Introduction
Kidney cancer is one of the most lethal cancer types
globally. In the United States, the 5-year survival rate of
metastatic kidney cancer was ~12% between 2014 and 2018 (1). The estimated number of newly diagnosed
kidney cancer cases annually is 73,820, and the projected number of
kidney cancer-associated deaths in 2019 in the United States was
14,770 (1). In Taiwan, in 2016,
there were 1,364 newly diagnosed kidney cancer cases, and 600
patients died of kidney cancer (2).
The incidence rate of renal cell carcinoma (RCC) in males and
females in Taiwan in 2016 was 7.75 and 3.86 per 100,000 population,
respectively, compared with 22.2 and 11.4 per 100,000 population,
respectively, in the US (1,2). Although the incidence of RCC in Taiwan
is not as high as that in Western countries, it is still an
important public health issue affecting patients with a median age
of diagnosis 61 and 62 years old in males and females, respectively
(2).
Clear cell RCC (ccRCC) is the most common
histological subtype of kidney cancer, which accounts for around
70–75% of all renal malignancies globally (3). The most distinct mutated driver gene of
the ccRCC is the VHL gene, which is found to be mutated in
51% of all patients with ccRCC globally (4). On the basis of the investigation of the
VHL pathway, the tyrosine kinase inhibitors (TKIs) that target this
pathway were established as the mainstay for systemic therapy for
metastatic ccRCC since the early 21st century (5). The VHL gene encodes the VHL
protein, an E3 ubiquitin ligase, which targets the hypoxia
inducible factors. One such example is hypoxia inducible factor-1α
(HIF-1α), which is the most researched target. When VHL is
mutated, HIF-1α cannot be degraded and accumulates, inducing the
expression of several angiogenesis-related factors, such as
vascular endothelial growth factor, platelet-derived growth factor
(PDGF) and TGF-β. This is an important process of tumorigenesis of
RCC (6). TKIs block the pathways of
angiogenesis and therefore inhibit tumor growth. Keeping this in
mind, it is important to understand the mutations underpinning the
pathogenesis of any cancer not only for diagnosis but also for
treatment.
Through the high throughput sequencing methods
developed in recent decades, gene alteration databases have been
developed within large-scale projects, such as The Cancer Genome
Atlas (TCGA) and Catalogue Of Somatic Mutations In Cancer (COSMIC)
(4). In addition to VHL, the
top ten mutated genes in ccRCC are the following: Protein
polybromo-1 (PBRM1), histone-lysine N-methyltransferase
SETD2, BRCA1-associated protein-1 (BAP1),
lysine-specific demethylase 5C (KDM5C), TP53, MTOR,
PTEN, low-density lipoprotein receptor-related protein 1B and
Lysine N-methyltransferase 2C (4).
Numerous epigenomic-related genes are mutated in ccRCC, which
suggests that epigenetic regulation plays an important role in the
molecular pathways underpinning ccRCC, hence leading to the
development of possible epigenetic therapies (7). However, most of the candidate genes in
these databases are based on Western populations. For example, only
seven (1.9%) Asian patients were included in TCGA database
(8). Comparing somatic mutations in
kidney cancer between patients in Asia and Western countries is
necessary since the incidence rate is different and there may be
some possible interethnic genetic differences.
In the present study, targeted gene sequencing was
used to evaluate gene alteration(s) in Taiwanese patients with
ccRCC. The top eight mutated genes in the COSMIC database were
targeted and the association between their gene mutation status and
clinical and pathological parameters and survival outcome of
patients was determined.
Material and methods
Patients
Patients were enrolled from the Chang Gung Memorial
(Taoyuan, Taiwan R.O.C.) between January 2006 and December 2010 and
National Taiwan University Hospital (Taipei, Taiwan R.O.C.) between
Jan 1st 2013 and Dec 31st 2014. The patients enrolled in the study
were subjective to the willing of participating and the
availability of tissue samples. The inclusion criteria of the study
were as follows: i) Patients who received radical/partial
nephrectomy, ii) Pathology diagnosis of clear cell RCC and iii)
Willing to participate and provided signed informed consent. The
exclusion criteria were: i) Histology types other than clear cell
RCC and ii) No adequate specimen available. All patients with a
renal tumor diagnosis received either partial or radical
nephrectomy according to clinical indications. The pathology of
each tumor was reviewed by pathologists specializing in kidney
cancer identification, and only those diagnosed as ccRCC were
included in the study. We randomly selected 96 patients with ccRCC
for this study. Clinical demographic parameters, cancer stage using
the American Joint Committee on Cancer (9) and pathological data including tumor
stage, lymph node status and Fuhrman grade were collected. The
overall survival time was determined as from the date of operation
to the date of death. If there was no date of death, the data would
be censored using the last date of follow-up at the outpatient
department.
Ethical statements
The study was approved by the Institutional Review
Board of Chang Gung Memorial Hospital (approval no. 106-3050C) and
National Taiwan University Hospital (approval no. 201312158RIND).
The retrospective genetic study and the treatment plan for the
patients was conducted according to clinical guidelines and
standard of care. The present genetic study results did not affect
the treatment plan of patients following surgery. Informed written
consent was provided by all patients.
Sample collection
After removal of the tumor, a specimen without
necrosis of ~5 mm3 at the central area of the tumor was
excised and packed in foil. Then, the samples were stored liquid
nitrogen tank (−196°C) within 1 h of collection. All procedures
were performed under aseptic conditions.
DNA extraction
DNA was extracted from the aforementioned samples
using the Qiagen blood and tissue DNeasy Blood & Tissue
extraction kit (Qiagen GmbH) according to the manufacturer's
protocol. Briefly, ~25 mg of tumor tissue was minced and
transferred to a 1.5-ml microcentrifuge tube, and 180 µl ATL buffer
and 20 µl proteinase K were added (all included in the
aforementioned kit). The tube was incubated at 56°C until totally
lysed, then 4 µl RNase A was added, and the sample was incubated
for 2 min at room temperature. After vortexing, 200 µl AL buffer
was added, followed by mixing and addition of 200 ml absolute
ethanol. The sample was transferred to a DNeasy Mini spin column
and centrifuged at 7,000 × g at room temperature for 1 min,
followed by washing and elution of DNA in nuclease-free water. The
nucleic acid concentration was measured with NanoDrop 1000 (Thermo
Fisher Scientific, Inc.), and 1% agarose gel electrophoresis with
ethidium bromide illumination was performed for quality
control.
Targeted genes
To compare gene alterations between Taiwanese
patients and patients in the COSMIC and TCGA databases, the top
eight most frequently mutated genes of ccRCC in the COSMIC database
were investigated (VHL, PBRM1, SETD2, BAP1, TP53, KDM5C,
MTOR and PTEN). A multiplex PCR target enrichment panel
for target-relevant genes was enriched with DNA GeneRead DNAseq
Custom panel V2 (cat. no. 181902 CNGHS-02735X-67). The DNA panel
was designed using the Qiagen GeneRead designer website (https://www.qiagen.com/us/shop/genes-and-pathways/custom-products/custom-array-products/generead-designer/).
