Introduction
Oral squamous cell carcinoma (OSCC) is the sixth
most common type of cancer in the world (1), and is the most common primary oral
cancer type in the oral maxillofacial region, with a 5-year
survival rate of 50–60% (2). Smoking
and alcohol are major risk factors for oral cancer, both exerting
synergistic effects (3).
Epithelial-mesenchymal transition (EMT) serves an important role in
tumorigenesis and tumor development. EMT is a process that involves
loss of cell polarity and cell-cell adhesion conferring tumor cells
the ability to migrate and metastasize (4).
mTORs, functioning as mechanistic targets, are
regulators of cell proliferation and metabolism (5). mTOR principally controls cell
metabolism by regulating the translation and transcription of
metabolic genes (6). It has been
revealed that mTOR can be activated in kidney cancer and
accelerates cancer progression (7).
Previous studies have reported that suppressing the mTOR-associated
signaling pathway can inhibit EMT (8–10).
Hypoxia inducible factor 1α (HIF-1α) leads to
insufficient blood supply and hypoxia in the tumor microenvironment
and affects tumor metabolism (11).
Moreover, the upregulation of HIF-1α serves a crucial role in
tumorigenesis, tumor angiogenesis, glycolysis and chemoresistance
(12). The downstream factor of
HIF-1α, pyruvate kinase M2 (PKM2), can interact with HIF-1α to
regulate cancer metabolism (13). In
addition, STAT3 can promote EMT progression (14), and HIF-1α, PKM2 and STAT3 are all
modulated by mTOR (15,16).
Metformin, used for treatment of type 2 diabetes,
has been associated with decreasing cancer incidence and mortality
(17). A previous study revealed
that metformin decreased the risk of liver, breast and colorectal
cancer (2). Other studies have
indicated that metformin inhibits EMT in prostate cancer, cervical
cancer and rectal cancer (18–20).
However, to the best of our knowledge, no reports have studied EMT
in OSCC. Moreover, the potential mechanism via which metformin
inhibits tumor growth is yet to be fully elucidated. Therefore, the
aims of the present study were to investigate the role of metformin
on inhibiting CoCl2-induced EMT in OSCC cells and to
examine whether EMT could be suppressed via the
mTOR/HIF-1α/PKM2/STAT3 signaling pathway.
Materials and methods
Cell lines and culture
The human OSCC CAL27 cell line was acquired from the
Department of Oral and Maxillofacial Surgery, Tooth Development and
Maxillary Reconstruction and Regeneration Laboratory at Jilin
University (Changchun, China). Cells were cultured in Dulbecco's
modified Eagle's (DMEM) medium supplemented with 10% FBS and 100
mg/ml penicillin/streptomycin (all purchased from Invitrogen;
Thermo Fisher Scientific, Inc.). Cells were cultured at 37°C in a
humid incubator with 5% CO2.
Metformin (MedChemExpress) and CoCl2
(Sigma-Aldrich; Merck KGaA) were dissolved in PBS (Invitrogen;
Thermo Fisher Scientific, Inc.) at a stock concentration of 160 mM
and 500 µM, respectively. Both were stored at −80°C.
Cell proliferation assay
Cells were seeded at a density of 1×104
cells/well in 96-well plates and cultured overnight at 37°C. After
treatment by indicated concentrations of CoCl2 (0, 50,
100, 200, 300, 400 and 500 µM) and metformin (0, 2.5, 5, 10, 20,
40, 80 and 160 mM) for 24 h at 37°C, 10 µl the Cell Counting Kit-8
(CCK-8) reagent (Invigentech) was added to each well according to
the manufacturer's protocol. After culturing for 2 h, the
absorbance was measured at 490 nm using a microplate reader (BioTek
Instruments, Inc.). After the screening process, the medium of
optimal concentrations (10 mM metformin with or without 300 µM
CoCl2) was used to culture cells for 24 h at 37°C. Then,
the above process were repeated. All results were measured three
times.
Cell migration assay
A wound-healing assay was performed to assess cell
migration, and 106 cells/well were seeded onto 6-well
plates for 24 h at 37°C. A wound was scraped using a 1,000-µl
pipette tip, and plates were washed three times with PBS. The cells
were cultured in fresh serum-free medium containing 300 µM
CoCl2, with or without 10 mM metformin, for 24 h at
37°C. Images were captured at the time points of 0 and 24 h after
wounding. The migration rate was quantified using the following
equation: (0-h scratching distance - 24-h scratching distance)/0-h
scratching distance. Representative images were obtained at ×40
magnification using an Olympus light microscope (Olympus
Corporation). All experiments were repeated at least three
times.
Cell invasion assay
Transwell assay was performed using 24-well
Transwell units (Corning, Inc.) with an 8-µm pore size
polycarbonate membrane which has been precoated with Matrigel
(Becton Dickinson) for 1 h at 37°C. Cells (1×105), 300
µM CoCl2, with or without 10 mM metformin, were
suspended in 100 µl DMEM without FBS were seeded into the upper
unit, while 600 µl DMEM with 10% FBS, was added to the lower units.
