Introduction
Colorectal cancer (CRC) is the second most common
cause of cancer-associated mortality in the United States,
according to the statistics update in 2020 (1). Systemic therapies, including
5-Fluorouracil (Fu)-based chemotherapy, molecular-targeted therapy
and immunotherapy, have improved the 5-year relative survival rate
of patients with CRC to 65%, according to statistics in 2019
(2). However, non-specific cytotoxic
antitumor agents usually induce side effects and decrease patient
tolerance to treatment (3).
Furthermore, drug resistance remains a challenge, leading to the
failure of CRC treatment (4). In
addition to gaining an understanding of the mechanism of intrinsic
and acquired therapy resistance, researchers have focused on
small-molecule compounds that induce less toxicity and have greater
efficacy for cancer treatment (5,6). In our
previous study, a chemical library obtained from ChemBridge
Corporation was screened for potential novel anticancer agents. The
results demonstrated that a synthetic compound,
2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol (2,3-DCPE), inhibits
cell proliferation and induces apoptosis and cell cycle arrest in
CRC cells (7).
The cell cycle is a process comprised of complex and
consecutive changes involved in cell proliferation (8). Cell cycle arrest, one of the DNA damage
responses (DDR) to DNA repair and apoptosis, is determined
according to severity of DNA damage (9). In response to DNA damage or DNA
replication blockage, cell cycle progression can be stalled in the
G1, S or G2 phase (9). This cell cycle arrest mechanism serves
as a protective system by which cells can repair damage and
maintain genomic stability (9).
Ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and
Rad3-related (ATR), members of phosphatidylinositol
3-kinase-related kinase family of proteins, are two important DDR
transducers that interact with p53, checkpoint kinase (Chk)1, Chk2
and CDK (9). Certain investigators
have reported S phase arrest in cancer cells treated with various
chemotherapeutic agents, including 5-Fu, mitomycin and cisplatin
(10,11). The ATM/ATR pathway is involved in S
phase arrest through the activation of Chk1 or Chk2 (12).
Our previous study demonstrated that 2,3-DCPE
induced S phase arrest, which was also mediated by activation of
the p53-independent ERK pathway in DLD-1 human colon cancer cells
(7). Additionally, 2,3-DCPE-induced
S phase arrest may be blocked by the ATM inhibitors wortmannin and
caffeine (7). These observations
indicated that, in addition to the function of the ERK pathway,
other mechanisms were involved in 2,3-DCPE-induced S phase arrest,
which prompted the investigation of the current study. The present
study aimed to investigate the molecular mechanism that is
associated with 2,3-DCPE-induced S phase arrest by in vitro
experiments.
Materials and methods
Cells and cell culture
The DLD-1 human colon cancer cell line was obtained
from the American Type Culture Collection. The cells were
maintained in RPMI-1640 supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.), 1% glutamine and 1% antibiotics, and
cultured at 37°C in a humidified incubator containing 5%
CO2.
Chemicals
DMSO was purchased from Sigma-Aldrich; Merck KGaA
and 2,3-DCPE was purchased from ChemBridge Corporation. 2,3-DCPE
was dissolved in DMSO at 20 mM to create a stock solution.
Wortmannin and caffeine were obtained from Sigma-Aldrich; Merck
KGaA.
Drug treatment
Exponentially proliferating DLD-1 cells were
continuously exposed to 2,3-DCPE. The cells were treated with 20 µM
2,3-DCPE for 8, 10, 12, 14, 16, 18, 24 and 32 h to investigate the
effect of cell cycle arrest and DDR-associated proteins. DMSO alone
was used as the control because it does not have any effect on
cells. Cells were pretreated in wortmannin (500 nM) or caffeine (2
mM) for 2 h and 2,3-DCPE (20 µM) was added and incubated for
another 24 h to investigate the effect of ATM/ATR inhibition on S
phase arrest. Cells cultured in DMSO were used as controls. Cells
were cultured under different pretreatment conditions (ATM/ATR
inhibitors or controls), as aforementioned, for 2 h and then
treated with 2,3-DCPE (20 µM) for a further 32 h to detect the
effect of ATM/ATR inhibitors on 2,3-DCPE-induced apoptosis. Each
experiment was performed, at least, in triplicate.
Flow cytometry assays
Following treatment, suspended DLD-1 cells were
collected separately and adherent cells were trypsinized. Then, the
cells were pooled and centrifuged at 2,000 × g at 4°C for 5 min
prior to being fixed in 70% ethanol overnight at 4°C. Following
this, the cells were stained with propidium iodide for analysis of
DNA content. Flow cytometry was performed at the Flow Cytometry
Core Laboratory at our institution as described previously
(7).
Western blotting
DLD-1 cells were rinsed with ice-cold PBS and lysed
in Laemmli lysis buffer (4% SDS, 20% glycerol, 10%
2-mercaptoethanol, 0.004% bromphenol blue, 0.125 M Tris HCl).
