Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most
aggressive primary pancreatic neoplasm and has the poorest
prognosis among solid tumors, with a 5-year-survival rate of only
9% (1). The management of pancreatic
cancer is notably difficult due to a poor response to available
therapeutic modalities, such as chemotherapy and radiotherapy
(2). Over the past decade,
gemcitabine has been demonstrated to improve median survival time
and quality of life in patients with advanced pancreatic cancer,
but the prognosis of the disease remains dismal (3). Gemcitabine resistance is one of the
main challenges, and the underlying mechanisms remain unclear.
It is known that numerous anticancer therapeutic
agents, including doxorubicin, cisplatin and camptotecin, typically
rely on causing DNA damage, which engages potent DNA damage
response signaling pathways that culminate in apoptosis or growth
arrest at checkpoints to allow for damage repair (4–6).
Cellular senescence, as a stress-responsive cell cycle arrest
program, is commonly considered as a tumor-suppressing response
(7) and has been demonstrated to
contribute to the outcomes of anticancer chemotherapy in
vivo, such as in lymphoma (8).
However, previous studies have suggested that senescence may
protect cancer cells from genotoxic treatment due to a
chemoprotective environment (9,10). In
addition, the senescence phenotype of pancreatic cancer cells
induced by gemcitabine, which is characterized by enhanced
senescence-associated β-galactosidase (SA-β-Gal) and increased
expression levels of senescence-associated secretory phenotype
(SASP), is associated with resistance to gemcitabine (11,12).
Thus, there is contrasting evidence on the tumor-suppressing and
tumor-promoting functions of therapy-induced senescence (TIS).
Therefore, the role of senescence in the chemotherapy of pancreatic
cancer and the underlying mechanisms remain unclear.
Emerging evidence has revealed that microRNAs
(miRNAs/miRs) are potential regulators of cellular senescence by
targeting genes involved in the production of reactive oxygen
species, shortening of telomeres, mitochondrial damage and cell
cycle arrest via production of tumor suppressor proteins (13). miR-7, which acts as a tumor
suppressor in various gastrointestinal types of cancer, including
pancreatic cancer (14), can
downregulate poly (ADP-ribose) polymerase 1 (PARP1), which
functions as a regulator of diverse biological processes, including
DNA repair and chromatin remodeling in the senescence program
(15,16). Thus, considering the role of cellular
senescence as a contributing factor to therapy resistance, the
present study aimed to investigate whether aberrant miR-7
expression modulated senescence to influence cancer response to
chemotherapy.
Materials and methods
Cell culture
The human pancreatic ductal adenocarcinoma PANC-1
cell line was obtained from The Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences. PANC-1 cells were
cultured in DMEM supplemented with 10% FBS (both Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in a 5% CO2 incubator.
A gemcitabine-resistant pancreatic cancer cell line was established
by treating PANC-1 cells with 0.25 µM gemcitabine for 4 weeks.
Constructs, oligonucleotides and
reagents
TargetScanHuman 7.2 (http://www.targetscan.org) was used to identify
potential target genes of miR-7. To confirm the molecular
interaction between miR-7 and PARP1, the total RNA of human
embryonic kidney 293T cells (purchased from The Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences and cultured
in DMEM with 10% FBS) was exacted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and then reverse
transcribed into cDNA using PrimeScript™ RT Master Mix according to
standard procedures (Takara Biotechnology Co., Ltd.). PCR
amplification was performed using PrimerSTAR HS DNA polymerase
(Takara Biotechnology Co., Ltd.) with initial denaturation at 98°C
for 10 sec, followed by 30 cycles of 5 sec denaturation at 55°C and
1 min extension at 72°C, and final extension at 72°C for 2 min. The
products were collected by 1% agarose gel electrophoresis using
Gel-red for visualization. The 3′-untranslated region (UTR; 769 bp)
of PARP1 was amplified by PCR using the following primers: forward,
5′-ACTGCTAGCGGTAATTGGGAGAGGTAGC-3′ and reverse,
5′-TGAGTCGACTAGAGAAGGCATCTGCATTTTTAATC-3′. The amplified 3′-UTR was
sequenced by Sangon Biotech Co., Ltd., double-digested with
NheI/SalI and inserted into the corresponding
digested reporter plasmid pmirGLO (7,350 bp; Promega Corporation).
Similarly, the putative counterparts [wild-type (WT) and mutant
(MT)] of the miR-7-target sequence in the 3′-UTR of PARP1 (GenBank
NM_001618.4) were also used to construct the corresponding reporter
plasmids, pmirGLO-PARP1-WT and pmirGLO-PARP1-MT, respectively.
Sequencing of the luciferase reporter constructs was performed by
Sangon Biotech Co., Ltd. Synthetic miR-7-5p mimics (50 nM),
miR-7-5p inhibitor (100 nM) and their negative control
oligonucleotides (non-targeting), purchased from Guangzhou RiboBio
Co., Ltd., were transfected into parent and gemcitabine-resistant
PANC-1 cells using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) for 15 min at room temperature.
Subsequent experiments were performed 24 h after transfection. The
PARP signaling inhibitor rucaparib (AG-014699) phosphate was
purchased from Selleck Chemicals. All the enzymes for molecular
cloning were purchased from New England Biolabs, Inc. The sequences
of the synthesized oligonucleotides used in the present study are
listed in Table SI.
Luciferase reporter assay
To determine the miR-7 target genes, PANC-1 cells
cultured in 24-well plates were co-transfected with reporter
plasmids and miR-7 mimics using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). Cells were harvested
and lysed 48 h after transfection. Luciferase assays were performed
using the Dual-Luciferase® Reporter Assay (cat. no.
E1910; Promega Corporation) according to the manufacturer's
protocol. Firefly luciferase activity was normalized to
Renilla luciferase activity, and the relative luciferase
score was calculated.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA from PANC-1 cells was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and 1 µg RNA was reverse transcribed into cDNA using
PrimeScript™ RT reagent kit according to the manufacturer's
protocol (Takara Biotechnology Co., Ltd.). qPCR was performed in
triplicate with the use of the SYBR® Premix Ex Taq™
(Takara Biotechnology Co., Ltd.) on the QuantStudio™ 6 Flex System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The primers
for the genes used in present study were synthesized by Sangon
Biotech Co., Ltd. The thermocycling conditions were as follows:
Initial denaturation at 95°C for 30 sec, followed by 40 cycles of 5
sec denaturation at 95°C and 34 sec extension at 60°C. U6 small
nuclear RNA and GAPDH were used as internal controls for miRNA and
mRNA assays, respectively. The 2−∆∆Cq method (17) was used to calculate the relative mRNA
expression level after normalization to U6 and GAPDH RNA. All the
RT-qPCR primer sequences are listed in Table SII.
Western blotting
RIPA lysis buffer (Thermo Fisher Scientific, Inc.)
was used to lyse and extract total protein from PANC-1
gemcitabine-resistant and parent cell lines. Protein concentration
was determined used a BCA protein assay and protein lysates (25
ng/µl; 6 µl/lane) were separated via 10 or 12% SDS-PAGE according
to molecular weight. Subsequently, separated protein bands were
transferred onto polyvinylidene fluoride membranes (PVDF) (EMD
Millipore) and blocked with 5% skimmed milk for 1 h at room
temperature. Next, the membranes were first probed with the
relevant primary antibodies overnight at 4°C and subsequently
incubated with HRP-labeled secondary antibodies for 2 h at room
temperature for visualization using Immobilon™ Western HRP
Substrate kit (EMD Millipore). The antibodies used are listed in
Table SIII.