After incubation for 24 h at 37°C, cells on the upper side of the
membrane were removed using PBS-soaked cotton swabs. The membrane
was fixed in paraformaldehyde for 30 min at 37°C and then stained
with 0.1% crystal violet for 30 min at room temperature. Cell
numbers under the membrane were counted using an Olympus light
microscope (magnification, ×400; Olympus Corporation).
RNA isolation and
reverse-transcription-quantitative (RT-q) PCR
Cells were cultured in DMEM with 10% FBS containing
300 µM CoCl2, with or without 10 mM metformin (metformin
and CoCl2 were added at the same time), for 48 h at
37°C. Total RNA was extracted using TRIzol® (TRIeasyTM
Total RNA Extraction reagent; Shanghai Yeasen Biotechnology Co.,
Ltd.) from the specified treated cells and maintained at −20°C for
12 h. Total RNA was reverse transcribed using Hifair™ II 1st Strand
cDNA Synthesis SuperMix (TRIeasy™ Total RNA Extraction reagent;
Shanghai Yeasen Biotechnology Co., Ltd.) for qPCR under the
recommended conditions: 25°C for 5 min, 42°C for 30 min, 85°C for 5
min and holding at 4°C (GeneAmp PCR System 9700; Thermo Fisher
Scientific, Inc.). cDNA corresponding to 25 ng RNA was used for
qPCR using a HieffTM qPCR SYBR® Green Master mix
(Shanghai Yeasen Biotechnology Co., Ltd.). The following
thermocycling conditions were used: Initial denaturation at 95°C
for 5 min and 40 cycles at 95°C for 10 sec and 60°C for 30 sec
(ProFlex PCR System; Thermo Fisher Scientific, Inc.). The
expression levels of human β-actin, E-cadherin, vimentin, snail
family transcriptional repressor 1 (Snail1), mTOR, HIF-1α, PKM2 and
STAT3 were detected. Gene expression normalized to β-actin was
calculated using the 2−ΔΔCq method (21). The RT-qPCR primers were as follows:
β-actin forward, 5′-CTCCATCCTGGCCTCGCTGT-3′ and reverse,
5′-GCTGTCACCTTCACCGTTCC-3′; E-cadherin forward,
5′-GCCTCCTGAAAAGAGAGTGGAAG-3′ and reverse,
5′-TGGCAGTGTCTCTCCAAATCCG-3′; vimentin forward,
5′-AGGCAAAGCAGGAGTCCACTGA-3′ and reverse,
5′-ATCTGGCGTTCCAGGGACTCAT-3′; Snail1 forward,
5′-ATCTGCGGCAAGGCGTTTTCCA-3′ and reverse,
5′-GAGCCCTCAGATTTGACCTGTC-3′; mTOR forward,
5′-AGCATCGGATGCTTAGGAGTGG-3′ and reverse,
5′-CAGCCAGTCATCTTTGGAGACC-3′; HIF-1α forward,
5′-TAGCCGAGGAAGAACTATGAAC-3′ and reverse,
5′-CTGAGGTTGGTTACTGTTGGTA-3′; PKM2 forward,
5′-ATGGCTGACACATTCCTGGAGC-3′ and reverse,
5′-CCTTCAACGTCTCCACTGATCG-3′; and STAT3 forward,
5′-CTTTGAGACCGAGGTGTATCACC-3′ and reverse,
5′-GGTCAGCATGTTGTACCACAGG-3′.
Xenograft mouse studies
To investigate whether metformin inhibited
CoCl2-induced EMT in vivo, the subcutaneous
xenografted growth of OSCC cells was monitored. For the experiment,
cells were cultured in DMEM with 20% FBS containing 300 µM
CoCl2, with or without 10 mM metformin, for 48 h at
37°C. There were 28 male BALB/C nude mice (Shanghai Vital River
Laboratory Animal Technology Co., Ltd.; age, 4–6 weeks; weight,
15–20 g) were housed under specific pathogen-free conditions, with
food and water provided ad libitum. After 1 week of
acclimation, the mice were randomly divided into four groups (seven
mice per group) and injected with 5×106 indicated OSCC
cells subcutaneously which was resuspended by PBS into the right
flank. Xenograft tumor volume and weight were measured every other
day. After 24 days, nude mice were euthanized by cervical
dislocation and tumors were collected. Tumor volume (mm3) was
calculated as follows: 1/2 × long diameter (mm) × short diameter
(mm)2. The present study was approved by the Animal
Research Ethics Committee of Jilin University. All animal
treatments were performed in accordance with the Regulations of the
Administration of Affairs Concerning Experimental Animals.