Protein concentration was determined using the BCA Assay kit (cat.
no. 23227, Pierce; Thermo Fisher Scientific). Equal amounts (20
µg/lane) of total cellular proteins were loaded onto a 10%
polyacrylamide gel, resolved using SDS-PAGE and subsequently
transferred to a PVDF membrane (Amersham; Cytiva). The membrane was
blocked for 1 h at room temperature in phosphate-buffered saline
containing 0.05% Tween-20 (PBST) supplemented with 5% non-fat dry
milk. The membrane was incubated overnight at 4°C with the
following primary antibodies: Phosphorylated (p)-Chk1(Ser317 and
Ser345) and p-Chk2 (Ser19, Ser33/35, Thr68) (Cell Signaling
Technology, Inc., cat. nos. 12302, 2348, 2666, 2665 and 2661,
respectively; 1:1,000 dilution), mouse anti-human Chk1/2 (Santa
Cruz Biotechnology, Inc., cat. no. sc-8408/sc-17747, 1:1,000
dilution), mouse anti-human Cdc25A (Santa Cruz Biotechnology, Inc.,
cat. no. sc-7389, 1:1,000 dilution) and mouse monoclonal
anti-phosphorylated-H2A histone family member X (p-H2A.X; Ser 139;
Upstate Biotechnology, Inc., cat. no. 613401, 1:1,000 dilution).
β-actin (Cell Signaling Technology, Inc., cat. no. 4970, 1:1,000
dilution) was used as the loading control. Following three washes
with PBST, the membrane was incubated for 1 h at room temperature
with the appropriate horseradish peroxidase-conjugated secondary
antibody (anti-rabbit/mouse IgG; Cell Signaling Technology, Inc.,
cat. no. 7074/7076, 1:2,000 dilution). After three washes with
PBST, immunoreactivity was observed using an ECL kit (Amersham;
Cytiva).
Statistical analysis
Statistical analysis was performed using SPSS
statistical software (version 21.0; IBM Corp.). All experiments
were performed in triplicate and data are presented as the mean ±
standard deviation. One-way ANOVA and Bonferroni's correction post
hoc analysis were used for comparison between multiple groups. The
histograms were plotted using Graph Pad Prism software (version
6.0; GraphPad Software, Inc.). P<0.05 was considered to indicate
a statistically significant difference.
Results
DNA damage is induced by 2,3-DCPE
To investigate the potential molecular mechanism of
2,3-DCPE as an anticancer treatment, DLD-1 colon cancer cells were
treated with 20 µM 2,3-DCPE for different durations (8, 10, 12, 14,
16 and 18 h) as a single agent. The cells were harvested and
changes in the cell cycle following treatment were analyzed using
flow. 2,3-DCPE induced an increase in the proportion of cells in
the S phase in a time-dependent manner (Fig. 1A). Additionally, the presence of DNA
damage was evaluated by measuring H2A.X levels. As shown in
Fig. 1B, p-H2A.X levels were
markedly increased in the cells treated with 20 μM 2,3-DCPE for 14,
16 and 18 h compared with the DMSO-treated group, while the
expression of total H2A.X did not exhibit a marked change. Since
H2A.X is phosphorylated in the initial stage of DNA double-strand
breaks (DSBs) (9), these findings
indicated that 2,3-DCPE may induce DSBs accompanied by cell cycle
arrest in the S phase.