Immunofluorescence
Cells were plated onto glass coverslips, fixed with
4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton
X-100 in phosphate-buffered saline (PBS) for 15 min at room
temperature. Next, 5% BSA (Invitrogen; Thermo Fisher Scientific,
Inc.) was applied for 1 h at room temperature, and then rabbit
anti-human phospho-NF-κB p65 (Ser536; Table SIII) was added and incubated at 4°C
overnight. Next, Alexa Fluor 594-conjugated goat anti-rabbit
secondary antibody was added and incubated for 2 h at room
temperature. Finally, the cells were stained with DAPI to stain
cell nuclei for 5 min at room temperature. Immunostaining signals
and DAPI-stained nuclei were visualized using a confocal microscope
at ×40 magnification (TCS SP8; Leica Microsystems, Inc.).
ALDEFLUOR™ assay
The ALDEFLUOR™ kit (Stemcell Technologies, Inc.) was
used to analyze the population with aldehyde dehydrogenase (ALDH)
enzymatic activity according to the manufacturer's protocol.
Briefly, cells were incubated in the ALDEFLUOR™ assay buffer
containing the ALDH substrate BAAA at 37°C for 45 min, whereas
control cells were incubated with 50 mM ALDH inhibitor
diethylamino-benzaldehyde under the same conditions. BAAA-stained
cells were analyzed using a FACSCalibur flow cytometer (Becton,
Dickinson and Company) with CellQuest software (version 4.0.2;
Becton, Dickinson and Company).
Flow cytometry assay
The ratio of CD24+CD326+
subpopulation was evaluated using flow cytometry assay. Briefly,
the cells were collected, resuspended in binding buffer and labeled
with 10 µl PE mouse anti-human CD24 antibody and 10 µl BB515 mouse
anti-human CD326 antibody (Table
SIII) in the dark at room temperature for 15 min. Cells were
then analyzed using a FACSCalibur flow cytometer (Becton, Dickinson
and Company) with CellQuest software (version 4.0.2; Becton,
Dickinson and Company) to determine the percentage of
CD24+CD326+ cells.
Tumorsphere culture
The tumorsphere culture method and medium
preparation were performed according to a previous study (18). PANC-1 cells were plated in 6-well
ultralow attachment plates (Corning Inc.) at a concentration of
10,000 cells/well. After 14 days, spheres with ≥50 µm in diameter
were counted under an inverted phase-contrast light microscope at
×10 magnification.
Cell viability assay
Cells were plated at a density of
2×105/well in 6-well plates with different
concentrations (0, 0.1, 0.25 and 1.0 µM) of gemcitabine at 37°C in
a 5% CO2 incubator for 24 h. After 2 days, cells were
collected and counted using a hemocytometer.
Cell senescence assay
Cell senescence was detected using a SA-β-Gal assay
kit (Cell Signaling Technology, Inc.), following the manufacturer's
protocol. The cells with different treatments were grown on 6-well
culture plates, washed and fixed for 15 min at room temperature
with 4% paraformaldehyde. Subsequently, cells were washed with PBS
three times and incubated with β-galactosidase staining solution
overnight at 37°C (pH 6.0). A light microscope at ×10 magnification
was used to collect the image files and the percentage of blue
cells was calculated using Image-Pro Plus software (version 6.0;
Media Cybernetics, Inc.).
EdU assay
Cell proliferation was analyzed using a Cell-Light
EdU DNA Cell Proliferation kit (Guangzhou RiboBio Co., Ltd.)
according to the manufacturer's protocol. PANC-1 parent and
gemcitabine-resistant cells were detected using a fluorescence
microscope (magnification, ×20), and cell proliferation was
determined as the ratio of EdU-positive cells.
Cell Counting Kit-8 (CCK-8) assay
The CCK-8 assay (Dojindo Molecular Technologies,
Inc.) was performed to assess gemcitabine-resistant PANC-1 cell
viability according to the manufacturer's protocol. Briefly, cells
were incubated in 96-well plates with 10 µl CCK-8 reagent per well
for 2 h at 37°C. Subsequently, cell viability was measured at a
wavelength of 450 nm using a microplate reader.
Tissue microarrays
The pancreatic tissue microarray HPan-Ade180Sur-02
was purchased from Shanghai Xinchao Biological Technology Co., Ltd.
HPan-Ade180Sur-02 incorporated 100 cases of pancreatic tumor and 80
cases of adjacent non-tumor tissues, of which 63 males and 37
females. The median age was 62 years (range, 34–85 years). All the
raw data including overall survival are available from Shanghai
Xinchao Biological Technology Co., Ltd.
Immunohistochemistry
Immunohistochemistry was performed on tissue
microarray chips (Shanghai Xinchao Biological Technology Co.,
Ltd.). Paraffin-embedded sections were immersed in xylene solution
to dewax for 10 min twice at room temperature and rehydrated in
100, 95, 85 and 75% gradient ethanol. Subsequently, 3% hydrogen
peroxide was used for quenching. After recovering the antigens and
blocking with 5% BSA (Invitrogen; Thermo Fisher Scientific, Inc.)
at 37°C for 1 h, the slides were probed with the primary antibody
rabbit anti-human histone H3 (trimethyl K9) at 4°C overnight and
then with an HRP-conjugated goat anti-rabbit secondary antibody for
2 h at room temperature (Table
SIII). The proteins were visualized in situ with DAB
chromogenic substrate. The sections were observed using light
microscopy at a magnification of ×20. The results of immunostaining
were evaluated as follows: The mean percentage of stained tumor
cells per specimen was determined semi-quantitatively and scored as
0 for no positive cells, 1 for <10% positive cells, 2 for 10–50%
positive cells and 3 for >50% positive cells. The intensity of
staining was determined as 0 for no staining, 1 for weak staining,
2 for moderate staining and 3 for strong staining. The staining
index (SI) for each specimen was calculated as the product of the
staining intensity by the percentage of positive tumor cells. A SI
of 0–3 represented negative triMeH3K9 expression, while a SI of 4–9
represented positive triMeH3K9 expression.
Bioinformatic analysis
Raw RNA-seq data and survival data for 175
pancreatic tumors were downloaded from The Cancer Genome Atlas
(TCGA) pancreatic ductal adenocarcinoma project (http://oncolnc.org). Data were analyzed for
cyclin-dependent kinase inhibitor 1A (CDKN1A/p21) and Ki67 gene
expression. The expression levels of CDKN1A were listed from the
lowest to the highest. The corresponding patients were numbered 1
to 175 in sequence. The expression levels of Ki67 were listed from
the highest to the lowest, and the patients were numbered as
aforementioned. The senescence score was the sum of the number
generated from the expression levels of CDKN1A and Ki67. The larger
summed number was associated with a more senescent state.
Kaplan-Meier analysis and log-rank test were used to assess the
association between the senescence score and survival. RNA-seq and
sample profiling dataset of GSE140077 (19) was obtained from the Gene Expression
Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) and used for
comparison of gene expression levels between the BxPC-3
gemcitabine-resistant cell line and the parental cell line. For
TCGA and GEO databases, the expression levels were transformed into
log2 (TPM + 1) for subsequent analyses.
Statistical analysis
Normally distributed data were presented as the mean
± SD of ≥3 independent experiments. The unpaired Student's t-test
was used to compare differences between two groups. Data were
analyzed via one-way ANOVA followed by Dunnett's post-hoc test for
multiple comparisons. Frequencies of categorical variables were
compared using χ2 test. Statistical analyses were
performed using SPSS 19.0 software (IBM Corp.). All statistical
tests were two-tailed, and P<0.05 was considered to indicate a
statistically significant difference.
Results
Association between cellular
senescence and prognosis in patients with pancreatic cancer
Cellular senescence is often observed in clinical
pancreatic cancer tissues (20).
