table II.Univariate and multivariate analysis
of clinical prognostic parameters of patients with protein tyrosine
phosphatase receptor type Z1-MET proto-oncogene receptor tyrosine
kinase fusion secondary glioblastoma multiforme in the Chinese
Glioma Genome Atlas RNA sequencing database. |
Table II.
Univariate and multivariate analysis
of clinical prognostic parameters of patients with protein tyrosine
phosphatase receptor type Z1-MET proto-oncogene receptor tyrosine
kinase fusion secondary glioblastoma multiforme in the Chinese
Glioma Genome Atlas RNA sequencing database.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variable | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Age at
diagnosis | 1.006 | 0.966–1.047 | 0.786 |
|
|
|
| Sex | 1.203 | 0.563–2.569 | 0.634 |
|
|
|
| MGMT | 0.468 | 0.176–1.243 | 0.127 |
|
|
|
| IDH | 0.369 | 0.163–0.836 | 0.017 | 1.087 | 0.392–3.018 | 0.392 |
| Radiotherapy | 0.503 | 0.223–1.133 | 0.097 |
|
|
|
| Chemotherapy | 0.364 | 0.144–0.924 | 0.033 | 0.369 | 0.114–1.194 | 0.096 |
| Risk score | 3.116 | 2.032–4.777 | <0.001 | 3.354 | 1.966–5.721 | <0.001 |
Functional pathway annotation in
patients with different risk scores
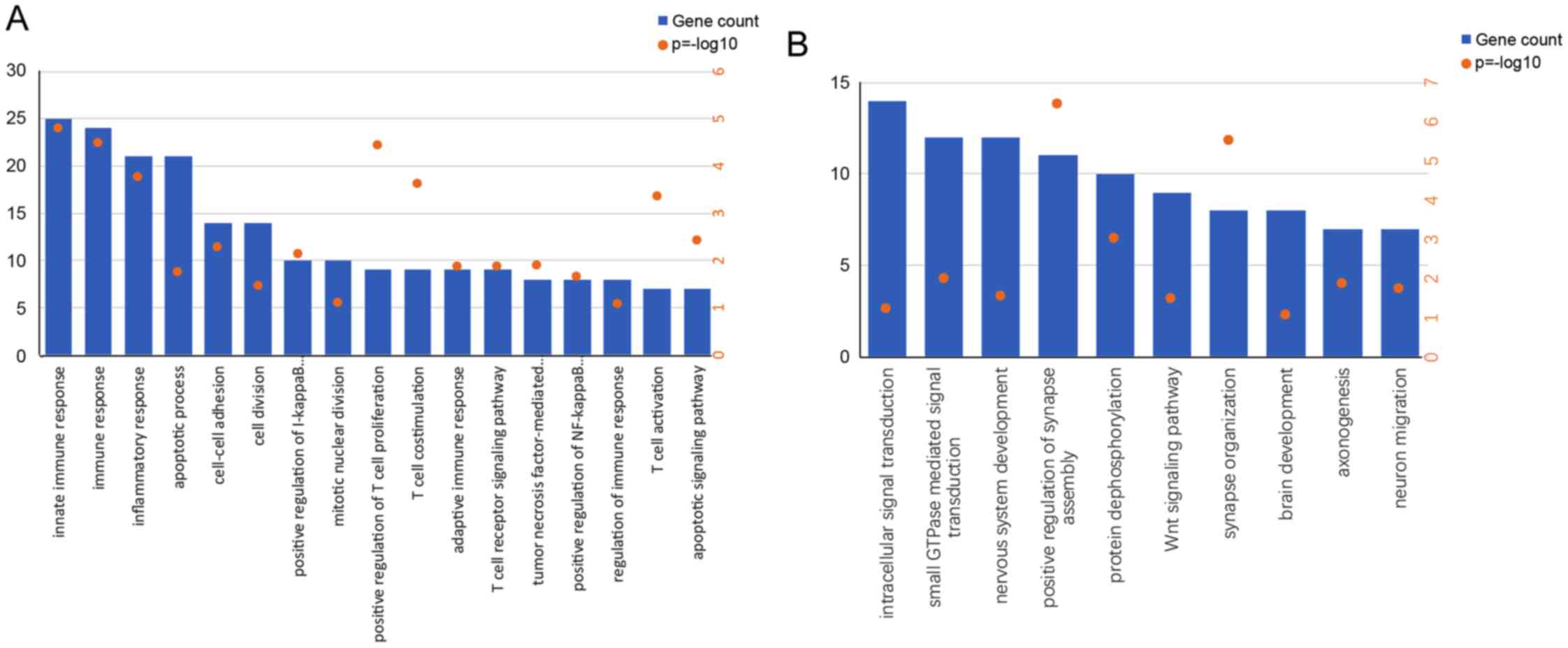
To investigate the potential functional pathways
activated in patients with high or low risk scores, GO analysis was
performed on genes positively or negatively associated with the
risk score. Genes enriched in ‘immune response’, ‘inflammatory