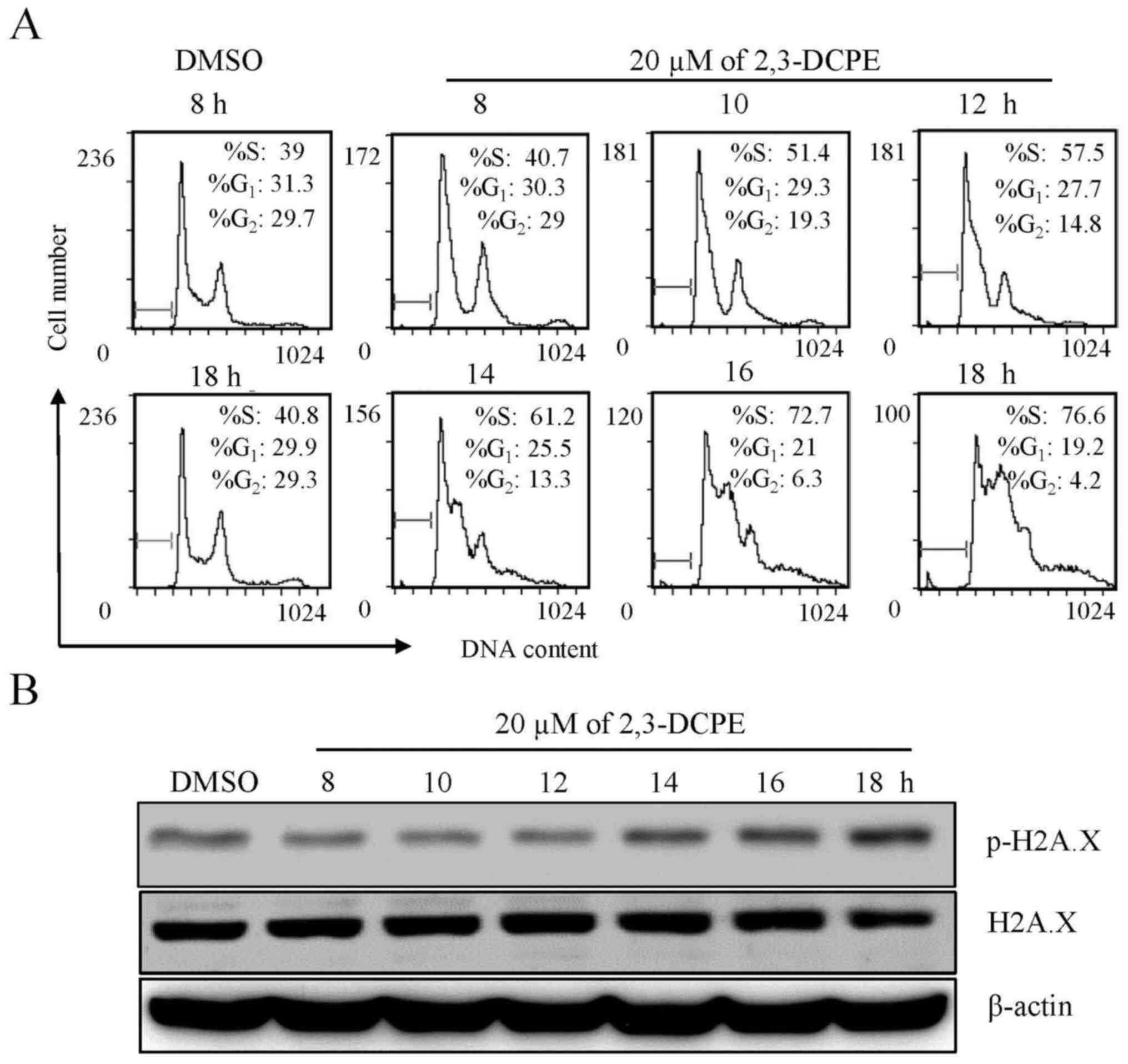 | Figure 1.Effect of 2,3-DCPE on S phase arrest
and p-H2A.X expression in the DLD-1 cell line. Cells were treated
with 20 μM 2,3-DCPE for 8, 10, 12, 14, 16 or 18 h. (A) Cell cycle
distribution is presented for each experimental condition. (B)
p-H2A.X and total H2A.X levels in the cellular extracts were
determined by western blotting with specific antibodies. β-actin
was used as an internal control. 2,3-DCPE,
2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol; p-, phosphorylated;
H2A.X, H2A histone family member X. |
2,3-DCPE-induced S phase arrest is
associated with the activation of Chk1 and the degradation of
Cdc25A
Subsequently, the expression levels of DDR-related
proteins, including Chk1 and Chk2, were evaluated by western
blotting. The results indicated that the expression level of p-Chk1
(Ser317 and Ser345) was markedly increased; however, the total
levels of Chk1 and Chk2 were markedly decreased in the cells
treated with 20 µM 2,3-DCPE for 16, 24 and 32 h compared with the
DMSO-treated group (Fig. 2A). There
was no difference between the different phosphorylation sites of
Chk2 following treatment with 20 µM 2,3-DCPE, including at sites
Ser19, Ser33/35, and Thr68 (data not shown). Cdc25A phosphatase is
one of the key targets of the checkpoint machinery that ensure
genetic stability (13). The most
important mechanism of Cdc25A function in regulating cell cycle
progression is the dephosphorylation of cyclin D-dependent kinases
(CDK4 and CDK6), which leads to the transition into S phase
(13). Therefore, the effects of
2,3-DCPE on the expression of Cdc25A in the DLD-1 cells were
examined. The downregulation of Cdc25A resulting from 2,3-DCPE
treatment is presented in Fig. 2B.
The results demonstrated that 2,3-DCPE decreased the expression of
Cdc25A in a time-dependent manner. Therefore, the data indicated
that DDR induced by 2,3-DCPE may involve the Chk1-Cdc25A signaling
pathway.
