Introduction
Macroautophagy, hereafter referred to as autophagy,
is a cellular process of self-digestion, by which cellular proteins
and organelles are removed and recycled. This process is important
in cell growth and development and for the maintenance of cellular
homeostasis (1,2). Autophagy begins with the formation of
double-membrane vesicles, known as autophagosomes, which fuse with
lysosomes to form autophagolysosomes; the lysosomal hydrolases then
degrade the vesicle contents for reuse (3). Autophagy is induced by intracellular
and extracellular stresses, including hypoxia, starvation,
oxidative stress, ischemia and chemotherapy (4–6).
Autophagy promotes cell survival and cell death in cancer,
according to the tumor microenvironment, stage, type and other
factors (7,8).
Numerous antifungal agents have shown cytotoxic
effects against human cancer cells (9,10),
although the associated mechanisms are not clearly understood.
Azoles are a main class of antifungal agents and are classified
into two groups, namely triazoles and imidazoles. Miconazole is an
imidazole derivative that is used as an antifungal agent to treat
superficial fungal infections, including athlete's foot, ringworm
and candidiasis. In addition, miconazole has been shown to inhibit
the growth of various solid tumors, including glioma, breast cancer
and osteosarcoma in humans (11–13). The
in vivo therapeutic efficacy of miconazole has also been
reported in human colon carcinoma xenografts in nude mice (14). Most previous studies have shown that
the antiproliferative effects of miconazole are mediated by the
induction of apoptosis (15,16). In addition, miconazole has been
reported to increase the production of reactive oxygen species
(ROS) and raise intracellular Ca2+ levels, thereby
killing rat embryonic cardiomyoblasts and human osteosarcoma cells
via oxidative stress (17,18). Moreover, miconazole has been shown to
inhibit the growth of many types of cancer cells. For example,
previous studies demonstrated that a combination of miconazole and
artemisinin effectively inhibited the growth of bladder and breast
cancer cell lines (11), and
miconazole alone inhibited the proliferation of bladder cancer
cells via the induction of G0/G1 cell cycle
arrest and apoptosis (15). Another
study showed that miconazole was non-cytotoxic to astrocytes and
microglial BV2 cells, but ameliorated the
neuroinflammation-mediated progression of Alzheimer's disease by
blocking the expression of inducible nitric oxide synthase
(19) which is also known to be
related in cancer growth (20,21).
However, the precise mechanism of the anticancer effects of
miconazole remain to be elucidated. Notably, another antifungal
agent, itraconazole, has been shown to inhibit the proliferation of
cancer cells by inducing autophagy (22).
Considering the abilities of miconazole and
itraconazole to induce ROS production and autophagy, the aim of the
present study was to examine whether miconazole induces autophagic
death in cancer cells. The findings of this study may lead to the
identification of a novel mechanism for the anticancer activity of
miconazole.
Materials and methods
Cell lines and reagents
Human glioblastoma U343MG, U87MG (ATCC®
HTB-14™; glioblastoma of unknown origin) and U251MG cells and human
breast cancer MDA-MB-231 cells (cat. no. HTB-26™; ATCC) and human
lung cancer A549 cells (cat. no. CCL-185™; ATCC) were obtained from
the American Type Culture Collection. All the cells were cultured
in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10%
fetal bovine serum (HyClone; Cytiva) and 1% penicillin/streptomycin
at 37°C in a humidified atmosphere of 5% CO2.
Miconazole, 3-methyladenine (3-MA), chloroquine (CQ),
4-phenylbutyric acid (4-PBA) and N-acetylcysteine (NAC) were
purchased from Sigma-Aldrich (Merck KGaA). Bafilomycin A1 (Baf A1)
was purchased from LC Laboratories. The pan-caspase inhibitor
z-VAD-fmk and human recombinant tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) were purchased from R&D
Systems, Inc.
Cell treatments
To examine the effect of z-VAD-fmk, a pan-caspase
inhibitor, on the cytotoxicity of miconazole, U251MG cells were
pre-treated with z-VAD-fmk for 30 min at 37°C, followed by
miconazole treatment for 24 h at 37°C. Autophagy inhibitors [3-MA
(1 mM), Baf A1 (20 nM) and CQ (50 µM)] were treated simultaneously
with miconazole for 24 h at 37°C in U251MG cells. In U251MG cells
an endoplasmic reticulum (ER) stress inhibitor 4-PBA was pretreated
for 30 min at 37°C followed by treatment with 20 µM miconazole for
24 h at 37°C. U251MG cells were pretreated with a ROS scavenger NAC
(5 mM) for 2 h at 37°C and then treated with miconazole 10 and 20
µM for 24 h at 37°C. U251 MG cells were treated with TRAIL (50
ng/ml) for 24 h at 37°C.
Western blotting
Anti-78-kDa glucose-regulated protein [also known as
binding immunoglobulin protein (BiP); sc-13968), anti-eukaryotic
translation initiation factor 2α (eIF2α; sc-133132), anti-CHOP
antibodies (sc-7351) and anti-pro caspase-3 (sc-7148) were
purchased from Santa Cruz Biotechnology, Inc.
Anti-inositol-requiring enzyme 1α (IRE1α; cat. no. 3294),
anti-phospho-eIF2α (p-eIF2α; cat. no. 9721), anti-cleaved caspase-3
(cat. no. 9661) and anti-autophagy protein 5 (ATG5; cat. no. 2630)
antibodies were purchased from Cell Signaling Technology, Inc. An
anti-microtubule-associated protein light chain 3 (LC3) antibody
(PD014) was purchased from Medical and Biological Laboratories Co.,
Ltd. These antibodies were used at a dilution of 1:1,000.
Anti-β-actin antibody (A5441) was acquired from Sigma-Aldrich
(Merck KGaA) and used at a dilution of 1:5,000. Secondary
antibodies [anti-rabbit IgG (1:2,000; sc-2004) and anti-mouse IgG
(1:2,000; sc-2005)] were purchased from Santa Cruz Biotechnology,
Inc. Harvested cells were washed twice with phosphate-buffered
saline (PBS) and then lysed in a cell lysis buffer [20 mM Tris-HCl,
pH 8.0, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM
Na3VO4, 1 mM NaF, 2 mM
ethylenediaminetetraacetic acid (EDTA), 200 nM aprotinin, 20 µM
leupeptin, 50 mM phenanthroline and 280 mM benzamidine-HCl]. The
protein concentration was determined using a bicinchoninic acid
protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). Western
blotting was performed as described previously (23). Equal amounts of protein (50 µg/lane)
were resolved by 13% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis; then, the proteins were transferred to
nitrocellulose membranes. The membranes were incubated overnight
with the primary antibodies at 4°C and then incubated with
secondary antibodies conjugated to horseradish peroxidase at room
temperature for 1 h. β-actin was used as a loading control.
