Introduction
Melanoma is the most serious type of skin cancer,
accounting for ~70% of skin cancer-associated deaths in the United
States in 2018 (1,2). The incidence of melanoma has risen
rapidly since 1975, and 95,710 newly diagnosed cases were predicted
for 2020 (3). With the development
of promising new treatments, particularly targeted therapies and
immunotherapy, among all patients with melanoma after diagnosis,
the 5-year survival rate of melanoma has increased to 92% in the
USA in 2019 (3–7). However, since up to 50% of patients
with melanoma do not respond to or acquire resistance to these
therapies (8,9), the overall mortality rate has not
markedly decreased. Melanoma-associated death decreased by ~6% in
the USA from 2013 to 2017 (3).
Therefore, it is important to investigate the molecular mechanisms
to identify new targets/strategies for the improved treatment of
melanoma.
Most melanomas arise from recurrent somatic
mutations, which lead to the dysregulation of oncogenic pathways
that regulate cell proliferation, apoptosis and invasion (10–12). The
most frequently mutated pathway in melanoma is the MAP kinase
(MAPK) signaling pathway, the mutation of which occurs in ~70% of
all melanomas and results in constitutive activation of MAPK
signaling. Notably, ~50% of melanomas harbor BRAF oncogenic
mutations, with V600E being the most common (13,14).
Therefore, targeting the BRAF-V600E mutation has attracted notable
research attention. Several inhibitors antagonizing this mutation
have been developed, two of which (vemurafenib and dabrafenib) have
been approved by the Food and Drug Association for treatment of
non-resectable BRAF-V600E/K mutant melanoma (15). Although the short-term response is
promising, most patients with melanoma acquire resistance to these
BRAF inhibitors and progress to more aggressive disease through
various mechanisms, such as other genetic alterations restoring the
MAPK pathway or activating the PI3K/Akt signaling pathway (16). However, ~40% of patients develop
resistance with unknown causes (17).
Yes-associated protein 1 (YAP) is an evolutionarily
conserved transcriptional coactivator that functions as a key
regulator of organ development, cancer progression and therapeutic
resistance (18). YAP governs cell
proliferation and apoptosis by controlling transcriptional programs
through interacting with transcription factors, such as TEAD and
β-catenin (19). In support of YAP
as an oncoprotein, overexpression of YAP has been associated with a
variety of human cancer types, including lung and breast cancer
(20,21). Previous studies have shown that YAP
promotes melanoma cell proliferation, invasion and resistance to
BRAF/MEK inhibitors (22–25). Consistently, patients with melanoma
with high expression of YAP display less responsiveness to RAF/MEK
inhibitors (25). However, the
molecular mechanism for the overexpression of YAP in melanoma is
largely unknown.
YAP is a major downstream effector of the Hippo
pathway, which is composed of a kinase cascade containing MAP4K,
MST1/2 and LATS1/2. In response to the changes in microenvironment,
such as cell-cell adhesions and mechanotransduction, activated
LATS1/2 kinases phosphorylate YAP at multiple Ser/Thr residues,
leading to YAP cytoplasmic translocation (26). Other post-translational modifications
also play a critical role in regulating YAP subcellular
localization and transcriptional activity (27). Notably, ubiquitination-mediated
proteolysis is a key mechanism controlling YAP protein levels.
Several E3 ubiquitin ligases of YAP have been identified.
β-transducin repeats-containing protein and F-box/WD
repeat-containing protein 7 promote YAP ubiquitination and
degradation in a phosphorylation-dependent manner (28,29).
Notably, SKP2-mediated K63-linked poly-ubiquitination of YAP is a
non-proteolytic signal, which enhances YAP nuclear accumulation
independent of the Hippo pathway (30). Similar to phosphorylation,
ubiquitination can be reversed by deubiquitinating enzymes (DUBs),
the dysregulation of which contributes to aberrant protein
expression. Several DUBs have been identified to regulate YAP
cytoplasmic/nuclear translocation and transcriptional activity,
such as OTUD1 and YOD1 (27);
however, the DUBs that directly control YAP protein stability in
melanoma remain to be identified.
The current study aimed to characterize ubiquitin
specific peptidase 22 (USP22) as a DUB controlling YAP abundance
and biological functions in melanoma.
Materials and methods
Melanoma specimens and
immunohistochemistry (IHC) analysis
The patient samples were collected by fixing into
formalin within 15–30 min after surgical resection between January
2015 and December 2018 under the protocol approved by the
Institutional Research Ethics Committee at Changxing People's
Hospital (Huzhou, China). The median age of patients was 60 years
(range, 15–85 years), and 56.7% of patients were male. All patients
with resectable melanoma were included. Patients were fully
informed, and written consent was obtained from all patients and/or
their guardians before sample collection. In total, 90 melanoma
tissue samples were fixed with 10% formalin for 24 h at room
temperature and embedded in paraffin. The sections were cut into
5-µm-thick sections and used for IHC staining with anti-USP22
antibody (1:500; cat. no. ab195289; Abcam) and YAP antibody (1:500;
cat. no. 14074; Cell Signaling Technology, Inc.). IHC analysis of
YAP and USP22 expression was based on visual inspection by two
members of our group of staining intensity, which is commonly used
by pathologists in the research field. To provide unbiased
analysis, the two researchers independently scored the samples as
low, medium and high using representative images as references. The
association between USP22 and YAP was analyzed using the
χ2 test. All procedures performed in the present study
involving human participants were approved and in accordance with
the ethical standards of the institutional and/or national research
committee, and in accordance with the 1964 Helsinki declaration and
its later amendments or comparable ethical standards.
Cell culture and reagents
Normal human epidermal melanocytes (NHEM; cat. no.
PCS-200-013), all melanoma cell lines (SK-MEL-3, SK-MEL-28, A375,
A2058 and G361) and 293T cells were obtained from the American Type
Culture Collection (ATCC) and cultured according to the suppliers'
instructions. The Dermal Cell Basal Medium (cat. no. PCS-200-030),
McCoy's 5a Medium (cat. no. 30-2007) was used for PCS-200-013,
SK-MEL-3 and G361 cells, Eagle's Minimum Essential Medium (cat. no.
30-2003) was used for SK-MEL-28 cells, and Dulbecco's Modified
Eagle's Medium (DMEM; cat. no. 30-2002) was used for A375, A2058
and 293T cells. All media were purchased from ATCC. FBS was
purchased from Thermo Fisher Scientific, Inc. (cat. no. 10438026),
and 10% FBS was added to the medium for all cells. The cells were
maintained in 5% CO2 incubator at 37°C. BRAF-V600E
inhibitor vemurafenib (cat. no. S1267) and proteasome inhibitor
MG-132 (cat. no. S2619) were purchased from Selleck Chemicals.
Cell transfection
Lipofectamine 3000 was used for transfection
following the manufacturer's instructions (cat. no. L3000008;
Invitrogen; Thermo Fisher Scientific, Inc.). Lentiviral packaging
and infection were carried out as previously described (31). Briefly, 293T cells were transfected
with 6 µg lentiviral vector [short hairpin (sh)GFP (shRNA targeting
GFP used as negative control), shYAP or shUSP22] and second
generation packaging plasmids, 2 µg psPAX2 (cat. no. 12260) and 4
µg pMD2.G (cat. no. 12259) (both Addgene, Inc.) using Lipofectamine
3000. After 18 h of transfection, DMEM was replaced with fresh DMEM
medium. Viruses were collected at 48 and 72 h and then filtered
with a 0.45-µm PES filter. The virus titer was measured using the
Lenti-X GoStix Plus kit (cat. no. 631280; Takara Bio, Inc.)
following the manufacturer's instructions. Targeted cells A375 and
A2058 were infected with the virus at MOI 5.0 and selected with 1
µg/ml puromycin for 3 days in a 5% CO2 incubator at 37°C
to eliminate the non-infected cells before harvesting.