ATM/ATR inhibition blocks
2,3-DCPE-induced S phase arrest
As an elementary investigation of the molecular
mechanism of 2,3-DCPE-induced S phase arrest, experiments with
ATM/ATR pathway inhibitors (wortmannin and caffeine) on the DLD-1
cell line were performed. The results demonstrated that neither
wortmannin nor caffeine as a single agent affected the cell cycle;
however, when cells were treated with wortmannin/caffeine +
2,3-DCPE, the inhibitors partially blocked 2,3-DCPE-induced S phase
arrest (Fig. 3A and B). Caffeine
reduced the proportion of S phase cells from 83% in the
2,3-DCPE/PBS-treated group to 39.6% in the caffeine/2,3-DCPE group,
and wortmannin reduced the proportion of S phase cells from 83 to
48.2% in the wortmannin/2,3-DCPE group. Moreover, wortmannin and
caffeine markedly inhibited the phosphorylation of Chk1 (Ser345 and
S317) and subsequently suppressed the degradation of Cdc25A
(Fig. 3C). These findings indicated
that the ATM-Chk1-Cdc25A pathway may be critical for
2,3-DCPE-induced S phase arrest in DLD-1 colon cancer cells.
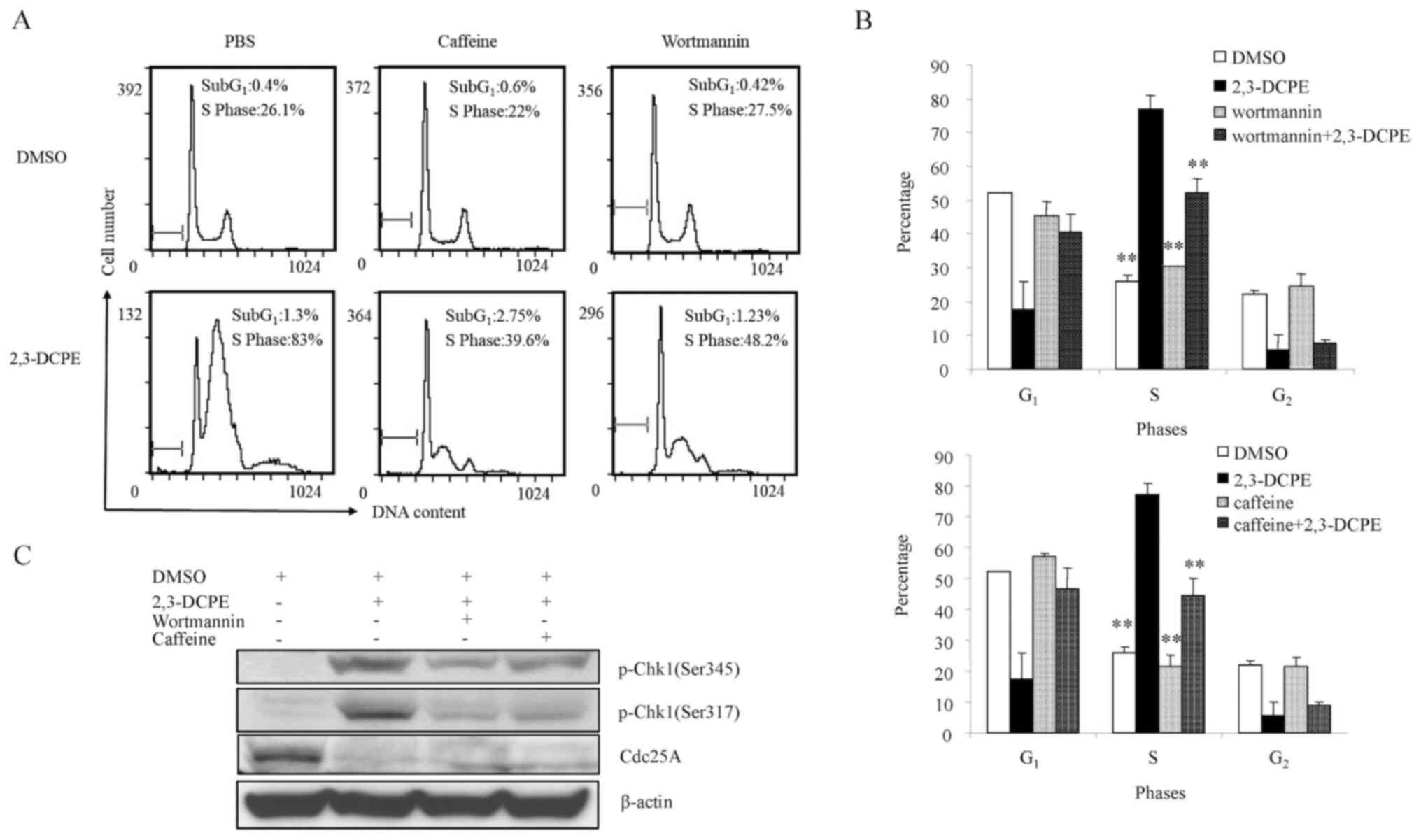 | Figure 3.Effect of ATM inhibitors on the
