Introduction
Colorectal cancer (CRC) is currently the second-most
lethal cancer globally, with an estimated 880,792 deaths occurring
in 2018, accounting for 9.2% of total cancer deaths worldwide
(1). In Vietnam, CRC is the fifth
most common cancer and the fourth leading cause of cancer death in
both sexes combined (2). Generally,
colorectal tumorigenesis undergoes a stepwise process of mutations
and clonal expansion, subsequently leading to invasive and
metastatic tumors (3–5). There are two major molecular pathways
contributing to CRC tumorigenesis, namely chromosomal instability
(CIN) and microsatellite instability (MSI) (6).
CIN appears to be the most common type of genetic
change in CRC, accounting for 85% of sporadic cases (5). CIN is often associated with copy number
variations (CNVs) and/or the mutations in cancer-associated genes,
including KRAS and BRAF (7,8). MSI, on
the other hand, accounts for 15% of all CRC (9), as a consequence of a post-replicative
DNA mismatch repair (MMR) deficiency mostly caused by germline
mutations (10) or epigenetic
silencing of MMR genes (11). MSI
high (MSI-H) CRC tends to be associated with a high frequency of
replication errors due to the slippage of DNA polymerase (12). MSI-low (MSI-L) tumors, however, seem
to occur through the CIN pathway, similar to microsatellite stable
(MSS) tumors (13). MSI-H CRC has
largely been recognized as a favorable prognostic factor (14–17).
One of the key early events during tumor progression
is the acquisition of mutations in KRAS oncogene, leading to
the activation of multiple epidermal growth factor receptor (EGFR)
signaling pathways, including the RAS-RAF-ERK-MAPK and the
PI3K-PTEN-AKT pathways (18).
Activation mutations of the KRAS gene have been detected in
30–50% of CRC cases, mostly affecting codons 12 and 13 (19), resulting in a substitution of glycine
with cysteine (p.G12C), valine (p.G12V), or aspartic acid (p.G12D,
p.G13D) (19–21). These mutations diminish the intrinsic
GTPase activity of KRAS and confer unresponsiveness to GTPase
activating proteins (GAPs), causing the accumulation of KRAS-GTP
and constitutively activating its downstream pathways, including
cell proliferation and survival (5,22). BRAF
is a serine/threonine protein kinase, which is associated with the
MEK/ERK signaling pathway (23).
Oncogenic mutations of BRAF have been detected in ~5–15% of
CRCs (23–25), mostly involving a substitution of
glutamic acid for valine at codon 600 (p.V600E) (26). Molecularly, BRAF mutations
trigger its kinase activity that activates ERK through its
effectors MEK1/2, which eventually induces cell proliferation
(27). Mutations in KRAS and
BRAF have been largely regarded as negative predictors for
anti-EGFR therapy among patients with CRC (28–33).
Detection of KRAS and BRAF mutations, therefore, has
been performed in a routine CRC diagnosis as an aid for prognosis
and therapeutic options (34–36).
Despite its significance, the MSI status and the
prevalence of KRAS and BRAF mutations and their
associations with the other tumor characteristics remain poorly
known for Vietnamese patients with CRC. The present study aimed to
determine the MSI status, mutation frequencies of KRAS exon
2 and BRAF exon 15, as well as the associations between MSI
status and other clinicopathological features in 151 Vietnamese
patients with CRC from two populous ethnic groups, Kinh and
Muong.
Materials and methods
Patients and clinicopathological
characteristics
Fifty pairs of primary CRC and adjacent normal
tissues (at least 3 cm from the tumor) were collected from an
unselected consecutive series of patients of the Kinh ethnic group
undergoing surgical resection of CRC at Hue Central Hospital (Hue,
Vietnam) between February 2017 and May 2018. These samples were
stored at −80°C after snap freezing in liquid nitrogen. The primary
formalin-fixed, paraffin-embedded (FFPE) colorectal samples (n=101)
of Kinh (n=37) and Muong (n=64) ethnic groups, who were surgically
treated and histologically diagnosed with CRC between May 2011 and
December 2016, were obtained from the pathology archives of the Hoa
Binh Hospital (Hoa Binh, Vietnam). None of the patients underwent
any other treatments before the surgery. Routine histopathologic
staging of the resected specimens was done by an experienced
pathologist, and all the cancer specimens contained at least 70%
carcinoma cells.
The clinicopathological characteristics of the
cohort, including the patient's sex, ethnicity, age at diagnosis,
Tumor-Node-Metastasis (TNM) stage, tumor site and tumor grade
(level of differentiation), are shown in Table I. The study was approved by the
Ethics Committee of Duy Tan University. Written informed consent
for the use of resected tissue and clinical data in research was
obtained from all patients.
 | Table I.Clinicopathological characteristics
of patients with colorectal cancer according to microsatellite
instability status. |
Table I.
Clinicopathological characteristics
of patients with colorectal cancer according to microsatellite
instability status.
|
|
| MSI sub-group | MSI-H vs.
MSI-L/MSS |
|---|
|
|
|
|
|
|---|
|
Characteristics | Cases, n=151 | MSI-H | MSI-L | MSS | P-value | MSI-L/MSS | P-value |
|---|
| Age, years |
|
|
|
| 0.122a |
| 0.066a |
| Mean ±
SD | 59.94±12.36 | 58.35±12.15 | 62.29±13.79 | 60.51±11.51 |
| 61.24±12.44 |
|
|
(range) | (23–90) | (27–87) | (32–90) | (23–84) |
| (23–90) |
|
|
≤50 | 32
(21.2) | 19 (27.9) | 7
(20.6) | 6
(12.2) |
| 13 (15.7) |
|
|
>50 | 119 (78.8) | 49 (72.1) | 27 (79.4) | 43 (87.8) |
| 70 (84.3) |
|
| Sex, n (%) |
|
|
|
| 0.008a |
| 0.170a |
|
Male | 84
(55.6) | 42 (61.8) | 11 (32.4) | 31 (66.3) |
| 42 (50.6) |
|
|
Female | 67
(44.4) | 26 (38.2) | 23 (67.6) | 18 (36.7) |
| 41 (49.4) |
|
| Ethnicity, n
(%) |
|
|
|
| 0.957a |
| 0.786a |
|
Kinh | 87
(57.6) | 40 (58.8) | 19 (55.9) | 28 (57.1) |
| 47 (56.6) |
|
|
Muong | 64
(42.4) | 28 (41.2) | 15 (44.1) | 21 (42.9) |
| 36 (43.4) |
|
| Location, n
(%) |
|
|
|
| 0.040a |
| 0.011a |
|
Colon | 104 (68.9) | 54 (79.4) | 20 (58.8) | 30 (61.2) |
| 50 (60.2) |
|
|
Rectum | 47
(31.1) | 14 (20.6) | 14 (41.2) | 19 (38.8) |
| 33 (39.8) |
|
| TNM stage, n
(%) |
|
|
|
| 0.536c |
| 0.772c |
| I | 7
(4.6) | 1 (1.5) | 1 (2.9) | 5
(10.2) |
| 6 (7.2) |
|
| II | 108 (71.5) | 51 (75.0) | 25 (73.5) | 32 (65.3) |
| 57 (68.7) |
|
|
III | 28
(18.5) | 12 (17.6) | 7
(20.6) | 9
(18.4) |
| 16 (19.3) |
|
| IV | 1
(0.7) | – | 1 (2.9) | – |
| 1 (1.2) |
|
|
Missing | 7
(4.6) | 4 (5.9) | – | 3 (6.1) |
| 3 (3.6) |
|
| T stage, n (%) |
|
|
|
| 0.056c |
| 0.016c |
| T2 | 10 (6.6) | 2 (2.9) | 2 (5.9) | 6
(12.2) |
| 8 (9.6) |
|
| T3 | 84
(55.6) | 34 (50.0) | 23 (67.6) | 27 (55.1) |
| 50 (60.2) |
|
| T4 | 57
(37.7) | 32 (47.1) | 9
(26.5) | 16 (32.7) |
| 25 (30.1) |
|
| Lymph node
invasion, n (%) |
|
|
|
| 0.962c |
| 0.797c |
| N0 | 115 (76.2) | 52 (76.5) | 26 (76.5) | 37 (75.5) |
| 63 (75.9) |
|
| N1 | 28
(18.5) | 13 (19.1) | 7
(20.6) | 8
(16.3) |
| 15 (18.1) |
|
| N2 | 2
(1.3) | – | – | 2 (4.1) |
| 2 (2.4) |
|
|
Missing | 6
(4.0) | 3 (4.4) | 1 (2.9) | 2 (4.1) |