response’, ‘positive regulation of T-cell proliferation’ and ‘cell
division’ were significantly positively associated with the risk
score in patients (Fig. 4A), whereas
those enriched in relatively normal GO terms were negatively
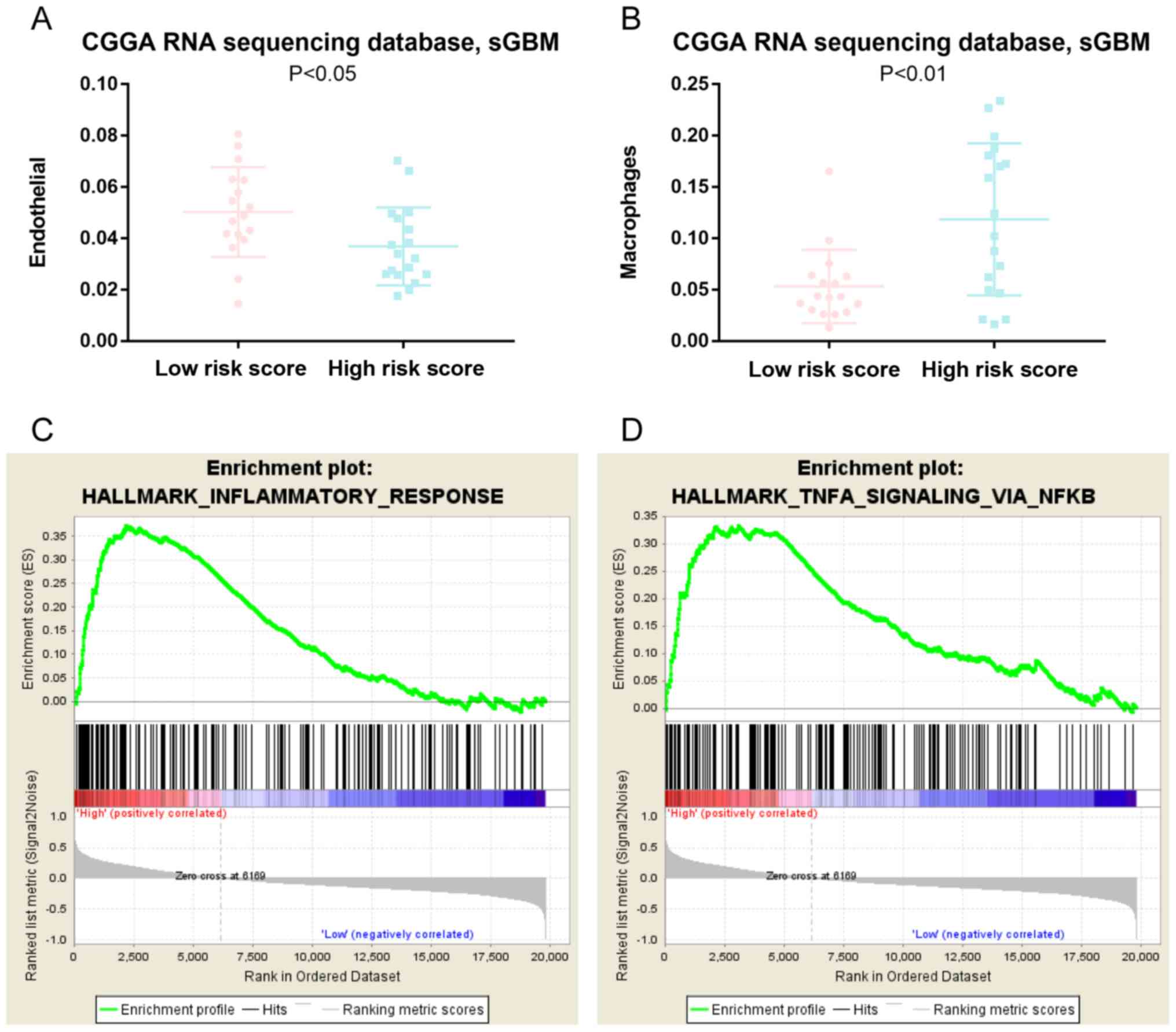
associated with the risk score in patients (Fig. 4B). To study the potential effect of
immune cell differences in the present cohort, reference gene
expression profiles of a major tumor-infiltrating immune cell type
(macrophages) were established and expression profiles were derived
from endothelial cells. The reference profiles were obtained as
cell type averages from the single-cell RNA sequencing data of
melanoma samples from Tirosh and colleagues (14). The reference cohort contained only
samples from primary tumors and non-lymphoid metastasis. The
reference gene expression profiles from each of the immune and
endothelial cells were used to predict the expression profile of
bulk tumor and to evaluate their association with the risk score of
patients with ZM-negative sGBM. The results demonstrated that
patients with sGBM without ZM fusion and a high risk score
exhibited a significantly higher macrophage signature expression
compared with that of patients with a low risk score (Fig. 5B). By contrast, patients with a high
risk score exhibited a lower endothelial cell signature expression
compared with that in patients with a low risk score (Fig. 5A), indicating that these pathways may