Plasmids
Flag-USP22 (OHu25420; used for ectopic expression of
USP22 in A375 and A2058 cells), HA-YAP (OHu15043; customized vector
pcDNA3.1+N-HA; used for ectopic expression of YAP in A375 and A2058
cells) and Myc-Ub (OHu28056; customized vector pcDNA3.1+N-Myc; used
for ectopic expression of ubiquitin in A375 cells for the in
vivo ubiquitination assay) were purchased from GenScript.
pcDNA3.1 (cat. no. V79020; Thermo Fisher Scientific, Inc.) was used
as an empty vector (EV) as a negative control. The Flag-USP22-C185S
mutation was generated using the QuikChange XL site-directed
mutagenesis kit (cat. no. 200521; Agilent Technologies, Inc.).
shYAP1-1# (cat. no. 42540) was purchased from Addgene, Inc.
shUSP22-1# (cat. no. TRCN0000291124) and shUSP22-2# (cat. no.
TRCN0000296867) were purchased from Sigma-Aldrich; Merck KGaA. To
ectopically express pcDNA 3.1 (EV), YAP or USP22, A375 and A2058
cells were transfected with the plasmids pcDNA 3.1, HA-YAP or
Flag-USP22 (5 µg each), respectively, using Lipofectamine 3000 at
37°C for 24 h. After 36 h of transfection, cells were used for
subsequent experiments.
Western blotting and
immunoprecipitation (IP)
All cells used for western blotting were lysed with
1X RIPA buffer (cat. no. 9806; Cell Signaling Technology, Inc.)
containing 1X protease inhibitor cocktail (cat. no. 78429; Thermo
Fisher Scientific, Inc.) and incubated for 15 min with rotation at
4°C. After centrifugation at 15,871 × g for 10 min at 4°C, the
supernatant was collected and the protein concentration was
measured using the BCA method. The samples were boiled at 95°C for
10 min. In total, 25–50 µg proteins in each lane were separated
using a 10% gel for SDS-PAGE and transferred to a PVDF membrane,
followed by blocking with 5% skimmed milk in 1X TBS containing 0.1%
Tween-20 (TBST) buffer (cat. no. BUF028; Bio-Rad Laboratories,
Inc.) at room temperature for 30 min. Next, the membrane was
incubated with primary antibodies at 4°C overnight. After washing
three times with 1X TBST buffer, the membrane was incubated with
secondary antibody at room temperature for 1 h, followed by washing
three times. All the primary antibodies were used at a dilution of
1:1,000 in 5% skimmed milk, and the secondary antibodies were used
at a dilution of 1:3,000 in 5% skimmed milk. The anti-YAP (cat. no.
14074), anti-GAPDH (cat. no. 5174), anti-ubiquitin (cat. no.
43124), anti-Myc-tag (cat. no. 2272), anti-cleaved PARP (cPARP;
cat. no. 5625), anti-HA (cat. no. 3724), anti-Flag (cat. no. 14793)
and anti-rabbit secondary antibody linked with HRP (cat. no. 7074)
were purchased from Cell Signaling Technology, Inc. The anti-USP22
antibody (cat. no. sc-390585) was purchased from Santa Cruz
Biotechnology, Inc. The western blot images were developed using
the chemiluminescence detection kit (cat. no. WBKLS0500; EMD
Millipore), the ChemiDoc Imaging system (Bio-Rad Laboratories,
Inc.) and the Image Lab 6.1 software (Bio-Rad Laboratories, Inc.).
For immunoprecipitation, 1,000 µg whole cell lysates were incubated
with 2 µg primary antibodies, including anti-USP22 (cat. no.
ab195289; Abcam), anti-YAP (cat. no. 14074; Cell Signaling
Technology, Inc.) or anti-HA (cat. no. 3724; Cell Signaling
Technology, Inc.), for 6 h and then incubated with 20 µl protein A
agarose (cat. no. 20333; Thermo Fisher Scientific, Inc.) for 1 h.
The immunoprecipitants were centrifuged at 15,871 × g for 1 min at
4°C and then washed with RIPA buffer (Cell Signaling Technology,
Inc.) three times, followed by boiling at 95°C for 10 min and
finally western blot analysis performed as aforementioned.
Cycloheximide-chase assay
Cells were plated in a 60-mm cell culture dish at
60% confluence for 24 h and then treated with 100 µg/ml
cycloheximide (cat. no. S7418; Selleck Chemicals) for 0, 2, 4, 6 or
8 h before harvesting for western blot analysis performed as
aforementioned.
In vivo ubiquitination analysis
A375 cells transfected with Myc-Ub, Flag-USP22
and/or HA-YAP were treated with 10 µM MG-132 for 12 h before
harvesting with 1X cell lysis buffer (Cell Signaling Technology,
Inc.) and incubating for 15 min with rotation at 4°C. After
centrifugation at 15,871 × g for 10 min at 4°C, 1,000 µg
supernatant was incubated with 2 µg primary antibodies, including
anti-YAP (cat. no. 14074; Cell Signaling Technology, Inc.) or
anti-HA (cat. no. 3724; Cell Signaling Technology, Inc.), for 4 h,
followed by addition of 20 µl protein A agarose (cat. no. 20333;
Thermo Fisher Scientific, Inc.) for 1 h or incubation with 20 µl
anti-HA agarose (cat. no. 26181; Thermo Fisher Scientific, Inc.)
for 5 h at 4°C with rotation. The immunoprecipitants were washed
three times with 1X cell lysis buffer. The samples were boiled at
95°C for 10 min. Ubiquitinated YAP protein was detected using
anti-ubiquitin or anti-Myc-tag and western blot analysis as
aforementioned.
Reverse transcription-quantitative
(RT-qPCR)
Total RNA was extracted from A375 cells transfected
with shUSP22 and/or shYAP using the RNeasy mini kit following the
manufacturer's instructions (cat. no. 74104; Qiagen, Inc.). In
total, 1 µg RNA was used for cDNA synthesis using the iScript™
Reverse Transcription Supermix according to the manufacturer's
instructions (cat. no. 1708841; Bio-Rad Laboratories, Inc.). The
mRNA levels were examined by SYBR Green Supermix (cat. no. 1725270;
Bio-Rad Laboratories, Inc.) using the following thermocycling
conditions: 95°C for 30 sec, then 40 cycles of 95°C for 10 sec and
60°C for 30 sec. GAPDH was used as an internal control. The
relative mRNA levels were quantified using the 2−ΔΔCq
method as described previously (32). The primers were as follows:
Connective tissue growth factor (CTGF) forward,
5′-CCAATGACAACGCCTCCTG-3′ and reverse, 5′-TGGTGCAGCCAGAAAGCTC-3′;
cysteine-rich angiogenic inducer 61 (Cyr61) forward,
5′-AGCCTCGCATCCTATACAACC-3′ and reverse,
5′-TTCTTTCACAAGGCGGCACTC-3′; YAP forward,
5′-CAGGAATTATTTCGGCAGGA-3′ and reverse, 5′-CATCCTGCTCCAGTGTAGGC-3′;
and GAPDH forward, 5′-GTCTCCTCTGACTTCAACAGCG-3′ and reverse,
5′-ACCACCCTGTTGCTGTAGCCAA-3′.
Dual-luciferase reporter assays
A375 cells with depleted YAP and/or USP22 were
co-transfected with 5X UAS-luciferase reporter (cat. no. 46756),
Gal4-TEAD4 (cat. no. 24640) (both Addgene, Inc.) and pRL
Renilla luciferase control reporter (cat. no. E2261; Promega
Corporation) using Lipofectamine 3000. After 36 h of transfection,
luciferase activity was measured using the Dual-Glo Luciferase
Assay kit (cat. no. E2920; Promega Corporation) following the
manufacturer's instructions. All luciferase activities were
normalized to Renilla luciferase activity.