2,3-DCPE-induced cell cycle arrest in the S phase. (A) Cell cycle
distribution was determined by flow cytometry assays and (B) the
quantitative analysis is presented in the bar chart. Data are
expressed as the mean ± standard deviation. **P<0.01 vs. the
2,3-DCPE group. (C) Western blotting was performed to investigate
the p-Chk1 and Cdc25A expression after cells were treated with
2,3-DCPE and wortmannin or caffeine. 2,3-DCPE,
2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol; p-, phosphorylated;
Chk, checkpoint kinase; ATM, ataxia-telangiectasia mutated; Cdc25A,
M-phase inducer phosphatase 1; DMSO, dimethyl sulfoxide. |
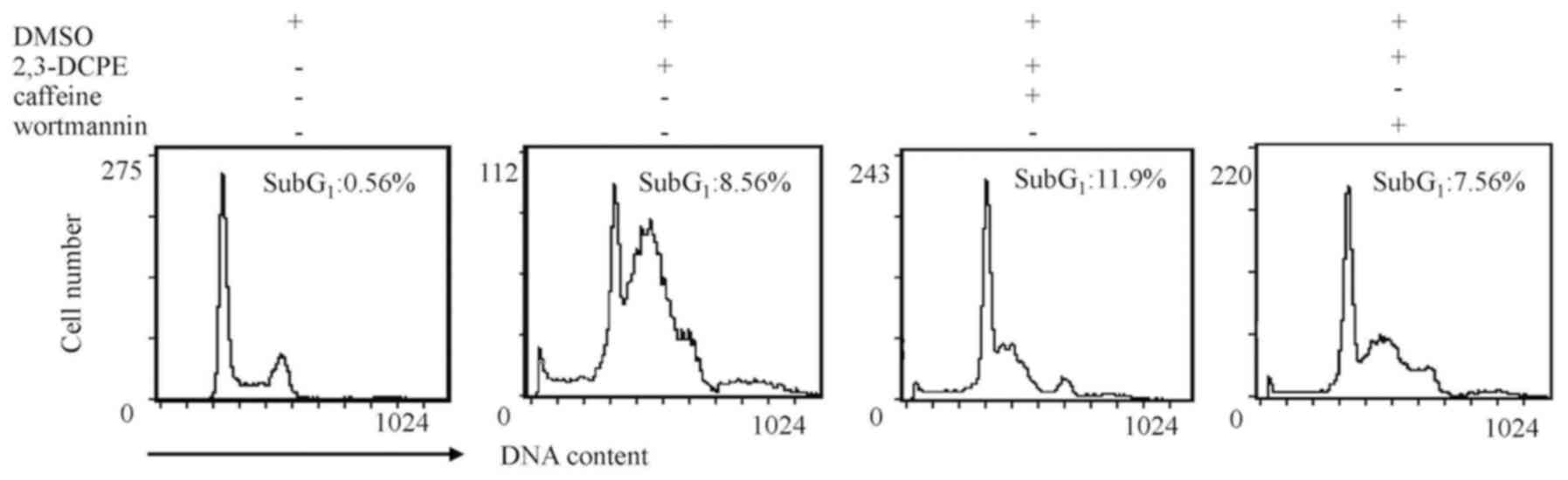
ATM/ATR inhibitors have a limited
effect on apoptosis
To further verify the apoptotic effects of ATM/ATR
inhibitors on DLD-1 cells, apoptosis rates were analyzed using flow
cytometry. As presented by Fig. 4,
caffeine increased the percentage of cells in sub-G1
from 8.56 to 11.9%; however, this difference was not notably
different. The apoptotic sub-G1 peak was not notably
different in cells treated with wortmannin. These data indicated
that, under these conditions, ATM/ATR inhibitors do not affect
2,3-DCPE-induced apoptosis.