Library preparation and targeted gene
sequencing
Targeted sequencing was performed according to a
previously described protocol (10).
Briefly, DNA libraries were prepared using components from TruSeq
DNA Sample Preparation kits (Illumina, Inc.). For each sample, 80
ng DNA was used as starting material. The DNA was enzymatically
fragmented and end-repaired, and the reaction was carried out at
4°C for 1 min, 32°C for 24 min and 65°C for 30 min. Immediately
after the reaction, ligation of barcoded adapters was performed,
and the reaction continued at 20°C for 15 min. Purification was
carried out to remove the free barcoded adapters, with subsequent
PCR enrichment for the targeted genes under the following
conditions: 95°C For 13 min, 98°C for 2 min, then six cycles of
98°C for 15 sec and 65°C for 15 min, and finally 72°C for 5 min.
Each reaction was cleaned up using 0.9× Ampure beads (Beckman
Coulter, Inc.) to remove unbound primers. For library preparation,
the NEBNext Multiplex Oligos kit was used (New England BioLabs,
Inc.). The enriched DNA was combined with universal primers,
identical index primers and a PCR master mix supplied in the kit.
The universal PCR conditions were as follows: 95°C For 13 min, 98°C
for 2 min, 20 cycles of 98°C for 15 sec and 60°C for 2 min, and
72°C for 5 min. Gel electrophoresis was performed to ensure that
the fragmental DNA library length was between 400 and 500 base
pairs, and the appropriate band was excised and purified using the
QIAquick Gel Extraction kit (Qiagen GmbH). All libraries were
sequenced on an Illumina MiSeq sequencer (pair-end, 2 × 300 bp)
following the manufacturer's instructions (Illumina, Inc.).
Data processing and analysis
The smCounter was used to generate data as
previously described (10). At each
target locus, posterior probabilities of the alleles (including
possible indels) were first calculated on the barcode level, noted
as P (Allele|BCk) for the kth
barcode. Assuming that the locus is covered by N mutually
independent barcodes, a prediction index I=-∑k=1Nlog10
[1-P(Allele|BCk)] is assigned to each allele,
representing the likelihood that the allele exists in at least one
DNA molecule. If a non-reference allele's prediction index exceeds
the preselected threshold, this allele is considered a candidate
variant. Candidate variants were confirmed only if they passed all
of the post-processing filters. The analysis process was as
follows: i) Raw reads QC as adapter trimming and quality filtering,
ii) Reference alignment using the Burrows-Wheeler Transform
algorithm (11), iii) Variant
calling using the Genome Analysis Toolkit (12), iv) Somatic mutation detection using
Mutect (13) and v) Variant
annotation using VEP (14).
Sanger sequencing validation
Sanger sequencing was used to validate the mutated
genes uncovered within the targeted sequencing data. The primers
were designed using Primer3 software (http://frodo.wi.mit.edu). Purified PCR products were
sequenced in both forward and reverse directions using ABI PRISM
BigDye Terminator Cycle Sequencing Ready Reaction kits (version 3;
Applied Biosystems; Thermo Fisher Scientific, Inc.) and an ABI
PRISM 3730 Genetic Analyzer (Thermo Fisher Scientific, Inc.).
Statistics
The χ2 test was used to validate the
association between mutated genes and clinicopathological
parameters. Data are presented as mean ± standard deviation.
Fisher's exact test was used to validate the relationship between
factors with sample sizes less than five. A Kaplan-Meier log-rank
test model was used to evaluate the relationship between mutated
genes and survival of patients. P<0.05 was considered to
indicate a statistically significant difference. There was no
repeat for the targeted sequencing and every sample was sequenced
once only. All statistics were performed using SPSS version 22 (IBM
Corp).
Genetic database comparison
The mutational percentage of the eight
targeted-sequenced genes were compared to the data from the COSMIC
and TCGA databases. To access the COSMIC database, the following
search terms were used: ‘Kidney’ in the tissue selection section,
‘include all’ in the sub-tissue selection section, ‘carcinoma’ in
the histology selection section and ‘clear cell renal cell
carcinoma’ in the sub-histology selection section. The top 20 genes
were reported. The top mutated genes of kidney cancer in TCGA
database were previously published (8), so the COSMIC and TCGA results were
compared directly.
Results
Demographic data and
clinicopathological parameters
A total of 96 patients with sporadic kidney cancer
who fulfilled the inclusion criteria were randomly selected for the
present study. The operations and sample collection were performed
between 2006 and 2014. The mean follow-up time was 39.42±29.85
months (range, 1–124 months). In total, 12 patients (12.5%) died
during follow-up. A summary of the demographic data is shown in
Table I. Among 96 patients, 64.6 and
34.4% were male and female, respectively. The mean age at diagnosis
was 57.64±14.73 years old (data not shown). Approximately 72% of
patients received radical nephrectomy. A tumor size >4 cm
accounted for 71.9% of all included tumors. This was compatible
with the percentage of patients having received radical
nephrectomy, as partial nephrectomy is usually performed for
patients with T1a renal tumors. Approximately two-thirds of
patients exhibited localized disease (stages I and II), and locally
advanced or metastatic disease (stages III and IV) occurred in
34.4% of patients. Twelves (12.5%) patients had metastatic disease
and received TKIs as systemic treatments.
 | Table I.Demographic and clinicopathological
parameters and the association with gene mutation status. |
Table I.
Demographic and clinicopathological
parameters and the association with gene mutation status.