Cell proliferation assay
A375 cells transfected with shUSP22 and/or shYAP
(1×104 per well) were seeded in 6-well plates in
triplicates and counted manually using a light microscope
(magnification, ×20) every day for 5 days. Data are shown as mean
cell number derived from three biological replicates.
Cell viability assay
A375 and A2058 cells, either ectopically expressing
USP22 or YAP, or depleted of USP22 or YAP (2,000-3,000 per well)
were seeded in 96-well plates in triplicates for 24 h and then
treated with 1 µM vemurafenib at 37°C for 24 h. The viable cells
were determined using CellTiter-Glo luminescent cell viability
reagent according to the manufacturer's instructions (cat. no.
G7570; Promega Corporation). Briefly, the aforementioned cells were
incubated with CellTiter-Glo solution at room temperature for 10
min. The luminescent signal was detected by GloMax Microplate
Luminometer (cat. no. GM2000; Promega Corporation). Data are shown
as percentage of the control cells from three biological
replicates.
Mouse tumor xenograft assay
All animal experiments were performed under a
protocol approved by the Institutional Animal Care and Use
Committee in Changxing People's Hospital. All mice were purchased
from the Model Animal Research Center of Nanjing University
(Nanjing, China) and housed in a room with conditions of 22°C,
50–60% humidity and a 12-h light/12-h dark cycle. Water and food
were always accessible and were checked every day. A375 cells
depleted of USP22 or USP22/YAP (2×106/each site) were
resuspended in 100 µl PBS and mixed with Matrigel (cat. no. 354234;
1:1; Corning, Inc.), and were injected subcutaneously into the two
front flanks of 5-week-old immunodeficient NOD-SCID male mice with
a body weight of 20–22 g. In total, 15 mice were used, with 5 mice
in each group, including group I (shGFP; negative control), group
II (shUSP22) and group III (shUSP22+shYAP). Tumor size was measured
every 3 days and tumor volumes were calculated using the following
equation: L × W2 × 0.52 (where L is length and W is
width). At the endpoint, the mice were euthanized with
CO2 in the chamber for 5 min with a displacement rate at
40% of the chamber volume per min, followed by decapitation to
confirm death before being placed in the freezer.
Statistical analysis
The RT-PCR assays, luciferase reporter assays, cell
proliferation and cell viability assays were performed three times.
The data are shown as mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference
between groups, which was analyzed by one-way ANOVA followed by
Tukey's post hoc test for multiple comparisons or Dunnett's test
for comparisons against a single control, using GraphPad Prism 8
(GraphPad Software, Inc.). In total, 90 melanoma tissue samples
were used in the IHC staining assay and data are shown as the
relative percentage of cases containing different staining
intensities. P<0.05 was considered to indicate significant
differences, as analyzed by Fisher's exact test using GraphPad
Prism 8. For the xenograft assay, the data are shown as mean ±
standard error of the mean.
Results
Expression of USP22 and YAP is
associated in melanoma
USP22 is frequently overexpressed and associated
with poor prognosis in various human cancer types (33–36).
Notably, metastatic melanoma displays much higher expression of
USP22 compared with the primary tumor, indicating it may play an
important role in melanoma progression (37). Given that the protein levels of YAP
are also increased in most melanomas (38), the present study investigated whether
there was a possible link between USP22 and YAP. First, the protein
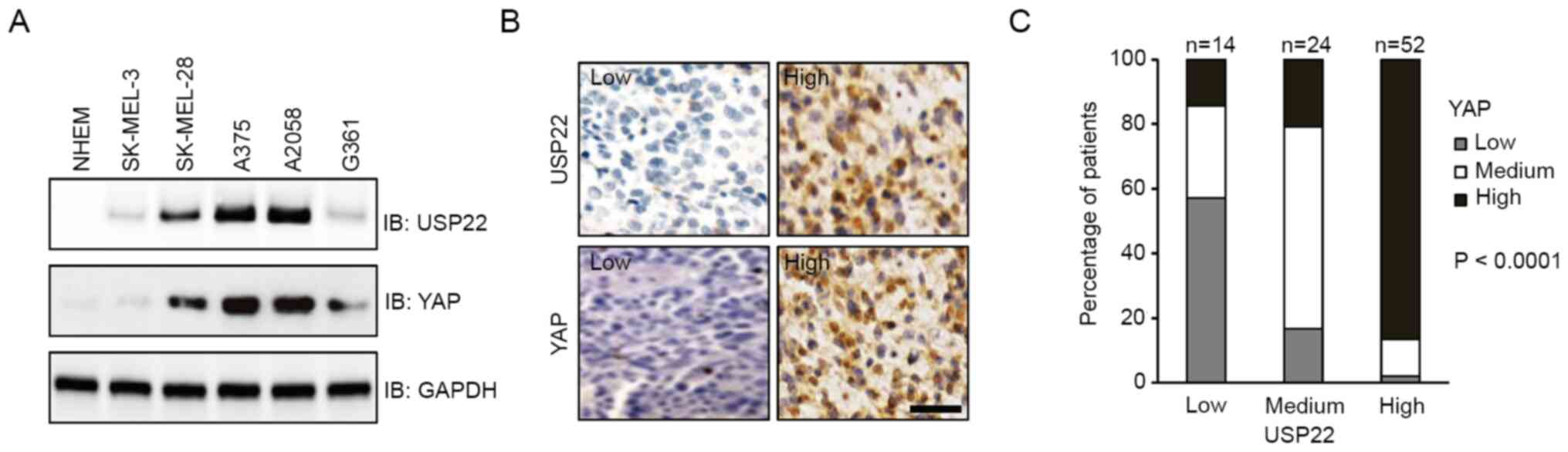
levels were analyzed in melanoma cell lines. Compared with NHEM,
all melanoma cell lines examined exhibited higher expression of
USP22 and YAP. Notably, cells with higher protein levels of USP22
also displayed higher expression of YAP (Fig. 1A). Consistent with this result, IHC
staining showed that both USP22 and YAP were highly expressed in
~50% of patient samples. Moreover, high expression of USP22 was
associated with high expression of YAP (Fig. 1B and C; P<0.0001). These results
suggested that elevated expression of USP22 and YAP coexisted in
melanoma.
USP22 interacts with and
deubiquitinates YAP
As a DUB, USP22 interacts with its substrates to
remove the ubiquitin moieties (39).
Having demonstrated crosstalk between USP22 and YAP, it was
investigated whether USP22 functioned as a YAP deubiquitinase.