Discussion
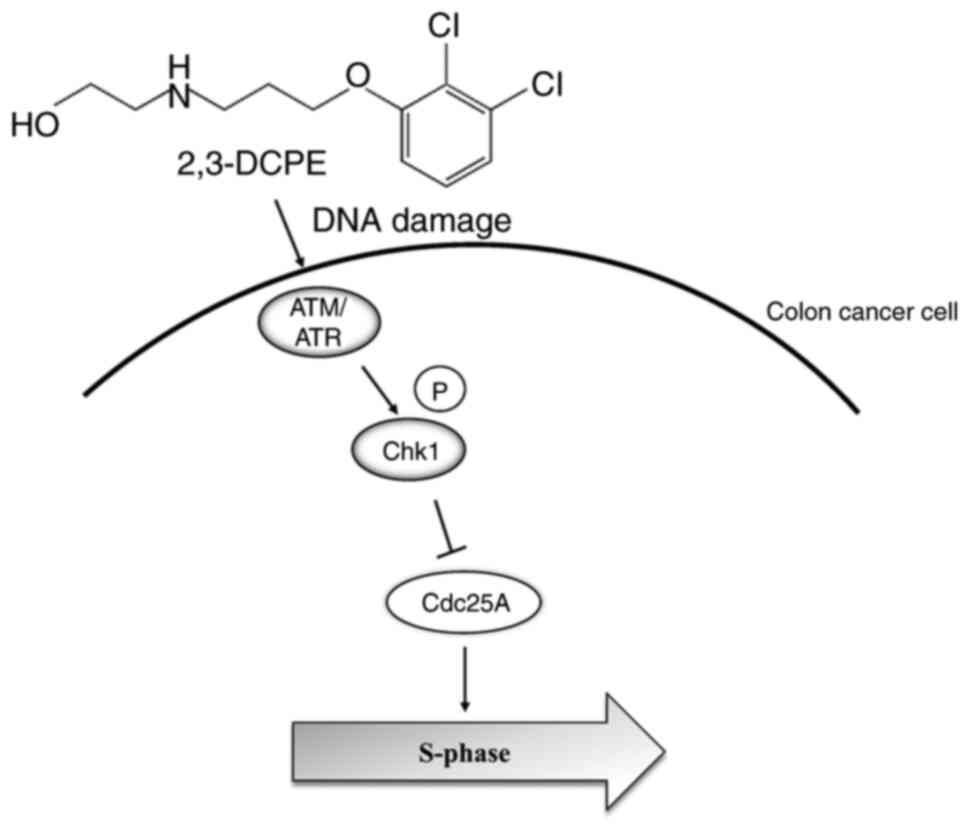
Our previous study demonstrated that 2,3-DCPE
induced S phase arrest and p21 overexpression; these results was
also observed in ATM-defective cancer cells (7). These findings indicated that the
antitumor effect of 2,3-DCPE may not depend on ATM. However,
molecular mechanisms underlying ATM and the corresponding signaling
pathway in cell cycle arrest remain to be elucidated. The present
study verified that 2,3-DCPE induced DNA damage and S phase arrest
via the ATM-Chk1-Cdc25A pathway (Fig.
5).
Common therapies for colon cancer, including
chemotherapy and radiotherapy, effectively kill cancer cells by
inducing DNA damage, the most deleterious type of which being the
DNA DSB (14). The phosphorylation
of H2A.X is considered an initial event in DSBs that leads to the
subsequent DDR (15). Certain agents
utilized for cancer therapy, including traditional chemotherapeutic
drugs and small-molecule compounds, increase H2A.X levels. The
increase in H2A.X is associated with the susceptibility of cancer
cells to treatment options (16–18). In
the present study, 2,3-DCPE increased p-H2A.X levels in DLD-1 cells
in a time-dependent manner, indicating its DNA-damaging effect.
To maintain the normal process of the cell cycle,
the DDR system is activated to modulate and repair DNA damage, in
which the cell cycle checkpoint is the key regulatory mechanism
(8). The G1/S checkpoint
is controlled by Chk2, while the G2/M checkpoint is
regulated by Chk1; thereafter, checkpoint kinases regulate cyclins
or cell division cycle genes to induce corresponding cell cycle
arrest (19). Numerous agents used
for CRC therapy regulate checkpoints and affect the cell cycle
(20). For instance, cisplatin
activates the G2/M checkpoint and decelerates the
replication phase, whereas oxaliplatin regulates the
G1/S checkpoint and blocks the G2/M
transition (21). Our previous study
demonstrated that 2,3-DCPE-induced S phase arrest may be mediated
by the activation of the p53-independent ERK/p21 pathway in DLD-1
human colon cancer cells (7). The
data of the current study demonstrated that p-Chk1 (Ser317 and
Ser345) was upregulated and Cdc25A was downregulated in the DLD-1
cells treated with 2,3-DCPE in a time-dependent manner. However,
the expression of p-Chk2 was not significantly altered. It has been
reported that Chk1 serves an important role in cell proliferation,
cell cycle and apoptosis in colon cancer (22,23).
Numerous therapeutic agents act on Chk1. For example, lidamycin, an
enediyne antibiotic, acts as an antitumor agent on colon tumor
cells by increasing the phosphorylation of Chk1 and Cdc25C, and the
expression of cyclin B, causing cell arrest in the G2
phase (24). Loratadine damages cell
DNA in colon cancer cells, thereby activating Chk1 and promoting
arrest in G2/M (25).
Similar to the results of the current study, it was reported that
5-Fu induces S phase arrest by activating Chk1. Furthermore, Chk1
downregulation abrogates this arrest and sensitizes colon cancer
cells to 5-Fu treatment (26). The
inhibition of Chk1 inducing chemosensitivity will be a novel focus
for future studies.