Statistical analysis
Statistical analysis was performed using SPSS v21
(IBM Corp.) and GraphPad Prism 8 (GraphPad Software, Inc.). Data
are presented as the mean ± SD of three independent experiments.
One-way ANOVA was used for comparisons among multiple groups (with
Tukey's post-hoc test), and unpaired t-test was used for
comparisons between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
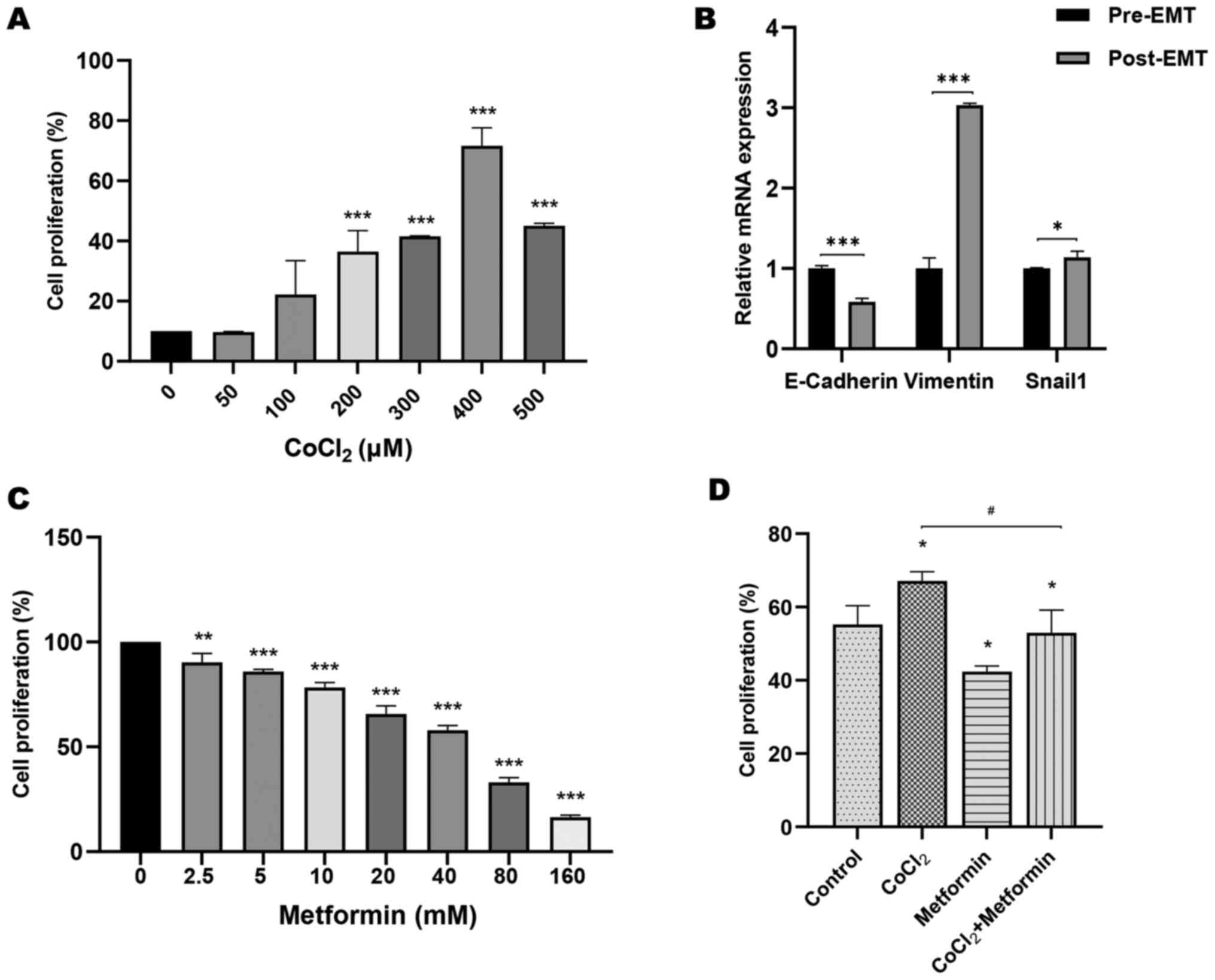
CoCl2 promotes
proliferation and induces EMT of OSCC cells
Cells were treated with 0, 50, 100, 200, 300, 400
and 500 µM CoCl2 to evaluate the potential effect of
CoCl2 in OSCC cells. CoCl2 at concentrations
≥100 µM could induce cell proliferation (Fig. 1A). The concentration of 300 µM
CoCl2 with the lowest error bar was chosen for
subsequent experiments. Moreover, the expression levels of
E-cadherin were significantly upregulated in the absence of
CoCl2 (pre-EMT). After cells were stimulated with
CoCl2, E-cadherin expression was significantly decreased
(post-EMT). Compared with the pre-EMT state, vimentin and Snail1
expression was significantly increased post-EMT by CoCl2
treatment (Fig. 1B). These data
indicated that CoCl2 promoted cell proliferation and
induced EMT when OSCC cells were stimulated with
CoCl2.