|
|
| VHL
mutation |
| PBRM1 mutation |
| SETH2 mutation |
| BAP1 mutation |
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Characteristic | Patient n (%) | Yes | No | P-value | Yes | No | P-value | Yes | No | P-value | Yes | No | P-value |
|---|
| Total | 96 | 48 | 48 |
| 25 | 71 |
| 21 | 75 |
| 9 | 87 |
|
| Sex |
|
|
| 0.010a |
|
| 0.007b |
|
| 0.120 |
|
| 1.000 |
|
Male | 62 (64.6) | 37 | 25 |
| 22 | 40 |
| 4 | 30 |
| 6 | 56 |
|
|
Female | 34 (35.4) | 11 | 23 |
| 3 | 31 |
| 17 | 45 |
| 3 | 31 |
|
| Age, years |
|
|
| 0.289 |
|
| 0.018a |
|
| 0.026a |
|
| 0.720 |
|
<65 | 61 (63.5) | 28 | 33 |
| 11 | 50 |
| 9 | 52 |
| 5 | 56 |
|
|
≥65 | 35 (36.5) | 20 | 15 |
| 14 | 21 |
| 12 | 23 |
| 4 | 31 |
|
| Tumor location |
|
|
| 0.525 |
|
| 0.307 |
|
|
|
|
| 1.000 |
|
Right | 35 (36.5) | 7 | 28 |
| 7 | 28 |
| 5 | 30 | 0.173 | 3 | 32 |
|
|
Left | 61 (63.5) | 18 | 43 |
| 18 | 43 |
| 16 | 45 |
| 6 | 55 |
|
| Type of
operation |
|
|
| 0.418 |
|
| 0.441 |
|
| 0.169 |
|
| 0.102 |
| Radical
nephrectomy | 69 (71.9) | 33 | 36 |
| 16 | 53 |
| 12 | 57 |
| 8 | 61 |
|
| Partial
nephrectomy | 26 (27.1) | 15 | 11 |
| 9 | 17 |
| 9 | 17 |
| 0 | 26 |
|
| Missing
data | 1 | 0 | 1 |
| 0 | 1 |
| 0 | 1 |
|
|
|
|
| Tumor stage |
|
|
| 0.681 |
|
| 0.101 |
|
| 0.314 |
|
| 0.007b |
| T1 | 57 (59.4) | 27 | 30 |
| 16 | 41 |
| 16 | 41 |
| 1 | 56 |
|
| T2 | 10 (10.4) | 4 | 6 |
| 0 | 10 |
| 1 | 9 |
| 3 | 7 |
|
| T3 | 26 (27.1) | 15 | 11 |
| 7 | 19 |
| 3 | 22 |
| 4 | 22 |
|
| T4 | 3 (3.1) | 2 | 1 |
| 2 | 1 |
| 1 | 3 |
| 1 | 2 |
|
| Tumor size, cm |
|
|
| 0.581 |
|
| 0.767 |
|
| 0.388 |
|
| 0.020a |
| ≤4 | 27 (28.1) | 12 | 15 |
| 7 | 20 |
| 8 | 19 |
| 0 | 27 |
|
| 4<
size ≤7 | 41 (42.7) | 23 | 18 |
| 12 | 29 |
| 9 | 32 |
| 3 | 38 |
|
|
>7 | 28 (29.2) | 13 | 15 |
| 6 | 22 |
| 4 | 24 |
| 6 | 22 |
|
| TNM stage |
|
|
| 0.628 |
|
| 0.286 |
|
| 0.296 |
|
| 0.012a |
| I | 56 (58.3) | 27 | 29 |
| 16 | 40 |
| 16 | 40 |
| 1 | 55 |
|
| II | 7 (7.3) | 3 | 4 |
| 0 | 7 |
| 1 | 6 |
| 2 | 5 |
|
|
III | 21 (21.9) | 13 | 8 |
| 7 | 14 |
| 3 | 18 |
| 3 | 18 |
|
| IV | 12 (12.5) | 5 | 7 |
| 2 | 10 |
| 1 | 11 |
| 3 | 9 |
|
| Fuhrman grade |
|
|
| 0.505 |
|
| 0.085 |
|
| 0.876 |
|
| 0.233 |
| 1 | 9 (9.4) | 3 | 6 |
| 0 | 9 |
| 2 | 7 |
| 0 | 9 |
|
| 2 | 47 (49) | 25 | 22 |
| 17 | 30 |
| 12 | 35 |
| 3 | 44 |
|
| 3 | 27 (28.1) | 15 | 12 |
| 7 | 20 |
| 4 | 23 |
| 5 | 22 |
|
| 4 | 8 (8.3) | 4 | 4 |
| 1 | 7 |
| 2 | 6 |