Moreover, the data of the current study demonstrated
that ATM/ATR inhibitors, wortmannin and caffeine, partially blocked
2,3-DCPE-induced S phase arrest and inhibited the phosphorylation
of Chk1 and the degradation of Cdc25A. The role of the ATM/ATR
signaling pathway in DDR has been extensively investigated. This
pathway is activated when intracellular DNA is damaged and leads to
cell cycle arrest in the S phase by the subsequent phosphorylation
of Chk1 and the degradation of Cdc25A (19,27).
Additionally, a previous study reported that ATM inhibition induces
chemoresistance to 5-Fu therapy (9).
The data from the current study indicated that the
ATM/ATR-Chk1-Cdc25A pathway was involved in 2,3-DCPE-induced S
phase arrest in the DLD-1 colon cancer cells.
The western blotting results in our previous study
and flow cytometry analysis in the current study demonstrated
2,3-DCPE-induced apoptosis in colon cancer cells (28). Notably, in the present study, when
cells are further treated with ATM/ATR inhibitors, 2,3-DCPE-induced
S phase arrest is partially blocked without a notable effect on
apoptosis. It was hypothesized that cell cycle arrest is a
protective response to DNA damage. Abrogation of the S phase
checkpoint causes excessive accumulation of DNA damage and induces
apoptosis (29). In the present
study, caffeine and wortmannin had limited effects on
2,3-DCPE-induced apoptosis. These results may be partially
explained by the fact that caffeine and wortmannin are non-specific
inhibitors with low potency and by the intricate mechanisms of
2,3-DCPE-induced cell cycle arrest and apoptosis; however, these
outcomes require further exploration.
There were certain limitations in the current study.
Other proteins related to ATM/ATR pathway, including cyclin B and
cdk2, were not detected. Whether ATM/ATR is the direct target of
2,3-DCPE requires more detailed elaboration by western blotting and
mass spectrometry.
In conclusion, the results of the current study
demonstrated that 2,3-DCPE induced S phase arrest via activation of
the ATM/ATR-Chk1-Cdc25A pathway in DLD-1 colon cancer cells,
furthering our understanding of 2,3-DCPE in colon cancer
therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81272681).
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
BB, LS, JW, JH, WZ, YL, KC, DX and HZ contributed to
the conception and design of the current study. Material
preparation, data collection was performed by BB, LS and JW. Data
analysis and the first draft of the manuscript was written by JH,
WZ and YL. Writing review and editing were performed by KC, DX and
HZ. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miller KD, Nogueira L, Mariotto AB,
Rowland JH, Yabroff KR, Alfano CM, Jemal A, Kramer JL and Siegel
RL: Cancer treatment and survivorship statistics, 2019. CA Cancer J
Clin. 69:363–385. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van der Jeught K, Xu HC, Li YJ, Lu XB and
Ji G: Drug resistance and new therapies in colorectal cancer. World
J Gastroenterol. 24:3834–3848. 2018. View Article : Google Scholar
|
|
5
|
Vidimar V, Licona C, Cerón-Camacho R,
Guerin E, Coliat P, Venkatasamy A, Ali M, Guenot D, Le Lagadec R,
Jung AC, et al: A redox ruthenium compound directly targets PHD2
and inhibits the HIF1 pathway to reduce tumor angiogenesis
independently of p53. Cancer Lett 440–441. 145–155. 2019.
View Article : Google Scholar
|
|
6
|
Tripodi F, Dapiaggi F, Orsini F, Pagliarin
R, Sello G and Coccetti P: Synthesis and biological evaluation of
new 3-amino-2-azetidinone derivatives as anti-colorectal cancer
agents. MedChemComm. 9:843–852. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu H, Zhang L, Wu S, Teraishi F, Davis
JJ, Jacob D and Fang B: Induction of S-phase arrest and p21
overexpression by a small molecule
2[[3-(2,3-dichlorophenoxy)propyl] amino]ethanol in correlation with
activation of ERK. Oncogene. 23:4984–4992. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hartwell LH and Kastan MB: Cell cycle
control and cancer. Science. 266:1821–1828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mirza-Aghazadeh-Attari M, Darband SG,
Kaviani M, Mihanfar A, Aghazadeh Attari J, Yousefi B and Majidinia
M: DNA damage response and repair in colorectal cancer: Defects,
regulation and therapeutic implications. DNA Repair (Amst).