Therefore, the present study investigated whether there was an
association between cellular senescence and prognosis of pancreatic
cancer. First, the clinical relevance of cellular senescence was
assessed with classification based on CDKN1A/p21 positivity in
combination with Ki67 negativity in pancreatic cancer samples from
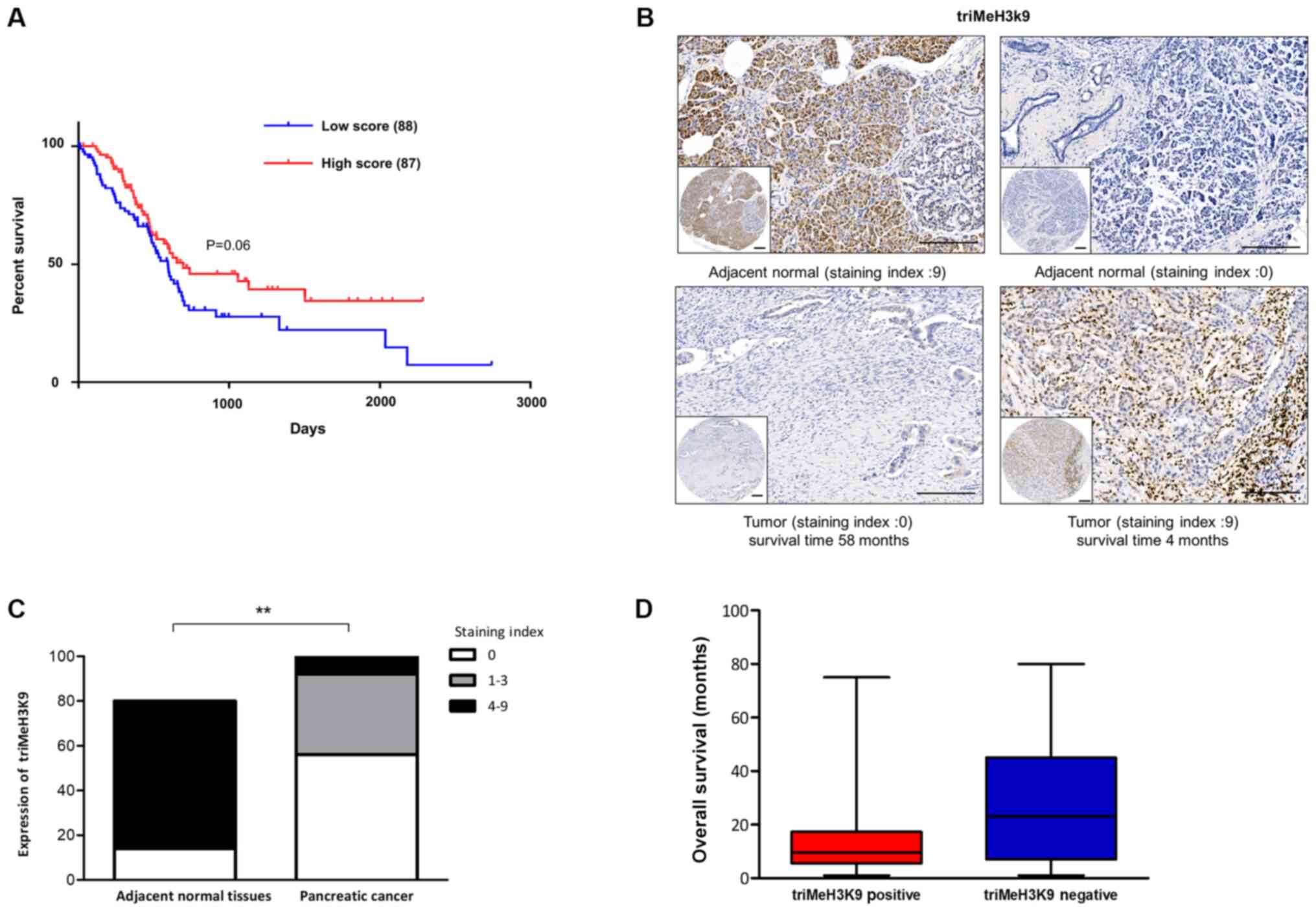
TCGA dataset. The results demonstrated that a higher senescence
level tended to be associated with improved patient prognosis
(Fig. 1A).
Considering that the formation of
senescence-associated heterochromatic foci specifically enriched
for H3K9me3 has been implicated in cellular senescence (21), the present study further examined the
expression levels of triMeH3K9 by immunohistochemistry in tissue
microarrays containing 100 cases of pancreatic tumors and 80 cases
of adjacent non-tumor tissues (Fig.
1B). It was observed that low triMeH3K9 expression (staining
index, 0–3) was detected in 92 of the examined PDAC tumors versus
14 of the adjacent non-tumor tissues (Fig. 1C). However, when the 100 cases of
PDAC tissues were divided into two groups, namely a positive and a
negative triMeH3K9 expression group, it was observed that the
average overall survival of the positive triMeH3K9 group was lower
than that of the negative group (Fig.
1D). These results indicated that cellular senescence
functioned as a potent tumor-suppressive process, as well as
exerting deleterious effects in patients with pancreatic
cancer.
Gemcitabine-induced senescent cells
present novel stem-cell features in gemcitabine-resistant
pancreatic cancer cells
The association between senescence and
gemcitabine-resistance in pancreatic cancer was further explored.
It is well known that gemcitabine is a first-line chemotherapeutic
agent in pancreatic cancer. One anticancer activity of gemcitabine
results from blocking DNA polymerase and causing DNA chain
termination (22). As a consequence
of DNA damage, senescence is activated in response to gemcitabine
to prevent the propagation of pancreatic cancer cells (21). In the present study, PDAC PANC-1
cells were exposed to different doses of gemcitabine, and it was
observed that gemcitabine induced senescence in PANC-1 cells, as
measured by decreased cell proliferation (Fig. 2A), increased SA-β-Gal activity
(Fig. 2B), elevated
senescence-associated signaling (Fig.
2C) and increased expression levels of several SASP factors
(Fig. 2D) mainly in a dose-dependent
manner.
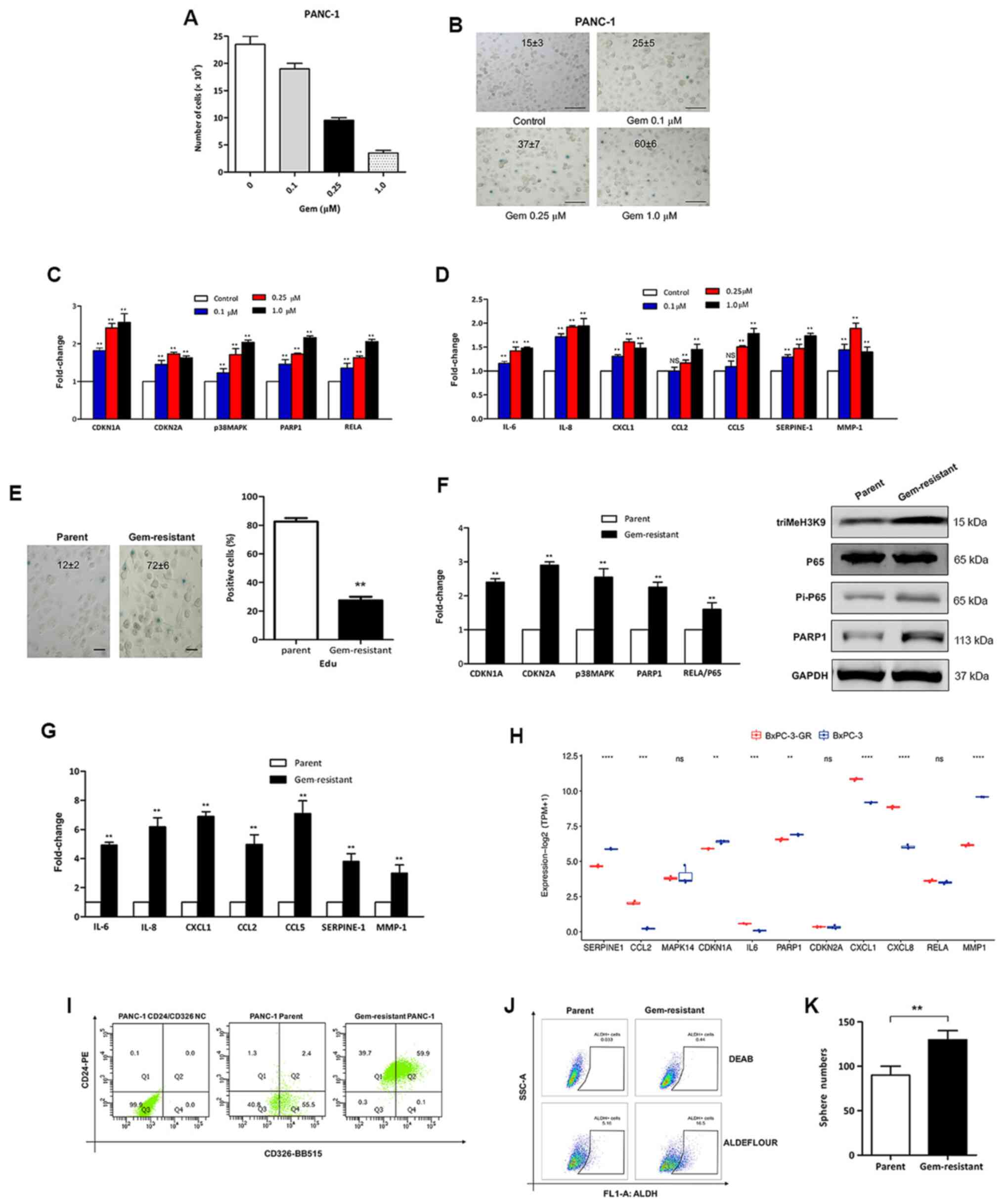 | Figure 2.Gem-induced senescent pancreatic
cancer cells possess phenotypic and functional stemness features.