| 3 (3.6) |
|
| Distant metastasis,
n (%) |
|
|
|
| 0.193c |
| 0.364c |
| M0 | 145 (96.0) | 66 (96.0) | 33 (97.1) | 46 (93.9) |
| 79 (95.2) |
|
| M1 | 1
(0.7) | – | 1 (2.9) | – |
| 1 (1.2) |
|
|
Missing | 5
(3.3) | 2 (2.9) | – | 3 (6.1) |
| 3 (3.6) |
|
| Differentiation, n
(%) |
|
|
|
| 0.076c |
| 0.103c |
|
Well | 25
(16.6) | 6 (8.8) | 5
(14.7) | 14 (28.6) |
| 19 (22.9) |
|
|
Moderately | 17
(11.3) | 9
(13.2) | 4
(11.8) | 4 (8.2) |
| 8 (9.6) |
|
|
Poorly | 5
(3.3) | 2 (2.9) | 2 (5.9) | 1 (2.0) |
| 3 (3.6) |
|
|
Missing | 104 (68.9) | 51 (75.0) | 23 (67.6) | 30 (61.2) |
| 53 (63.9) |
|
| BRAF, n
(%) |
|
|
|
| 0.350b |
| 0.327b |
|
Mutant | 4
(2.6) | 3 (4.4) | 1 (2.9) | – |
| 1 (1.2) |
|
|
Wild-type |
147(97.4) | 65 (95.6) | 33 (97.1) | 49
(100.0) |
| 82 (98.8) |
|
| KRAS, n
(%) |
|
|
|
| 0.516a |
| 0.546a |
|
Mutant | 56
(37.1) | 27 (39.7) | 14 (41.2) | 15 (30.6) |
| 29 (34.9) |
|
|
Wild-type | 95
(62.9) | 41 (60.3) | 20 (58.8) | 34 (69.4) |
| 54 (65.1) |
|
DNA extraction
For the FFPE CRC specimens (n=101), five to eight
sections with a thickness of ~10 µm per sample were cut using a
rotary microtome (HM 325 Rotary Microtome; Thermo Fisher
Scientific, Inc.) and immediately collected into a sterile 2 ml
microcentrifuge tube. After xylene/ethanol deparaffinization,
samples were lysed with ~500 µl of lysis buffer (10 mM NaCl, 20 mM
Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0), 0.5% SDS and 200 µg/ml
proteinase K) at 56°C for ~3 h. DNA was then isolated using a
mixture of phenol-chloroform, followed by ethanol precipitation, as
previously described (37).
For the fresh-frozen paired samples (n=50), a
pea-size tissue sample was cut and collected into a sterile 2 ml
microcentrifuge tube containing 500 µl of ice-cold lysis buffer and
a 2.5-mm diameter iron ball. The sample was homogenized by a
TissueLyser LT (Qiagen Inc.) according to the manufacturer's
instructions, and DNA was extracted as aforementioned. DNA samples
were then dissolved in Tris-EDTA buffer or ddH2O and
kept at −20°C for later use. DNA purity and concentration were
assessed by a NanoDrop 2000 Spectrophotometer (ND-2000; Thermo
Fisher Scientific, Inc.).
MSI analysis
Multiplex PCR amplification of short
tandem repeat (STR) loci
Typing of MSI was performed using the standard
Bethesda marker panel (38) and
CAT25 (39), as previously described
(40) (Table SI). Multiplex PCR reaction was done
in a total volume of 25 µl using 12.5 µl Phusion U Multiplex PCR
Master mix (Thermo Fisher Scientific, Inc.), 0.4 mM of each primer
pairs (Integrated DNA Technologies, Inc.), and 50 ng of genomic DNA
(gDNA). For set 1 (BAT25, BAT26 and CAT25), the cycling parameters
were as follows: 30 sec at 98°C; 30 cycles for fresh-frozen and 32
cycles for FFPE samples of 10 sec at 98°C, 30 sec at 57°C, 30 sec
at 72°C, and a final extension step at 72°C for 7 min. For set 2
(D2S123, D5S346 and D17S250), the annealing temperature was
adjusted to 52°C.
Fragment analysis
The PCR products were subjected to the ABI 3500
Genetic Analyzer (Thermo Fisher Scientific, Inc.) for fragment
analysis. The sizes and patterns of the PCR products were then
resolved using GeneMapper 5.0 software (Thermo Fisher Scientific,
Inc.). The occurrence of MSI in a CRC sample was specified by the
presence of novel alleles as compared with the corresponding normal
sample and/or the appearance of allelic imbalance at heterozygous
loci. Tumors were considered as MSI-H if instability was detected
in at least two out of six tested markers; as MSI-L if instability
was observed in only one of the markers; and as MSS if no
instabilities were detected (38,39).
Determination of allelic
imbalance
For dinucleotide makers, allelic ratios (R) were
calculated based on the fluorescent intensity peak height, as
previously reported (41,42). In normal tissues, the peak height
ratio of <70% could be considered as evidence of allelic
imbalance (AI) or partial loss of heterozygosity (pLOH) (43). In practice, the AI thresholds were
generally set from 70% to as low as 59% (42,44). In
the present study, the AI thresholds were set at <59% or
>125%. For simplicity, the smaller allele (in size) was always
made the numerator, and the site with the allelic ratio of >1.7
or <0.8 was recognized as a site of AI or pLOH and will be
reckoned as MSI (Fig. S1).
KRAS and BRAF mutation detection
PCR amplification
The amplification mixture consisted of 12.5 µl 2X
DreamTaq PCR Master mix (Thermo Fisher Scientific, Inc.), 0.4 mM of
each primer pairs (Table SII), and
50–100 ng gDNA. For KRAS, the PCR conditions were 3 min at
95°C, followed by 32 (for fresh-frozen samples) to 35 cycles (for
FFPE samples) of 30 sec at 95, 50 and 72°C, sequentially. For
BRAF, the annealing temperature was adjusted to 48°C. To
avoid a cross-sample contamination, at least one negative control
was included for each round of PCR amplification. PCR products were
examined by 2% agarose gel electrophoresis to confirm the existence
of amplified fragments before sequencing.
Sanger sequencing
All the successfully amplified fragments were
subjected to unidirectional sequence analysis based on the Sanger
method on an ABI 3500 Genetic Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) after being purified using the GeneJet PCR
Purification kit (Thermo Fisher Scientific, Inc.).
Data analysis
Tumors were classified by MSI or KRAS
mutation status. The clinicopathological features were assessed and
compared between groups. Anatomic location was distinguished as the
colon (from cecum to sigmoid colon) or rectum (rectum and anus).
Tumor grade was categorized as: Well-differentiated (G1),
moderately differentiated (G2), poorly differentiated (G3), and
undifferentiated (G4) tumors. The node metastasis (N) in the TNM
staging system was assigned by the number of metastatic lymph nodes
as N1 (1–3 affected nodes) and N2 (≥4 affected nodes).
Statistical analysis
SPSS 22.0 (SPSS, Inc.) was used for all statistical
analyses. Continuous variables were presented as the mean±standard
deviation. The associations between MSI, KRAS and
BRAF mutational status and the clinicopathological factors
were determined by the χ2, Fisher's exact, or
Kruskal-Wallis test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinicopathological features of
patients
Of the 151 patients, 84 (55.6%) were male, and 67
(44.4%) were female; 87 (57.6%) were Kinh, and 64 (42.4%) were
Muong ethnicity. There were 104 (68.9%) tumors at the colon and 47
(31.1%) tumors at the rectum. The pathological stage was defined
according to the TNM stage classification; 7 patients (4.6%) had
stage I, 108 patients (71.5%) had stage II, 28 patients (18.5%) had
stage III, 1 patient (0.7%) had stage IV, and 7 patients (4.6%) had
an undetermined stage tumor. Tumor grade was available for only 47
patients, of which 25 tumors (53.2%) were well-differentiated, 17
tumors (36.2%) were moderately differentiated, and 5 tumors (10.6%)
were poorly differentiated. The patients' age ranged from 23 to 90
years (mean age, 59.94±12.36 years) (Table I).