induce immune cells and suppress endothelial cells in patients with
a high risk score. GSEA was performed with the risk score ranging
from low to high; ZM-negative patients with higher risk scores
tended to exhibit a higher inflammatory response and tumor necrosis
factor (TNF)-α signaling activation by NF-κB (Fig. 5C and D). These results may provide
further therapeutic strategies for patients with ZM-negative
sGBM.
Discussion
Based on whole-transcriptome sequencing on 42 sGBM
samples from our own internal data, six genes were identified as an
independent signature with the most significant survival-associated
mRNAs and the best AUC in sGBMs without ZM fusion.
SIRPD is a member of a signal regulatory protein
family involved in signal transduction and cell adhesion (15). No evidence has been confirmed in
association with cancer initiation or progression.
The ZNF117 gene encodes a protein containing
multiple C2H2-type zinc finger motifs. It has
been demonstrated to participate in the maturation and
differentiation of adipocytes and may control fat accumulation
(16). The association between
ZNF117 and human cancer requires further investigation.
KIF11, which is located on chromosome 22, encodes a
kinesin-like motor protein that participates in chromosome
positioning, centrosome separation and bipolar spindle formation
during cell mitosis (17).
Interactions between KIF11, MCF7 and phosphatase and tensin homolog
regulate chromosome stability (18).