(A) PANC-1 cells were treated with the indicated doses of gem for
24 h, and the cells were counted after 3 days. (B) PANC-1 cells
exposed to gem were stained for SA-β-Gal activity. The values shown
in the panels represent the percentage ± SD of SA-β-Gal-positive
cells. Scale bar, 100 µm. Cells were harvested and subjected to
RT-qPCR to detect (C) senescence-associated signaling and (D)
secretion of molecular mRNA. **P<0.01 vs. control. (E)
Gem-resistant PANC-1 cells were either fixed and stained for
SA-β-Gal (left), or incubated for 24 h with EdU and then fixed and
stained (right). The results are shown as the percentage of
positive cells (>100 cells scored; n=3 independent experiments).
**P<0.01 vs. parent. Scale bar, 50 µm. (F) Relative expression
levels of senescence-associated signaling molecules detected by
qPCR (left panel) and western blotting (right) panel in
gem-resistant PANC-1 cells. **P<0.01 vs. parent. (G) RT-qPCR
analysis of mRNA levels encoding the senescence-associated
secretory phenotype in gem-resistant PANC-1 cells. **P<0.01 vs.
parent. (H) Expression profile of senescence-associated genes in
gem-resistant and parental BxPC-3 cells, obtained from the
GSE140077 dataset from the Gene Expression Omnibus database.
**P<0.01; ***P<0.001; ****P<0.0001; ns, not significant.
Ratio and the potential of pancreatic cancer stem cells were
increased in gem-resistant PANC-1 cells. (I)
CD24+CD326+ subpopulation, (J)
ALDH+ subpopulation and (K) sphere numbers in parental
and gem-resistant PANC-1 cells. **P<0.01. SA-β-Gal,
senescence-associated β-galactosidase; RT-qPCR, reverse
transcription-quantitative PCR; Gem, gemcitabine; triMeH3K9,
histone H3 trimethyl K9; PARP1, poly (ADP-ribose) polymerase 1;
CDKN, cyclin-dependent kinase inhibitor; NC, negative control;
ALDH, aldehyde dehydrogenase. |
Furthermore, gemcitabine-resistant pancreatic cancer
cells exhibited upregulated SA-β-Gal activity (Fig. 2E, left panel) and a significant
decrease in DNA synthesis (Fig. 2E,
right panel). In addition, senescence was confirmed by increased
expression levels of CDKN1A/p21, CDKN2A/p16 and p38MAPK/PARP1/NF-κB
senescent signaling, and of triMeH3K9 proteins by qPCR or western
blot analysis (Fig. 2F), as well as
elevated mRNA levels of SASP components (Fig. 2G) using qPCR. In addition, the
transcriptional levels of senescence-associated genes in
gemcitabine-resistant and parental BxPC-3 cells were determined by
analyzing the GEO GSE140077 dataset. Notably, most key
senescence-associated genes were significantly higher in
gemcitabine-resistant BxPC-3 cells compared with their levels in
parental BxPC-3 cells (Fig. 2H).
These data indicated that gemcitabine induced senescence in
pancreatic cancer cells, and that gemcitabine-resistant pancreatic
cells had an increased senescence response.
It is known that cancer stem cells possess the
ability to survive therapeutic intervention. Since a previous study
reported that there was a cell-intrinsic association between the
senescence program and the acquisition of self-renewing properties
(23), the present study
investigated whether the senescence condition promoted cancer
stemness to induce resistance to gemcitabine in pancreatic cancer.
Thus, stemness-associated membrane markers were analyzed in
senescent PANC-1 cells with gemcitabine resistance. Acquisition of
the stemness-associated markers CD24 and CD326 was observed in
gemcitabine-resistant pancreatic cancer (Fig. 2I). In addition, as shown in Fig. 2J and K, ALDH+
subpopulations were markedly increased in gemcitabine-resistant
pancreatic cancer, as revealed using the ALDEFLUOR™ assay, and the
number of spheres of pancreatic cancer cells was significantly
increased in the gemcitabine-resistant group compared with that in
the parental group. Hence, pancreatic cancer cells acquired novel
stem-cell features upon chemotherapy-induced cellular
senescence.
Gemcitabine-induced senescence is
repressed by miR-7 via targeting PARP1/NF-κB signaling in
pancreatic cancer cells
In our previous study, it was observed that miR-7
expression was decreased in pancreatic cancer, as well as in
patients with gemcitabine-resistant pancreatic cancer, thus
potentially being a biomarker for gemcitabine sensitivity in
pancreatic cancer (24), and this
may be associated with the regulation of senescence. The present
study further explored whether miR-7 was involved in gemcitabine
resistance via regulation of senescence in pancreatic cancer cells.
Compared with that in the parental group, miR-7 exhibited a
significantly decreased expression in the gemcitabine-resistant
group of PANC-1 cells (Fig. 3A).