Large proportion of Vietnamese
patients with CRC are classed as MSI-H
All tumors were typed for MSI and classified as
MSI-H, MSI-L or MSS based on their number of unstable markers
(Fig. 1A-C). Of these 151 patients,
there were 68 patients (45.0%) with MSI-H, 34 patients (22.5%) with
MSI-L, and 49 patients (32.5%) with MSS (Fig. 1D). The mean age of patients at
diagnosis in the MSI-H group (58.35±12.15 years) was lower compared
with MSS (60.51±11.51) and MSI-L (62.29±13.79) groups. There was a
significant association between MSI status and the patients' sex
(P=0.008), and tumor location (P=0.040) (Table I).
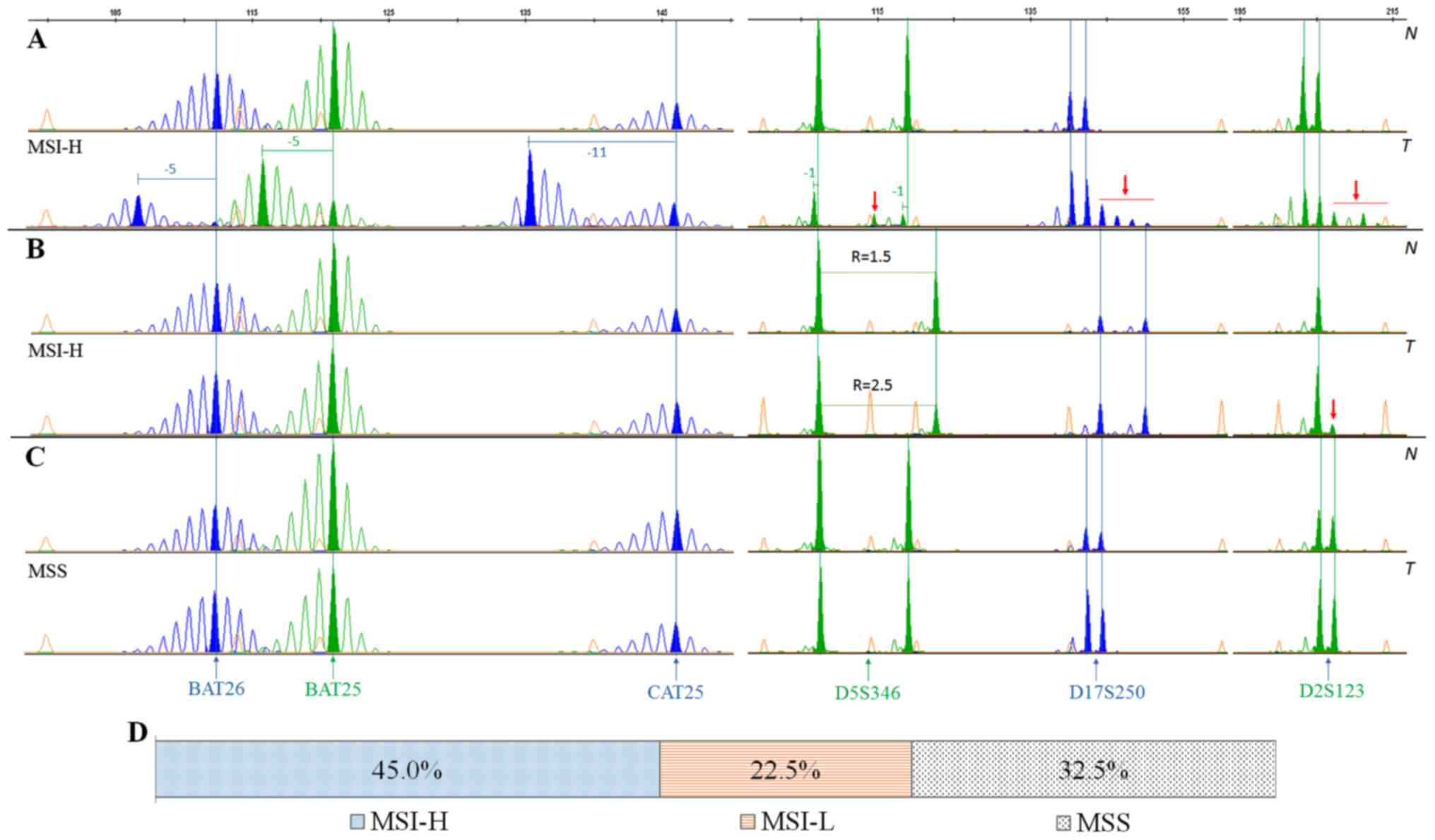 | Figure 1.MSI detection by multiplex PCR using
6 primer pairs, comprising 3 mononucleotide markers (BAT26, BAT25
and CAT25) and three dinucleotide markers (D2S123, D5S346 and
D17S250). (A) A colorectal tumor with MSI-H; all six tested markers
showed instability. (B) A MSI-H colorectal tumor with a minimum
number of unstable markers (two out of six). (C) A MSS colorectal
tumor where no unstable markers were detected. (D) The frequency of
MSI-H, MSI-L and MSS in Vietnamese patients with colorectal cancer.
Orange peaks represent the internal length standard of 600 LIZ.
Filled peaks span the largest area and are defined as the main
products. Shift lengths are denoted above the corresponding product
peaks. N, tumor-matched adjacent normal tissue samples; T, paired
tumor samples; MSI-H, microsatellite instability-high; MSS,
microsatellite stability; R, allelic ratio of a heterozygous locus,
as calculated by dividing the peak height of the smaller allele by
larger allele. |
MSI-H vs. MSI-L/MSS CRC showed significant
differences in the tumor location and T stage. Accordingly, MSI-H
tumors were often found to be located at the colon (54/68; 79.4%)
compared with MSI-L/MSS groups (50/83; 60.2%) (P=0.011).
Furthermore, MSI-H was positively associated with more advanced T
stages (P=0.016), especially for T4 stage tumors, which has been
detected in 32 out of 68 (47.1%) patients with MSI-H compared with
25 out of 83 (30.1%) patients with MSI-L/MSS (Table I). In parallel, pairwise comparisons
between MSI groups also revealed the significant differences
(P≤0.05) between MSI-H vs. MSI-L and MSI-H vs. MSS tumors in both
tumor location and T stage (Table
SIII). Additionally, the results revealed significant
differences in sex between MSI-H vs. MSI-L (P=0.005) and MSI-L vs.
MSS (P=0.006) tumors (Table SIII).
Specifically, male were more often in the MSI-H (42/68, 61.8%) and
MSS (31/49, 66.3%) groups compared with the MSI-L tumors (11/34;
32.4%) (Table I). Moreover, the
statistical analyses have also uncovered associations between MSI
status and the tumor grade and age at onset (Table SIII). Particularly, MSI-H (P=0.027)
tumors appeared to be significantly less differentiated and younger
onset (P=0.041) compared with MSS tumors (Table SIII).
Taken together, the data revealed that a substantial
proportion of Vietnamese patients with CRC were MSI-H. MSI-H tumors
exhibited with more advanced T stage tumors and colon-located
tumors. Furthermore, MSI-H tumors occurred more in male compared
with MSI-L tumors, and less differentiated and younger age at onset
compared with MSS tumors. These distinct clinicopathological
features of MSI-H tumors specified that the carcinogenic pathway
underlying MSI-H tumors might be different from that of MSI-L/MSS
tumors.