69:34–52. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang B, Cui B, Du J, Shen X, Wang K, Chen
J, Xiao L, Sun C and Li Y: ATR activated by EB virus facilitates
chemotherapy resistance to cisplatin or 5-fluorouracil in human
nasopharyngeal carcinoma. Cancer Manag Res. 11:573–585. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu FY, Wu YH, Zhou SJ, Deng YL, Zhang ZY,
Zhang EL and Huang ZY: Minocycline and cisplatin exert synergistic
growth suppression on hepatocellular carcinoma by inducing S phase
arrest and apoptosis. Oncol Rep. 32:835–844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yao J, Huang A, Zheng X, Liu T, Lin Z,
Zhang S, Yang Q, Zhang T and Ma H: 53BP1 loss induces
chemoresistance of colorectal cancer cells to 5-fluorouracil by
inhibiting the ATM-CHK2-P53 pathway. J Cancer Res Clin Oncol.
143:419–431. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen T and Huang S: The role of Cdc25A in
the regulation of cell proliferation and apoptosis. Anticancer
Agents Med Chem. 12:631–639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuo LJ and Yang LX: Gamma-H2AX - a novel
biomarker for DNA double-strand breaks. In Vivo. 22:305–309.
2008.PubMed/NCBI
|
|
16
|
Zhang X, Huang Q, Wang X, Xu Y, Xu R, Han
M, Huang B, Chen A, Qiu C, Sun T, et al: Bufalin enhances
radiosensitivity of glioblastoma by suppressing mitochondrial
function and DNA damage repair. Biomed Pharmacother. 94:627–635.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shin SY, Ahn S, Koh D and Lim Y:
p53-dependent and -independent mechanisms are involved in
(E)-1-(2-hydroxyphenyl)-3-(2-methoxynaphthalen-1-yl)prop-2-en-1-one
(HMP)-induced apoptosis in HCT116 colon cancer cells. Biochem
Biophys Res Commun. 479:913–919. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
DE Wever O, Sobczak-Thépot J,
Vercoutter-Edouart AS, Michalski JC, Ouelaa-Benslama R, Stupack DG,
Bracke M, Wang JYJ, Gespach C and Emami S: Priming and potentiation
of DNA damage response by fibronectin in human colon cancer cells
and tumor-derived myofibroblasts. Int J Oncol. 39:393–400.
2011.
|
|
19
|
Solier S, Zhang YW, Ballestrero A, Pommier
Y and Zoppoli G: DNA damage response pathways and cell cycle
checkpoints in colorectal cancer: Current concepts and future
perspectives for targeted treatment. Curr Cancer Drug Targets.
12:356–371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
William-Faltaos S, Rouillard D, Lechat P
and Bastian G: Cell cycle arrest and apoptosis induced by
oxaliplatin (L-OHP) on four human cancer cell lines. Anticancer
Res. 26((3A)): 2093–2099. 2006.PubMed/NCBI
|
|
21
|
Voland C, Bord A, Péleraux A, Pénarier G,
Carrière D, Galiègue S, Cvitkovic E, Jbilo O and Casellas P:
Repression of cell cycle-related proteins by oxaliplatin but not
cisplatin in human colon cancer cells. Mol Cancer Ther.
5:2149–2157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fang Y, Yu H, Liang X, Xu J and Cai X:
Chk1-induced CCNB1 overexpression promotes cell proliferation and
tumor growth in human colorectal cancer. Cancer Biol Ther.
15:1268–1279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Greenow KR, Clarke AR, Williams GT and
Jones R: Wnt-driven intestinal tumourigenesis is suppressed by Chk1
deficiency but enhanced by conditional haploinsufficiency.
Oncogene. 33:4089–4096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Bian C, Ren K, Jin H, Li B and Shao
RG: Lidamycin induces marked G2 cell cycle arrest in human colon
carcinoma HT-29 cells through activation of p38 MAPK pathway. Oncol
Rep. 17:597–603. 2007.PubMed/NCBI
|
|
25
|
Soule BP, Simone NL, DeGraff WG, Choudhuri
R, Cook JA and Mitchell JB: Loratadine dysregulates cell cycle
progression and enhances the effect of radiation in human tumor
cell lines. Radiat Oncol. 5:82010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiao Z, Xue J, Sowin TJ, Rosenberg SH and
Zhang H: A novel mechanism of checkpoint abrogation conferred by
Chk1 downregulation. Oncogene. 24:1403–1411. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pauklin S, Kristjuhan A, Maimets T and
Jaks V: ARF and ATM/ATR cooperate in p53-mediated apoptosis upon
oncogenic stress. Biochem Biophys Res Commun. 334:386–394. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu S, Zhu H, Gu J, Zhang L, Teraishi F,
Davis JJ, Jacob DA and Fang B: Induction of apoptosis and
down-regulation of Bcl-XL in cancer cells by a novel small
molecule, 2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol. Cancer
Res. 64:1110–1113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carrassa L and Damia G: DNA damage
response inhibitors: Mechanisms and potential applications in
cancer therapy. Cancer Treat Rev. 60:139–151. 2017. View Article : Google Scholar : PubMed/NCBI
|