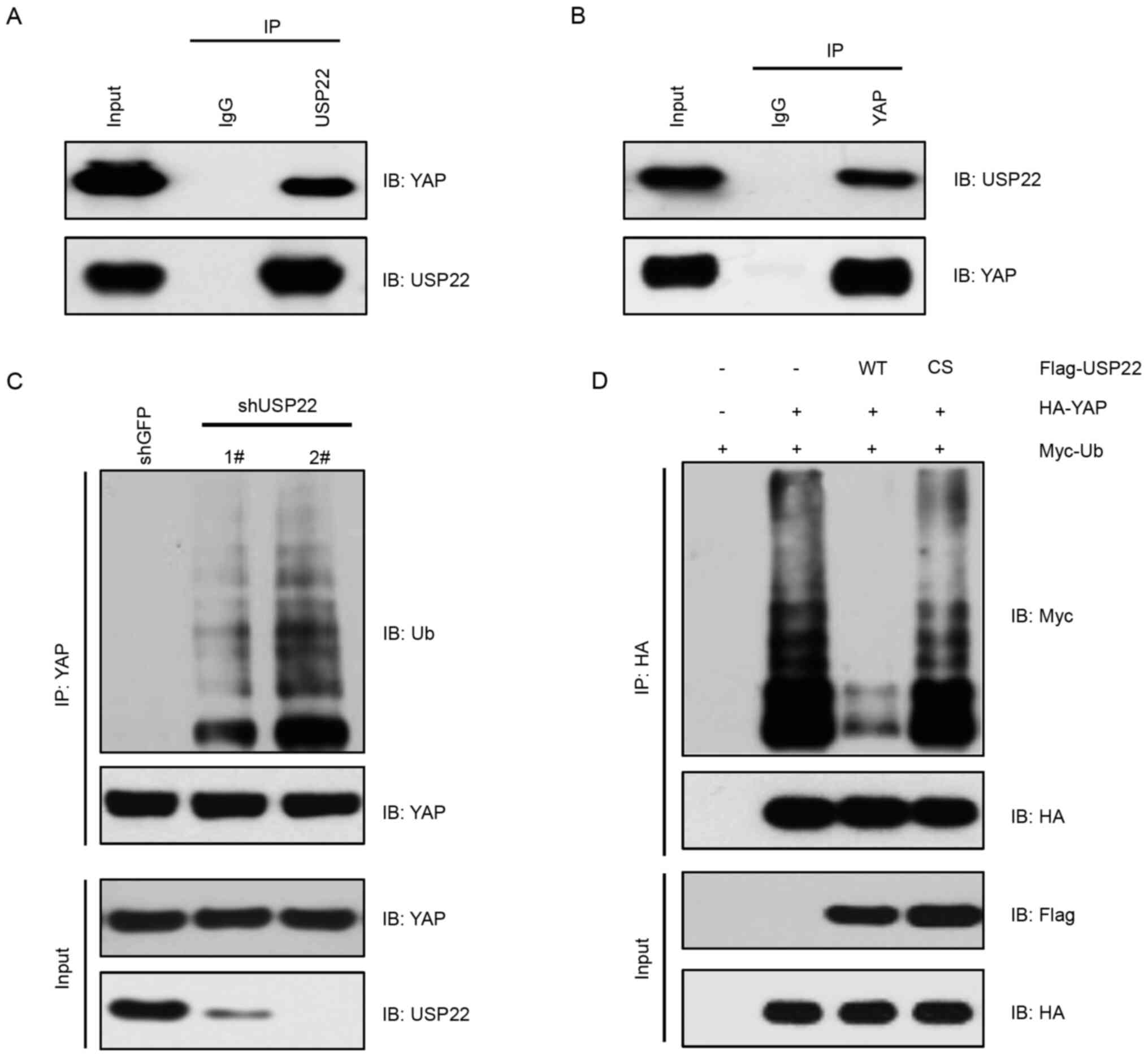
Using IP assays, USP22 interaction with YAP at the endogenous level
in A375 melanoma cells was observed (Fig. 2A and B). Consistently, knockdown of
USP22 by shRNA in A375 cells markedly increased YAP ubiquitination
(Fig. 2C). On the other hand,
overexpression of USP22-WT, but not the enzymatic-dead form
USP22-C185S, decreased YAP ubiquitination (Fig. 2D). These results suggested that USP22
was a deubiquitinase of YAP, which decreased YAP ubiquitination in
melanoma cells.
USP22 stabilizes YAP protein
For most of its substrates, USP22 increases the
protein stability by antagonizing ubiquitination-mediated protein
degradation (40). To determine the
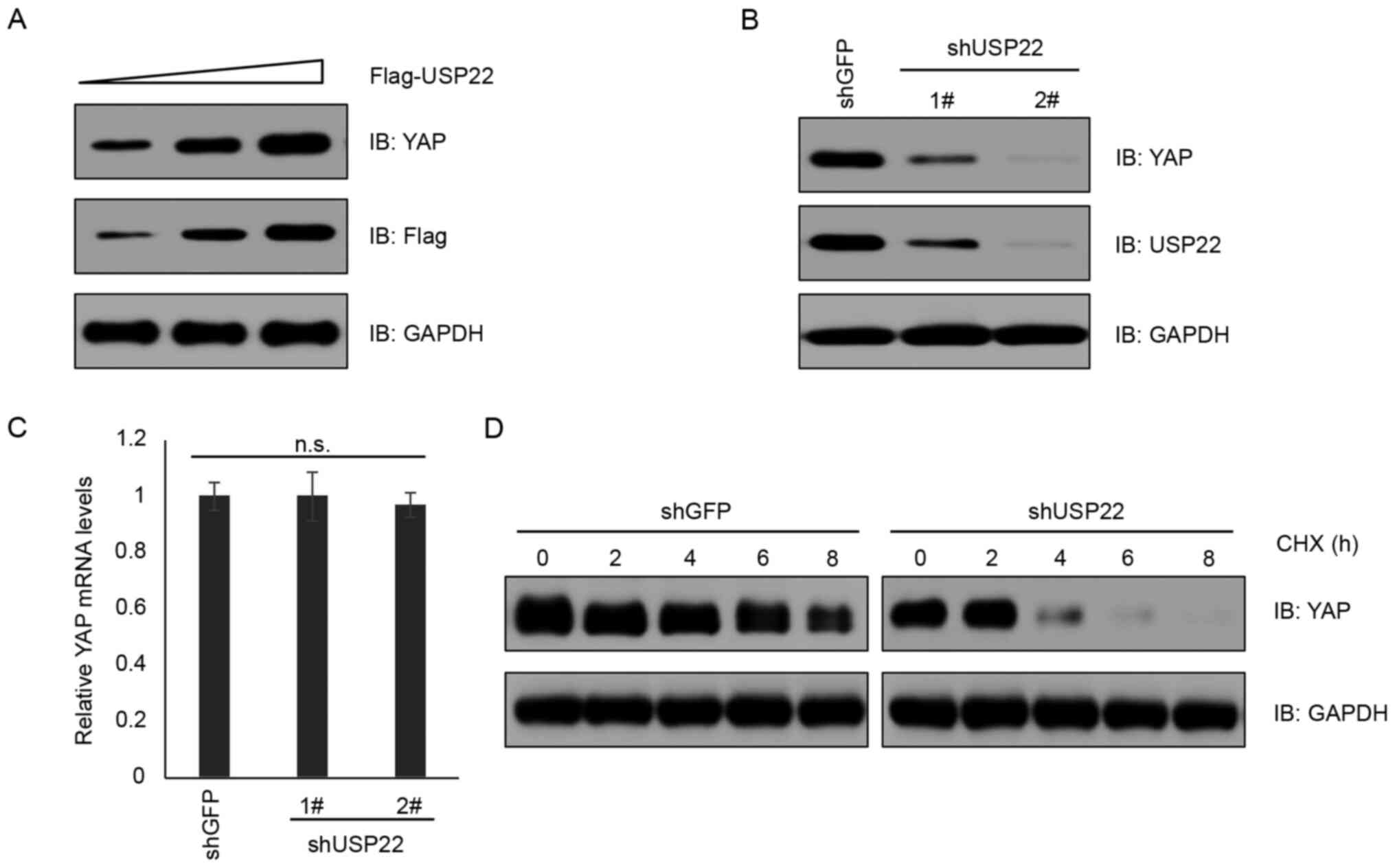
important role of USP22 in governing YAP abundance, USP22 was
overexpressed in A375 cells and it was found that YAP expression
was elevated in a USP22 dose-dependent manner (Fig. 3A). By contrast, knockdown of USP22 by
shRNA decreased YAP protein levels, but not mRNA levels (Fig. 3B and C; P>0.05). Consistently,
knockdown of USP22 shortened the half-life of YAP protein in a
cycloheximide chase assay (Fig. 3D).