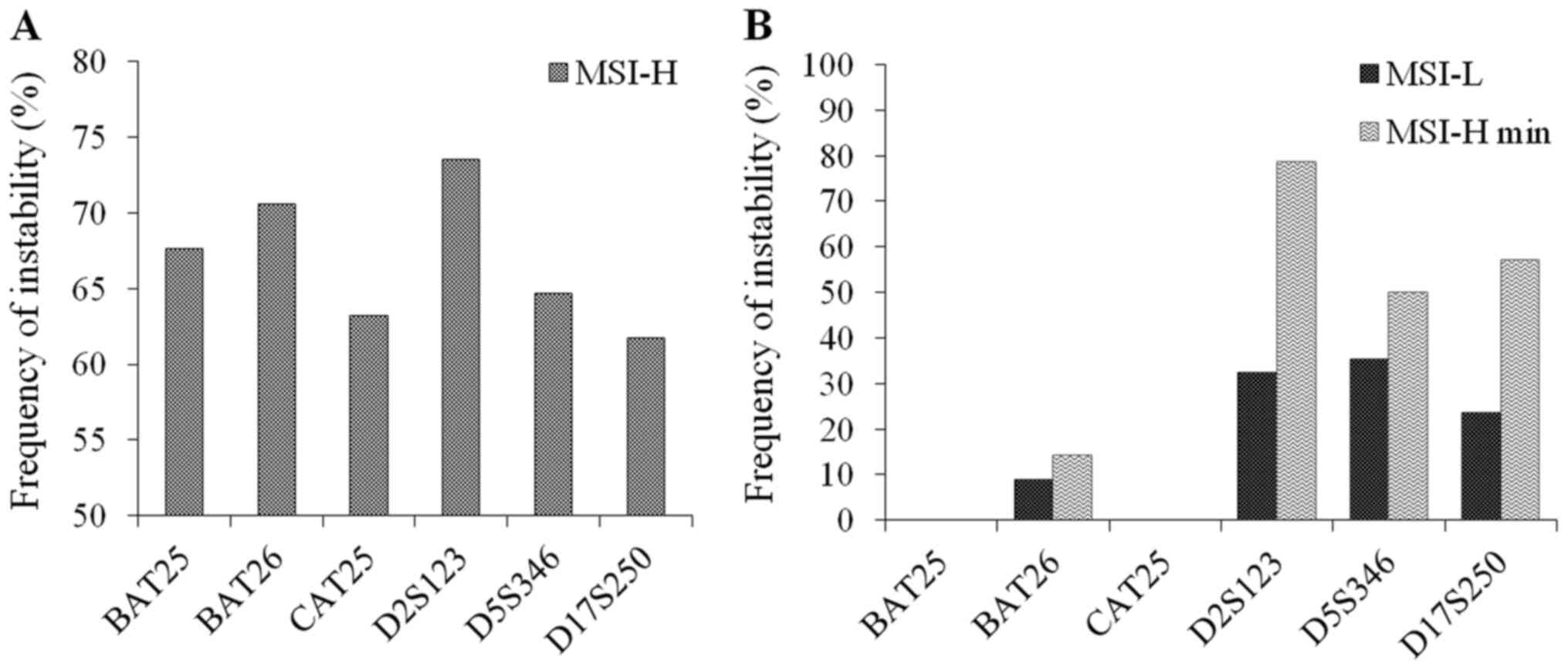
Diagnostic role of each individual
marker
The diagnostic role of microsatellite markers was
assessed by analyzing the frequency of instability of these markers
on three groups: MSI-H, MSI-L and MSI-H cases with a minimum number
(2 out of 6) of unstable markers (MSI-H min, Table SIV). Noticeably, the frequency of
instability was similar between mononucleotide and dinucleotide
markers within MSI-H group. Of which, D2S123 and BAT26 were the
most unstable markers that showed evidence of instability in 73.5
and 70.6% of the MSI-H cases, respectively (Fig. 2A). However, for MSI-L and especially
for MSI-H min group, the dinucleotide markers appeared to be much
more sensitive in the detection of instability compared with
mononucleotide markers. Particularly, while BAT25 and CAT25 did not
show any evidence of instability in MSI-L and MSI-H min CRC, the
frequency of instability detected in BAT26, D2S123, D5S346 and
D17S250 in MSI-L and MSI-H min were 8.8, 32.4, 35.3, 23.5%, and
14.3, 78.6, 50, 57.1%, respectively (Fig. 2B).
KRAS and BRAF mutations and their
co-existence with MSI-H
Electropherograms of KRAS and BRAF
gene sequences carrying hotspot substitution mutations were
illustrated in Fig. S2A and B.
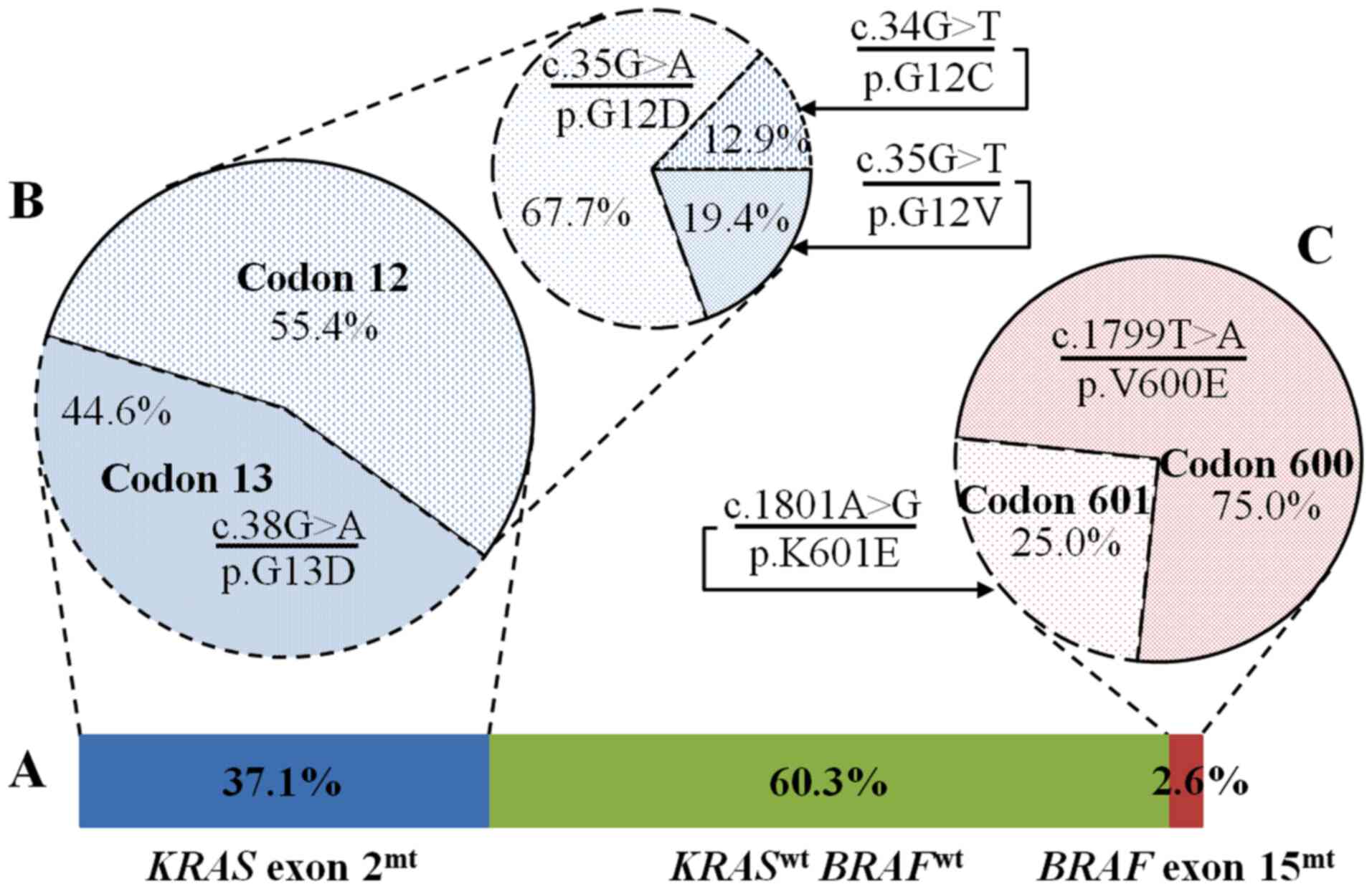
Sequence analysis of KRAS exon 2 showed
single point mutations in 56 of 151 tested samples (37.1%)
(Fig. 3A), of which 31 tumors
(55.4%) had mutations in codon 12 and 25 tumors (44.6%) had
mutations in codon 13 (Fig. 3B). The
most frequent alteration in codon 12 was c.35G>A (21/31; 67.7%)
transition, causing a replacement of glycine with aspartic acid
(p.G12D). Two other mutations at codon 12 were c.35G>T (6/31;
19.4%) and c.34G>T (4/31; 12.9%) transversion, leading to the
substitution of valine (p.G12V) or cysteine (p.G12C) for glycine,
respectively (Fig. 3B). Mutations in
codon 13 were exclusively c.38G>A (25/25) transition, resulting
in an exchange aspartic acid for glycine (p.G12D) (Fig. 3B).