KIF11 has been identified as a novel prognostic biomarker and a
treatment target for various types of cancer, including lung
squamous cell carcinoma, prostate cancer and chronic myeloid
leukemia (19–22). In addition, previous studies
demonstrated that KIF11 was an important regulator of activity in
tumor stem cells of esophageal and colorectal cancer (23) and that upregulation of KIF11 was
associated with high-grade astrocytoma (24). Targeting KIF11 also altered the cell
fate and reduced glioma cell invasion (25). Thus, KIF11 may be a potential
therapeutic target in glioma treatment.
The FHDC1 gene is located on human chromosome 4q31.3
(26) and encodes a protein product
involved in the developmental stages of the mouse brain and the
actin- and microtubule-dependent regulation of Golgi morphology of
human cells (27). However, a
limited number of studies have demonstrated an association between
the FHDC1 gene and cancer development and progression. Further
clinical studies on FHDC1-induced glioma initiation and progression
require to be performed.
DDAH2 encodes a dimethylarginine
dimethylaminohydrolase, which is important in nitric oxide
generation by regulating the cellular concentrations of
methylarginines. DDAH2 has been demonstrated to be associated with
various types of disease, including hypertension (28), type 2 diabetes (29) and cancer. Upregulation of DDAH2 is
associated with the invasiveness of lung adenocarcinoma by inducing
proliferation and capillary-like tube formation of vascular
endothelial cells (30). Further
studies may focus on the therapeutic applications and the
tumorigenic mechanisms of DDAH2 in other types of human cancer.
C7orf60, also known as the base methyltransferase of
25S rRNA 2 homolog gene, was identified as a genomic event in human
complementary DNA (31,32) and associated with smoking cessation
(33) and Saccharomyces
cerevisiae infection (34).
However, C7orf60 has not been demonstrated to be associated with
cancer initiation or development.
In the present study, these six genes were assigned
as an independent signature with the most significant
survival-associated mRNAs and the best AUC. Patients with
ZM-negative sGBM and a high risk score exhibited a shorter OS and
PFS compared with those with a low risk score, revealing prognostic
value independent of other clinical features, including age,
gender, IDH mutation and MGMT promoter methylation status.
Over the past decades, the tumor microenvironment
and a number of tumor-associated cell types, including endothelial
cells, pericytes, immune inflammatory cells, cancer-associated
fibroblasts and stem and progenitor cells of the tumor stroma, have
increasingly been demonstrated to contribute to the biology of
numerous tumors and to regulate signaling that controls their
development and progression. Immune cells differ in their gene
expression profiles depending on their state and site of origin
(for example blood or tumors) (35).
Tumor-associated endothelial cell phenotypes have been implicated
in cancer development and tumor-associated angiogenesis (36–38).
These endothelial cells participate in establishing lymphatic
vessels (39) that serve as channels
for the seeding of metastases commonly observed in a number of
cancer types. Tumor-promoting inflammatory cells include various
macrophage subtypes that serve diverse and crucial roles in giving
rise to tumorigenesis and malignancy (40). Qian and Pollard (41) demonstrated that tumor-associated
macrophages suppressed cytotoxic T-cell and natural killer cell
activity, which have been independently identified as
myeloid-derived suppressor cells.
The results of the present study demonstrated that
ZM-negative sGBM patients with a high risk score exhibited
significantly higher macrophage expression compared with that of
patients with a low risk score, whereas patients with a high risk
score exhibited a lower endothelial cell signature expression
compared with those with a low risk score, indicating increased
immune cells and suppressed endothelial cells in samples from
patients with a high risk score. In addition, NF-κB-induced TNF-α
signaling activation tended to be enriched in patients with a high
risk score, implying potential therapeutic targets for treatment in
these patients.
The major advantage of the present study was the use
of next-generation sequencing and systematic analysis under
multidimensional conditions. The results identified a risk score
that was associated with immune response in patients with
ZM-negative sGBM and may guide further clinical management.