Metformin prevents proliferation,
migration, invasion and EMT of OSCC cells induced by
CoCl2
Cells were treated with 0, 2.5, 5, 10, 20, 40, 80
and 160 mM metformin to investigate the underlying
anti-proliferative effect of metformin in OSCC. Proliferation of
cells treated with metformin decreased significantly in a
dose-dependent manner compared with that of untreated cells
(Fig. 1C). A concentration of 10 mM
metformin was used for CCK-8, wound-healing, Transwell, RT-qPCR and
nude mice xenograft assays. Moreover, compared with the group
treated with CoCl2, cell proliferation in the
CoCl2 + metformin group was significantly attenuated by
the addition of metformin (Fig. 1D).
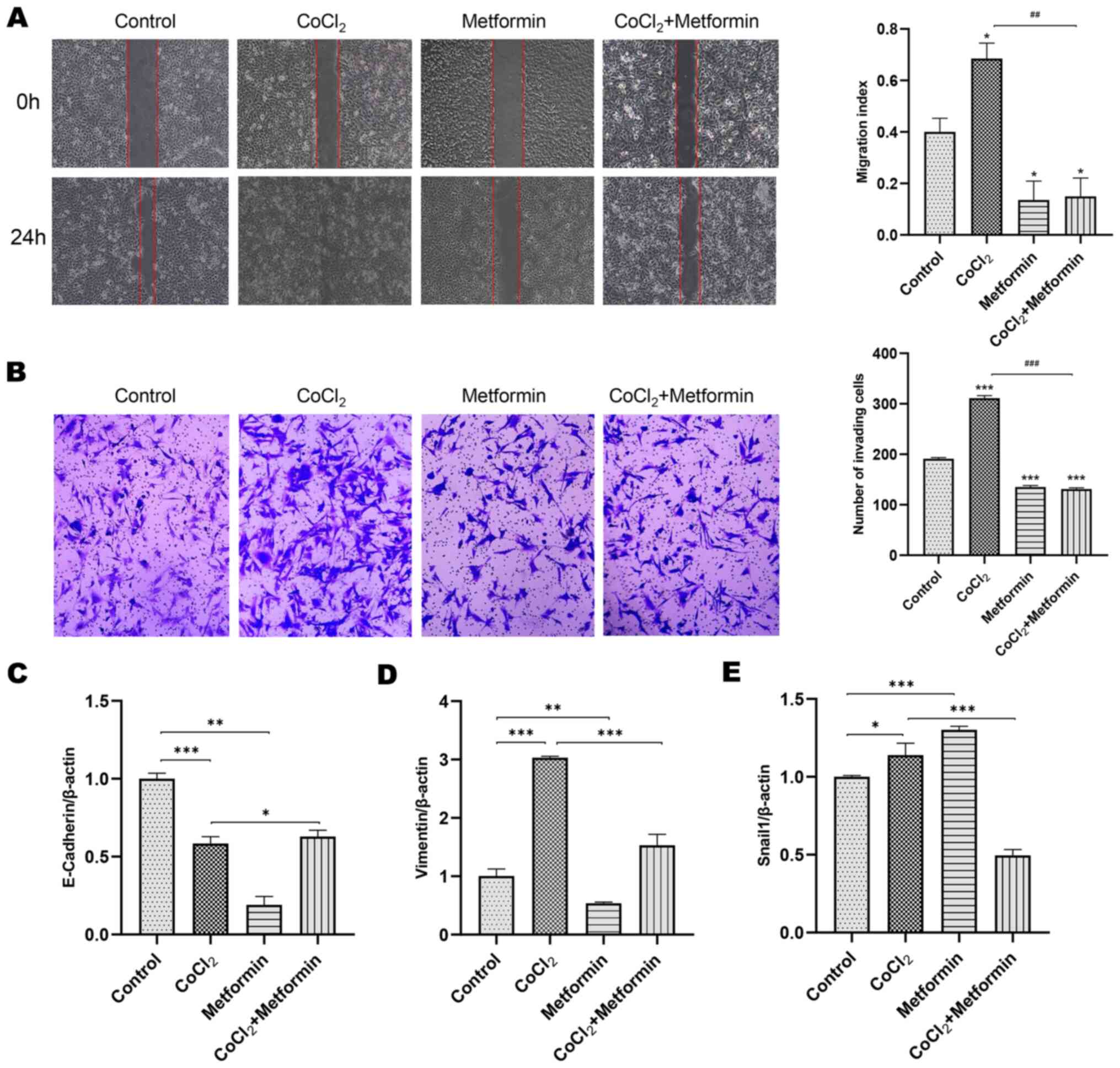
Additionally, cells were treated with 300 µM CoCl2, with
or without 10 mM metformin and the result revealed that
CoCl2 significantly increased the migration of cells
compared with the control group. This phenomenon could be abolished
by the addition of metformin (Fig. 2A
and B).