Four of 151 tumors (2.6%) comprised a point mutation
in BRAF exon 15 (Fig. 3A).
Three out of four mutations (75.0%) were c.1799T>A transversion
replacing glutamic acid for valine in codon 600 (p.V600E). The
remained mutation (25.0%) was c.1801A>G transition that switches
a lysine to glutamic acid (p.K601E) (Fig. 3C). Noticeably, there was a
substantial co-existence of mutant KRAS and MSI-H (27/151,
17.9%) compared with MSI-L (14/151; 9.3%) and MSS (15/151; 9.9%)
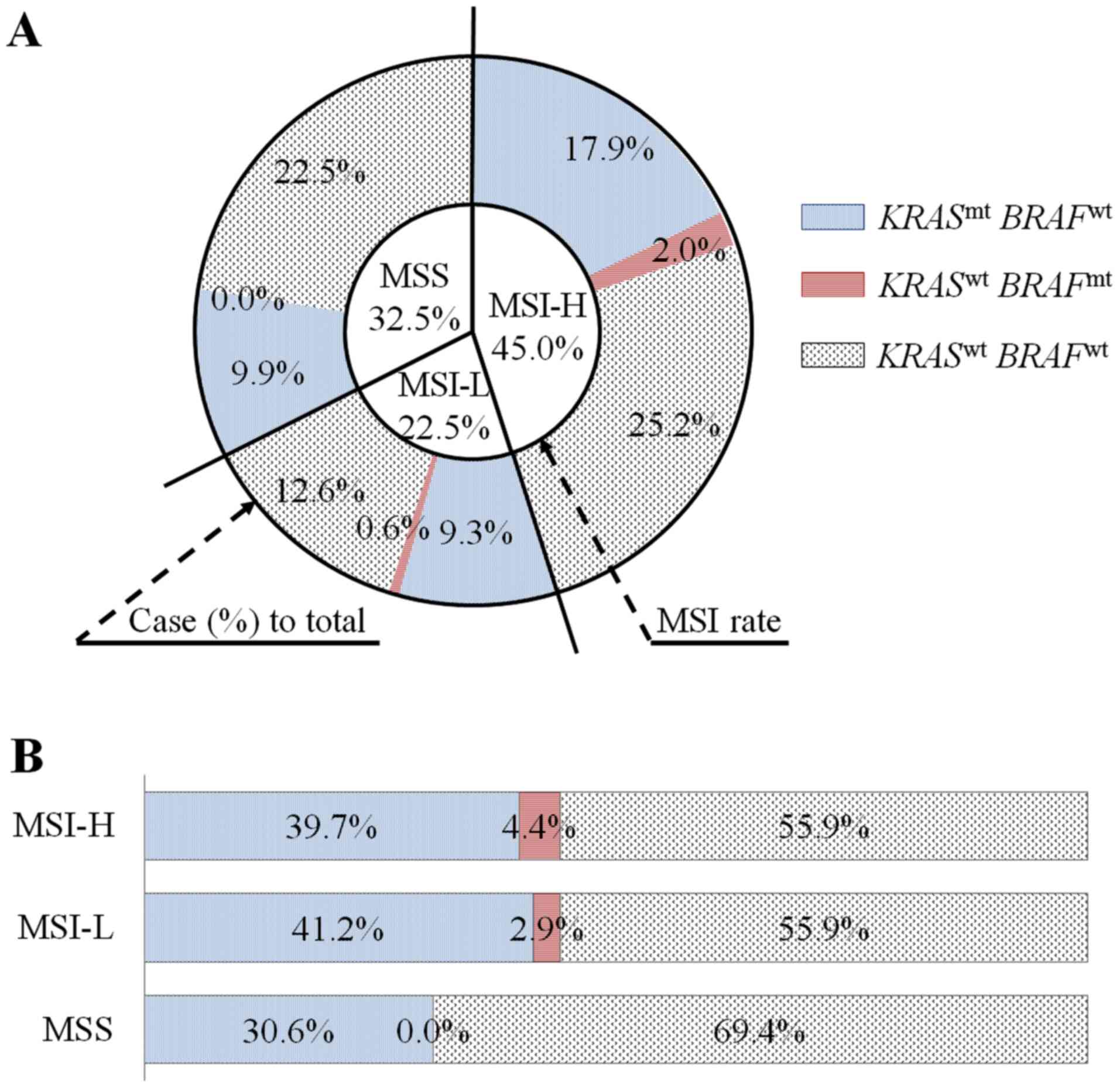
(Fig. 4A). Within MSI-H group,
mutant KRAS and mutant BRAF accounted for 39.7 and
4.4% (Fig. 4B); while within mutated
KRAS and mutated BRAF groups, MSI-H accounted for
48.2% (27/56) and 75% (3/4), respectively (Table II).
| Table II.Association of KRAS and
BRAF mutational status with clinicopathological features in
colorectal cancer. |
Table II.
Association of KRAS and
BRAF mutational status with clinicopathological features in
colorectal cancer.
|
| BRAF | KRAS |
|---|
|
|
|
|
|---|
| Clinicopathological
features |
BRAFmt n (%) |
BRAFwt n (%) | P-value |
KRASmt n (%) |
KRASwt n (%) | P-value |
|---|
| Age, years |
|
| 1.000b |
|
| 0.379a |
|
≤50 | 1
(250.0) | 31
(21.1) |
| 14 (25.0) | 18 (18.9) |
|
|
>50 | 3 (75.0) | 116 (78.9) |
| 42 (75.0) | 77 (81.1) |
|
| Sex |
|
| 1.000b |
|
| 0.465a |
|
Male | 2 (50.0) | 82
(55.8) |
| 29 (51.8) | 55 (57.9) |
|
|
Female | 2 (50.0) | 65
(44.2) |
| 27 (48.2) | 40 (42.1) |
|
| Ethnicity |
|
| 0.638b |
|
| 0.013a |
|
Kinh | 3 (75.0) | 84
(57.1) |
| 25 (44.6) | 62 (65.3) |
|
|
Muong | 1 (25.0) | 63
(42.9) |
| 31 (55.4) | 33 (34.7) |
|
| Location |
|
| 1.000b |
|
| 0.194a |
|
Colon | 3 (75.0) | 101 (68.7) |
| 35 (62.5) | 69 (72.6) |
|
|
Rectum | 1 (25.0) | 46
(31.3) |
| 21 (37.5) | 26 (27.4) |
|
| TNM stage |
|
| 0.699c |
|
| 0.086c |
| I | – | 7
(4.8) |
| 4 (7.1) | 3 (3.2) |
|
| II | 3 (75.0) | 105 (71.4) |
| 41 (73.2) | 67 (70.5) |
|
|
III | 1 (25.0) | 27
(18.4) |
| 6
(10.7) | 22 (23.2) |
|
| IV | – | 1
(0.7) |
| 1 (1.8) | – |
|
|
Missing | – | 7
(4.8) |
| 4 (7.1) | 3 (3.2) |
|
| T stage |
|
| 0.216 |
|
|
>0.999c |
| T2 | – | 10 (6.8) |
| 5 (8.9) | 5 (5.3) |
|
| T3 | 4
(100.0) | 80
(54.4) |
| 29 (51.8) | 55 (57.9) |
|
| T4 | – | 57
(38.8) |
| 22 (39.3) | 35 (36.8) |
|
| Lymph node
metastasis |
|
| 0.843c |
|
| 0.048c |
| N0 | 3 (75.0) | 112 (76.2) |
| 45 (80.4) | 70 (73.7) |
|
| N1 | 1 (25.0) | 27
(18.4) |
| 6
(10.7) | 22 (23.2) |
|
| N2 | – | 2
(1.4) |
| – | 2 (2.1) |
|
|
Missing | – | 6
(4.1) |
| 5 (8.9) | 1 (1.1) |
|
| Tumor grade
(differentiation) |
|
| 0.046c |
|
| 0.114c |
|
Well | – | 25
(17.0) |
| 44 (78.6) | 21 (22.1) |
|
|
Moderately | 2 (50.0) | 15
(10.2) |
| 4 (7.1) | 11 (11.6) |
|
|
Poorly | 1 (25.0) | 4
(2.7) |
| 6
(10.7) | 3 (3.2) |
|
|
Missing | 1 (25.0) | 103 (70.1) |
| 2 (3.6) | 60 (63.2) |
|
| BRAF |
|
|
|
|
| 0.297b |
|
Mutant |
|
|
| – | 4 (4.2) |
|
|
Wild-type |
|
|
| 56 (100) | 91 (95.8) |
|
| MSI |
|
| 0.151c |
|
| 0.356c |
|
High | 3 (75.0) | 65
(44.2) |
| 27 (48.2) | 41 (43.2) |
|
|
Low | 1 (25.0) | 33
(22.4) |
| 14 (25.0) | 20 (21.1) |
|
|
Stable | – | 49
(33.3) |
| 15 (26.8) | 34 (35.8) |
|
The statistical tests disclosed significant
differences in the ethnicity and lymph node metastasis rate between
KRAS mutant and wild-type tumors. Accordingly, there were
significantly more Muong (55.4%) than Kinh patients (44.6%) within
the KRAS mutated tumors (P=0.013). Moreover, KRAS
mutations were significantly associated with none (N0) or fewer
number lymph node metastasis (N1) compared with KRAS
wild-type (P=0.048). Lastly, the Kruskal-Wallis test has unveiled
that BRAF mutant tumors were significantly less
differentiated (P=0.046) compared with BRAF wild-type
tumors. KRAS and BRAF mutations were mutually
exclusive (Table II).