Immunoblots were visualized on an X-ray film using enhanced
chemiluminescence reagent (cat. no. WBKLS0100; MilliporeSigma). The
blots were quantified by densitometric analysis using ImageJ v4.0
software (National Institutes of Health), and the relative
expression level of each target protein was normalized using the
internal standard β-actin.
Cell counting assay
Cells were seeded in 6-well culture plates
(5×104/well) and treated with vehicle control (DMSO;
0.06%) or miconazole (10, 20 or 30 µM) for 24 h at 37°C. Afterward,
the cells were detached using 0.25% trypsin/EDTA and resuspended in
DMEM. Trypan blue was added to the cell suspension at 37°C for 5
min and viable cells were counted using a hemocytometer.
Flow cytometric analysis
Cell cycle analysis was performed by flow cytometry.
For DNA content analysis, ~1×106 cells were fixed in 80%
ethanol at 4°C for 24 h. The ethanol-fixed cells were stained with
PI staining solution (50 µg/ml PI, 0.1 mg/ml RNase A, 0.1% NP-40,
0.1% trisodium citrate) for 30 min at 4°C, and then analyzed using
a FACSCanto™ II flow cytometer (BD Biosciences).
Detection of LC3 translocation
To investigate the localization of green fluorescent
protein-fused LC3 (GFP-LC3), U87MG cells were grown on two-well
chamber slides and transfected with 1μg of GFP-LC3 plasmid, using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The GFP-LC3 plasmid was provided by Professor
Tamotsu Yoshimori (Department of Cellular Regulation Research,
Institute for Microbial Diseases, Osaka University, Japan)
(24). After 24 h, the medium was
changed to complete growth medium, and positive stable clones were
selected by incubating the cells in the presence of G418 (1 mg/ml)
for 2 weeks. Stable transfected cells were grown on two-well
chamber slides and treated with 20 µM miconazole for 24 h at 37°C.
After this, the cells were washed with PBS three times and then
fixed with 4% paraformaldehyde at room temperature for 10 min. The
fixed cells were washed with PBS three times and mounted with a
fluorescent mounting medium (Dako; Agilent Technologies, Inc.).
Images were obtained using a fluorescence microscope (Leica DM
3000; Leica Microsystems, GmbH). The GFP fluorescence intensity was
analyzed using LSM 5 Image Browser software (Zeiss GmbH).
Measurement of ROS production
Intracellular ROS production was measured using a
fluorescent probe, 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCFDA; Sigma-Aldrich; Merck KGaA). U251MG cells were
pretreated with miconazole at a concentration of 10 and 20 µM for
10 min at 37°C, followed by the addition of 10 µM
H2DCFDA and incubation for 30 min at room temperature.
Subsequently, the cells were trypsinized, resuspended in PBS and
transferred to Falcon® FACS tubes. The fluorescence
emission from H2DCFDA was analyzed using a FACSCanto™ II
flow cytometer (BD Biosciences) at an excitation wavelength of 488
nm and emission wavelength of 520 nm. The data were analyzed using
FACSDiva software version 6.0 (BD Biosciences).
Small interfering RNA (siRNA)
transfection
siRNAs against ATG5 (cat. no. sc-41445) and BiP
(cat. no. sc-29338), and a scrambled siRNA (cat. no. sc-37007) were
obtained from Santa Cruz Biotechnology, Inc. The sequence
information of these siRNAs was not provided by Santa Cruz
Biotechnology, Inc. U251MG cells were seeded in 6-well plates at a
density of 2×105 cells per well and transfected with the
50 nM siRNAs using Lipofectamine 2000. After 5 h of transfection,
the medium was changed to complete growth medium, and then