| 0 | 8 |
|
| No grade | 5 (5.2) | 1 | 4 |
| 0 | 5 |
| 1 | 4 |
| 1 | 4 |
|
Summary of somatic mutations
A total of 6,516 non- synonymous mutations in exons
and 565 mutations at splice junctions were observed within the 96
samples. Among the non-synonymous mutations, there were 5,908
missense mutations, 323 frameshift mutations, 11 in-frame deletions
or insertions, and two start-loss and 278 stop-gain mutations. The
most frequent single nucleotide substitution in missense mutations
was T:A to G:C (data not shown). The percentage and type of gene
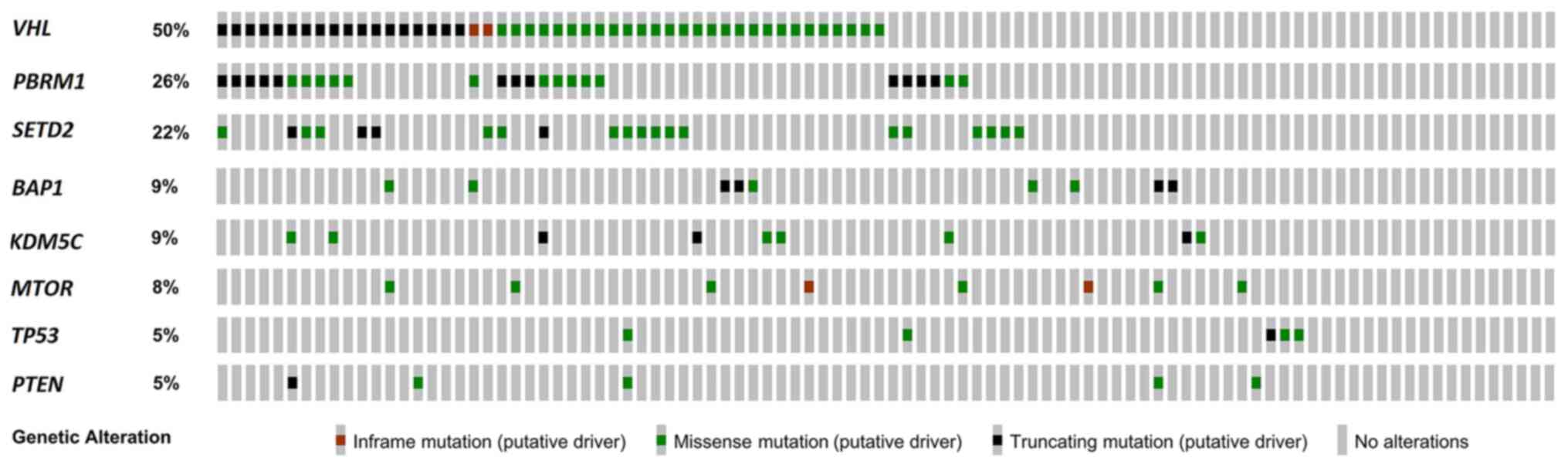
mutation are shown in Fig. 1. There
were three genes that exceeded 20% of the mutation rate: VHL,
PBRM1 And SETD2. The somatic mutations mapped to genes
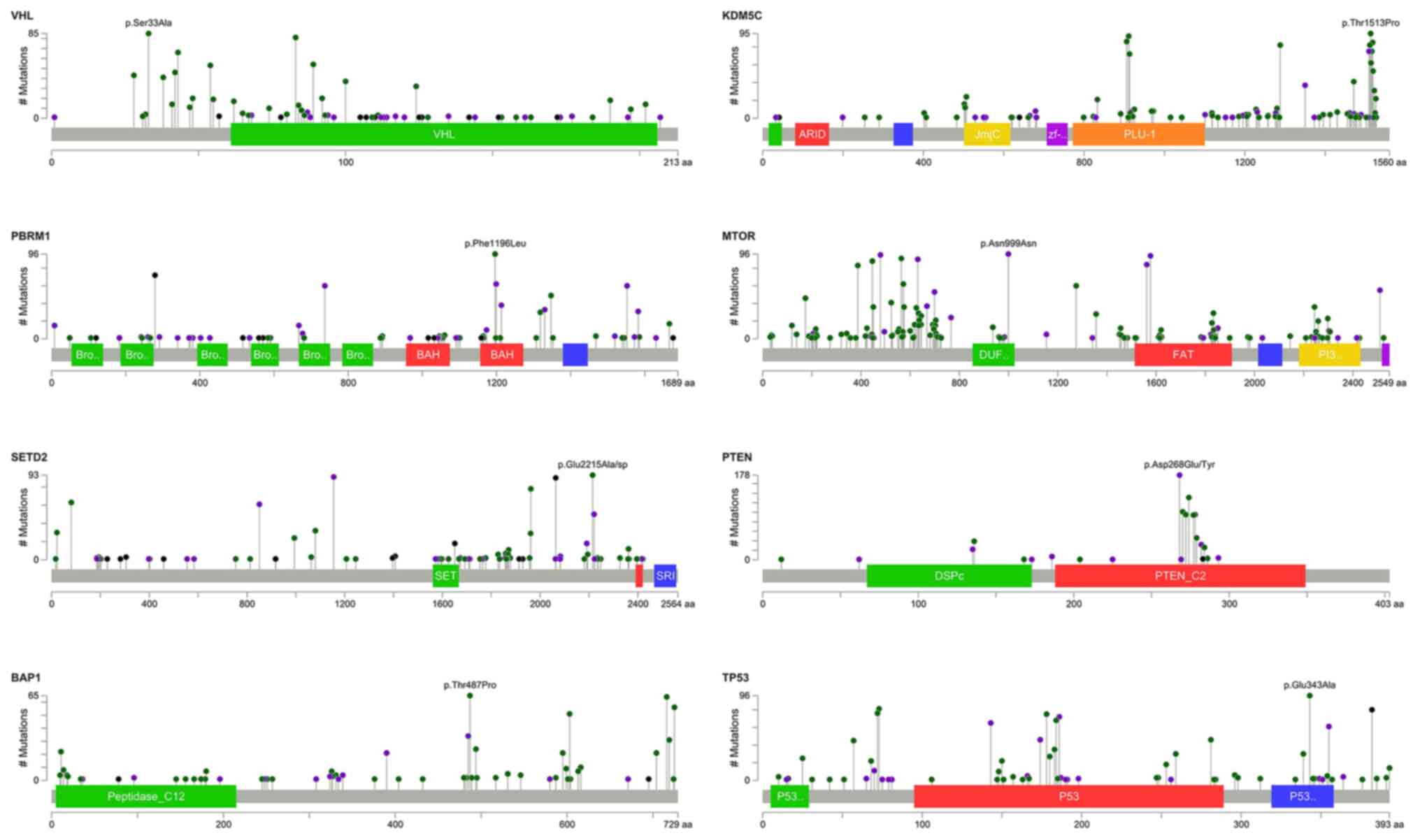
are shown in Fig. 2.
Sanger sequencing validation
Sanger sequencing was used to validate the accuracy
of targeted sequencing for the three targeted genes VHL,
PBRM1 and BAP1 since we only sequenced the samples once.
The consensus rates of each gene were 93.8, 93.3 and 100% for
VHL, PBRM1 and BAP1, respectively (data not shown).
The high consensus rates indicated that the targeted sequencing
data were reliable, and the mutations were true mutations. Some
selected results of the Sanger sequencing were shown in the
supplementary figures. Each panel in the supplementary figures
indicated individual samples, which were labeled as NTCG or RCC
followed by digits. Fig. S1
presents the selected results of Sanger sequencing validation for
VHL indels. Panels (A) to (H) reveal the position of
mutation and the resulted frameshift mutation of associated amino
acids. Fig. S2 shows the selected
results of Sanger sequencing validation for VHL SNVs. Panels
(A) to (C) show the position of mutation and the resulted
associated amino acid changes. Fig.
S3 presents selected results of Sanger sequencing validation
for PBRM1 indels. Panel (A) to (I) reveal the position of
mutation and the resulted frameshift mutation of associated amino
acids. Fig. S4 shows the selected
results of Sanger sequencing validation for PBRM1 SNVs.
Panels (A) to (E) show the position of mutation and the resulted
associated amino acid changes. Fig.
S5 reveals the selected results of Sanger sequencing validation
for BAP1 indels. Panels (A) to (C) reveal the position of
mutation and the resulted frameshift mutation of associated amino
acids.