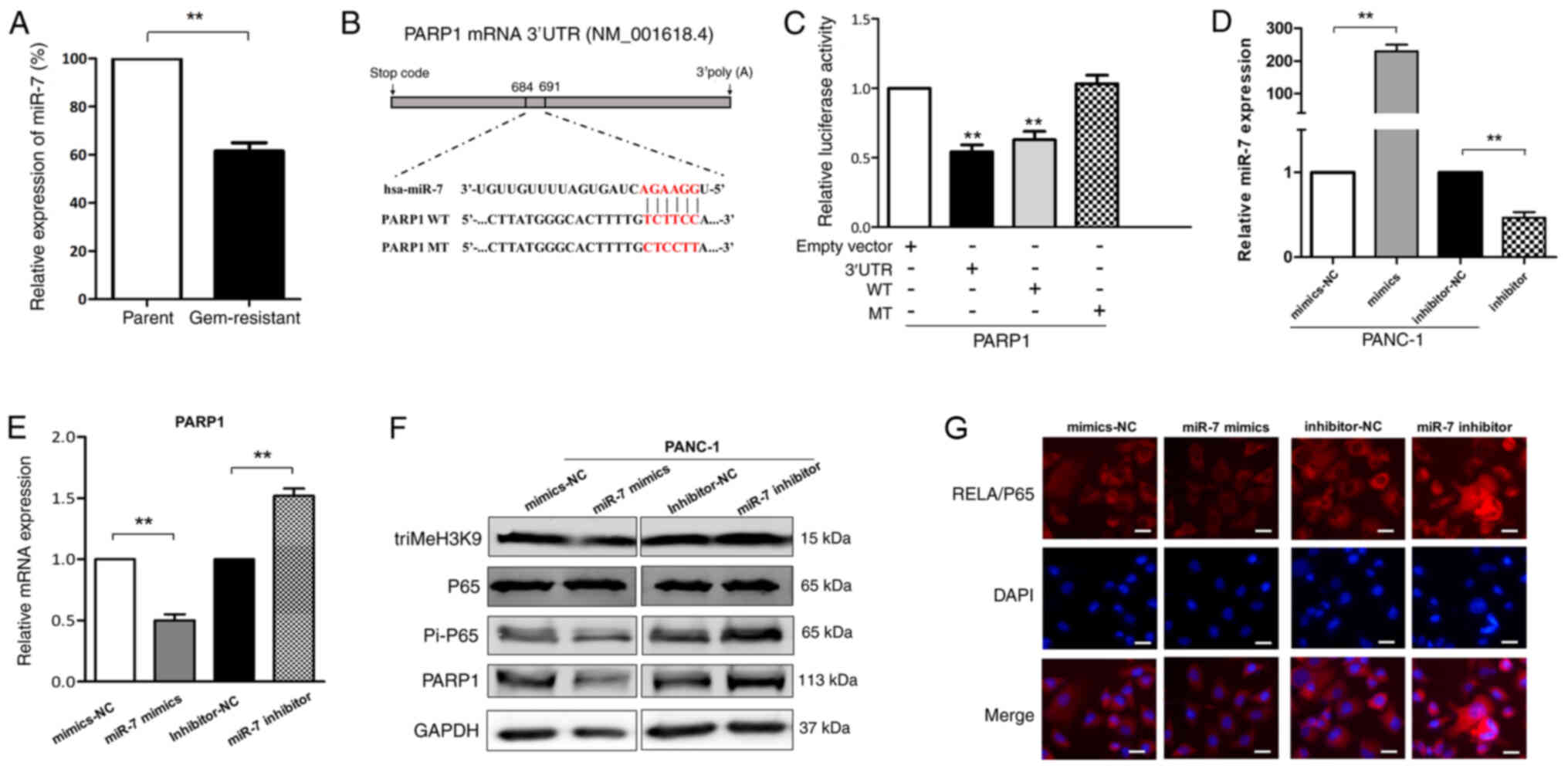 | Figure 3.Gem-induced senescence is repressed
by miR-7 via targeting PARP1/NF-κB signaling in pancreatic cancer
cells. (A) miR-7 expression by qPCR assay in parental and
gem-resistant PANC-1 cells. **P<0.01. (B) Predicted
miR-7-binding site sequence in the 3′-UTR of PARP1 and its WT or MT
counterparts. (C) Luciferase reporter assays were performed at 48 h
post-transfection to measure the relative luciferase activity on
the corresponding constructs of 3′-UTR of PARP1 and their WT or MT
counterparts for determination of PARP1 as miR-7 target in PANC-1
cells. **P<0.01 vs. empty vector. (D) Expression levels of miR-7
were analyzed by RT-qPCR in PANC-1 cells after transfection with
miR-7 mimics or inhibitor. **P<0.01. (E) PARP1 mRNA expression
in PANC-1 cells with overexpressed (mimics) or inhibited
(inhibitor) miR-7 was detected by RT-qPCR. **P<0.01. (F) Western
blot analysis of miR-7 mimics- or inhibitor-transfected PANC-1 cell
lysates probed with antibodies against triMeH3K9, p65, Pi-p65 and
PARP1. (G) Stimulation of NF-κB activity in pancreatic cancer
cells. Immunofluorescent staining with Pi-p65/RelA of PANC-1 cells
transfected with miR-7 mimics or inhibitor. Nuclei were
counterstained with DAPI. Scale bar, 50 µm. miR, microRNA; PARP1,
poly (ADP-ribose) polymerase 1; WT, wild-type; MT, mutant; UTR,
untranslated region; RT-qPCR, reverse transcription-quantitative
PCR; Gem, gemcitabine; triMeH3K9, histone H3 trimethyl K9; NC,
negative control; Pi, phospho. |
To further explore how miR-7 may be involved in
gemcitabine resistance through the regulation of senescence,
TargetScanHuman 7.2 was used, which identified PARP1 (GenBank
NM_001618.4) as a potential target gene of miR-7. PARP1 is a
genotoxic sensor involved in the activation of NF-κB and it
mediates the pro-invasive capacity of senescence-associated
secretome (25). Thus, the 3′-UTR of
PARP1 (PPU), and the corresponding PPU-WT or PPU-MT sequence of the
putative miR-7-binding site were cloned into the pmirGLO
Dual-luciferase reporter vector (Fig.
3B). As a result, the luciferase activity in PANC-1 cells was
significantly decreased after co-transfection of miR-7 mimics with
pmirGLO-PPU or pmirGLO-PPU-WT for 48 h, while the luciferase
activity upon transfection with pmirGLO-PPU-MT was not affected by
the inhibition of miR-7 mimics (Fig.
3C). Furthermore, when miR-7 was ectopically expressed
(Fig. 3D), both the mRNA and protein
levels of PARP1 were decreased using miR-7 mimics and increased
using miR-7 inhibitors in PANC-1 cells (Fig. 3E and F).
Additionally, the effects of miR-7 on PARP1/NF-κB
signaling were further evaluated. It is well known that NF-κB is
present as an inactive cytoplasmic form, while activated NF-κB is
translocated into the nucleus and binds to specific κB enhancer
elements in the promoter of targets genes. Thus, the present study
investigated the expression levels of the p65/RelA subunit of NF-κB
by western blot analysis and immunofluorescence. The results
revealed that, after inhibiting PARP1 expression using miR-7
mimics, the phosphorylation of p65/RelA decreased in PANC-1 cells
(Fig. 3F). Additionally, under
control conditions, p65/RelA localized mainly in the cytoplasm,
while in the miR-7 inhibitor group, p65/RelA localized in the
nucleus (Fig. 3G). A marked nuclear
accumulation of p65/RelA in pancreatic cancer cells was observed
upon miR-7 inhibitor treatment, which was stronger than that
induced by miR-7 mimics (Fig. 3G).
Furthermore, as expected, miR-7 negatively regulated triMeH3K9
formation (Fig. 3F). Overall, the
current data demonstrated that PARP1 may be a target of miR-7, and
that overexpression of miR-7 resulted in a marked decrease in
cellular PARP1 and subsequent NF-κB inactivation, further
repressing the senescence-associated phenotype.
miR-7 restores sensitivity to
gemcitabine by restraining cellular senescence in pancreatic cancer
cells
The present study demonstrated that
gemcitabine-resistant pancreatic cancer cells had increased
chemotherapy-induced senescence, and senescence-associated
reprogramming promoted pancreatic cancer stemness. Additionally, it
revealed that miR-7 expression was decreased in
gemcitabine-resistant PANC-1 cells, and that miR-7 may be an
important regulator of cellular senescence via targeting
PARP1/NF-κB signaling. Therefore, miR-7 expression was restored in
gemcitabine-resistant PANC-1 cells to determine if this could
sensitize pancreatic cancer cells to gemcitabine by repressing
cellular senescence and decreasing the population of cancer stem
cells.
Gemcitabine-resistant PANC-1 cells were transfected
with miR-7 mimics or treated with the PARP inhibitor rucaparib.
After 48 h, it was observed that the overexpression of miR-7 in
gemcitabine-resistant PANC-1 cells markedly decreased cellular
senescence, and comparable results were also obtained in the
rucaparib group, including decreased SA-β-Gal activity (Fig. 4A), decreased expression levels of
SASP (Fig. 4B) and downregulation of
PARP1/NF-κB senescent signaling (Fig.