transfected cells were cultured for 20 h at 37°C, followed by
incubation with miconazole (20 µM) for 24 h at 37°C. The cells were
then harvested for viability measurements and western blot
analysis.
Statistical analysis
Data represent the mean ± standard deviation of
three independent experiments. Differences among groups were
evaluated by one-way ANOVA followed by the post hoc Dunnett's test
using SPSS 11.5 (SPSS, Inc.) software. P<0.05 was considered to
indicate a statistically ssignificant difference.
Results
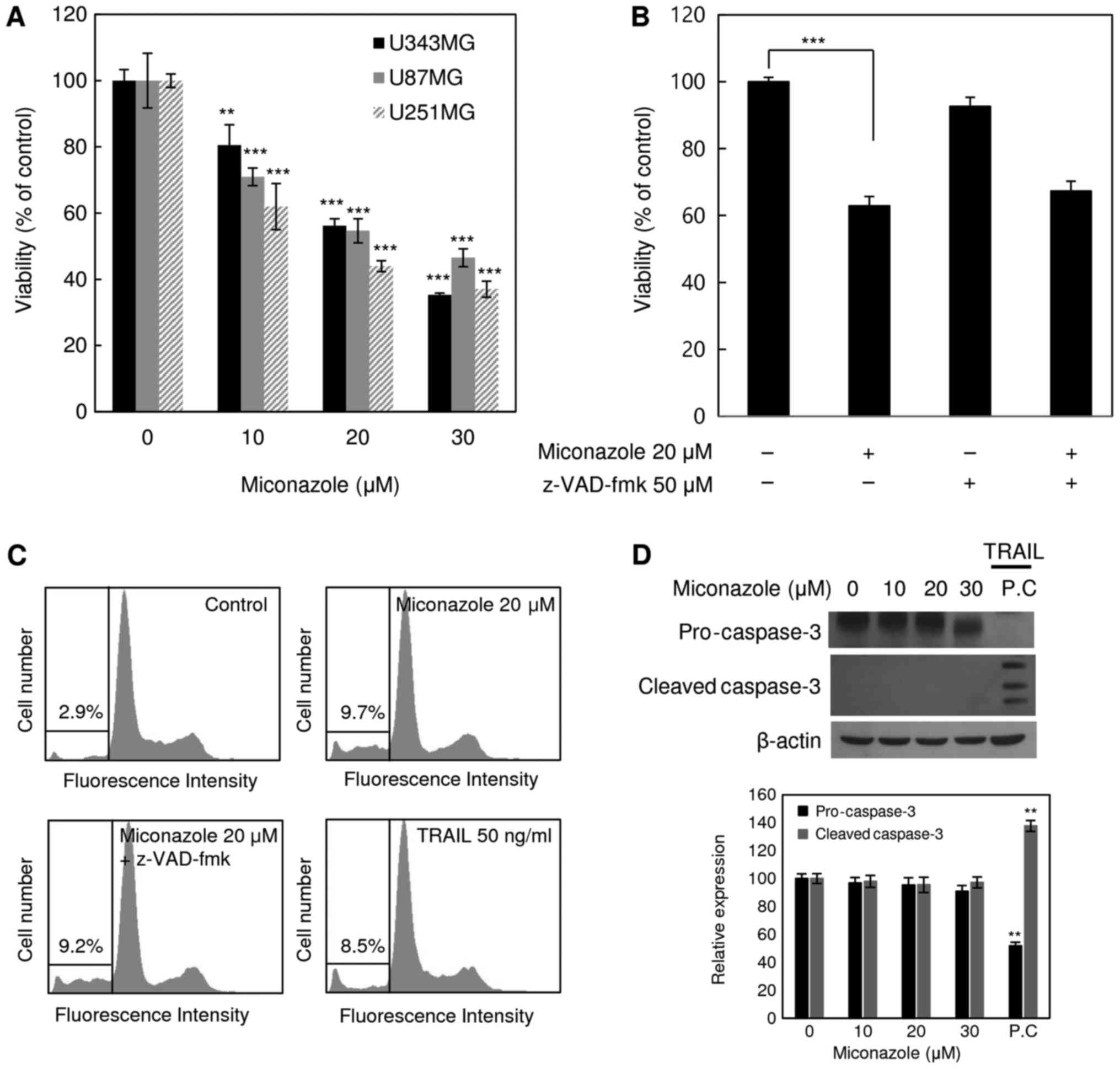
Miconazole increases cell death
The cytotoxicity of miconazole against the human
glioblastoma cell lines U343MG, U87MG and U251MG was examined. The
cell death-inducing effect of miconazole was measured by the direct
counting of viable cells following treatment with a range of
miconazole concentrations (0–30 µM) for 24 h. As shown in Fig. 1A, miconazole reduced the viability of
all three cell lines in a concentration-dependent manner. At a
miconazole concentration of 20 µM, the viability of the U343MG,
U87MG and U251MG cells was 44, 46 and 56% lower, respectively, than
that of the cells treated with the vehicle. These results suggest
that miconazole exerted cytotoxic effects against the human
glioblastoma cells. The U251MG cell line, which exhibited the
lowest viability in response to miconazole treatment, was selected
for subsequent analyses. To determine whether the reduced cell
viability was due to the induction of apoptosis, U251MG cells were
pretreated with the pan-caspase inhibitor, z-VAD-fmk, for 30 min,
followed by miconazole treatment for 24 h. Subsequently, cell
viability and cell cycle analyses were performed (Fig. 1B and C). Treatment with z-VAD-fmk did
not have any significant effect on the miconazole-mediated
reduction of cell viability. Similarly, treatment with z-VAD-fmk
did not affect the miconazole-mediated accumulation of the sub-G1
fraction, suggesting that the cytotoxicity of miconazole was not
associated with the induction of apoptosis. Cleaved caspase-3 was
not detected in U251MG cells treated with miconazole (Fig. 1D). These data suggest that the
cytotoxicity of miconazole was not due to caspase-dependent
apoptotic cell death.
Miconazole induces autophagy in human
glioblastoma cells
As autophagy is a major mode of cell death that
occurs in response to adverse cellular stress, whether miconazole
induces autophagy was investigated using the autophagic marker
LC3-II. Treatment with miconazole significantly increased the level
of LC3-II in U251MG cells in a time- and concentration-dependent
manner (Fig. 2A and B,
respectively), suggesting that miconazole induces autophagy. The
examination of cell lines not of glioblastoma origin, namely
MDA-MD-231 breast cancer and A549 lung cancer cell lines, revealed
that they also exhibited reduced viability and a marked increase in
the level of LC3-II following miconazole treatment (Fig. S1). The immunofluorescent expression
pattern of LC3 in stable GFP-LC3-transfected U87MG cells was also
examined. The number of GFP-LC3 puncta was significantly increased
in the miconazole-treated cells (Fig.
S2). To confirm the involvement of autophagy in the cytotoxic
effect of miconazole, the autophagy inhibitor 3-MA and an
ATG5-specific siRNA were used to suppress autophagy. 3-MA inhibited
the LC3-I-to-LC3-II conversion and cell death in miconazole-treated
cells (Fig. 2C). In addition,
transfection of the cells with ATG5 siRNA resulted in the
significant inhibition of miconazole-induced LC3-II accumulation
and cell growth (Fig. 2D).