Gene alteration in the Taiwanese
cohort
The most frequently mutated gene was VHL
(50%), followed by PBRM1 (26%) and SETD2 (22%)
(Fig. 1). Concurrence of VHL
and PBRM1 mutations was found in 19 (19.79%) patients in our
cohort. Only one (1.04%) patient had both PBRM1 and
BAP1 mutations. None of the 96 patients had both
SETD2 and BAP1 mutation (data not shown). The
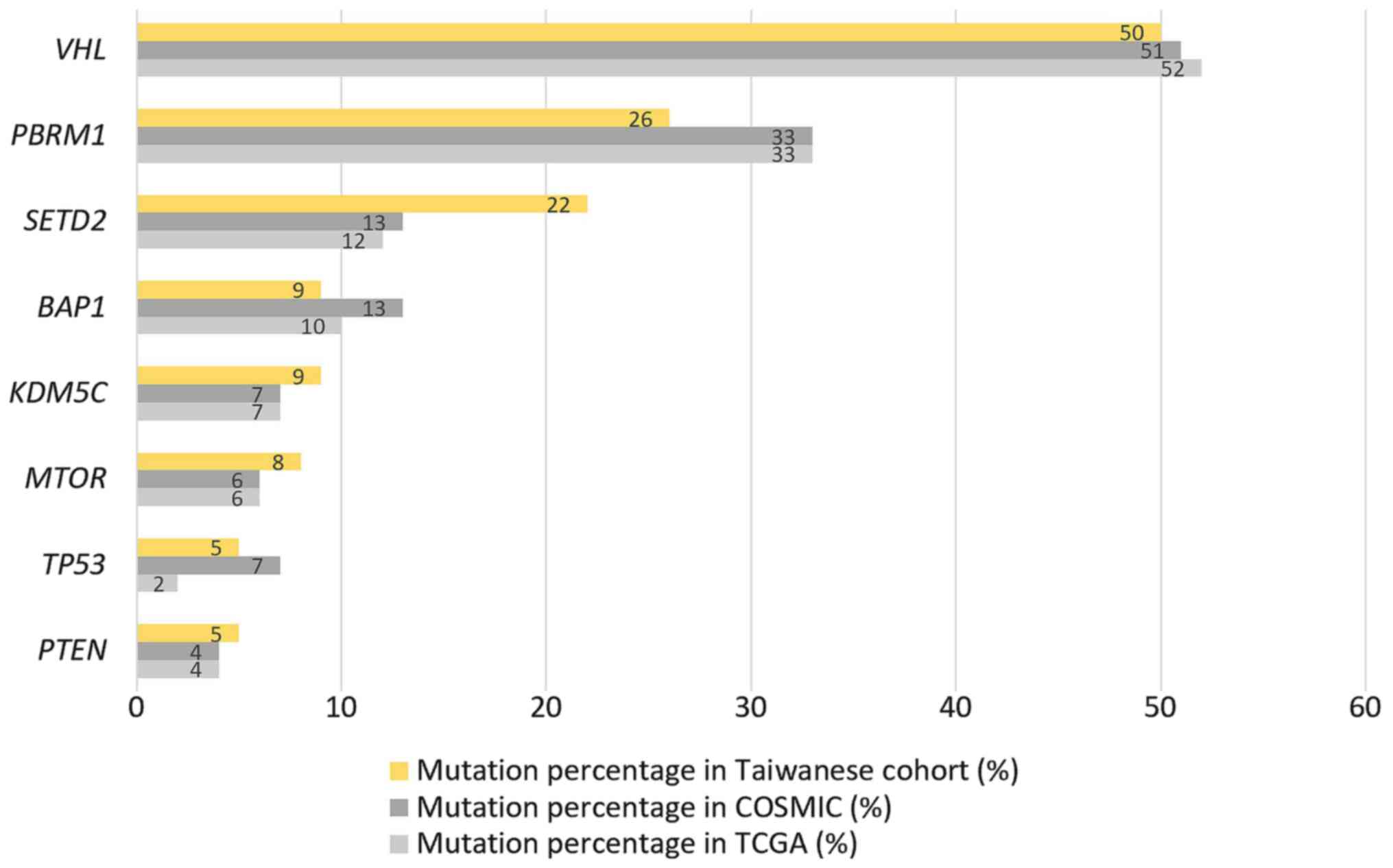
comparison of the gene alterations between the Taiwanese cohort and
COSMIC/TCGA databases illustrated in Fig. 3 shows that the order of the
mutational frequency between these cohorts was similar. However,
the Taiwanese cohort had lower mutation rates in PBRM1 (26
vs. 33/33%) and BAP1 (9 vs. 13/10%) and higher mutation
rates in SETD2 (22 vs. 13/12%) and KDM5C (9 vs.
7/7%).
Gene alteration and
clinicopathological parameters
The association between the mutation statuses of
VHL, PBRM1, SETD2 and BAP1 and clinicopathological
parameters were determined (Table
I). Patient sex was significantly associated with mutations in
VHL (P=0.01) and PBRM1 (P=0.007), with higher
frequencies of mutations in each gene in males. Age was
significantly associated with mutations in PBRM1 (P=0.018)
and SETD2 (P=0.026), with higher mutation rates in patients
≥65 years old. The BAP1 mutation status was significantly
different between the tumor size (P=0.020), tumor stage (P=0.007)
and Tumor-Node-Metastasis stage (P=0.012). None of these top four
genes were associated with the Fuhrman grade of the tumors. The
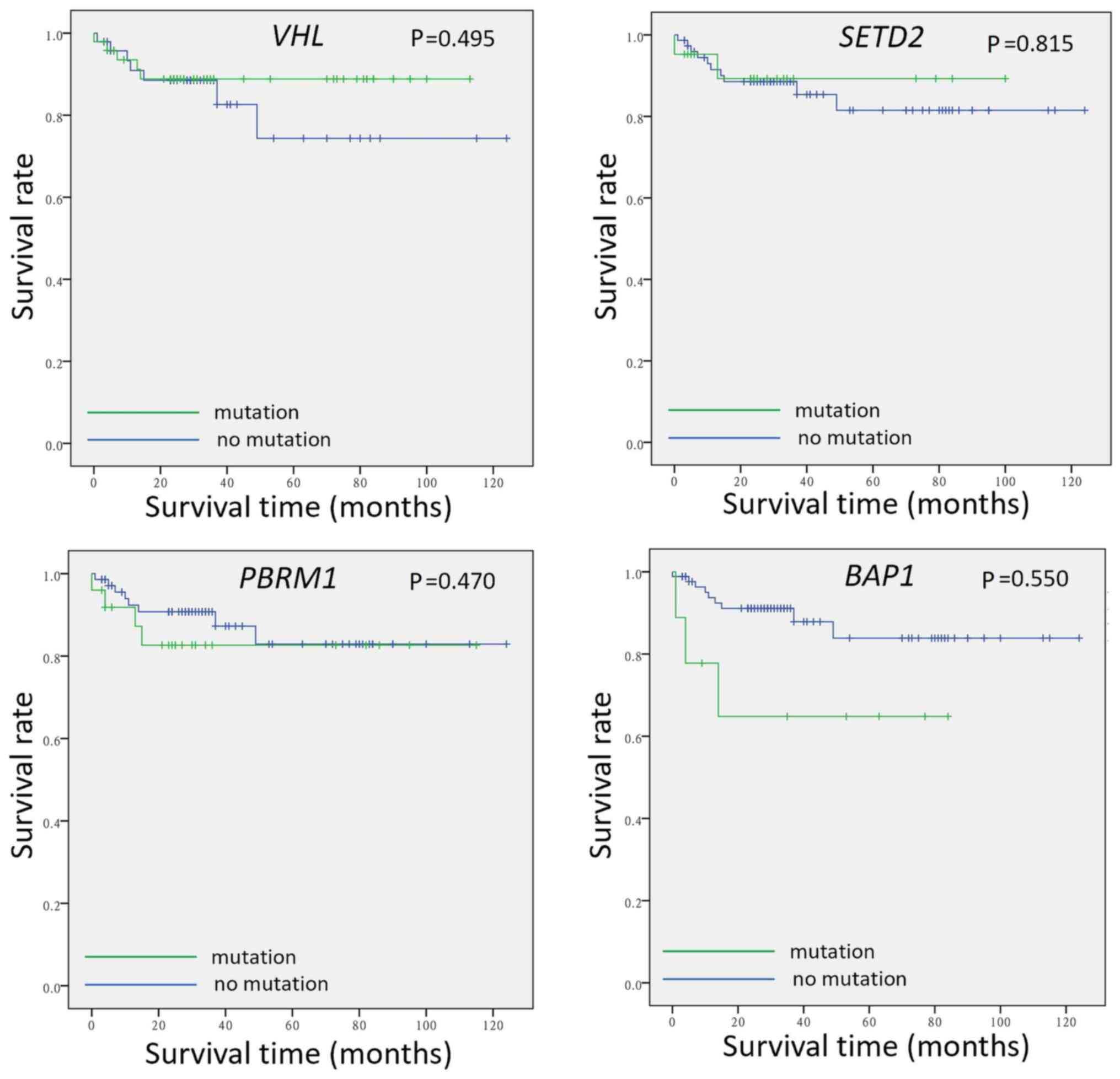
association between the mutation status and survival of patients
was validated for VHL, PBRM1, SETD2 and BAP1. There
was no significant association between patient survival and
mutational status of these four genes (Fig. 4).