Discussion
Currently, it is well accepted that certain
alterations at the molecular level favor CRC tumorigenesis and are
used as prognostic markers. MSI phenotype, for example, was
commonly proposed as a favorable prognostic biomarker for CRC
(14,15,45–47) and
mutations in KRAS and BRAF genes were decisively used
as predictors of resistance to monoclonal antibodies targeting
epidermal growth factor receptors (EGFRs) (48–50).
Therefore, defining the status of MSI and the hotspot mutations in
KRAS and BRAF is of utmost importance to support the
prognosis and selection of treatment methods.
Among 151 patients with CRC, 45.0% of patients were
classed as MSI-H, 22.5% were classed as MSI-L, and 32.5% as MSS.
The proportion of MSI-H in Vietnamese patients with CRC in the
present study is similar to that of African American (45%; 10/22)
(51), substantially higher than
Singaporean (30%; 32/109) patients with CRC (52), and strikingly higher than that of
Japanese (4.5–6%) (53,54), Korean (5.5–9%) (55), and Western (10–20%) patients with CRC
(56,57). These differences could be due to the
genetic basis of the patient cohorts, microsatellite markers used
for MSI detection, and/or the interpretation of the results
(15,58). Several previous studies have
suggested that BAT26 was a fairly good indicator of what would have
been seen with the entire panel (16,59).
However, in the present study, all six tested markers showed
similar sensitivity in detecting MSI-H tumors, with ~70% of the
cases, in which BAT26 and D2S123 were the most unstable markers
(Fig. 2A). Particularly, in MSI-L
and MSI-H min tumors, dinucleotides markers were found to be more
sensitive than mononucleotide markers in detecting MSI (Fig. 2B). This finding was in concordance
with one previous study in other Asian patients with CRC (52), indicating the significance of these
dinucleotide markers in assessing MSI in patients with CRC. On the
other hand, CAT25, the additional marker suggested by Findeisen
et al (39), did not seem to
have added any value to the MSI diagnostics of this set of tumors
(Fig. 2B).
Moreover, the current study showed significant
associations between MSI-H and colon tumor location, males, younger
onset, and more advanced T stages. The association between MSI-H
and proximal tumor sites was noted by a number of previous studies
(55,60), but contradictory findings that
highlighted the association of MSI-H to female (51,61,62) and
lower T stages (55,63) were also reported. These results
specified that MSI-H tumors possessed distinct clinicopathological
features and possibly have been derived from a distinct
carcinogenic pathway compared with MSI-L/MSS ones.
The sequencing data disclosed 37.1% patients with
CRC carried a KRAS exon 2 mutation, which typically affects
codon 12 (55.4%) and codon 13 (44.6%). This observation was in
accordance with previous studies (64–66), and
marginally lower than several other studies (50,53,67–69),
where KRAS mutations were detected in ~35-37 and 40–42% of
the tested patients with CRC, respectively. The present study also
presented a significant difference in the frequency of this
mutation in two ethnic groups by showing that 48.4% of Muong and
28.7% of Kinh patients with CRC had this mutation, suggesting a
possible variation in the frequency and types of KRAS
mutations among populations. Moreover, the present data also
indicated that mutant KRAS was reversely associated with the
rate of lymph node metastasis, which was in accordance with a study
in the Chinese population (69).
Mutations in KRAS exon 2 have been largely used as a
predictive marker of unresponsiveness to EGFR therapies (48–50).
However, more than 50% of CRC patients with a wild-type KRAS
exon 2 are also resistant to this approach, possibly due to other
‘rare mutations’ in other exons of this gene or in other genes of
the RAS/RAF family (25,67,69–72).
These findings indicated the need for expanding the genetic test in
CRC patients with a wild-type KRAS exon 2 before anti-EGFR
therapy.
BRAF exon 15 mutations, on the other hand,
were detected in only 2.6% Vietnamese patients with CRC. This ratio
was similar to that of Greek patients with CRC (21), substantially lower than other Western
countries (~9%) (25,50), and marginally lower than other Asian
populations (~4.5–7%), including Japan (53,66) and
China (69,73). In agreement with the previous
studies, the present study also presented the mutual exclusivity of
BRAF and KRAS mutations (23,66).
Remarkably, the substantial rate of co-existence of MSI with
KRAS and BRAF mutations found in the present study
may indicate the poor prognosis of these cases (16,74). A
future study on the associations between overall survival and/or
recurrence-free survival and MSI with KRAS and BRAF
mutations will gain some insight into this issue.
In summary, the present results revealed the typical
molecular features and subgroups of Vietnamese patients with CRC.
Particularly, the strikingly high proportion of MSI-H tumors and
their substantial co-existence with KRAS and BRAF
mutations should be taken into account for future diagnosis and
clinical treatments for this type of cancer.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Adam F. Johnson
(Institute of Research and Development, Duy Tan University, Danang,
Vietnam) for the critical reading of the manuscript.
Funding
This study was supported by the National Foundation
for Science and Technology Development (NAFOSTED; grant no.
106-YS.01-2015.12).
Availability of data and materials
Data and materials available on request from the
authors
Authors' contributions
HTN conceived the project, conducted most of the
experiments and data analysis, and wrote the manuscript. DTL
designed/selected primers and prepared figures. QHD and VBT helped
with sample preparation and DNA extraction. BVN collected samples
and patients' data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Duy Tan University (approval no. DTU-IRB/2019.15). Written informed
consent for the use of resected tissue and clinical data in
research was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The other authors report no conflicts of
interest.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pham T, Bui L, Kim G, Hoang D, Tran T and
Hoang M: Cancers in Vietnam-burden and control efforts: a narrative
scoping review. Cancer Contr. 26:1073274819863802. 2019.
|
|
3
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nguyen HT and Duong HQ: The molecular
characteristics of colorectal cancer: Implications for diagnosis
and therapy (Review). Oncol Lett. 16:9–18. 2018.PubMed/NCBI
|
|
6
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grady WM and Carethers JM: Genomic and
epigenetic instability in colorectal cancer pathogenesis.