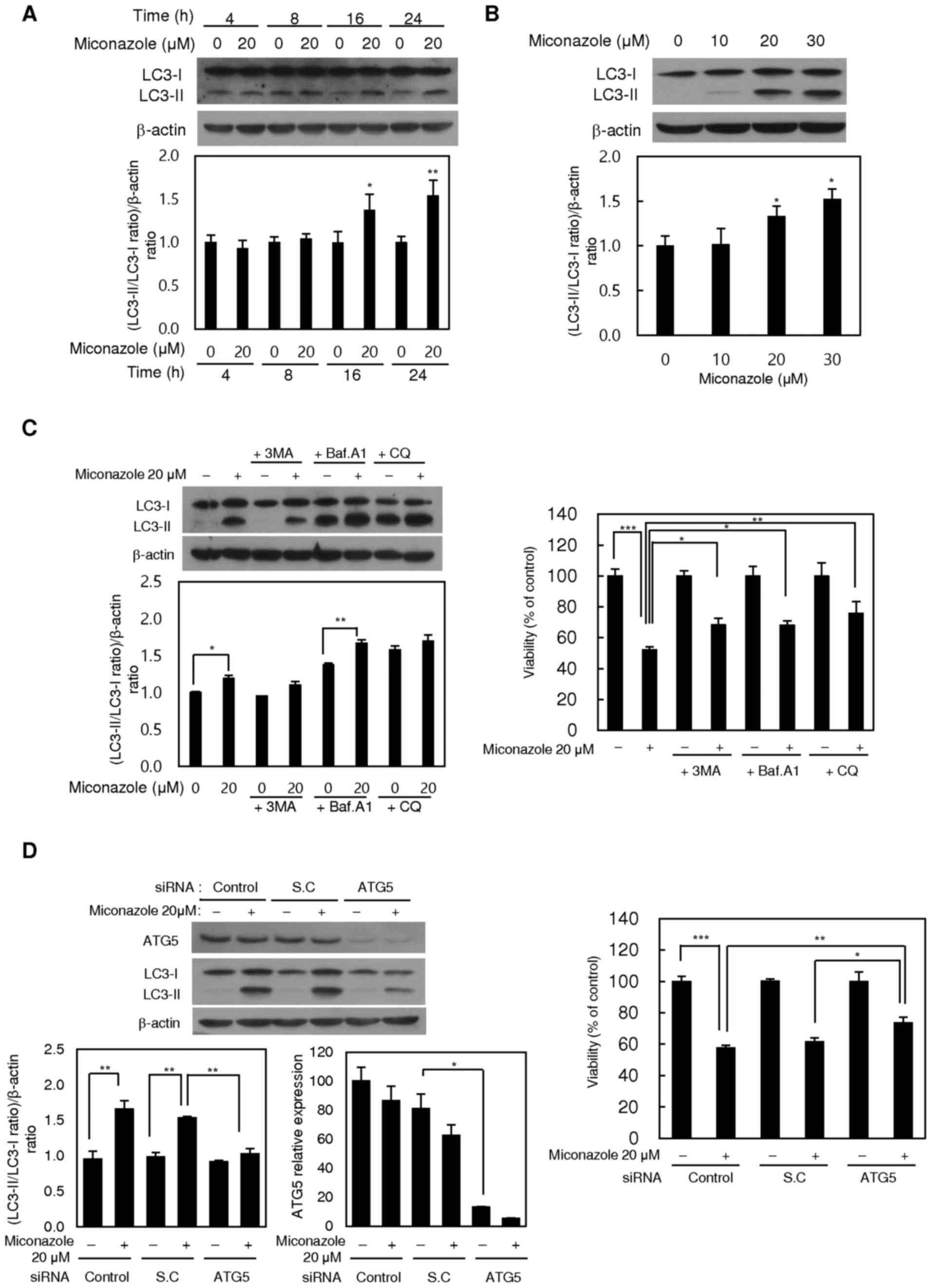 | Figure 2.Autophagy-inducing effects of
miconazole. Expression of the LC3 protein in U251MG cells treated
with (A) 20 µM miconazole for the indicated times or (B) various
miconazole concentrations for 24 h, as determined by western
blotting. β-actin was used as a loading control. Densitometric
analysis of the western blots is shown in the bar graphs. (C)
Expression of LC3 protein in U251MG cells treated with 20 µM
miconazole alone or cotreated with 1 mM 3-MA, 20 nM Baf A1 or 50 µM
CQ for 24 h, and the viability of these cells. LC3 expression was
determined by western blotting using β-actin as a loading control.
Densitometric analysis of the western blots is shown in the bar
graph. (D) Expression of ATG5 and LC3 proteins in U251MG cells
transiently transfected with an ATG5 siRNA or SC siRNA, and treated
with 20 µM miconazole or vehicle for 24 h, and the viability of
these cells. ATG5 and LC3 expression was determined by western
blotting using β-actin as a loading control. Densitometric analysis
of the western blots is shown in the bar graph. All graphical data
are the mean ± standard deviation of three independent experiments.
*P<0.05, **P<0.01 and ***P<0.001 vs. 0 µM miconazole or as
indicated. LC3, microtubule-associated protein light chain 3; 3-MA,
3-methyladenine; Baf A1, bafilomycin A1; CQ, chloroquine; siRNA,
small interfering RNA; SC, scrambled control. |
The miconazole-induced autophagic flux was further
investigated in the presence or absence of the
autophagosome-lysosome fusion inhibitors, Baf A1 and CQ.
Pretreatment with Baf A1 or CQ alone increased the level of LC3-II,
and the addition of miconazole caused a further increase in LC3-II,
suggesting that miconazole induced autophagic flux. Pretreatment
with Baf A1 and CQ also diminished the miconazole-induced cell
death (Fig. 2C). These data
demonstrate that autophagy plays a crucial role in
miconazole-induced cell death.
Endoplasmic reticulum (ER) stress is
involved in miconazole-induced autophagy
To determine whether miconazole induces autophagy
via the activation of ER stress in human glioblastoma cells, the
levels of ER stress response markers, namely BiP, IRE1α, p-eIF2α
and CHOP, were examined. Miconazole increased the levels of these
ER stress response proteins in a concentration-dependent manner
(Fig. 3A). To further investigate
whether ER stress is involved in miconazole-induced autophagy,
4-PBA, a chemical inhibitor of ER stress, was used. U251MG cells
were pretreated with 0.5 or 1 mM 4-PBA for 30 min, followed by 20
µM miconazole treatment for 24 h. The increase in the LC3-II level
that was observed after miconazole treatment was significantly
inhibited by pretreatment with 4-PBA in a concentration-dependent
manner. Pretreatment with 4-PBA also attenuated miconazole-induced
cell death (Fig. 3B). Transfection
with BiP siRNA attenuated the miconazole-induced increase in LC3-II
levels in U251MG cells. The inhibition of ER stress was associated
with a simultaneous reduction in miconazole-induced cell death
(Fig. 3C). These data suggest that
ER stress plays a key role in miconazole-induced autophagic cell
death.