However, the extent of the immune response and the type of
macrophage activation that may promote glioma progression in
ZM-negative sGBM still requires to be clarified. The cellular
components within the tumor microenvironment requires to be studied
further to reveal their clinical implications. In addition to the
requirement for exploration of the immunoregulatory role of glioma
cells, the results of the present study require validation in
independent cohorts. Another limitation of the present stud is the
relatively low number of sGBM samples and future studies may be
performed in other, larger datasets, including The Cancer Genome
Atlas.
In conclusion, the present study demonstrated the
clinical utility of next-generation sequencing-based cancer gene
profiling in sGBM. The results revealed the diagnostic value of a
six gene-based risk score signature and clinical features of
patients with ZM-negative sGBM. An increase in immune cells and a
decrease in endothelial cells were identified in patients with a
high risk score; potential therapeutic strategies were identified
as NF-κB-induced TNF-α signaling activation in patients with
ZM-negative sGBM with a high risk score.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81502606), Beijing Municipal
Administration of Hospitals' Youth Program (grant no. QML20160502),
General Technological Program of Peking Education Commission (grant
no. KM201810025023) and Beijing Nova Program (grant no.
Z171100001117022).
Authors' contributions
BC and KW designed the study and drafted the
manuscript. SY revised the manuscript. SY, CZ, GL and ZW performed
the data analyses. ZB contributed to the conception of the study
and drafted the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Beijing
Tiantan Hospital institutional review board (Beijing, China) and
informed consent was obtained from all individual participants
included in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ZM-fusion
|
PTPRZ1-MET fusion
|
|
PTPRZ1
|
protein tyrosine phosphatase receptor
type Z1
|
|
MET
|
MET proto-oncogene receptor tyrosine
kinase
|
|
SIRPD
|
signal regulatory protein δ
|
|
ZNF117
|
zinc finger protein 117
|
|
KIF11
|
kinesin family member 11
|
|
FHDC1
|
FH2 domain containing 1
|
|
DDAH2
|
dimethylarginine
dimethylaminohydrolase 2
|
|
C7orf60
|
base methyltransferase of 25S rRNA 2
homolog
|
|
IDH
|
isocitrate dehydrogenase
|
|
sGBM
|
secondary glioblastoma multiforme
|
|
CGGA
|
Chinese Glioma Genome Atlas
|
|
GO
|
Gene Ontology
|
|
GSEA
|
Gene Set Enrichment Analysis
|
|
ROC
|
receiver operating characteristic
|
|
AUC
|
area under the curve
|
|
HR
|
hazard ratio
|
|
PFS
|
progression-free survival
|
|
OS
|
overall survival
|
References
|
1
|
Wang Y and Jiang T: Understanding high
grade glioma: Molecular mechanism, therapy and comprehensive
management. Cancer Lett. 331:139–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mertens F, Johansson B, Fioretos T and
Mitelman F: The emerging complexity of gene fusions in cancer. Nat
Rev Cancer. 15:371–381. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh D, Chan JM, Zoppoli P, Niola F,
Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S,
et al: Transforming fusions of FGFR and TACC genes in human
glioblastoma. Science. 337:1231–1235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bandopadhayay P, Ramkissoon LA, Jain P,
Bergthold G, Wala J, Zeid R, Schumacher SE, Urbanski L, O'Rourke R,
Gibson WJ, et al: MYB-QKI rearrangements in angiocentric glioma
drive tumorigenicity through a tripartite mechanism. Nat Genet.
48:273–282. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bao ZS, Chen HM, Yang MY, Zhang CB, Yu K,
Ye WL, Hu BQ, Yan W, Zhang W, Akers J, et al: RNA-seq of 272
gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in
secondary glioblastomas. Genome Res. 24:1765–1773. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang C, Cheng W, Ren X, Wang Z, Liu X, Li
G, Han S, Jiang T and Wu A: Tumor Purity as an Underlying Key
Factor in Glioma. Clin Cancer Res. 23:6279–6291. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chai R, Zhang K, Wang K, Li G, Huang R,
Zhao Z, Liu Y and Chen J: A novel gene signature based on five
glioblastoma stem-like cell relevant genes predicts the survival of
primary glioblastoma. J Cancer Res Clin Oncol. 144:439–447. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li
G, Liang J, Hu H, Liu Y, Zhang W and Jiang T: Molecular and
clinical characterization of PD-L1 expression at transcriptional
level via 976 samples of brain glioma. Oncoimmunology.