The markers of EMT were detected using RT-qPCR. In
the CoCl2 group, E-cadherin expression was decreased,
while vimentin and Snial1 expression was increased, which all could
be reversed by metformin (Fig.
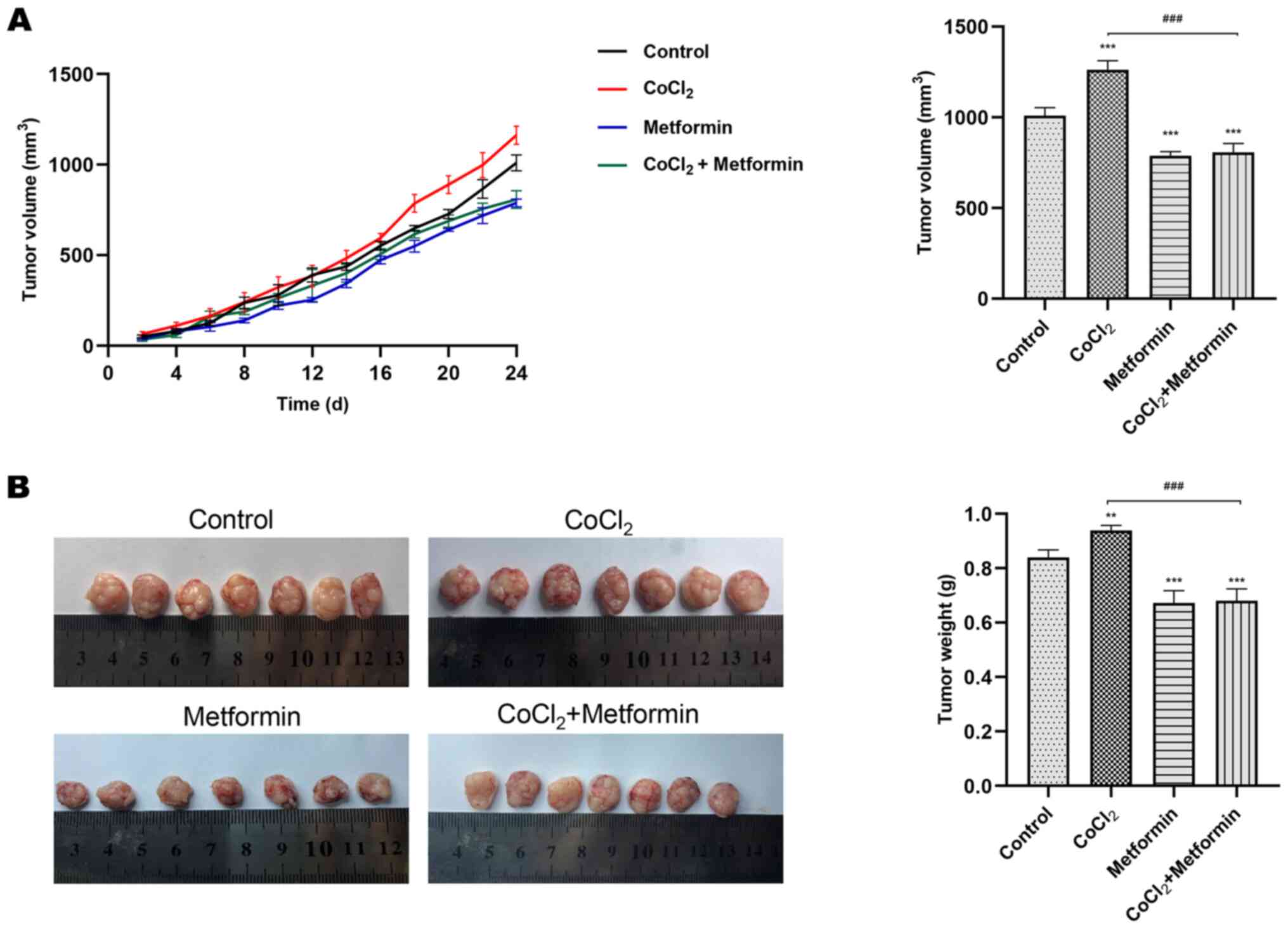
2C-E). In vivo compared with the control group, the
volume and weight of xenografts in the metformin group were
reduced. Using CoCl2 alone promoted tumor growth, which
could be inhibited by the addition of metformin (Fig. 3A and B). These findings suggested
that metformin inhibited the cell proliferative, migratory and
invasive abilities, as well as reversed CoCl2-induced
EMT.