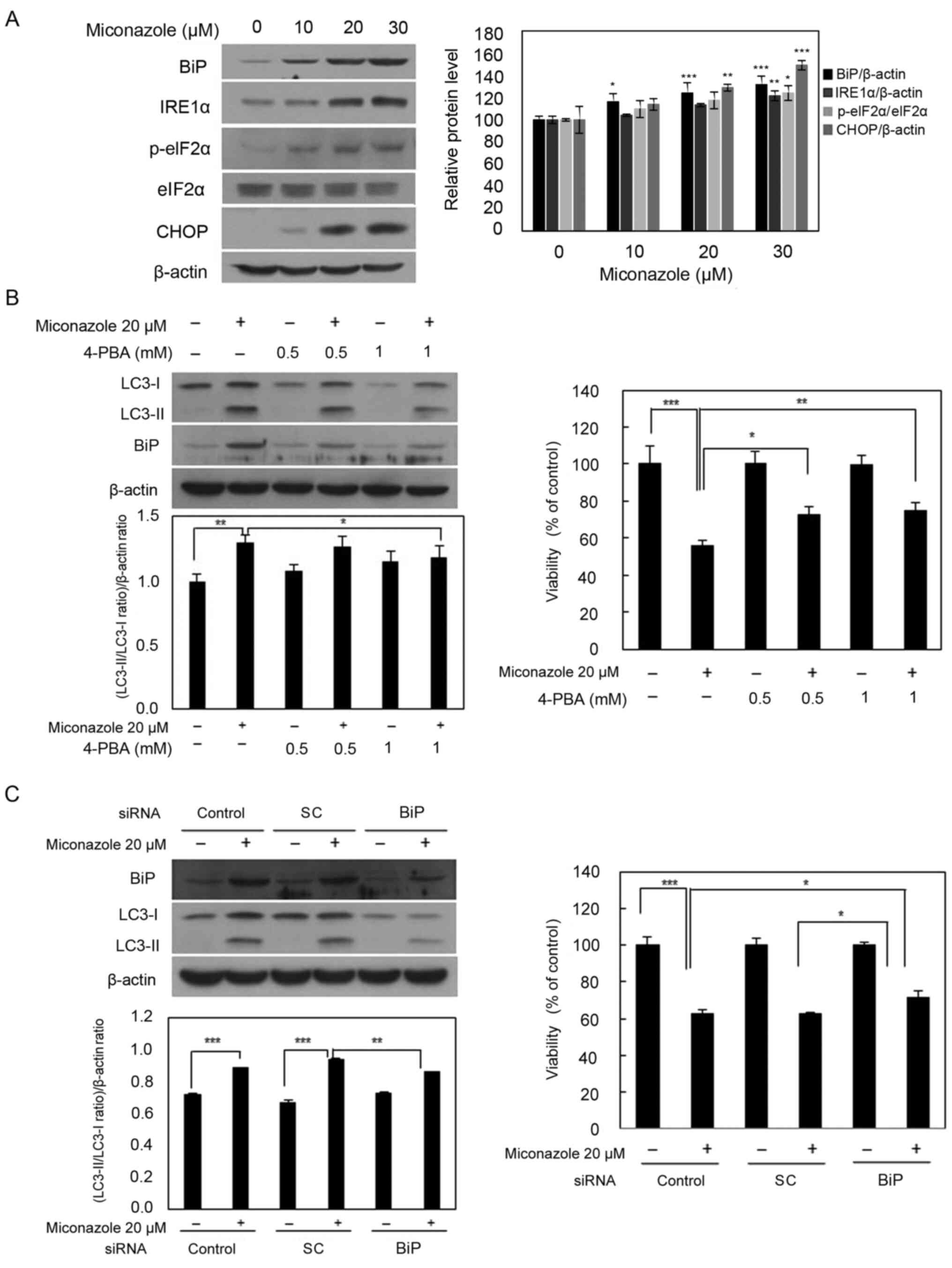 | Figure 3.ER stress-inducing effects of
miconazole. (A) Expression of ER stress-associated proteins in
U251MG cells exposed to the indicated concentrations of miconazole
for 24 h, as determined by western blotting. Densitometric analysis
of the western blots is shown in the bar graph. Data are the mean ±
SD of three independent experiments. (B) Levels of BiP and LC3-I
and II proteins in U251MG cells pretreated with 4-PBA for 30 min
and then treated with 20 µM miconazole for 24 h. Densitometric
analysis of the western blots is shown in the bar graph (n=3). Cell
viability is expressed as the percentage of control ± SD for pooled
data from three experiments, each performed in triplicate. (C)
Levels of BiP and LC3-I and II proteins in cells transfected with
BiP siRNA or SC siRNA and treated with miconazole for 24 h, as
determined by western blotting. Densitometric analysis of the
western blots is shown in the bar graph (n=3). Cell viability data
are the mean ± SD of three independent experiments (n=3).
*P<0.05, **P<0.01 and ***P<0.001 vs. 0 µM miconazole or as
indicated. ER, endoplasmic reticulum; BiP, binding immunoglobulin
protein; LC3, microtubule-associated protein light chain 3; 4-PBA,
4-phenylbutyric acid; siRNA, small interfering RNA; SC, scrambled
control; IRE1α, inositol-requiring enzyme 1α; p-, phospho-; eIF2α,
eukaryotic initiation factor 2α. |
Miconazole induces ROS production in
human glioblastoma cells
Whether miconazole induces ROS production in human
glioblastoma cells was tested using flow cytometry. As shown in
Fig. 4A, the treatment of U251MG
cells with miconazole for 10 min caused a marked increase in the
emission of fluorescence by H2DCFDA, suggesting that
miconazole induces ROS production. To further confirm the role of
ROS in miconazole-induced autophagic cell death, cells were
pretreated with 5 mM NAC, a ROS scavenger, for 2 h, followed by
treatment with 10 or 20 µM miconazole for 24 h at 37°C. As shown in
Fig. 4B, miconazole treatment
increased cell death in a concentration-dependent manner, while
cotreatment with NAC attenuated the miconazole-induced cell death.
Furthermore, cotreatment with miconazole and NAC led to significant
reductions in LC3-II levels. These results indicate that ROS are
involved in the miconazole-induced autophagic cell death of U251MG
cells.