5:e11963102016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang K, Huang R, Li G, Zeng F, Zhao Z, Liu
Y, Hu H and Jiang T: CKAP2 expression is associated with glioma
tumor growth and acts as a prognostic factor in high-grade glioma.
Oncol Rep. 40:2036–2046. 2018.PubMed/NCBI
|
|
13
|
Pogorelyy MV, Fedorova AD, McLaren JE,
Ladell K, Bagaev DV, Eliseev AV, Mikelov AI, Koneva AE, Zvyagin IV,
Price DA, et al: Exploring the pre-immune landscape of
antigen-specific T cells. Genome Med. 10:682018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tirosh I, Izar B, Prakadan SM, Wadsworth
MH II, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G,
et al: Dissecting the multicellular ecosystem of metastatic
melanoma by single-cell RNA-seq. Science. 352:189–196. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deloukas P, Matthews LH, Ashurst J, Burton
J, Gilbert JG, Jones M, Stavrides G, Almeida JP, Babbage AK,
Bagguley CL, et al: The DNA sequence and comparative analysis of
human chromosome 20. Nature. 414:865–871. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ambele MA, Dessels C, Durandt C and Pepper
MS: Genome-wide analysis of gene expression during adipogenesis in
human adipose-derived stromal cells reveals novel patterns of gene
expression during adipocyte differentiation. Stem Cell Res.
16:725–734. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Ree JH, Nam HJ, Jeganathan KB,
Kanakkanthara A and van Deursen JM: Pten regulates spindle pole
movement through Dlg1-mediated recruitment of Eg5 to centrosomes.
Nat Cell Biol. 18:814–821. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feizabadi MS, Jun Y and Reddy JN:
Distinctions between dynamic characteristics of the single EG5
motor protein along neural vs. Cancerous microtubules. Biochem
Biophys Res Commun. 478:1630–1633. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu M, Zhu H, Wang X, Zhang D, Xiong L, Xu
L and You Y: The prognostic role of Eg5 expression in laryngeal
squamous cell carcinoma. Pathology. 48:214–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin Y, Sun H, Xu J, Xiao F, Wang H, Yang
Y, Ren H, Wu CT, Gao C and Wang L: Kinesin spindle protein
inhibitor SB743921 induces mitotic arrest and apoptosis and
overcomes imatinib resistance of chronic myeloid leukemia cells.
Leuk Lymphoma. 56:1813–1820. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wissing MD, De Morree ES, Dezentje VO,
Buijs JT, De Krijger RR, Smit VT, Van Weerden WM, Gelderblom H and
van der Pluijm G: Nuclear Eg5 (kinesin spindle protein) expression
predicts docetaxel response and prostate cancer aggressiveness.