Metformin prevents EMT of OSCC induced
by CoCl2 via the mTOR/HIF-1α/PKM2/STAT3 signaling
pathway
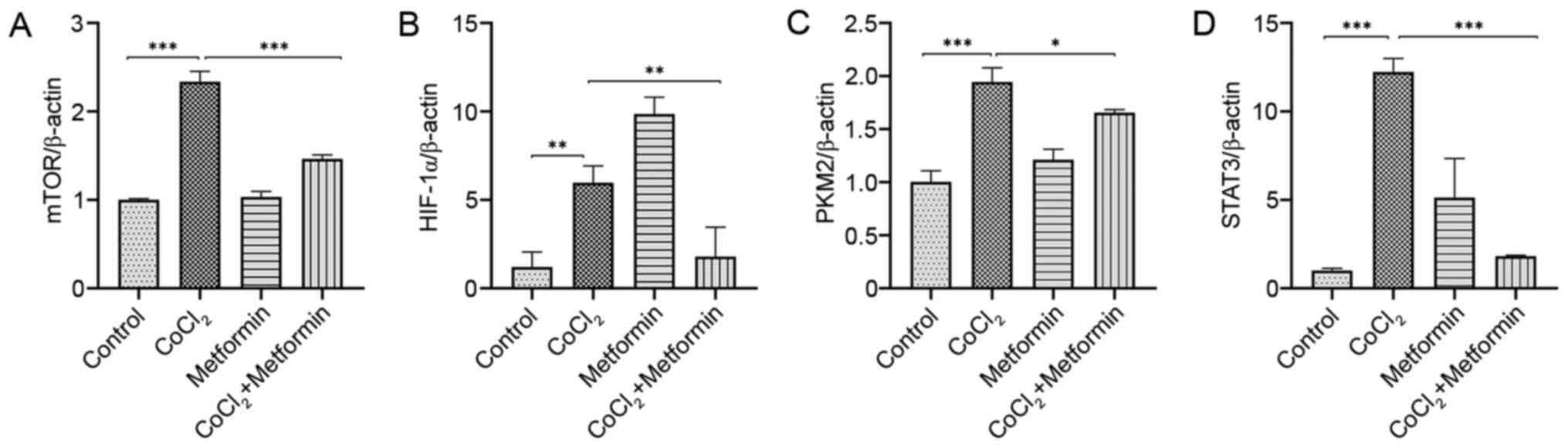
The expression levels of mTOR, HIF-1α, PKM2 and
STAT3 in the EMT process were detected via RT-qPCR. High expression
levels of mTOR and PKM2 in CoCl2-induced EMT were
identified, which were inhibited by metformin (Fig. 4A and C). Moreover, HIF-1α was
upregulated in the CoCl2 group compared with the control
group, but its expression was highest in the metformin group. In
the CoCl2 + metformin group, HIF-1α's expression was
decreased compared with the CoCl2 group (Fig. 4B). Thus, it was suggested that
metformin suppressed CoCl2-induced EMT, but using
metformin independently did not exert this effect. STAT3 expression
was significantly increased in the CoCl2 group, but was
significantly decreased by the addition of metformin (Fig. 4D).
Discussion
EMT is a major process in tumor metastasis, as well
as a vital factor involved in mortality in patients with OSCC
(22). Previous studies have
reported that CoCl2 at an appropriate concentration
promotes cell proliferation and induces EMT in liver and mammary
gland cancer (23,24). The present study used
CoCl2 to induce EMT and then investigated the underlying
mechanism of metformin inhibition on CoCl2-induced EMT
in OSCC.
Metformin, as an antidiabetic drug, has been
revealed to exert effects to decrease cancer incidence and
mortality rates in various types of human cancer (17). The use of metformin in diabetic
patients is associated with a decreased incidence in cancer types,
including pancreatic, liver and colon cancer, and can decrease
cancer-associated mortality (25).
Moreover, several studies have reported that metformin inhibits
tumor growth via multiple mechanisms, including by suppressing
tumor cell proliferation (26) and
EMT, affecting tumor autophagy and metabolism (12,27), and
inducing apoptosis of cancer stem cells (28). Although no studies on the effects of
metformin on EMT in OSCC, previous studies have demonstrated that
metformin suppresses EMT in other types of cancer, including
cervical and breast carcinoma (4,18). The
present study demonstrated that metformin inhibited
CoCl2-induced proliferation, migration, invasion and EMT
in OSCC.
The inhibitory effect of metformin on EMT in
cervical carcinoma via the mTOR-p70s6k relative signaling pathway
has been observed (19). mTOR can
accelerate cell proliferation and adjust cellular energy
homeostasis (29). In EMT,
overactivation of mTOR is closely associated with tumor progression
(30). Thus, targeting the
mTOR-associated pathway is the key to research.
mTOR regulates HIF-1α, and overexpression of HIF-1α
facilitates tumor metastasis and EMT in colorectal cancer (15). Moreover, PKM2 is an important
downstream executor of HIF-1α; the expression and function of PKM2
is associated with HIF-1α in feedforward loops, and HIF-1 is also a
target gene of the PKM2/STAT3 signaling pathway (16). Metformin inhibits the expression
levels of HIF-1α and PKM2 in gastric cancer (12), as wel as preventing the activation of
STAT3 in breast cancer (31) and
colorectal cancer (20). The
aforementioned studies suggest that the mechanism of metformin
suppressing CoCl2-induced EMT in OSCC may also occur by
inhibiting the mTOR-regulated HIF-1α/PKM2/STAT3 signaling pathway.