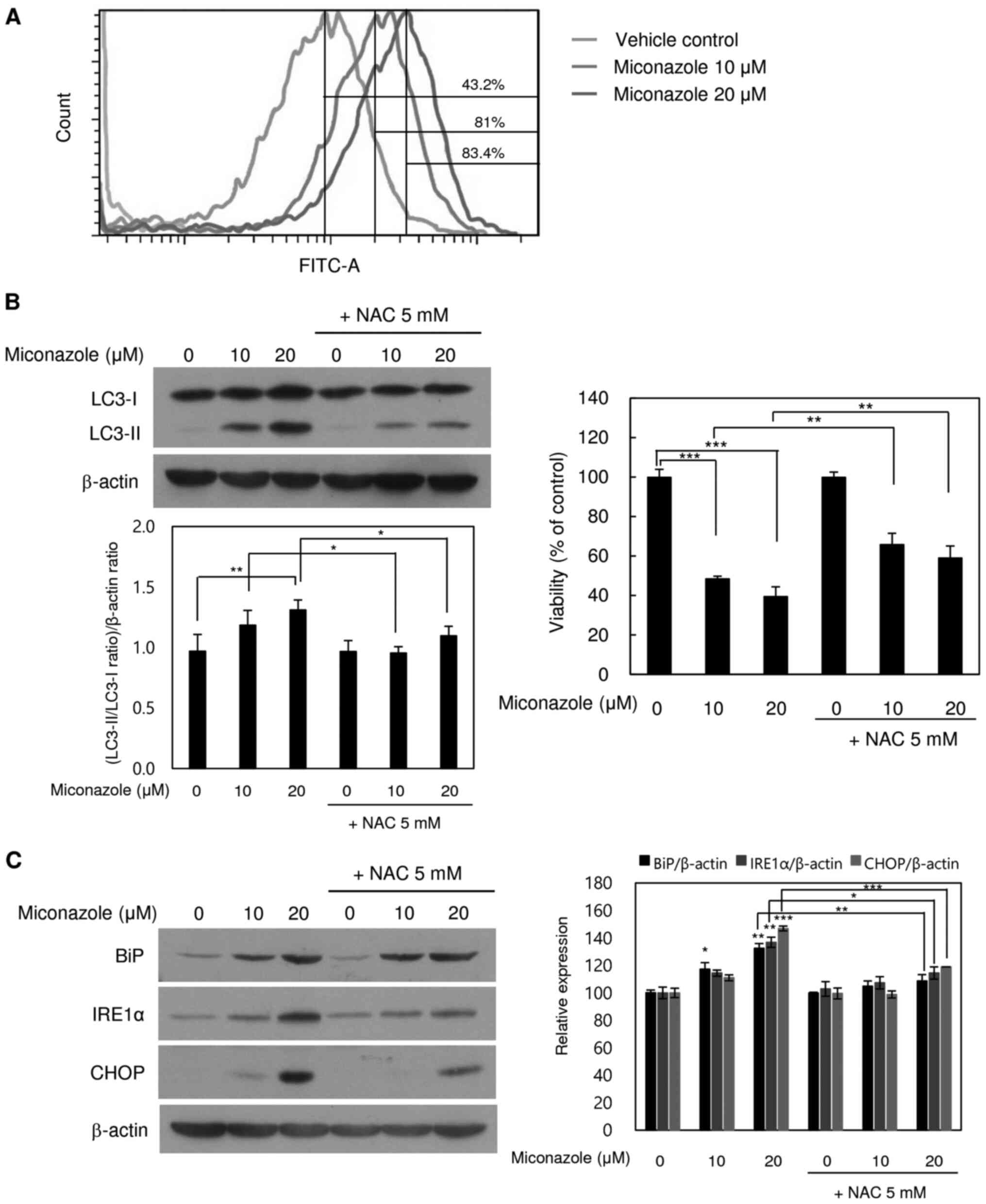 | Figure 4.ROS- and ER stress-inducing effects
of miconazole in relation to autophagic cell death. (A) ROS
production in cells treated with miconazole for 10 min, as measured
by flow cytometry. Fluorescence intensity is directly proportional
to the quantity of ROS species in the cells. (B) Levels of LC3-I
and LC3-II proteins in U251MG cells treated with miconazole in the
presence or absence of 5 mM NAC, as determined by western blotting
and densitometry using β-actin as a loading control. (C) Levels of
BiP, IRE1α and CHOP proteins in cells pretreated with 5 mM NAC for
2 h and then treated with miconazole at the indicated
concentrations for 24 h, as determined by western blotting using
β-actin as a loading control. Densitometric analysis of the western
blots is shown in the bar graph. Densitometric and cell viability
data are the mean ± standard deviation of three independent
experiments. *P<0.05, **P<0.01 and ***P<0.001. ROS,
reactive oxygen species; ER, endoplasmic reticulum; LC3,
microtubule-associated protein light chain 3; NAC,
N-acetylcysteine; BiP, binding immunoglobulin protein;
IRE1α, inositol-requiring enzyme 1α |
To further examine whether miconazole-induced ROS
production induces ER stress, cells were treated with miconazole
alone or with NAC for 24 h. The cotreatment with NAC significantly
blocked ER stress, as indicated by lower levels of the ER stress
markers BiP and IRE1α (Fig. 4C).
These findings are consistent with the increase in viability of
miconazole-treated U251MG cells observed when the cells were
cotreated with NAC. These results indicate that ROS production and
ER stress are involved in the miconazole-induced autophagic death
of U251MG cells.
Discussion
Miconazole has been shown to inhibit the
proliferation of human cancer cells via cell cycle arrest, the
induction of ROS production and the release of intracellular
Ca2+ (13). To date,
apoptosis has been implicated as the main mechanism of
miconazole-induced cell death (14,25). To
the best of our knowledge, the present study is the first to report
miconazole-induced autophagy in human glioblastoma cells.
Mechanistically, the results indicate that miconazole induced
autophagy via the ROS-mediated activation of ER stress. Autophagy
is a dynamic process comprising the sequestration of cytoplasmic
organelles and proteins into autophagosomes, which fuse with
lysosomes, enabling the degradation of target proteins by lysosomal
hydrolases. However, increased autophagy can directly induce cell
death (3,7). It has been reported that excessive
autophagic activity, which causes the overconsumption of critical
cellular components, is responsible for type II nonapoptotic
programmed cell death, also known as autophagic cell death
(26). The present study
demonstrated that miconazole triggered autophagy-mediated cell
death in U251MG cells, as well as in MDA-MB-231 and A549 cells, as
indicated by an increase in LC3-II levels. Cleaved caspase-3, a
marker of apoptosis, was not detected in U251MG cells following
miconazole treatment, and no change in the cytotoxicity of
miconazole was observed when the cells were cotreated with
z-VAD-fmk, a pan-caspase inhibitor. However, although miconazole
induced LC3-II expression, ATG5 expression was unaffected. Other
studies have also observed no change in the protein expression
level of ATG5 in experimentally induced autophagy (27,28). In
addition, the activity of ATG5 is reportedly regulated by
phosphorylation (29). By contrast,
the inhibition of miconazole-mediated autophagy via the knockdown
of ATG5 or using autophagy inhibitors resulted in the decreased
conversion of LC3-I to LC3-II and the restoration of cell
viability. These results suggest that autophagy may be an essential
mechanism in miconazole-induced cell death but do not eliminate the
potential existence of other mechanisms. For example, miconazole
has been reported to inhibit fumarate hydratase activity and
enhance cisplatin-induced cell death in gastric cancer (30). In addition, miconazole has been
demonstrated to induce cell death via the accumulation of
Ca2+ in human breast cancer (31). Caspase-independent mechanisms of cell
death also exist and may play roles in alternative anticancer
therapeutic strategies, such as alteration of the mitochondrial
permeability transition (32) or
cytotoxic T-lymphocyte granule exocytosis via the Fas/Fas ligand
pathway (33). Although the
occurrence of caspase-independent apoptosis was not investigated in
the present study, cell death did not increase when miconazole was
supplemented with z-VAD-fmk.