These results supported the notion that USP22 functions as a
deubiquitinase to stabilize YAP protein.
USP22 promotes melanoma mainly through
YAP
As a transcriptional activator, YAP exerts its
oncogenic functions by promoting the transcription of downstream
target genes, such as CTGF and Cyr61 (41). To assess the effect of USP22 on YAP
transactivation, luciferase assays were performed using a
YAP-responsive luciferase reporter system (42), and it was found that the reporter was
significantly suppressed in USP22-knockdown cells (Fig. 4A and B; P<0.01). Moreover, the
reporter activity was similar in cells depleted with USP22 alone or
both USP22 and YAP (Fig. 4A and B;
P>0.05). Consistently, USP22-knockdown significantly suppressed
the mRNA levels of YAP downstream target genes (Fig. 4C; P<0.01), including CTGF and
Cyr61, while there was no further reduction when combined with YAP
depletion (Fig. 4C; P>0.05).
These results indicated that USP22 controlled YAP-dependent
transcription. The biological function of USP22-mediated
stabilization of YAP in melanoma was investigated. It was found
that silencing USP22 in A375 cells significantly suppressed cell
proliferation (Fig. 4D; P<0.01).
Moreover, there was no synergistic effect on cell proliferation
when both USP22 and YAP were depleted (Fig. 4D; P>0.05), suggesting that USP22
promoted cell proliferation largely through YAP. To obtain in
vivo evidence supporting this notion, mouse xenograft
experiments were performed using A375 cells. Notably, depletion of
USP22 alone displayed similar tumor inhibition effects as depletion
of both USP22 and YAP (Fig. 4E and
F). Together, these results suggested that YAP was a major
downstream effector mediating the oncogenic function of USP22 in
melanoma progression.
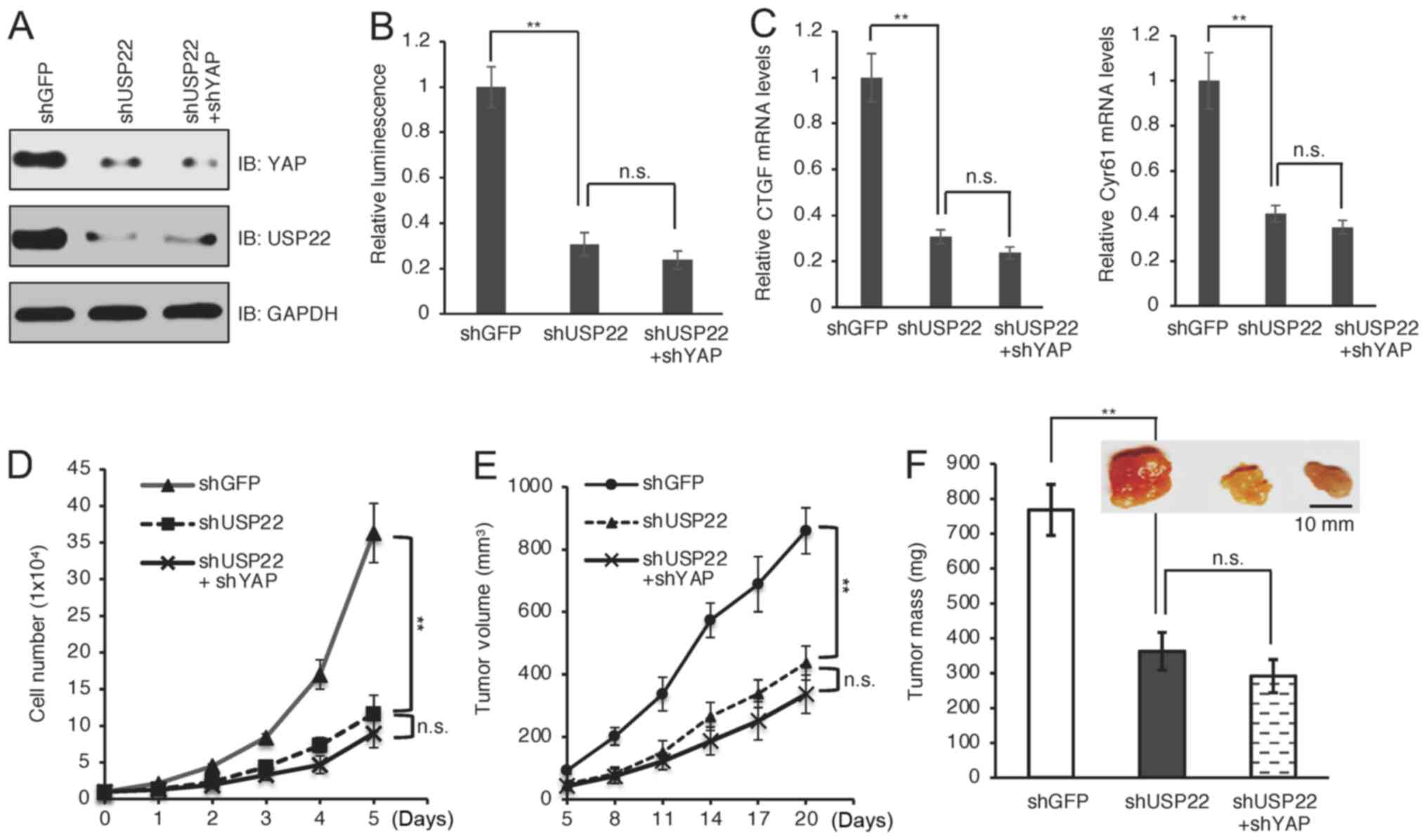 | Figure 4.Depletion of USP22 suppresses
YAP-dependent transcription, cell proliferation and tumor growth.
(A) A375 cells were infected with YAP and/or USP shRNA and
subjected to western blotting. (B) YAP- and/or USP22-depleted A375
cells were transfected with a luciferase reporter system and
subjected to luciferase activity analysis. (C) Reverse
transcription PCR analysis of mRNA levels of YAP downstream
targets, including CTGF and Cyr61 in A375 cells depleted of USP22
and/or YAP. (D) Cell proliferation of indicated cells. (E)
USP22-depleted A375 cells were used for mouse xenograft assays. (F)
Representative subcutaneous tumors were shown and statistical
analysis of tumor weight. Scale bar, 10 mm. **P<0.01 shUSP22 vs.