Oncotarget. 5:7357–7367. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martens-de Kemp SR, Nagel R, Stigter-van
Walsum M, van der Meulen IH, van Beusechem VW, Braakhuis BJ and
Brakenhoff RH: Functional genetic screens identify genes essential
for tumor cell survival in head and neck and lung cancer. Clin
Cancer Res. 19:1994–2003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Imai T, Oue N, Sentani K, Sakamoto N,
Uraoka N, Egi H, Hinoi T, Ohdan H, Yoshida K and Yasui W: KIF11 Is
Required for Spheroid Formation by Oesophageal and Colorectal
Cancer Cells. Anticancer Res. 37:47–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu L, Liu X, Mare M, Dumont AS, Zhang H,
Yan D and Xiong Z: Overexpression of Eg5 correlates with high grade
astrocytic neoplasm. J Neurooncol. 126:77–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Venere M, Horbinski C, Crish JF, Jin X,
Vasanji A, Major J, Burrows AC, Chang C, Prokop J, Wu Q, et al: The
mitotic kinesin KIF11 is a driver of invasion, proliferation, and
self-renewal in glioblastoma. Sci Transl Med. 7:304ra1432015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Katoh M and Katoh M: Identification and
characterization of human FHDC1, mouse Fhdc1 and zebrafish fhdc1
genes in silico. Int J Mol Med. 13:929–934. 2004.PubMed/NCBI
|
|
27
|
Copeland SJ, Thurston SF and Copeland JW:
Actin- and microtubule-dependent regulation of Golgi morphology by
FHDC1. Mol Biol Cell. 27:260–276. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Z, Chen S, Zhang L, Lu G, Zhou C,
Wang DW, Wang L, Badengmu B, Zhai Z and Qin L: Association between
variation in the genes DDAH1 and DDAH2 and hypertension among
Uygur, Kazakh and Han ethnic groups in China. Sao Paulo Med J.
134:205–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yuan Q, Hu CP, Gong ZC, Bai YP, Liu SY, Li
YJ and Jiang JL: Accelerated onset of senescence of endothelial
progenitor cells in patients with type 2 diabetes mellitus: Role of
dimethylarginine dimethylaminohydrolase 2 and asymmetric
dimethylarginine. Biochem Biophys Res Commun. 458:869–876. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shiozawa T, Iyama S, Toshima S, Sakata A,
Usui S, Minami Y, Sato Y, Hizawa N and Noguchi M: Dimethylarginine
dimethylaminohydrolase 2 promotes tumor angiogenesis in lung
adenocarcinoma. Virchows Arch. 468:179–190. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kimura K, Wakamatsu A, Suzuki Y, Ota T,
Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K,
Wakaguri H, et al: Diversification of transcriptional modulation:
Large-scale identification and characterization of putative
alternative promoters of human genes. Genome Res. 16:55–65. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ota T, Suzuki Y, Nishikawa T, Otsuki T,
Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, et al:
Complete sequencing and characterization of 21,243 full-length
human cDNAs. Nat Genet. 36:40–45. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rose JE, Behm FM, Drgon T, Johnson C and
Uhl GR: Personalized smoking cessation: interactions between
nicotine dose, dependence and quit-success genotype score. Mol Med.
16:247–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sharma S, Watzinger P, Kotter P and Entian
KD: Identification of a novel methyltransferase, Bmt2, responsible
for the N-1-methyl-adenosine base modification of 25S rRNA in
Saccharomyces cerevisiae. Nucleic Acids Res. 41:5428–5443. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Racle J, de Jonge K, Baumgaertner P,
Speiser DE and Gfeller D: Simultaneous enumeration of cancer and
immune cell types from bulk tumor gene expression data. Elife.
6:e264762017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pasquale EB: Eph receptors and ephrins in
cancer: Bidirectional signalling and beyond. Nat Rev Cancer.
10:165–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahmed Z and Bicknell R: Angiogenic
signalling pathways. Methods Mol Biol. 467:3–24. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dejana E, Orsenigo F, Molendini C, Baluk P
and McDonald DM: Organization and signaling of endothelial
cell-to-cell junctions in various regions of the blood and
lymphatic vascular trees. Cell Tissue Res. 335:17–25. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tammela T and Alitalo K:
Lymphangiogenesis: Molecular mechanisms and future promise. Cell.
140:460–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Coffelt SB, Lewis CE, Naldini L, Brown JM,
Ferrara N and De Palma M: Elusive identities and overlapping
phenotypes of proangiogenic myeloid cells in tumors. Am J Pathol.
176:1564–1576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qian BZ and Pollard JW: Macrophage
diversity enhances tumor progression and metastasis. Cell.
141:39–51. 2010. View Article : Google Scholar : PubMed/NCBI
|