Previous studies have shown that the ER plays an
essential role in the process of autophagy and that autophagy is
critical for the maintenance of ER homeostasis (34,35). In
mammalian cells, ER stress promotes autophagy, enabling the removal
of misfolded proteins (36–39). The mechanism of miconazole-induced ER
stress is unknown. In the present study, ER stress signaling was
shown to participate in the miconazole-induced autophagic cell
death. To confirm the role of ER stress in miconazole-induced cell
death, 4-PBA and a BiP siRNA were used, and the inhibition of ER
stress was demonstrated to prevent the miconazole-mediated
induction of autophagic cell death.
The results of the present study also demonstrated
that ROS, known as regulators of autophagy (40), are involved in miconazole-induced
autophagy. The excessive production of ROS, including superoxide
radicals, hydrogen peroxide and hydroxyl radicals, causes oxidative
stress and, ultimately, cell death (41). ROS also act as second-messenger
signaling molecules, regulating multiple cell functions, including
autophagy and apoptosis (42–44). The
induction of intracellular oxidative stress by miconazole has been
demonstrated in rat neonatal cardiomyocytes and bladder cancer
cells, as well as in Candida albicans and other fungi
(18,25,45–48). The
present study revealed that the NAC-mediated scavenging of
miconazole-induced ROS decreased the levels of LC3-II and molecular
markers of ER stress. These data suggest that the increased
production of intracellular ROS in miconazole-treated cells causes
ER stress, which in turn induces autophagic cell death.
In summary, the present study indicate that
miconazole induced autophagy-mediated cell death in glioblastoma
cell lines and that the induction of intracellular ROS production
and ER stress might be the underlying mechanism by which autophagy
was activated. The results support the chemotherapeutic potential
of miconazole and provide the first evidence of the involvement of
autophagy in miconazole-induced cell death.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Research
Foundation of Korea (grants nos. 2016R1A2B1012055,
2018R1A2B5A01023660 and 2020R1H1A2102379), funded by the Korean
Government (MSIP).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HJJ performed the experiments and analyzed the
results. WKB analyzed and interpreted the data and wrote the
manuscript. IS, BKJ and SIS were involved in project development,
data analysis and editing the manuscript. HJJ and SIS confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
3-MA
|
3-methyladenine
|
|
4-PBA
|
4-phenylbutyric acid
|
|
ATG5
|
autophagy protein 5
|
|
Baf A1
|
bafilomycin A1
|
|
BiP
|
binding immunoglobulin protein
|
|
CQ
|
chloroquine
|
|
DMEM
|
Dulbecco's Modified Eagle's medium
|
|
EDTA
|
ethylenediaminetetraacetic acid
|
|
eIF2α
|
eukaryotic translation initiation
factor 2α
|
|
ER
|
endoplasmic reticulum
|
|
H2DCFDA
|
2,7-dichlorodihydrofluorescein
diacetate
|
|
IRE1α
|
inositol-requiring enzyme 1α
|
|
LC3
|
microtubule-associated protein light
chain 3
|
|
NAC
|
N-acetylcysteine
|
|
p-
|
phospho-
|
|
PBS
|
phosphate-buffered saline
|
|
ROS
|
reactive oxygen species
|
|
siRNA
|
small interfering RNA
|
References
|
1
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mazure NM and Pouysségur J:
Hypoxia-induced autophagy: Cell death or cell survival? Curr Opin
Cell Biol. 22:177–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Descloux C, Ginet V, Clarke PG, Puyal J
and Truttmann AC: Neuronal death after perinatal cerebral
hypoxia-ischemia: Focus on autophagy-mediated cell death. Int J Dev
Neurosci. 45:75–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Furtado CM, Marcondes MC, Sola-Penna M, de
Souza ML and Zancan P: Clotrimazole preferentially inhibits human
breast cancer cell proliferation, viability and glycolysis. PLoS
One. 7:e304622012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gupta A, Unadkat JD and Mao Q:
Interactions of azole antifungal agents with the human breast
cancer resistance protein (BCRP). J Pharm Sci. 96:3226–3235. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shahbazfar AA, Zare P, Ranjbaran M,
Tayefi-Nasrabadi H, Fakhri O, Farshi Y, Shadi S and Khoshkerdar A:
A survey on anticancer effects of artemisinin, iron, miconazole,
and butyric acid on 5637 (bladder cancer) and 4T1 (Breast cancer)
cell lines. J Cancer Res Ther. 10:1057–1062. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park JY, Jung HJ, Seo I, Jha BK, Suh SI,
Suh MH and Baek WK: Translational suppression of HIF-1α by
miconazole through the mTOR signaling pathway. Cell Oncol (Dordr).
37:269–279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang HT, Chen WC, Chen JS, Lu YC, Hsu SS,
Wang JL, Cheng HH, Cheng JS, Jiann BP, Chiang AJ, et al: Effect of
miconazole on intracellular Ca2+ levels and
proliferation in human osteosarcoma cells. Life Sci. 76:2091–2101.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu CH, Jeng JH, Wang YJ, Tseng CJ, Liang
YC, Chen CH, Lee HM, Lin JK, Lin CH, Lin SY, et al: Antitumor
effects of miconazole on human colon carcinoma xenografts in nude
mice through induction of apoptosis and G0/G1 cell cycle arrest.
Toxicol Appl Pharmacol. 180:22–35. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan SY, Shiau MY, Ou YC, Huang YC, Chen
CC, Cheng CL, Chiu KY, Wang SS and Tsai KJ: Miconazole induces
apoptosis via the death receptor 5-dependent and
mitochondrial-mediated pathways in human bladder cancer cells.
Oncol Rep. 37:3606–3616. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chengzhu WU, Gao M, Shen L, Bohan LI, Bai
X, Gui J, Hongmei LI, Huo Q and Tao MA: Miconazole triggers various
forms of cell death in human breast cancer MDA-MB-231 cells.
Pharmazie. 74:290–294. 2019.PubMed/NCBI
|
|
17
|
Kobayashi D, Kondo K, Uehara N, Otokozawa
S, Tsuji N, Yagihashi A and Watanabe N: Endogenous reactive oxygen
species is an important mediator of miconazole antifungal effect.