4C). Additionally, assessment of the mRNA expression levels of
the cancer stem cell transcription factors OCT3/4 and NANOG, which
are involved in pluripotent cell maintenance and differentiation
(26), in gemcitabine-resistant
PANC-1 cells revealed that miR-7 mimics or rucaparib treatment
decreased the expression levels of these molecules compared with
those observed after gemcitabine treatment alone in
gemcitabine-resistant PANC-1 cells (Fig.
4D). Similarly, following ALDEFLOUR™ staining, the population
of ALDH+ cells in the miR-7 mimics group was decreased
by ~62% compared with that in the miR-7 mimics-NC group (Fig. 4E). Next, to evaluate the role of
miR-7 in the gemcitabine sensitivity of pancreatic cancer cells,
gemcitabine-resistant cells were treated with gemcitabine for 24 h
after being transfected with miR-7 mimics or being incubated with
rucaparib, and cell viability was evaluated 3 days later by CCK-8
assay. The results revealed that overexpression of miR-7 or
treatment with the PARP inhibitor rucaparib significantly increased
the sensitivity of resistant PANC-1 cells to gemcitabine (Fig. 4F). The current findings suggested
that PARP1 depletion by miR-7 may attenuate senescence and decrease
the stem cell population, which may enhance the sensitivity of
PANC-1 cells to gemcitabine.
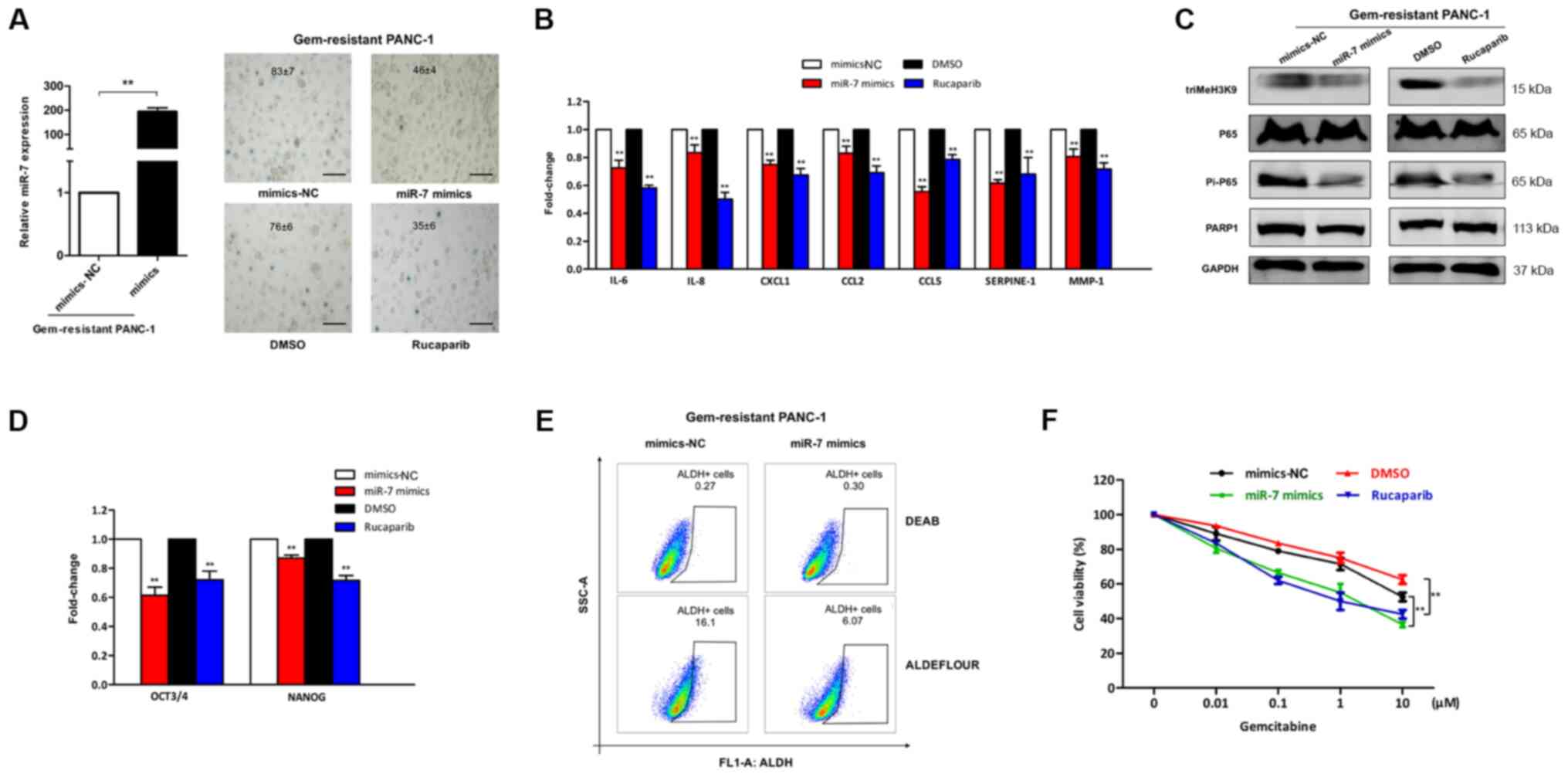 | Figure 4.miR-7/PARP1/NF-κB axis is engaged in
the chemo-sensitivity of PANC-1 cells to gemcitabine. (A) After
being transfected with miR-7 mimics or treated with the PARP
inhibitor rucaparib in gem-resistant PANC-1 cells, the relative
miR-7 expression was detected by RT-qPCR (left; **P<0.01) and
gem-resistant cells were subjected to senescence-associated
β-galactosidase staining to determine the number of positive cells,
which is shown as the mean percentage ± SD (right). Scale bar, 100
µm. (B) mRNA transcripts of senescence-associated secretory
phenotype and (C) protein levels of the PARP1/NF-κB senescent axis
were respectively analyzed by RT-qPCR and western blotting in
gem-resistant PANC-1 cells transfected with miR-7 mimics or treated
with rucaparib. **P<0.01 vs. mimics-NC or DMSO. (D) Expression
levels of the indicated stem cell-associated genes in gem-resistant
PANC-1 cells treated with miR-7 mimics or rucaparib was assessed by
RT-qPCR. **P<0.01 vs. mimics-NC or DMSO. (E) ALDH activity with
or without miR-7 mimics in gem-resistant PANC-1 cells was analyzed
by flow cytometry. (F) Gem-resistant cancer cells were treated with
gem for 24 h after being transfected with miR-7 mimics or incubated
with rucaparib. The viability of the cells was determined by Cell
Counting Kit-8 assay. **P<0.01. RT-qPCR, reverse
transcription-quantitative PCR; ALDH, aldehyde dehydrogenase; Gem,
gemcitabine; triMeH3K9, histone H3 trimethyl K9; PARP1, poly
(ADP-ribose) polymerase 1; miR, microRNA; NC, negative control. |
Discussion
Pancreatic cancer is an aggressive disease
associated with major morbidity and mortality, and chemo-resistance
is a major obstacle for PDAC treatment (27). In the last decade, gemcitabine has
been considered as the standard treatment, and has been widely
utilized as the first-line drug for advanced pancreatic cancer
(3). However, development of
chemo-resistance to gemcitabine severely limits the effectiveness
of this chemotherapy (28).
Therefore, it is urgent to clarify the underlying mechanism of the
development of gemcitabine resistance.
Recent studies have demonstrated that cellular
senescence may be involved in the acquisition of tumor cell
resistance to chemotherapy (29).