Discussion
Kidney cancer incidence varies around the world and
is highest in Northern America, Europe, Australia and New Zealand,
with lower incidences in Asia and Africa (15). These differences in incidence may
reflect varying diets and lifestyle, and interethnic genetic
profiling may play a role as well. The present study demonstrated
that the top eight most frequently mutated genes of ccRCC in COSMIC
and Taiwan are similar, except for some differences in the mutation
rate of particular genes. The analysis was focused on the top four
genes VHL, PBRM1, SETD2 and BAP1, which are all
located on chromosome 3p (16).
Chromosome 3p deletion is frequent in ccRCC, resulting in high
mutation rates of these genes (16).
VHL was still the most frequently mutated gene, with lower
mutation rates of PBRM1 and BAP1 within the Taiwanese
cohort and higher levels of SETD2 mutation compared with the
COSMIC and TCGA databases.
The elevated mutation rate of VHL in the Taiwanese,
TCGA and COSMIC cohorts suggested that the VHL pathway is the main
pathogenic pathway in ccRCC globally. With normal oxygen levels and
an intact VHL gene, HIF-1α binds to the VHL protein and is
degraded via ubiquitylation. When VHL is mutated, HIF-1α
accumulates and increases the transcription of genes containing the
hypoxia response element (6). This
would increase the expression of downstream proteins, such as the
vascular endothelial growth factor, platelet-derived growth factor
and transforming growth factor α, thereby enhancing neoangiogenesis
and carcinogenesis of ccRCC (17).
Through the investigation of the role of the VHL pathway in the
carcinogenesis of ccRCC, TKIs have become the mainstay of systemic
treatments for metastatic ccRCC since the early 2000s (18). In a Japanese comprehensive mutational
analysis study of the VHL gene in patients with ccRCC, Kondo
et al revealed that VHL mutation is not associated
with the clinicopathological parameters including tumor diameter,
stage, grading, distant metastasis and lymph node metastasis.
However, VHL is less frequently mutated in patients >55
years old (19). The present study
also showed similar results, which suggested that there is no
significant difference in the role of the VHL gene in ccRCC
in Taiwanese patients.
PBRM1 is the second most frequently mutated
gene associated with ccRCC. PBRM1 encodes the BAF180
protein, which is a subunit of the SWI/SNF chromatin-remodeling
complex (20). The SWI/SNF complex
is a tumor suppressor, and mutations on the subunit-coding genes
are found in numerous malignancies, such as lung, colorectal,
pancreatic, head and neck and kidney cancer (20), especially in RCC (21). Nargund et al used a mouse
model to show that the PBRM1 protein can inhibit the HIF1/STAT3
signaling pathway in vhl−/− cells. The loss of
Pbrm1 function would position the mTORC1 activation at the
third driver event of ccRCC (22).
The present study provided evidence of sequential driver gene
mutations in the pathogenesis of ccRCC. Concurrence of VHL
and PBRM1 mutations was found in 19 (19.79%) patients in our
cohort.
The SETD2 gene encodes the SETD2 protein, a
histone methyltransferase specific for lysine 36 located on histone
H3 (H3K36). Methylation of H3K36 is associated with active
chromatin; H3K36 trimethylation is required for homologous
recombination repair and genome stability, which depends on the
methyltransferase function of SETD2 (23). Haploinsufficiency of the SETD2
gene has been shown to drive genomic instability in the early phase
of RCC (24). SETD2
loss-of-function also promotes renal cancer branched evolution
through DNA repair impairment and replication stress (25). In the present cohort, the rate of
SETD2 mutations was higher compared with that reported in
the COSMIC/TCGA database. Therefore, further downstream validation
of the role of SETD2 in ccRCC within Taiwanese patients is
necessary.
The BAP1 gene encodes the deubiquitinating
enzyme BRCA1-associated protein-1, which acts with other co-factors
to epigenetically regulate genes targeted by polycomb repressive
complex 1, regulates gene transcription and deubiquitylates target
substrates, such as BRCA1-associated RING domain 1, ubiquitylation
of histone 2A and O-glucosyltransferase (26). In RCC, a mutation in BAP1
causes disruption of the host cell factor-1 binding motif of BAP1
and impairs BAP1-mediated suppression of cell proliferation
(27). Notably, BAP1 loss is
mutually exclusive with PBRM1 in the literature (4,8). In the
present cohort, there were 25 (26.04%) patients with PBRM1
mutation and 9 (9.37%) with BAP1 mutation(s), but only one
(1.04%) patient had both PBRM1 and BAP1 mutations
(data not shown), which is compatible to the literature. In
addition, the mutation of SETD2 and BAP1 in the
present Taiwanese cohort also showed mutually exclusive trend. None
of the 96 patients had both SETD2 and BAP1 mutation.
Mutual exclusive mutations are important to develop synthetic
lethality therapies (28). Further
studies to investigate the interaction of these genes may discover
the potential therapeutic role for ccRCC.
The genetic landscapes of several cancer types have
been described through the development of high-throughput
sequencing technology (4,8). Thereafter, the genetic biomarkers that
can be used to predict the survival outcomes of patients with
specific cancer types have been widely investigated. VHL is
the key driver gene in ~50% of patients with ccRCC. However, a
recently published meta-analysis showed that VHL mutation
status is not associated with clinicopathological parameters, such
as nuclear grade, disease stage or OS (29). Nevertheless, a specific type of
VHL dysregulation, such as VHL methylation combined
with other VHL pathway-associated markers like HIF1-α and ERK5
protein, can help in predicting disease-specific survival for all
stages of ccRCC (30). The results
from various studies evaluating the prognostic value of PRBM1,
BAP1 and SETD2 are inconsistent. The lack of expression
of PBRM1 has been shown to be associated with poor recurrence-free
as well as cancer-specific survival (31,32).