shGFP. YAP, yes-associated protein; USP22, ubiquitin-specific
peptidase 22; sh, short hairpin; n.s., non-significant; CTGF,
connective tissue growth factor; Cyr61, cysteine-rich angiogenic
inducer 61; IB, immunoblot. |
USP22 confers resistance to
vemurafenib in melanoma cells
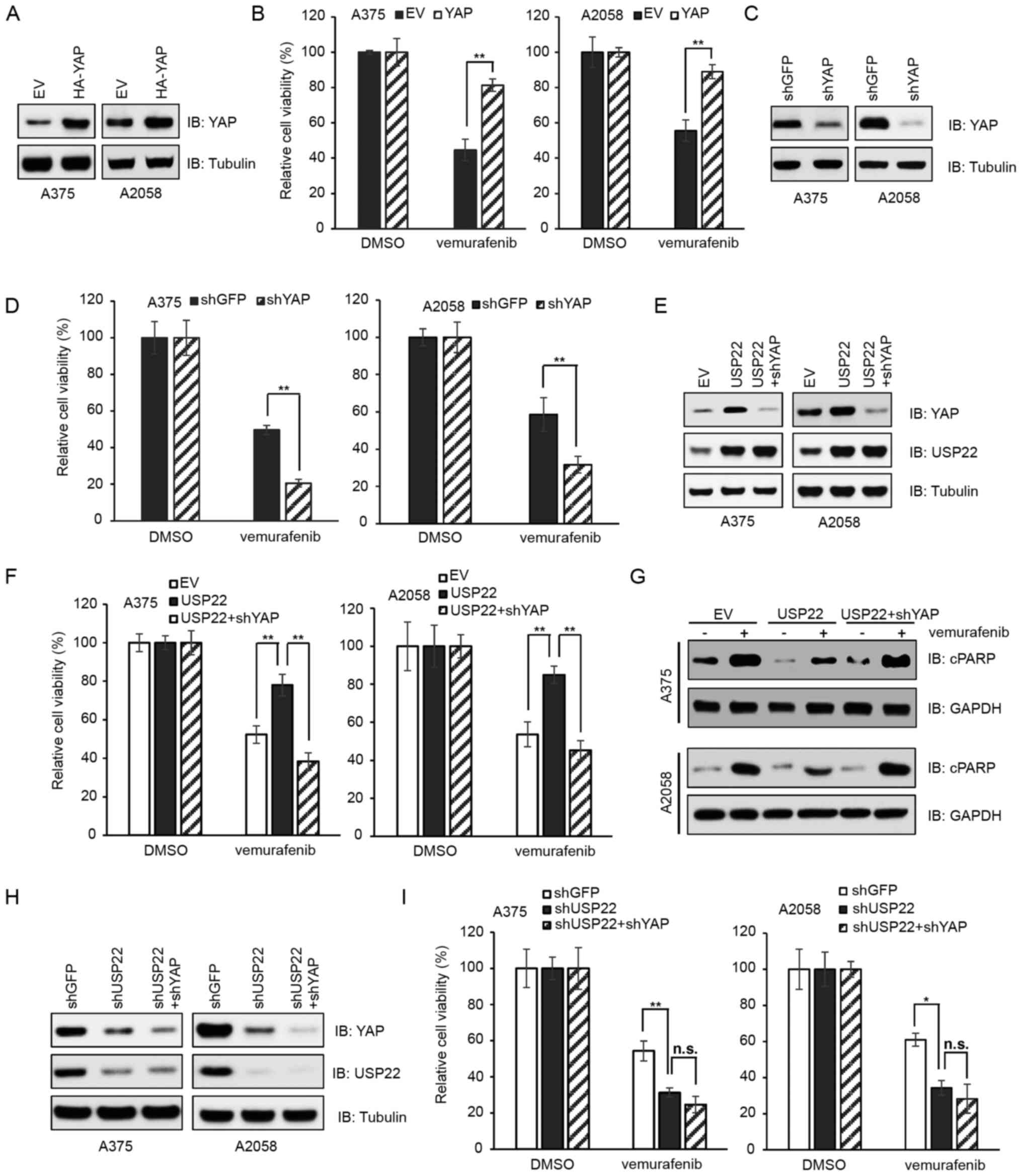
Different cell lines were generated to test the
contribution of USP22 and YAP in vemurafenib resistance. These
included cells transfected with EV (pcDNA3.1) or HA-YAP (Fig. 5A), cells infected with shGFP or shYAP
virus (Fig. 5C), cells expressing EV
(pcDNA3.1) or USP22 and/or shYAP (Fig.
5E) and cells infected with shGFP, shUSP22 or shUSP22 + shYAP
(Fig. 5H). Consistent with earlier
reports where the elevation of YAP expression contributed to
resistance to BRAF-targeted therapy in melanoma (24,25), the
present study reported that compared with ectopic expression of EV,
ectopic expression of YAP promoted cell survival following
vemurafenib treatment (Fig. 5B;
P<0.01). By contrast, depletion of YAP decreased viability when
treated with vemurafenib (Fig. 5D;
P<0.01). Notably, compared with ectopic expression of EV
(pcDNA3.1), ectopic expression of USP22 suppressed
vemurafenib-induced cell death, which could be reversed by
YAP-silencing (Fig. 5F;
P<0.01). Moreover, upon vemurafenib treatment, the protein
levels of apoptosis marker cPARP were lower in USP22-overexpressed
cells compared with EV-treated cells (Fig. 5G). Furthermore, cells depleted of
USP22 or depleted of USP22 plus YAP exhibited similar sensitivity
to vemurafenib (Fig. 5I). These data
suggested that USP22 dictated cell sensitivity to the BRAF
inhibitor vemurafenib via YAP.
Discussion
Ubiquitination can be reversed by deubiquitinating
enzymes that remove ubiquitin from targets to regulate protein
activity and stability. In humans, there are ~100 DUBs, which
belong to five sub-families: USPs (the largest subfamily), ovarian
tumor proteases, ubiquitin carboxy-terminal hydrolases,
Machado-Joseph disease protein domain proteases and JAB1/MPN/Mov34
metalloenzyme domain metalloproteases (43). As the antagonist of ubiquitination,
DUBs play important roles in numerous cellular processes, including
transcription, DNA repair, protein degradation and signaling
transduction. DUBs can be an oncogenes or tumor suppressors
depending on their substrates or context. Dysregulation of DUBs is
associated with various human diseases ranging from cancer to
neurological disorders (44,45).
USP22 is a member of the USP family, which is
evolutionarily conserved from yeast to humans; it contains a zinc
finger domain at the N-terminus that binds other three components
to form the deubiquitinating module. The catalytic domain is
located at the C-terminus (46).
USP22 was initially reported to promote deubiquitylation of
histones H2A and H2B, leading to transcription activation (47). USP22 also deubiquitinates non-histone
proteins, including telomeric repeat-binding factor 1 (TRF1),
sirtuin 1 (SIRT1), cyclin B1 and others (40), leading to protein stabilization by
preventing proteasome-mediated degradation. For example, USP22
promotes TRF1 deubiquitylation to enhance TRF1 protein stability
and maintain telomere integrity (48). In the present study, it was revealed
that USP22 specifically interacted with and deubiquitinated YAP.
Moreover, depletion of USP22 decreased YAP protein expression. As a
result, USP22-knockdown significantly suppressed melanoma cell
proliferation and tumor growth in a xenograft mouse model, which
was similar to YAP-knockdown. Therefore, the current findings
indicated that YAP may be a critical downstream mediator of USP22
in tumorigenesis. However, the substrate and ubiquitin chain
specificity for USP22 is unknown, which will be an interesting
research topic in the future. The present identification of YAP as
its novel substrate might provide insight into this puzzle.
USP22 is largely considered as an oncoprotein, which
is frequently overexpressed in several types of cancer, including
lung cancer, pancreatic cancer, salivary adenoid cystic carcinoma,
prostate cancer and liver cancer, and associated with poor overall
survival (49–52). It was reported that USP22 promoted
lung cancer by regulating pathways of ubiquitination and
immunosuppression (33). USP22 also
deubiquitinated and stabilized CDC274 (PD-L1) to decrease the
efficacy of CD274-targeted immunotherapy in mice (53). Furthermore, USP22 facilitated
prostate cancer progression through enhancing androgen receptor-
and Myc-driven oncogenic signaling pathways (34). Similarly, overexpression of YAP was
associated with poor patient survival in numerous types of cancer,
including colon cancer, lymph node metastatic melanoma and
pancreatic cancer (54–56). Moreover, the Human Protein Atlas
project also showed that higher expression of USP22 and YAP is
associated with decreased survival in patients with melanoma
(57). The present study also
observed that USP22 was highly expressed in patients with melanoma
via unknown molecular mechanisms. Moreover, higher USP22 expression
was positively associated with higher YAP expression in melanoma
cell lines and patient samples. The current findings provide a
possible molecular mechanism for aberrant YAP expression in
melanoma. USP22 has been reported to be regulated at both
transcriptional and post-translational levels. For example, SP1 and
protein kinase A/cAMP response element-binding protein could bind
to the USP22 promoter to suppress or promote USP22 transcription,
respectively (58,59). Phosphorylation of T147 and S237 by
cyclin-dependent kinase 1 or acetylation of K129 leads to enhanced
USP22 deubiquitinase activity (60,61).