The present results confirmed that metformin significantly
inhibited CoCl2-induced EMT via the mTOR/HIF-1α/PKM2
signaling pathway in OSCC cells. However, there are certain
limitations to the current study, and additional cell lines are
required to further validate the present results. In addition, the
expression levels of markers including mTOR/HIF-1α/PKM2/STAT3
should be further analyzed using western blotting or
immunohistochemical staining. Additionally, rapamycin, an mTOR
inhibitor, should be used to confirm that metformin inhibits EMT
via the mTOR-associated HIF-1α/PKM2/STAT3 signaling pathway.
In conclusion, the present study demonstrated that
EMT served an important role in the proliferation, migration and
invasion of OSCC cells. Metformin was capable of reversing
CoCl2-induced EMT. Furthermore, it was identified that
metformin exerted its effects by suppressing mTOR, HIF-1α, PKM2 and
STAT3 activation. The current results were obtained using OSCC cell
models in vitro and xenograft nude-mice models in
vivo, and indicated that metformin may offer a novel strategy
for the treatment of patients with OSCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from
Jilin Provincial Development and Reform Commission high-tech
industrialization project (grant no. 2014G075).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WY and ZY conceived the experiments. YL and BS
designed the experiments. WY, YL, XL, XM, BS and ZY performed the
experiments. WY and YL analyzed the data and wrote the manuscript.
ZY and BS reviewed and edited the manuscript, and acquired the
funds. All authors read and approved the final manuscript, and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Animal
Research Ethics Committee of Jilin University (Changchun, China).
All animal treatments were performed in accordance with the
Regulations of the Administration of Affairs Concerning
Experimental Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Z, Lai ST, Xie L, Zhao JD, Ma NY, Zhu
J, Ren ZG and Jiang GL: Metformin is associated with reduced risk
of pancreatic cancer in patients with type 2 diabetes mellitus: A
systematic review and meta-analysis. Diabetes Res Clin Pract.
106:19–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galeone C, Edefonti V, Parpinel M,
Leoncini E, Matsuo K, Talamini R, Olshan AF, Zevallos JP, Winn DM,
Jayaprakash V, et al: Folate intake and the risk of oral cavity and
pharyngeal cancer: A pooled analysis within the International Head
and Neck Cancer Epidemiology Consortium. Int J Cancer. 136:904–914.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang R, Zhang P, Wang H, Hou D, Li W,
Xiao G and Li C: Inhibitory effects of metformin at low
concentration on epithelial-mesenchymal transition of
CD44(+)CD117(+) ovarian cancer stem cells. Stem Cell Res Ther.
6:2622015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hua H, Kong Q, Zhang H, Wang J, Luo T and
Jiang Y: Targeting mTOR for cancer therapy. J Hematol Oncol.
12:712019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de la Cruz López KG, Toledo Guzmán ME,
Sánchez EO and García Carrancá A: mTORC1 as a Regulator of
Mitochondrial Functions and a Therapeutic Target in Cancer. Front
Oncol. 9:13732019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Doan H, Parsons A, Devkumar S, Selvarajah
J, Miralles F and Carroll VA: HIF-mediated Suppression of DEPTOR
Confers Resistance to mTOR Kinase Inhibition in Renal Cancer.
iScience. 21:509–520. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baek SH, Ko JH, Lee JH, Kim C, Lee H, Nam
D, Lee J, Lee SG, Yang WM, Um JY, et al: Ginkgolic acid inhibits
invasion and migration and TGF-β-induced EMT of lung cancer cells
through PI3K/Akt/mTOR inactivation. J Cell Physiol. 232:346–354.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng J, Bai X, Feng X, Ni J, Beretov J,
Graham P and Li Y: Inhibition of PI3K/Akt/mTOR signaling pathway
alleviates ovarian cancer chemoresistance through reversing
epithelial-mesenchymal transition and decreasing cancer stem cell
marker expression. BMC Cancer. 19:6182019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi D, Kim CL, Kim JE, Mo JS and Jeong
HS: Hesperetin inhibit EMT in TGF-β treated podocyte by regulation
of mTOR pathway. Biochem Biophys Res Commun. 528:154–159. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo Y, Xiao Z, Yang L, Gao Y, Zhu Q, Hu L,
Huang D and Xu Q: Hypoxia-inducible factors in hepatocellular
carcinoma (Review). Oncol Rep. 43:3–15. 2020.PubMed/NCBI
|
|
12
|
Chen G, Feng W, Zhang S, Bian K, Yang Y,
Fang C, Chen M, Yang J and Zou X: Metformin inhibits gastric cancer
via the inhibition of HIF1α/PKM2 signaling. Am J Cancer Res.