BAP1 expression has been associated with high Fuhrman grade,
advanced pathological Tumor stage, sarcomatoid dedifferentiation
and significantly worsened disease-free survival and OS for
patients with non-metastatic ccRCC (33). However, another study revealed that
BAP1 and SETD2 mutations are associated with
decreased cancer-specific survival (CSS), but the same was not true
of PBRM1 for all stages of ccRCC (34). Furthermore, one study employing an
immunohistochemistry microarray to evaluate the association between
different markers with OS, CSS and progression-free survival (PFS)
for localized ccRCC showed that there was no association with BAP1
and PBRM1 expression (35). Another
study integrated recurrent somatic mutations with clinical outcomes
for >1,000 patients with ccRCC at varying cancer stages and
reported that BAP1 mutation is associated with large tumor
size, TP53 mutation is associated with poor CSS and
SETD2 mutation is associated with poor PFS (36). The present data indicated that
VHL, PBRM1, SETD2 and BAP1 are not associated with OS
in all stages of ccRCC. The diverse conclusions of these genetic
prognostic biomarkers indicate that pathogenesis and cancer
progression are associated with multiple gene dysregulations and
that a single gene mutation is less likely to be a strong
predictor.
Notably, half of the top eight highly mutated genes
are epigenetic modifiers (SETD2, PBRM1, BAP1 and
KDM5C). Indeed, as more techniques for epigenomic studies
are quickly developed, comprehensive genomic and epigenomic studies
are being introduced. A comprehensive molecular characterization of
RCC using TCGA database published in 2018 demonstrated that somatic
alteration of BAP1, PBRM1 and metabolic pathways correlates
with subtype-specific decreased survival, and cyclin-dependent
kinase inhibitor 2A alteration, DNA hypermethylation and T helper 2
immune signature are correlated with decreased survival within all
subtypes of RCC (37). As the
chromatin accessibility landscape of numerous types of primary
human cancer is developed (38),
further investigation of the genomic and epigenomic interactions
and improved understanding of the fundamental regulatory basis of
carcinogenesis is expected.
In the present study only 12 patients had metastatic
disease who received TKIs as systemic treatments. This number was
not sufficient to analyze the association of gene mutations with
response to the systemic treatments. However, in the recent reports
published by the National Health Insurance Administration of
Taiwan, the response rate (~35%) of Taiwanese patients with RCC to
immune checkpoint inhibitors was higher compared with patients in
the clinical trials in Western countries (39). Further comprehensive genetic and
epigenetic studies as well as gene expression and downstream
validation are necessary to resolve the possible mechanisms
underlying these differences.
There were some limitations to the present study.
First, paired normal tissues were not sequenced as controls. Thus,
copy number alteration and deep analysis could not be performed.
Second, targeted sequencing was used, and only the top eight genes
associated with ccRCC in COSMIC were included. In this manner, some
potential unique gene alterations in the Taiwanese cohort might
have been missed. Third, downstream validation of each mutated gene
was not performed due to insufficient remaining tissue material.
Therefore, the expression changes of affected protein(s) and
association with clinicopathological parameters could not be
evaluated. However, most of the mutations of the target genes were
non-synonymous mutations that would cause the alteration of protein
expression. At last, the mean follow-up time was not long enough,
and subsequent adjuvant or systemic treatments were not evaluated.
This may have affected the results of gene mutation impact on
survival. Nevertheless, the present study still provided
information concerning the commonly mutated gene status associated
with ccRCC in a Taiwanese cohort.
Overall, the current data showed that the highly
frequently mutated genes associated with ccRCC in Taiwan are
similar to those reported in the COSMIC/TCGA databases. However,
the concurrence of VHL and PBRM1 mutation was ≤20% in
the present cohort, and the SETD2 mutation rate was also
higher compared with the COSMIC/TCGA cohorts. SETD2 mutation
was mutually exclusive to BAP1 mutation, in addition to
PBRM1. These results indicated that role of SETD2
mutation may be distinct in Taiwanese cohort. Further comprehensive
genetic and epigenetic studies such as somatic mutations, DNA
methylation assay, chromatin immunoprecipitation sequencing, assay
for Transposase-Accessible Chromatin using sequencing as well as
downstream validations with gene expression and functional study
are necessary to validate the function and interaction of these
somatic mutations.
Supplementary Material
Supporting Data
Acknowledgments
The authors would like to thank Mr. Yu-Sin Chang and
Dr Chin-Hsuan Hsieh (Lab of Uro-Oncology, Chang Gung Memorial
Hospital) for their assistance.
Funding
This study was supported by The Chang Gung Medical
Research Program (grant nos. CORPG3F0291, CMRPG3E1941-2 and
CORPG3J0121).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request. The additional datasets analyzed during the current study
are available in the COSMIC and TCGA database.
Authors' contributions
PHL conducted the study design, analyzed and
interpretated the data and wrote the manuscript. CYH conducted the
study design and analyzed the data. KJY and HCK did the DNA
extraction and performed the targeted sequencing. YCL collected the
samples and extracted the DNA. CKC and CYL collected and processed
the samples. YHC and IHS assisted to collect and process the
samples and managed the administrative work and funds. STP
conceived and designed the study and revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Institutional Review
Board of Chang Gung Memorial Hospital (approval no. 106-3050C) and
National Taiwan University Hospital (approval no. 201312158RIND).
Written informed consent was provided by all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller K and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2018. View Article : Google Scholar
|
|
2
|
2016 Taiwan Cancer Registry. simplehttps://www.hpa.gov.tw/Pages/Detail.aspx?nodeid=269&pid=10227April
4–2020
|
|
3
|
Shuch B, Amin A, Armstrong AJ, Eble JN,
Ficarra V, Lopez-Beltran A, Martignoni G, Rini BI and Kutikov A:
Understanding pathologic variants of renal cell carcinoma:
Distilling therapeutic opportunities from biologic complexity. Eur
Urol. 67:85–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
COSMIC Cancer Browser. simplehttps://cancer.sanger.ac.uk/cosmic/browse/tissue?wgs=off&sn=kidney&ss=NS&hn=carcinoma&sh=clear_cell_renal_cell_carcinoma&in=t&src=tissue&all_data=nFebruary
15–2020
|
|
5
|
Motzer RJ, Hutson TE, Tomczak P,
Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik
C, Kim ST, et al: Sunitinib versus interferon alfa in metastatic
renal-cell carcinoma. N Engl J Med. 356:115–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim WY and Kaelin WG: Role of VHL gene
mutation in human cancer. J Clin Oncol. 15:4991–5004. 2004.
View Article : Google Scholar
|
|
7
|
Xing T and He H: Epigenomics of clear cell
renal cell carcinoma: Mechanisms and potential use in molecular
pathology. Chin J Cancer Res. 28:80–91. 2016.PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Research Network, .
Comprehensive molecular characterization of clear cell renal cell
carcinoma. Nature. 499:43–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Edge SB and Compton CC: The American joint
committee on cancer: The 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu C, Ranjbar MR, Wu Z, DiCarlo J and Wang
Y: Detecting very low allele fraction variants using targeted DNA
sequencing and a novel molecular barcode-aware variant caller. BMC
Genomics. 18:52017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A mapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McLaren W, Pritchard B, Rios D, Chen Y,
Flicek P and Cunningham F: Deriving the consequences of genomic
variants with the ensembl API and SNP effect predictor.