Antimicrob Agents Chemother. 46:3113–3117. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee KP, Kim JE and Park WH: Cytoprotective
effect of rhamnetin on miconazole-induced H9c2 cell damage. Nutr
Res Pract. 9:586–591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yeo IJ, Yun J, Son DJ, Han SB and Hong JT:
Antifungal drug miconazole ameliorated memory deficits in a mouse
model of LPS-induced memory loss through targeting iNOS. Cell Death
Dis. 11:6232020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen LD, Liu ZH, Zhang LF, Yao JN and Wang
CF: Sanggenon C induces apoptosis of colon cancer cells via
inhibition of NO production, iNOS expression and ROS activation of
the mitochondrial pathway. Oncol Rep. 38:2123–2131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Holotiuk VV, Kryzhanivska AY, Churpiy IK,
Tataryn BB and Ivasiutyn DY: Role of nitric oxide in pathogenesis
of tumor growth and its possible application in cancer treatment.
Exp Oncol. 41:210–215. 2019.PubMed/NCBI
|
|
22
|
Liu R, Li J, Zhang T, Zou L, Chen Y, Wang
K, Lei Y, Yuan K, Li Y, Lan J, et al: Itraconazole suppresses the
growth of glioblastoma through induction of autophagy: Involvement
of abnormal cholesterol trafficking. Autophagy. 10:1241–1255. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jung HJ, Seo I, Casciello F, Jacquelin S,
Lane SW, Suh SI, Suh MH, Lee JS and Baek WK: The anticancer effect
of chaetocin is enhanced by inhibition of autophagy. Cell Death
Dis. 7:e20982016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Won KJ, Lin HY, Jung S, Cho SM, Shin HC,
Bae YM, Lee SH, Kim HJ, Jeon BH and Kim B: Antifungal miconazole
induces cardiotoxicity via inhibition of APE/Ref-1-related pathway
in rat neonatal cardiomyocytes. Toxicol Sci. 126:298–305. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Portt L, Norman G, Clapp C, Greenwood M
and Greenwood MT: Anti-apoptosis and cell survival: A review.
Biochim Biophys Acta. 1813:238–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanaka S, Hikita H, Tatsumi T, Sakamori R,
Nozaki Y, Sakane S, Shiode Y, Nakabori T, Saito Y, Hiramatsu N, et
al: Rubicon inhibits autophagy and accelerates hepatocyte apoptosis
and lipid accumulation in nonalcoholic fatty liver disease in mice.
Hepatology. 64:1994–2014. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Basu A: Regulation of autophagy by protein
kinase C-ε in breast cancer cells. Int J Mol Sci. 21:42472020.
View Article : Google Scholar
|
|
29
|
Keil E, Höcker R, Schuster M, Essmann F,
Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schulze-Osthoff K
and Schmitz I: Phosphorylation of Atg5 by the Gadd45β-MEKK4-p38
pathway inhibits autophagy. Cell Death Differ. 20:321–332. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu HE, Wang F, Yu F, Zeng ZL, Wang Y, Lu
YX, Jin Y, Wang DS, Qiu MZ, Pu HY, et al: Suppression of fumarate
hydratase activity increases the efficacy of cisplatin-mediated
chemotherapy in gastric cancer. Cell Death Dis. 10:4132019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roan CJ, Chou CT, Liang WZ, Chang HT, Kuo
DH, Kuo CC, Chen FA, Shieh P and Jan CR: Effect of miconazole on
[Ca2+]i and cytotoxicity in ZR-75-1 human breast cancer
cells. Chin J Physiol. 58:377–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hirsch T, Marchetti P, Susin SA,
Dallaporta B, Zamzami N, Marzo I, Geuskens M and Kroemer G: The
apoptosis-necrosis paradox. Apoptogenic proteases activated after
mitochondrial permeability transition determine the mode of cell
death. Oncogene. 15:1573–1581. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sarin A, Williams MS, Alexander-Miller MA,
Berzofsky JA, Zacharchuk CM and Henkart PA: Target cell lysis by
CTL granule exocytosis is independent of ICE/Ced-3 family
proteases. Immunity. 6:209–215. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yorimitsu T and Klionsky DJ: Endoplasmic
reticulum stress: A new pathway to induce autophagy. Autophagy.
3:160–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alpha phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yin JJ, Xie G, Zhang N and Li Y:
Inhibiting autophagy promotes endoplasmic reticulum stress and the
ROS induced nod like receptor 3 dependent proinflammatory response
in HepG2 cells. Mol Med Rep. 14:3999–4007. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz
DB, Ron D and Yin XM: Linking of autophagy to ubiquitin-proteasome
system is important for the regulation of endoplasmic reticulum
stress and cell viability. Am J Pathol. 171:513–524. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hwang MS and Baek WK: Glucosamine induces
autophagic cell death through the stimulation of ER stress in human
glioma cancer cells. Biochem Biophys Res Commun. 399:111–116. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen Y, Azad MB and Gibson SB: Superoxide
is the major reactive oxygen species regulating autophagy. Cell
Death Differ. 16:1040–1052. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Miki H, Uehara N, Kimura A, Sasaki T, Yuri
T, Yoshizawa K and Tsubura A: Resveratrol induces apoptosis via
ROS-triggered autophagy in human colon cancer cells. Int J Oncol.
40:1020–1028. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Poillet-Perez L, Despouy G,
Delage-Mourroux R and Boyer-Guittaut M: Interplay between ROS and
autophagy in cancer cells, from tumor initiation to cancer therapy.
Redox Biol. 4:184–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhou DR, Eid R, Miller KA, Boucher E,
Mandato CA and Greenwood MT: Intracellular second messengers
mediate stress inducible hormesis and programmed cell death: A
review. Biochim Biophys Acta Mol Cell Res. 1866:773–792. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Delattin N, Cammue BP and Thevissen K:
Reactive oxygen species-inducing antifungal agents and their
activity against fungal biofilms. Future Med Chem. 6:77–90. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kumar CG and Poornachandra Y: Biodirected
synthesis of Miconazole-conjugated bacterial silver nanoparticles
and their application as antifungal agents and drug delivery
vehicles. Colloids Surf B Biointerfaces. 125:110–119. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu CX, Yan L, Wang XJ, Miao Q, Li XX,
Yang F, Cao YB, Gao PH, Bi XL and Jiang YY: Transposition of the
Zorro2 retrotransposon is activated by miconazole in Candida
albicans. Biol Pharm Bull. 37:37–43. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tsai TF, Chen PC, Lin YC, Chou KY, Chen
HE, Ho CY, Lin JF and Hwang TI: Miconazole contributes to NRF2
activation by noncanonical P62-KEAP1 pathway in bladder cancer
cells. Drug Des Devel Ther. 14:1209–1218. 2020. View Article : Google Scholar : PubMed/NCBI
|