Cellular senescence is a state of permanent cell growth arrest that
is often one of the terminal outcomes of chemotherapy (29). Traditionally, senescence induction is
considered to be an important mechanism of cancer prevention and
cellular aging (30). However, a
recent study has revealed that senescence is a prominent solid
tumor response to therapy in which cancer cells evade apoptosis and
instead enter into a stable and prolonged cell cycle arrest
(31). The present study detected
TIS in a gemcitabine-resistant PDAC cell line through β-Gal
activity assay, assessment of SASP expression and senescence
signaling. As a result, it was demonstrated that senescence
protected tumors from gemcitabine-induced cytotoxicity. Thus, the
present study suggested that senescence may be involved in poor
chemotherapy responses in pancreatic cancer.
Compared with tumors undergoing apoptosis, there are
several problems with tumors becoming senescent. Firstly,
senescence can send cancer cells into a state similar to extended
sleep, in which cells are alive but not dividing; therefore, tumor
growth is arrested and does not regress (32). Senescence helps cancer cells avoid
death and turns them into cancer stem cells. Secondly, a
cell-cycle-targeting chemotherapeutic agent such as gemcitabine
(33) will lose efficiency in
senescent cells. Thirdly, the senescence-evoked cell-intrinsic
factors can reprogram cancer cells into a stem-like state (23), and the subpopulation of
chemotherapy-induced senescent cancer cells have demonstrated the
capability of escaping, thus leading to relapse (29). Lastly, senescent tumor cells are
metabolically active, and can secrete cytokines and other paracrine
factors, which feed and stimulate the growth of adjacent cells
(34). Thus, senescence is viewed as
a potentially important target in cancer therapy.
Recently, the ectopic expression of miRNAs in the
regulation and induction of senescence has been investigated
(13). Our previous study
demonstrated that miR-7 served as a marker for gemcitabine
sensitivity in pancreatic cancer (24). In the present study, miR-7 expression
was downregulated in accelerated senescent gemcitabine-resistant
pancreatic cancer cells, implying that decreased miR-7 expression
may be an important cause of TIS in gemcitabine-resistant
pancreatic cancer cells. To explore the potential mechanism
underlying the involvement of miR-7 on senescent signaling in
gemcitabine-resistant PDAC cells, bioinformatics analysis and
dual-luciferase reporter assays were utilized to predict the gene
targets of miR-7. The results revealed that miR-7 directly targeted
PARP1 and suppressed PARP1/NF-κB signaling in gemcitabine-resistant
PDAC cells. PARP1, which detects DNA damage (such as that mediated
by chemotherapeutics) and drives DNA repair, acts upstream of
NF-κB, while the PARP1/NF-κB signaling cascade activated during
senescence drives the formation of a secretome endowed with
pro-tumoral and pro-metastatic properties (25). The current results revealed that
miR-7-mediated suppression of cellular senescence in pancreatic
cancer cells was attributed to the effect of chemotherapy by
blockade of the PARP1/NF-κB signaling cascade, which is consistent
with our previous study that miR-7 enhances the sensitivity to
gemcitabine chemotherapy (24).
Next, it was demonstrated that the influence of miR-7 co-treatment
with gemcitabine on the viability of gemcitabine-resistant
pancreatic cancer cells affected the stemness phenotype and
senescence of the cells. The present results strongly suggested
that miR-7 exerted a regulatory effect on chemo-sensitivity via
inhibition of senescence-like phenotypes in pancreatic cancer.
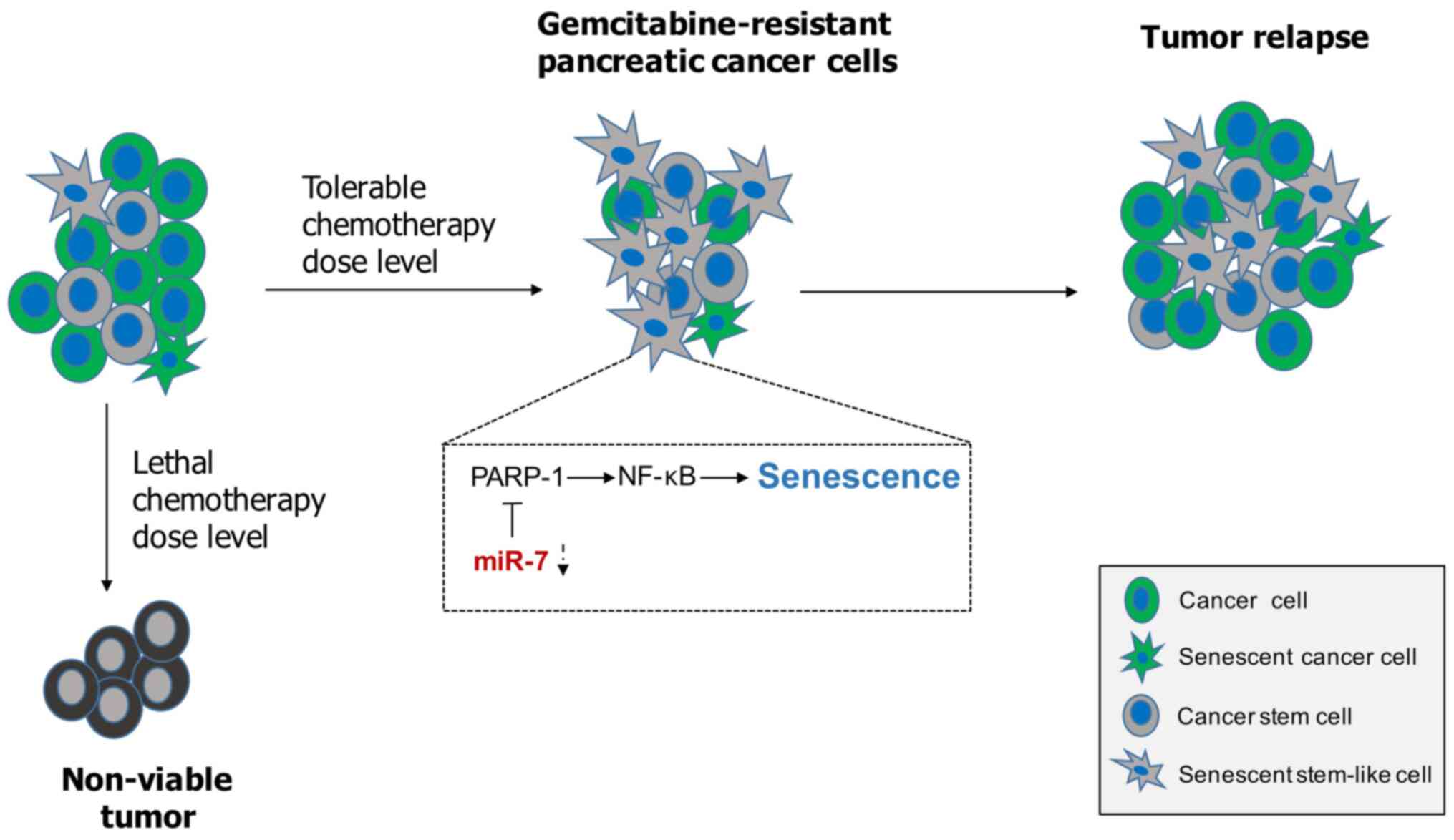
In conclusion, cellular senescence appears to be
primarily a beneficial response by keeping tumor growth in check.
However, activating a senescence program within tumor stem cells
may exert potential detrimental effects on chemotherapy resistance
(Fig. 5). In the present study,
miR-7 significantly inhibited senescence-like changes to sensitize
pancreatic cancer to gemcitabine through PARP1/NF-κB signaling.
Attenuating senescence via miR-7 may contribute to fight against
gemcitabine resistance in pancreatic cancer in vitro.