Gastroenterology. 135:1079–1099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Markowitz SD and Bertagnolli MM: Molecular
origins of cancer: Molecular basis of colorectal cancer. N Engl J
Med. 361:2449–2460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sinicrope FA and Sargent DJ: Molecular
pathways: microsatellite instability in colorectal cancer:
prognostic, predictive, and therapeutic implications. Clin Cancer
Res. 18:1506–1512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Worthley DL and Leggett BA: Colorectal
cancer: Molecular features and clinical opportunities. Clin Biochem
Rev. 31:31–38. 2010.PubMed/NCBI
|
|
11
|
Weisenberger DJ, Siegmund KD, Campan M,
Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D,
Buchanan D, et al: CpG island methylator phenotype underlies
sporadic microsatellite instability and is tightly associated with
BRAF mutation in colorectal cancer. Nat Genet. 38:787–793. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abdel-Rahman WM and Peltomäki P: Molecular
basis and diagnostics of hereditary colorectal cancers. Ann Med.
36:379–388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pawlik TM, Raut CP and Rodriguez-Bigas MA:
Colorectal carcinogenesis: MSI-H versus MSI-L. Dis Markers.
20:199–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Benatti P, Gafà R, Barana D, Marino M,
Scarselli A, Pedroni M, Maestri I, Guerzoni L, Roncucci L,
Menigatti M, et al: Microsatellite instability and colorectal
cancer prognosis. Clin Cancer Res. 11:8332–8340. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Popat S, Hubner R and Houlston RS:
Systematic review of microsatellite instability and colorectal
cancer prognosis. J Clin Oncol. 23:609–618. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Samowitz WS, Albertsen H, Herrick J, Levin
TR, Sweeney C, Murtaugh MA, Wolff RK and Slattery ML: Evaluation of
a large, population-based sample supports a CpG island methylator
phenotype in colon cancer. Gastroenterology. 129:837–845. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sinicrope FA and Sargent DJ: Clinical
implications of microsatellite instability in sporadic colon
cancers. Curr Opin Oncol. 21:369–373. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wee P and Wang Z: Epidermal growth factor
receptor cell proliferation signaling pathways. Cancers (Basel).
9:1–45. 2017.
|
|
19
|
Fernández-Medarde A and Santos E: Ras in
cancer and developmental diseases. Genes Cancer. 2:344–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Neumann J, Zeindl-Eberhart E, Kirchner T
and Jung A: Frequency and type of KRAS mutations in routine
diagnostic analysis of metastatic colorectal cancer. Pathol Res
Pract. 205:858–862. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kosmidou V, Oikonomou E, Vlassi M,
Avlonitis S, Katseli A, Tsipras I, Mourtzoukou D, Kontogeorgos G,
Zografos G and Pintzas A: Tumor heterogeneity revealed by KRAS,
BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor
mutation profile differences and their therapeutic implications.
Hum Mutat. 35:329–340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schubbert S, Shannon K and Bollag G:
Hyperactive Ras in developmental disorders and cancer. Nat Rev
Cancer. 7:295–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rajagopalan H, Bardelli A, Lengauer C,
Kinzler KW, Vogelstein B and Velculescu VE: Tumorigenesis: RAF/RAS
oncogenes and mismatch-repair status. Nature. 418:9342002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yuen ST, Davies H, Chan TL, Ho JW, Bignell
GR, Cox C, Stephens P, Edkins S, Tsui WW, Chan AS, et al:
Similarity of the phenotypic patterns associated with BRAF and KRAS
mutations in colorectal neoplasia. Cancer Res. 62:6451–6455.
2002.PubMed/NCBI
|
|
25
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF, NRAS,
and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy
in chemotherapy-refractory metastatic colorectal cancer: A
retrospective consortium analysis. Lancet Oncol. 11:753–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Calistri D, Rengucci C, Seymour I,
Leonardi E, Truini M, Malacarne D, Castagnola P and Giaretti W:
KRAS, p53 and BRAF gene mutations and aneuploidy in sporadic
colorectal cancer progression. Cell Oncol. 28:161–166.
2006.PubMed/NCBI
|
|
27
|
Cantwell-Dorris ER, O'Leary JJ and Sheils
OM: BRAFV600E: Implications for carcinogenesis and molecular
therapy. Mol Cancer Ther. 10:385–394. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Di Nicolantonio F, Martini M, Molinari F,
Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L,
Frattini M, Siena S, et al: Wild-type BRAF is required for response
to panitumumab or cetuximab in metastatic colorectal cancer. J Clin
Oncol. 26:5705–5712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karapetis CS, Khambata-Ford S, Jonker DJ,
O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD,
Robitaille S, et al: K-ras mutations and benefit from cetuximab in
advanced colorectal cancer. N Engl J Med. 359:1757–1765. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bardelli A and Siena S: Molecular
mechanisms of resistance to cetuximab and panitumumab in colorectal
cancer. J Clin Oncol. 28:1254–1261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
van Brummelen EMJ, de Boer A, Beijnen JH
and Schellens JHM: BRAF mutations as predictive biomarker for
response to anti-EGFR monoclonal antibodies. Oncologist.
22:864–872. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao B, Wang L, Qiu H, Zhang M, Sun L,
Peng P, Yu Q and Yuan X: Mechanisms of resistance to anti-EGFR
therapy in colorectal cancer. Oncotarget. 8:3980–4000. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Borràs E, Jurado I, Hernan I, Gamundi MJ,
Dias M, Martí I, Mañé B, Arcusa A, Agúndez JAG, Blanca M, et al:
Clinical pharmacogenomic testing of KRAS, BRAF and EGFR mutations
by high resolution melting analysis and ultra-deep pyrosequencing.
BMC Cancer. 11:4062011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mohamed Suhaimi NA, Foong YM, Lee DYS,
Phyo WM, Cima I, Lee EXW, Goh WL, Lim WY, Chia KS, Kong SL, et al:
Non-invasive sensitive detection of KRAS and BRAF mutation in
circulating tumor cells of colorectal cancer patients. Mol Oncol.
9:850–860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sclafani F, Chau I, Cunningham D, Hahne
JC, Vlachogiannis G, Eltahir Z, Lampis A, Braconi C, Kalaitzaki E,
De Castro DG, et al: KRAS and BRAF mutations in circulating tumour
DNA from locally advanced rectal cancer. Sci Rep. 8:14452018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nguyen HT, Geens M, Mertzanidou A, Jacobs
K, Heirman C, Breckpot K and Spits C: Gain of 20q11.21 in human
embryonic stem cells improves cell survival by increased expression
of Bcl-xL. Mol Hum Reprod. 20:168–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Boland CR, Thibodeau SN, Hamilton SR,
Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA,
Fodde R, Ranzani GN and Srivastava S: A National Cancer Institute
Workshop on Microsatellite Instability for cancer detection and
familial predisposition: development of international criteria for
the determination of microsatellite instability in colorectal
cancer. Cancer Res. 58:5248–5257. 1998.PubMed/NCBI
|
|
39
|
Findeisen P, Kloor M, Merx S, Sutter C,
Woerner SM, Dostmann N, Benner A, Dondog B, Pawlita M, Dippold W,
et al: T25 repeat in the 3′ untranslated region of the CASP2 gene:
A sensitive and specific marker for microsatellite instability in
colorectal cancer. Cancer Res. 65:8072–8078. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nguyen HT, Markouli C, Geens M, Barbé L,
Sermon K and Spits C: Human embryonic stem cells show low-grade
microsatellite instability. Mol Hum Reprod. 20:981–989. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Skotheim RI, Diep CB, Kraggerud SM,
Jakobsen KS and Lothe RA: Evaluation of loss of
heterozygosity/allelic imbalance scoring in tumor DNA. Cancer Genet
Cytogenet. 127:64–70. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Heaphy CM, Hines WC, Butler KS, Haaland
CM, Heywood G, Fischer EG, Bisoffi M and Griffith JK: Assessment of
the frequency of allelic imbalance in human tissue using a
multiplex polymerase chain reaction system. J Mol Diagn. 9:266–271.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gilder JR, Inman K, Shields W and Krane
DE: Magnitude-dependent variation in peak height balance at
heterozygous STR loci. Int J Legal Med. 125:87–94. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Leclair B, Frégeau CJ, Bowen KL and
Fourney RM: Systematic analysis of stutter percentages and allele
peak height and peak area ratios at heterozygous STR loci for
forensic casework and database samples. J Forensic Sci. 49:968–980.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sinicrope FA, Foster NR, Thibodeau SN,
Marsoni S, Monges G, Labianca R, Kim GP, Yothers G, Allegra C,
Moore MJ, et al: DNA mismatch repair status and colon cancer
recurrence and survival in clinical trials of 5-fluorouracil-based
adjuvant therapy. J Natl Cancer Inst. 103:863–875. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Roth AD, Delorenzi M, Tejpar S, Yan P,
Klingbiel D, Fiocca R, d'Ario G, Cisar L, Labianca R, Cunningham D,
et al: Integrated analysis of molecular and clinical prognostic
factors in stage II/III colon cancer. J Natl Cancer Inst.
104:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Klingbiel D, Saridaki Z, Roth AD, Bosman
FT, Delorenzi M and Tejpar S: Prognosis of stage II and III colon
cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation
to microsatellite status: Results of the PETACC-3 trial. Ann Oncol.
26:126–132. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Allegra CJ, Jessup JM, Somerfield MR,
Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF and
Schilsky RL: American Society of Clinical Oncology provisional
clinical opinion: Testing for KRAS gene mutations in patients with
metastatic colorectal carcinoma to predict response to
anti-epidermal growth factor receptor monoclonal antibody therapy.
J Clin Oncol. 27:2091–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Van Cutsem E, Köhne CH, Láng I, Folprecht
G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D,
Tejpar S, et al: Cetuximab plus irinotecan, fluorouracil, and
leucovorin as first-line treatment for metastatic colorectal
cancer: Updated analysis of overall survival according to tumor
KRAS and BRAF mutation status. J Clin Oncol. 29:2011–2019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Douillard JY, Oliner KS, Siena S,
Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham
D, Jassem J, et al: Panitumumab-FOLFOX4 treatment and RAS mutations
in colorectal cancer. N Engl J Med. 369:1023–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ashktorab H, Smoot DT, Carethers JM,
Rahmanian M, Kittles R, Vosganian G, Doura M, Nidhiry E, Naab T,
Momen B, et al: High incidence of microsatellite instability in
colorectal cancer from African Americans. Clin Cancer Res.
9:1112–1117. 2003.PubMed/NCBI
|
|
52
|
Salto-Tellez M, Tan SY, Chiu LL and Koay
ESC: Dinucleotide microsatellite repeats are essential for the
diagnosis of microsatellite instability in colorectal cancer in
Asian patients. World J Gastroenterol. 11:2781–2783. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Asaka S, Arai Y, Nishimura Y, Yamaguchi K,
Ishikubo T, Yatsuoka T, Tanaka Y and Akagi K: Microsatellite
instability-low colorectal cancer acquires a KRAS mutation during
the progression from Dukes' A to Dukes' B. Carcinogenesis.
30:494–499. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yamada K, Kanazawa S, Koike J, Sugiyama H,
Xu C, Funahashi K, Boland CR, Koi M and Hemmi H: Microsatellite
instability at tetranucleotide repeats in sporadic colorectal
cancer in Japan. Oncol Rep. 23:551–561. 2010.PubMed/NCBI
|
|
55
|
Kim CG, Ahn JB, Jung M, Beom SH, Kim C,
Kim JH, Heo SJ, Park HS, Kim JH, Kim NK, et al: Effects of
microsatellite instability on recurrence patterns and outcomes in
colorectal cancers. Br J Cancer. 115:25–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Woerner SM, Benner A, Sutter C, Schiller
M, Yuan YP, Keller G, Bork P, Doeberitz M and Gebert JF:
Pathogenesis of DNA repair-deficient cancers: A statistical
meta-analysis of putative Real Common Target genes. Oncogene.
22:2226–2235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yamamoto H, Adachi Y, Taniguchi H,
Kunimoto H, Nosho K, Suzuki H and Shinomura Y: Interrelationship
between microsatellite instability and microRNA in gastrointestinal
cancer. World J Gastroenterol. 18:2745–2755. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Poynter JN, Siegmund KD, Weisenberger DJ,
Long TI, Thibodeau SN, Lindor N, Young J, Jenkins MA, Hopper JL,
Baron JA, et al Colon Cancer Family Registry Investigators, :
Molecular characterization of MSI-H colorectal cancer by MLHI
promoter methylation, immunohistochemistry, and mismatch repair
germline mutation screening. Cancer Epidemiol Biomarkers Prev.
17:3208–3215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kim GP, Colangelo LH, Wieand HS, Paik S,
Kirsch IR, Wolmark N and Allegra CJ; National Cancer Institute, :
Prognostic and predictive roles of high-degree microsatellite
instability in colon cancer: a National Cancer Institute-National
Surgical Adjuvant Breast and Bowel Project Collaborative Study. J
Clin Oncol. 25:767–772. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Thibodeau SN, Bren G and Schaid D:
Microsatellite instability in cancer of the proximal colon.
Science. 260:816–819. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cho YK, Kim HC, Kim SH, Park JH, Yun HR,
Cho YB, Yun SH, Lee WY and Chun HK: Location-related differences in
sporadic microsatellite unstable colorectal cancer. Dig Liver Dis.
42:611–615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yang Y, Wang G, He J, Ren S, Wu F, Zhang J
and Wang F: Gender differences in colorectal cancer survival: A
meta-analysis. Int J Cancer. 141:1942–1949. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Gryfe R, Kim H, Hsieh ETK, Aronson MD,
Holowaty EJ, Bull SB, Redston M and Gallinger S: Tumor
microsatellite instability and clinical outcome in young patients
with colorectal cancer. N Engl J Med. 342:69–77. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Van Cutsem E, Köhne CH, Hitre E, Zaluski
J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G,
et al: Cetuximab and chemotherapy as initial treatment for
metastatic colorectal cancer. N Engl J Med. 360:1408–1417. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Roth AD, Tejpar S, Delorenzi M, Yan P,
Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C,
et al: Prognostic role of KRAS and BRAF in stage II and III
resected colon cancer: Results of the translational study on the
PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 28:466–474.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yokota T: Are KRAS/BRAF mutations potent
prognostic and/or predictive biomarkers in colorectal cancers?
Anticancer Agents Med Chem. 12:163–171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Vaughn CP, Zobell SD, Furtado LV, Baker CL
and Samowitz WS: Frequency of KRAS, BRAF, and NRAS mutations in
colorectal cancer. Genes Chromosomes Cancer. 50:307–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Peeters M, Kafatos G, Taylor A, Gastanaga
VM, Oliner KS, Hechmati G, Terwey JH and van Krieken JH: Prevalence
of RAS mutations and individual variation patterns among patients
with metastatic colorectal cancer: A pooled analysis of randomised
controlled trials. Eur J Cancer. 51:1704–1713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Guo F, Gong H, Zhao H, Chen J, Zhang Y,
Zhang L, Shi X, Zhang A, Jin H, Zhang J, et al: Mutation status and
prognostic values of KRAS, NRAS, BRAF and PIK3CA in 353 Chinese
colorectal cancer patients. Sci Rep. 8:60762018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Loupakis F, Ruzzo A, Cremolini C, Vincenzi
B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E,
et al: KRAS codon 61, 146 and BRAF mutations predict resistance to
cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type
metastatic colorectal cancer. Br J Cancer. 101:715–721. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Peeters M, Douillard JY, Van Cutsem E,
Siena S, Zhang K, Williams R and Wiezorek J: Mutant KRAS codon 12
and 13 alleles in patients with metastatic colorectal cancer:
Assessment as prognostic and predictive biomarkers of response to
panitumumab. J Clin Oncol. 31:759–765. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hsu HC, Thiam TK, Lu YJ, Yeh CY, Tsai WS,
You JF, Hung HY, Tsai CN, Hsu A, Chen HC, et al: Mutations of
KRAS/NRAS/BRAF predict cetuximab resistance in metastatic
colorectal cancer patients. Oncotarget. 7:22257–22270. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Li HT, Lu YY, An YX, Wang X and Zhao QC:
KRAS, BRAF and PIK3CA mutations in human colorectal cancer:
Relationship with metastatic colorectal cancer. Oncol Rep.
25:1691–1697. 2011.PubMed/NCBI
|
|
74
|
Hu J, Yan WY, Xie L, Cheng L, Yang M, Li
L, Shi J, Liu BR and Qian XP: Coexistence of MSI with KRAS mutation
is associated with worse prognosis in colorectal cancer. Medicine
(Baltimore). 95:e56492016. View Article : Google Scholar : PubMed/NCBI
|