Moreover, anaphase-promoting complex cell division cycle protein 20
(APCCDC20), an E3 ubiquitin ligase, promotes USP22
degradation in a cell cycle-dependent manner (61). It will be interesting to determine
whether APCCDC20 or other mechanisms contribute to USP22
and YAP overexpression in melanoma.
Another notable finding in the current study was
that overexpression of USP22 resulted in resistance to the BRAF
inhibitor vemurafenib. One of the milestones in the melanoma
research field was the discovery of the BRAF-V600E mutation,
leading to the rapid development of targeted therapies to improve
overall survival rate for patients with melanoma (14,62).
However, acquired resistance to these targeted therapies also
emerged due to different mechanisms. For example, additional
genetic alteration was considered the main mechanism, which led to
the recovery of the MAPK pathway and activation of PI3K/Akt/mTOR
signaling (63). Notably, in a
genetic screen that silenced >5,000 targets, YAP stood out to be
a critical determinant of BRAF inhibitor resistance. Deletion of
YAP resulted in the best response to vemurafenib treatment in
various types of cancer cells harboring the BRAF-V600E mutation
(25). Consistently, the present
study determined that USP22 is a key regulator of YAP expression
and BRAF inhibitor resistance in melanoma.
Overall, the current study provided evidence showing
that USP22 functions as the deubiquitinase to govern YAP
ubiquitination and protein stability. Overexpression of USP22 lead
to elevation of YAP protein levels and subsequent resistance to
vemurafenib in melanoma. Therefore, the present study provided a
molecular mechanism and rationale for combined inhibition of
USP22/YAP and BRAF as an option for melanoma treatment. However, a
limitation of the current study was that most experiments were
performed using melanoma cells. Further studies using animal
models, such as USP22-knockout mice, are required to demonstrate
the critical role of the USP22/YAP axis in melanoma and BRAF
inhibitor resistance.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YW and ZJ performed the experiments. YW and JL
analyzed the data, confirmed its authenticity and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Research Ethics Committee at Changxing People's Hospital (Huzhou,
China). Written informed consent was obtained from patients and/or
their guardians.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shain AH and Bastian BC: From melanocytes
to melanomas. Nat Rev Cancer. 16:345–358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davis LE, Shalin SC and Tackett AJ:
Current state of melanoma diagnosis and treatment. Cancer Biol
Ther. 20:1366–1379. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rodriguez-Cerdeira C, Carnero Gregorio M,
Lopez-Barcenas A, Sánchez-Blanco E, Sánchez-Blanco B, Fabbrocini G,
Bardhi B, Sinani A and Guzman RA: Advances in immunotherapy for
melanoma: A comprehensive review. Mediators Inflamm.
2017:32642172017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Domingues B, Lopes JM, Soares P and Populo
H: Melanoma treatment in review. Immunotargets Ther. 7:35–49. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weiss SA, Wolchok JD and Sznol M:
Immunotherapy of melanoma: Facts and hopes. Clin Cancer Res.
25:5191–5201. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhandaru M and Rotte A: Monoclonal
antibodies for the treatment of melanoma: Present and future
strategies. Methods Mol Biol. 1904:83–108. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kozar I, Margue C, Rothengatter S, Haan C
and Kreis S: Many ways to resistance: How melanoma cells evade
targeted therapies. Biochim Biophys Acta Rev Cancer. 1871:313–322.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lugowska I, Teterycz P and Rutkowski P:
Immunotherapy of melanoma. Contemp Oncol (Pozn). 22:61–67.
2018.PubMed/NCBI
|
|
10
|
Paluncic J, Kovacevic Z, Jansson PJ,
Kalinowski D, Merlot AM, Huang ML, Lok HC, Sahni S, Lane DJ and
Richardson DR: Roads to melanoma: Key pathways and emerging players
in melanoma progression and oncogenic signaling. Biochim Biophys
Acta. 1863:770–784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Haluska FG, Tsao H, Wu H, Haluska FS,
Lazar A and Goel V: Genetic alterations in signaling pathways in
melanoma. Clin Cancer Res. 12:2301s–2307s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y and Chen Z: Mutation detection and
molecular targeted tumor therapies. STEMedicine. 1:e112020.
View Article : Google Scholar
|
|
13
|
Mehnert JM and Kluger HM: Driver mutations
in melanoma: Lessons learned from bench-to-bedside studies. Curr
Oncol Rep. 14:449–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Savoia P, Fava P, Casoni F and Cremona O:
Targeting the ERK signaling pathway in melanoma. Int J Mol Sci.
20:14832019. View Article : Google Scholar
|
|
15
|
Fujimura T, Fujisawa Y, Kambayashi Y and
Aiba S: Significance of BRAF kinase inhibitors for melanoma
treatment: From bench to bedside. Cancers (Basel). 11:13422019.
View Article : Google Scholar
|
|
16
|
Luebker SA and Koepsell SA: Diverse
mechanisms of BRAF inhibitor resistance in melanoma identified in
clinical and preclinical studies. Front Oncol. 9:2682019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johnson DB, Menzies AM, Zimmer L, Eroglu
Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, et al:
Acquired BRAF inhibitor resistance: A multicenter meta-analysis of
the spectrum and frequencies, clinical behaviour, and phenotypic
associations of resistance mechanisms. Eur J Cancer. 51:2792–2799.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma S, Meng Z, Chen R and Guan KL: The
hippo pathway: Biology and pathophysiology. Annu Rev Biochem.
88:577–604. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim MK, Jang JW and Bae SC: DNA binding
partners of YAP/TAZ. BMB Rep. 51:126–133. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moroishi T, Hansen CG and Guan KL: The
emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 15:73–79.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zanconato F, Cordenonsi M and Piccolo S:
YAP/TAZ at the roots of cancer. Cancer Cell. 29:783–803. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiong H, Yu Q, Gong Y, Chen W, Tong Y,
Wang Y, Xu H and Shi Y: Yes-Associated protein (YAP) promotes
tumorigenesis in melanoma cells through stimulation of low-density
lipoprotein receptor-related protein 1 (LRP1). Sci Rep.
7:155282017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nallet-Staub F, Marsaud V, Li L, Gilbert
C, Dodier S, Bataille V, Sudol M, Herlyn M and Mauviel A:
Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in
cutaneous melanoma. J Invest Dermatol. 134:123–132. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fisher ML, Grun D, Adhikary G, Xu W and
Eckert RL: Inhibition of YAP function overcomes BRAF inhibitor
resistance in melanoma cancer stem cells. Oncotarget.
8:110257–110272. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin L, Sabnis AJ, Chan E, Olivas V, Cade
L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, et al: The Hippo
effector YAP promotes resistance to RAF- and MEK-targeted cancer
therapies. Nat Genet. 47:250–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meng Z, Moroishi T and Guan KL: Mechanisms
of Hippo pathway regulation. Genes Dev. 30:1–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan F, Qian M, He Q, Zhu H and Yang B: The
posttranslational modifications of Hippo-YAP pathway in cancer.
Biochim Biophys Acta Gen Subj. 1864:1293972020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao B, Li L, Tumaneng K, Wang CY and Guan
KL: A coordinated phosphorylation by Lats and CK1 regulates YAP
stability through SCF (beta-TRCP). Genes Dev. 24:72–85. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C,
Yao Y and Liu Q: Fbxw7 is an independent prognostic marker and
induces apoptosis and growth arrest by regulating YAP abundance in
hepatocellular carcinoma. Mol Cancer. 13:1102014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao F, Zhou Z, Kim J, Hang Q, Xiao Z, Ton
BN, Chang L, Liu N, Zeng L, Wang W, et al: SKP2- and
OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP
nuclear localization and activity. Nat Commun. 9:22692018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tiscornia G, Singer O and Verma IM:
Production and purification of lentiviral vectors. Nat Protoc.