Bioinformatics. 26:2069–2070. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Capitanio U, Bensalah K, Bex A, Boorjian
SA, Bray F, Coleman J, Gore JL, Sun M, Wood C and Russo P:
Epidemiology of renal cell carcinoma. Eur Urol. 75:74–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brugarolas J: Molecular genetics of
clear-cell renal cell carcinoma. J Clin Oncol. 32:1968–1976. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaelin WG Jr: The von hippel-lindau tumor
suppressor protein and clear cell renal carcinoma. Clin Cancer Res.
13:680s–684s. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Posadas EM, Limvorasak S and Figlin RA:
Targeted therapies for renal cell carcinoma. Nat Rev Nephrol.
13:496–511. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kondo K, Yao M, Yoshida M, Kishida T,
Shuin T, Miura T, Moriyama M, Kobayashi K, Sakai N, Kaneko S, et
al: Comprehensive mutational analysis of the VHL gene in sporadic
renal cell carcinoma: Relationship to clinicopathological
parameters. Genes Chromosomes Cancer. 34:58–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hodges C, Kirkland JG and Crabtree GR: The
many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold
Spring Harb Perspect Med. 6(pii): a0269302016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Varela I, Tarpey P, Raine K, Huang D, Ong
CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al: Exome
sequencing identifies frequent mutation of the SWI/SNF complex gene
PBRM1 in renal carcinoma. Nature. 469:539–542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nargund AM, Pham CG, Dong Y, Wang PI,
Osmangeyoglu HU, Xie Y, Aras O, Han S, Oyama T, Takeda S, et al:
The SWI/SNF protein PBRM1 restrains VHL-loss-driven clear cell
renal cell carcinoma. Cell Rep. 18:2893–2906. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pfister SX, Ahrabi S, Zalmas LP, Sarkar S,
Aymard F, Bachrati CZ, Helleday T, Legube G, La Thangue NB, Porter
AC and Humphrey TC: SETD2-Dependent histone H3K36 trimethylation is
required for homologous recombination repair and genome stability.
Cell Rep. 7:2006–2018. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chiang YC, Park IY, Terzo EA, Tripathi DN,
Mason FM, Fahey CC, Karki M, Shuster CB, Sohn BH, Chowdhury P, et
al: SETD2 haploinsufficiency for microtubule methylation is an
early driver of genomic instability in renal cell carcinoma. Cancer
Res. 78:3135–3146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanu N, Gronroos E, Martinez P, Burrell
RA, Yi Goh X, Bartkova J, Maya-Mendoza A, Mistrík M, Rowan AJ,
Patel H, et al: SETD2 loss-of-function promotes renal cancer
branched evolution through replication stress and impaired DNA
repair. Oncogene. 34:5699–5708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carbone M, Yang H, Pass HI, Krausz T,
Testa JR and Gaudino G: BAP1 and cancer. Nat Rev Cancer.
13:153–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peña-Llopis S, Vega-Rubín-de-Celis S, Liao
A, Leng N, Pavía-Jiménez A, Wang S, Yamasaki T, Zhrebker L,
Sivanand S, Spence P, et al: BAP1 loss defines a new class of renal
cell carcinoma. Nat Genet. 44:751–759. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang A, Garraway LA, Ashworth A and Weber
B: Synthetic lethality as an engine for cancer drug target
discovery. Nat Rev Drug Discov. 19:23–38. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim HS, Kim JH, Jang HJ, Han B and Zang
DY: Clinicopathologic significance of VHL gene alteration in
clear-cell renal cell carcinoma: An updated meta-analysis and
review. Int J Mol Sci. 19:E25292018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Salinas-Sanchez AS, Serrano-Oviedo L,
Nam-Cha SY, Roche-Losada O, Sánchez-Prieto R and Giménez-Bachs JM:
Prognostic value of the VHL, HIF-1α, and VEGF signaling pathway and
associated MAPK (ERK1/2 and ERK5) pathways in clear-cell renal cell
carcinoma. A long-term study. Clin Genitourin Cancer. 15:e923–e33.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
da Costa WH, Rezende M, Carneiro FC, Rocha
RM, da Cunha IW, Carraro DM, Guimaraes GC and de Cassio Zequi S:
Polybromo-1 (PBRM1), a SWI/SNF complex subunit is a prognostic
marker in clear cell renal cell carcinoma. BJU Int. 113:E157–E163.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pawlowski R, Muhl SM, Sulser T, Krek W,
Moch H and Schraml P: Loss of PBRM1 expression is associated with
renal cell carcinoma progression. Int J Cancer. 132:E11–E117. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kapur P, Christie A, Raman JD, Then MT,
Nuhn P, Buchner A, Bastian P, Seitz C, Shariat SF, Bensalah K, et
al: BAP1 immunohistochemistry predicts outcomes in a
multi-institutional cohort with clear cell renal cell carcinoma. J
Urol. 191:603–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hakimi AA, Ostrovnaya I, Reva B, Schultz
N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, et al:
Adverse outcomes in clear cell renal cell carcinoma with mutations
of 3p21 epigenetic regulators BAP1 and SETD2: A report by MSKCC and
the KIRC TCGA research network. Clin Cancer Res. 19:3259–3267.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SH, Park WS, Park EY, Park B, Joo J,
Joung JY, Seo HK, Lee KH and Chung J: The prognostic value of BAP1,
PBRM1, pS6, PTEN, TGase2, PD-L1, CA9, PSMA, and Ki-67 tissue
markers in localized renal cell carcinoma: A retrospective study of
tissue microarrays using immunohistochemistry. PLoS One.
12:e01796102017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Manley BJ, Zabor EC, Casuscelli J,
Tennenbaum DM, Redzematovic A, Becerra MF, Benfante N, Sato Y,
Morikawa T, Kume H, et al: Integration of recurrent somatic
mutations with clinical outcomes: A pooled analysis of 1049
patients with clear cell renal cell carcinoma. Eur Urol Focus.
3:421–427. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ricketts CJ, De Cubas AA, Fan H, Smith CC,
Lang M, Reznik ED, Bowlby R, Gibb EA, Akbani R, Beroukhim R, et al:
The cancer genome atlas comprehensive molecular characterization of
renal cell carcinoma. Cell Rep. 23:313–326. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Corces MR, Granja JM, Shams S, Louie BH,
Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al:
The chromatin accessibility landscape of primary human cancers.
Science. 362:eaav18982018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Data published by the Taiwanese National
Health Insurance Administration. simplehttps://www.nhi.gov.tw/Content_List.aspx?n=7157A9A3E2A3B110&topn=3FC7D09599D25979April
4–2020
|