However, there are several limitations in the present study,
including the use of only one pancreatic cancer cell line and
performing only in vitro experiments. Thus, further research
on more pancreatic cancer cell lines and in vivo studies are
required to fully clarify the molecular mechanism of miR-7 and
senescence in chemotherapy-resistant pancreatic cancer in the
future.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by funding from the
National Natural Science Foundation of China (grant nos. 81802328
and 81773073), the Wenzhou Science and Technology Plan Project
(grant nos. Y20180214 and Y20180846) and the Young Talents Program
of the First Affiliated Hospital of Wenzhou Medical University
(grant no. qnyc094).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request, and the GSE140077 dataset is available in the Gene
Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140077).
Authors' contributions
ZQY performed the experiments, analyzed the data and
wrote the manuscript. HBC and TYZ performed the experiments and
analyzed the data. ZC contributed to the conception, the design and
the draft of the study. LT designed and supervised the study,
interpreted the data and revised the manuscript. DNG designed and
supervised the study, interpreted the data and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neoptolemos JP, Kleeff J, Michl P,
Costello E, Greenhalf W and Palmer DH: Therapeutic developments in
pancreatic cancer: Current and future perspectives. Nat Rev
Gastroenterol Hepatol. 15:333–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mohammed S, Van Buren G II and Fisher WE:
Pancreatic cancer: Advances in treatment. World J Gastroenterol.
20:9354–9360. 2014.PubMed/NCBI
|
|
4
|
Marino Gammazza A, Campanella C, Barone R,
Caruso Bavisotto C, Gorska M, Wozniak M, Carini F, Cappello F,
D'Anneo A, Lauricella M, et al: Doxorubicin anti-tumor mechanisms
include Hsp60 post-translational modifications leading to the
Hsp60/p53 complex dissociation and instauration of replicative
senescence. Cancer Lett. 385:75–86. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li W, Wang W, Dong H, Li Y, Li L, Han L,
Han Z, Wang S, Ma D and Wang H: Cisplatin-induced senescence in
ovarian cancer cells is mediated by GRP78. Oncol Rep. 31:2525–2534.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JJ, Park IH, Rhee WJ, Kim HS and Shin
JS: HMGB1 modulates the balance between senescence and apoptosis in
response to genotoxic stress. FASEB J. 33:10942–10953. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perez-Mancera PA, Young AR and Narita M:
Inside and out: The activities of senescence in cancer. Nat Rev
Cancer. 14:547–558. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dorr JR, Yu Y, Milanovic M, Beuster G,
Zasada C, Däbritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K,
et al: Synthetic lethal metabolic targeting of cellular senescence
in cancer therapy. Nature. 501:421–425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaur A, Webster MR, Marchbank K, Behera R,
Ndoye A, Kugel CH III, Dang VM, Appleton J, O'Connell MP, Cheng P,
et al: sFRP2 in the aged microenvironment drives melanoma
metastasis and therapy resistance. Nature. 532:250–254. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun Y, Coppe JP and Lam EW: Cellular
senescence: The sought or the unwanted? Trends Mol Med. 24:871–885.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Y, Baba T and Mukaida N: Gemcitabine
induces cell senescence in human pancreatic cancer cell lines.
Biochem Biophys Res Commun. 477:515–519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hua YQ, Zhu YD, Xie GQ, Zhang K, Sheng J,
Zhu ZF, Ning ZY, Chen H, Chen Z, Meng ZQ and Liu LM: Long
non-coding SBF2-AS1 acting as a competing endogenous RNA to sponge
microRNA-142-3p to participate in gemcitabine resistance in
pancreatic cancer via upregulating TWF1. Aging (Albany NY).
11:8860–8878. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Williams J, Smith F, Kumar S, Vijayan M
and Reddy PH: Are microRNAs true sensors of ageing and cellular
senescence? Ageing Res Rev. 35:350–363. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gu DN, Jiang MJ, Mei Z, Dai JJ, Dai CY,
Fang C, Huang Q and Tian L: microRNA-7 impairs autophagy-derived
pools of glucose to suppress pancreatic cancer progression. Cancer
Lett. 400:69–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo H, Liang H, Chen Y, Chen S, Xu Y, Xu
L, Liu J, Zhou K, Peng J, Guo G, et al: miR-7-5p overexpression
suppresses cell proliferation and promotes apoptosis through
inhibiting the ability of DNA damage repair of PARP-1 and BRCA1 in
TK6 cells exposed to hydroquinone. Chem Biol Interact. 283:84–90.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lai J, Yang H, Zhu Y, Ruan M, Huang Y and
Zhang Q: MiR-7-5p-mediated downregulation of PARP1 impacts DNA
homologous recombination repair and resistance to doxorubicin in
small cell lung cancer. BMC Cancer. 19:6022019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li L, Hao X, Qin J, Tang W, He F, Smith A,
Zhang M, Simeone DM, Qiao XT, Chen ZN, et al: Antibody against
CD44s inhibits pancreatic tumor initiation and postradiation
recurrence in mice. Gastroenterology. 146:1108–1118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou J, Zhang L, Zheng H, Ge W, Huang Y,
Yan Y, Zhou X, Zhu W, Kong Y, Ding Y and Wang W: Identification of
chemoresistance-related mRNAs based on gemcitabine-resistant
pancreatic cancer cell lines. Cancer Med. 9:1115–1130. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cheng Q, Ouyang X, Zhang R, Zhu L and Song
X: Senescence-associated genes and non-coding RNAs function in
pancreatic cancer progression. RNA Biol. 17:1693–1706. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perez RF, Tejedor JR, Bayon GF, Fernández
AF and Fraga MF: Distinct chromatin signatures of DNA
hypomethylation in aging and cancer. Aging Cell. 17:e127442018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Milanovic M, Fan DNY, Belenki D, Däbritz
JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa
IA, et al: Senescence-associated reprogramming promotes cancer
stemness. Nature. 553:96–100. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ye ZQ, Zou CL, Chen HB, Jiang MJ, Mei Z
and Gu DN: MicroRNA-7 as a potential biomarker for prognosis in
pancreatic cancer. Dis Markers. 2020:27821012020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohanna M, Giuliano S, Bonet C, Imbert V,
Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B,
Hofman P, et al: Senescent cells develop a PARP-1 and nuclear
factor-{kappa}B-associated secretome (PNAS). Genes Dev.
25:1245–1261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Suresh B, Lee J, Kim KS and Ramakrishna S:
The importance of ubiquitination and deubiquitination in cellular
reprogramming. Stem Cells Int. 2016:67059272016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dauer P, Nomura A, Saluja A and Banerjee
S: Microenvironment in determining chemo-resistance in pancreatic
cancer: Neighborhood matters. Pancreatology. 17:7–12. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Binenbaum Y, Na'ara S and Gil Z:
Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug
Resist Updat. 23:55–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guillon J, Petit C, Toutain B, Guette C,
Lelièvre E and Coqueret O: Chemotherapy-induced senescence, an
adaptive mechanism driving resistance and tumor heterogeneity. Cell
Cycle. 18:2385–2397. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Calcinotto A, Kohli J, Zagato E,
Pellegrini L, Demaria M and Alimonti A: Cellular senescence: Aging,
cancer, and injury. Physiol Rev. 99:1047–1078. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Demaria M, O'Leary MN, Chang J, Shao L,
Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al:
Cellular senescence promotes adverse effects of chemotherapy and
cancer relapse. Cancer Discov. 7:165–176. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Craske MG and Barlow DH: Nocturnal panic.
J Nerv Ment Dis. 177:160–167. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hamed SS, Straubinger RM and Jusko WJ:
Pharmacodynamic modeling of cell cycle and apoptotic effects of
gemcitabine on pancreatic adenocarcinoma cells. Cancer Chemother
Pharmacol. 72:553–563. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Acosta JC, Banito A, Wuestefeld T,
Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka
F, Andrulis M, et al: A complex secretory program orchestrated by
the inflammasome controls paracrine senescence. Nat Cell Biol.
15:978–990. 2013. View Article : Google Scholar : PubMed/NCBI
|