1:241–245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han B, Sun Y, Yang D, Zhang H, Mo S, Chen
X, Lu H, Mao X and Hu J: USP22 promotes development of lung
adenocarcinoma through ubiquitination and immunosuppression. Aging
(Albany NY). 12:6990–7005. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schrecengost RS, Dean JL, Goodwin JF,
Schiewer MJ, Urban MW, Stanek TJ, Sussman RT, Hicks JL, Birbe RC,
Draganova-Tacheva RA, et al: USP22 regulates oncogenic signaling
pathways to drive lethal cancer progression. Cancer Res.
74:272–286. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang X, Zang H, Luo Y, Wu J, Fang Z, Zhu W
and Li Y: High expression of USP22 predicts poor prognosis and
advanced clinicopathological features in solid tumors: A
meta-analysis. Onco Targets Ther. 11:3035–3046. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCann JJ, Vasilevskaya IA, Poudel Neupane
N, Shafi AA, McNair C, Dylgjeri E, Mandigo AC, Schiewer MJ,
Schrecengost RS, Gallagher P, et al: USP22 functions as an
oncogenic driver in prostate cancer by regulating cell
proliferation and DNA repair. Cancer Res. 80:430–443. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Luise C, Capra M, Donzelli M, Mazzarol G,
Jodice MG, Nuciforo P, Viale G, Di Fiore PP and Confalonieri S: An
atlas of altered expression of deubiquitinating enzymes in human
cancer. PLoS One. 6:e158912011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang X, Tang JZ, Vergara IA, Zhang Y,
Szeto P, Yang L, Mintoff C, Colebatch A, McIntosh L, Mitchell KA,
et al: Somatic hypermutation of the YAP oncogene in a human
cutaneous melanoma. Mol Cancer Res. 17:1435–1449. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Komander D, Clague MJ and Urbe S: Breaking
the chains: Structure and function of the deubiquitinases. Nat Rev
Mol Cell Biol. 10:550–563. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Melo-Cardenas J, Zhang Y, Zhang DD and
Fang D: Ubiquitin-specific peptidase 22 functions and its
involvement in disease. Oncotarget. 7:44848–44856. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu FX, Zhao B, Panupinthu N, Jewell JL,
Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al:
Regulation of the Hippo-YAP pathway by G-protein-coupled receptor
signaling. Cell. 150:780–791. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim
J, Xie J, Ikenoue T, Yu J, Li L, et al: Inactivation of YAP
oncoprotein by the Hippo pathway is involved in cell contact
inhibition and tissue growth control. Genes Dev. 21:2747–2761.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nijman SM, Luna-Vargas MP, Velds A,
Brummelkamp TR, Dirac AM, Sixma TK and Bernards R: A genomic and
functional inventory of deubiquitinating enzymes. Cell.
123:773–786. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Reyes-Turcu FE, Ventii KH and Wilkinson
KD: Regulation and cellular roles of ubiquitin-specific
deubiquitinating enzymes. Annu Rev Biochem. 78:363–397. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu L, Yin S, Brobbey C and Gan W:
Ubiquitination in cancer stem cell: Roles and targeted cancer
therapy. STEMedicine. 1:e372020. View Article : Google Scholar
|
|
46
|
Samara NL, Datta AB, Berndsen CE, Zhang X,
Yao T, Cohen RE and Wolberger C: Structural insights into the
assembly and function of the SAGA deubiquitinating module. Science.
328:1025–1029. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhao Y, Lang G, Ito S, Bonnet J, Metzger
E, Sawatsubashi S, Suzuki E, Le Guezennec X, Stunnenberg HG,
Krasnov A, et al: A TFTC/STAGA module mediates histone H2A and H2B
deubiquitination, coactivates nuclear receptors, and counteracts
heterochromatin silencing. Mol Cell. 29:92–101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Atanassov BS, Evrard YA, Multani AS, Zhang
Z, Tora L, Devys D, Chang S and Dent SY: Gcn5 and SAGA regulate
shelterin protein turnover and telomere maintenance. Mol Cell.
35:352–364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ning J, Zhang J, Liu W, Lang Y, Xue Y and
Xu S: Overexpression of ubiquitin-specific protease 22 predicts
poor survival in patients with early-stage non-small cell lung
cancer. Eur J Histochem. 56:e462012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tang B, Liang X, Tang F, Zhang J, Zeng S,
Jin S, Zhou L, Kudo Y and Qi G: Expression of USP22 and Survivin is
an indicator of malignant behavior in hepatocellular carcinoma. Int
J Oncol. 47:2208–2216. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ning Z, Wang A, Liang J, Xie Y, Liu J,
Feng L, Yan Q and Wang Z: USP22 promotes the G1/S phase transition
by upregulating FoxM1 expression via beta-catenin nuclear
localization and is associated with poor prognosis in stage II
pancreatic ductal adenocarcinoma. Int J Oncol. 45:1594–1608. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dai W, Yao Y, Zhou Q and Sun CF:
Ubiquitin-specific peptidase 22, a histone deubiquitinating enzyme,
is a novel poor prognostic factor for salivary adenoid cystic
carcinoma. PLoS One. 9:e871482014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Huang X, Zhang Q, Lou Y, Wang J, Zhao X,
Wang L, Zhang X, Li S, Zhao Y, Chen Q, et al: USP22 Deubiquitinates
CD274 to suppress anticancer immunity. Cancer Immunol Res.
7:1580–1590. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang L, Shi S, Guo Z, Zhang X, Han S, Yang
A, Wen W and Zhu Q: Overexpression of YAP and TAZ is an independent
predictor of prognosis in colorectal cancer and related to the
proliferation and metastasis of colon cancer cells. PLoS One.
8:e655392013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee CK, Jeong SH, Jang C, Bae H, Kim YH,
Park I, Kim SK and Koh GY: Tumor metastasis to lymph nodes requires
YAP-dependent metabolic adaptation. Science. 363:644–649. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rozengurt E, Sinnett-Smith J and Eibl G:
Yes-associated protein (YAP) in pancreatic cancer: At the epicenter
of a targetable signaling network associated with patient survival.
Signal Transduct Target Ther. 3:112018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Uhlen M, Fagerberg L, Hallstrom BM,
Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C,
Sjöstedt E, Asplund A, et al: Proteomics. Tissue-based map of the
human proteome. Science. 347:12604192015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xiong J, Che X, Li X, Yu H, Gong Z and Li
W: Cloning and characterization of the human USP22 gene promoter.
PLoS One. 7:e527162012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xiong J, Zhou X, Gong Z, Wang T, Zhang C,
Xu X, Liu J and Li W: PKA/CREB regulates the constitutive promoter
activity of the USP22 gene. Oncol Rep. 33:1505–1511. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Armour SM, Bennett EJ, Braun CR, Zhang XY,
McMahon SB, Gygi SP, Harper JW and Sinclair DA: A high-confidence
interaction map identifies SIRT1 as a mediator of acetylation of
USP22 and the SAGA coactivator complex. Mol Cell Biol.
33:1487–1502. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lin Z, Tan C, Qiu Q, Kong S, Yang H, Zhao
F, Liu Z, Li J, Kong Q, Gao B, et al: Ubiquitin-specific protease
22 is a deubiquitinase of CCNB1. Cell Discov. 1:150282015.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Manzano JL, Layos L, Buges C, de Los
Llanos Gil M, Vila L, Martínez-Balibrea E and Martínez-Cardús A:
Resistant mechanisms to BRAF inhibitors in melanoma. Ann Transl
Med. 4:2372016. View Article : Google Scholar : PubMed/NCBI
|