5:1423–1434. 2015.PubMed/NCBI
|
|
13
|
Hasan D, Gamen E, Abu Tarboush N, Ismail
Y, Pak O and Azab B: PKM2 and HIF-1α regulation in prostate cancer
cell lines. PLoS One. 13:e02037452018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin C, Ren Z, Yang X, Yang R, Chen Y, Liu
Z, Dai Z, Zhang Y, He Y, Zhang C, et al: Nerve growth factor
(NGF)-TrkA axis in head and neck squamous cell carcinoma triggers
EMT and confers resistance to the EGFR inhibitor erlotinib. Cancer
Lett. 472:81–96. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lai HH, Li JN, Wang MY, Huang HY, Croce
CM, Sun HL, Lyu YJ, Kang JW, Chiu CF, Hung MC, et al: HIF-1α
promotes autophagic proteolysis of Dicer and enhances tumor
metastasis. J Clin Invest. 128:625–643. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li YH, Li XF, Liu JT, Wang H, Fan LL, Li J
and Sun GP: PKM2, a potential target for regulating cancer. Gene.
668:48–53. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heckman-Stoddard BM, DeCensi A,
Sahasrabuddhe VV and Ford LG: Repurposing metformin for the
prevention of cancer and cancer recurrence. Diabetologia.
60:1639–1647. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Q, Tong D, Liu G, Xu J, Do K, Geary K,
Zhang D, Zhang J, Zhang Y, Li Y, et al: Metformin reverses prostate
cancer resistance to enzalutamide by targeting TGF-β1/STAT3
axis-regulated EMT. Cell Death Dis. 8:e30072017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng K and Hao M: Metformin inhibits
TGF-β1-induced epithelial-to-mesenchymal transition via PKM2
relative-mTOR/p70s6k signaling pathway in cervical carcinoma cells.
Int J Mol Sci. 17:E20002016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park JH, Kim YH, Park EH, Lee SJ, Kim H,
Kim A, Lee SB, Shim S, Jang H, Myung JK, et al: Effects of
metformin and phenformin on apoptosis and epithelial-mesenchymal
transition in chemoresistant rectal cancer. Cancer Sci.
110:2834–2845. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Joseph JP, Harishankar MK, Pillai AA and
Devi A: Hypoxia induced EMT: A review on the mechanism of tumor
progression and metastasis in OSCC. Oral Oncol. 80:23–32. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kong D, Zhang F, Shao J, Wu L, Zhang X,
Chen L, Lu Y and Zheng S: Curcumin inhibits cobalt chloride-induced
epithelial-to-mesenchymal transition associated with interference
with TGF-β/Smad signaling in hepatocytes. Lab Invest. 95:1234–1245.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li S, Zhang J, Yang H, Wu C, Dang X and
Liu Y: Copper depletion inhibits CoCl2-induced aggressive phenotype
of MCF-7 cells via downregulation of HIF-1 and inhibition of
Snail/Twist-mediated epithelial-mesenchymal transition. Sci Rep.
5:124102015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Santi M, Baldelli G, Diotallevi A,
Galluzzi L, Schiavano GF and Brandi G: Metformin prevents cell
tumorigenesis through autophagy-related cell death. Sci Rep.
9:662019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abdel-Wahab AF, Mahmoud W and Al-Harizy
RM: Targeting glucose metabolism to suppress cancer progression:
Prospective of anti-glycolytic cancer therapy. Pharmacol Res.
150:1045112019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen H, Lin C, Lu C, Wang Y, Han R, Li L,
Hao S and He Y: Metformin-sensitized NSCLC cells to osimertinib via
AMPK-dependent autophagy inhibition. Clin Respir J. 13:781–790.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lei Y, Yi Y, Liu Y, Liu X, Keller ET, Qian
CN, Zhang J and Lu Y: Metformin targets multiple signaling pathways
in cancer. Chin J Cancer. 36:172017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Quan C, Sun J, Lin Z, Jin T, Dong B, Meng
Z and Piao J: Ezrin promotes pancreatic cancer cell proliferation
and invasion through activating the Akt/mTOR pathway and inducing
YAP translocation. Cancer Manag Res. 11:6553–6566. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Esparza-López J, Alvarado-Muñoz JF,
Escobar-Arriaga E, Ulloa-Aguirre A and de Jesús Ibarra-Sánchez M:
Metformin reverses mesenchymal phenotype of primary breast cancer
cells through STAT3/NF-κB pathways. BMC Cancer. 19:7282019.
View Article : Google Scholar : PubMed/NCBI
|