Introduction
Acute myeloid leukemia (AML) is a term used to
describe a group of genetically highly heterogeneous malignant
clonal diseases characterized by abnormal differentiation and
proliferation of immature myeloid protocells in the bone marrow.
With the continuous optimization of chemotherapy, hematopoietic
stem cell transplantation and supportive treatment, the prognosis
of AML has improved; however, the 5-year overall survival rate
remains low at 15-30% (1). Drug
resistance of leukemic cells is the main cause of relapsed and
refractory AML; thus, it is necessary to identify
resistance-related therapeutic targets (2).
AXL receptor tyrosine kinase (AXL), a member of the
receptor tyrosine kinase Tyro3, AXL and Mertk (TAM) family, is
activated by growth arrest-specific factor 6 (GAS6). GAS6 binding
leads to AXL dimerization, autophosphorylation and activation of
subsequent signaling pathways, such as the PI3K/AKT, MAPK, STAT and
NF-κB cascades (3). Upregulation and
activation of AXL have been demonstrated to promote cell
proliferation, chemotherapy resistance, invasion and metastasis in
several types of human cancer, thus becoming a therapeutic target
worthy of exploration (4).
Ben-Batalla et al (5)
reported that AXL-mRNA is expressed in 57% (64/112) of
newly-diagnosed normal karyotype genetic medium-risk AML cases and
is an independent adverse prognostic factor. Hong et al
(6) revealed that AXL-mRNA
expression is upregulated in relapse-resistant AML cases and
mediates resistance to a variety of chemotherapy drugs in U937
cells. Park et al (7)
demonstrated that AXL is constitutively activated in blast cells
from patients with AML and FMS-like tyrosine kinase 3
(FLT3)-internal tandem duplication (ITD)+ AML cells, and
the levels of total AXL and phosphorylated (p-)AXL protein are
markedly increased following treatment with FLT3 inhibitor
midostaurin (PKC412) or quizartinib (AC220) (8). These studies suggested that AXL is
associated with drug resistance of leukemic cells and may be used
as a therapeutic target for AML.
AXL-targeted therapies mainly include small-molecule
inhibitors, ligand decoy antibodies (9,10) and
monoclonal antibodies (11,12). BGB324 (R428) is the first selective
AXL small-molecule inhibitor to enter clinical research, and was
found to effectively inhibit the phosphorylation of AXL in AML
cells and AML blast cells, induce cell apoptosis, and increase
sensitivity of AML cells to doxorubicin and cytarabine (also known
as Ara-c) (5). A multicenter phase
Ib/II clinical study of BGB324 as a single agent or in combination
with cytarabine/decitabine for the treatment of high-risk
myelodysplastic syndromes and relapsed/refractory leukemia is under
way (NCT02488408). DAXL-88 is a novel human antibody targeting AXL,
which was constructed by Duan et al (13) by analyzing the spatial pattern of the
AXL-GAS6 interaction and panning through the entire human natural
phage antibody library. DAXL-88 blocks the interaction of AXL-GAS6
by binding to human and mouse AXL protein with a high affinity,
inhibits the migration and invasion of human ovarian cancer SKOV3
cells and non-small cell lung cancer A549 cells induced by GAS6,
and reverses the upregulation of p-AXL, p-AKT and p-ERK activated
by GAS6 (14). However, DAXL-88 has
no cytotoxic effect on these tumor cells. Duan et al
(13) further modified DAXL-88 by
conjugating it to monomethyl auristatin E (MMAE), a small molecule
microtubule interferant, to form an antibody-drug conjugate termed
DAXL-88-MMAE. After DAXL-88-MMAE binds to AXL, the antibody is
internalized, and MMAE is released by lysosomal protease cleavage,
which prevents microtubulin polymerization, causes cell cycle
arrest and induces apoptosis.
The present study aimed to solve the problem of drug
resistance in the clinical treatment of AML, and proposed AXL as a
therapeutic target. By comparing AXL antigen expression among
drug-sensitive and drug-resistant human AML cell lines, and AML
blast cells from patients with different clinical characteristics,
FLT3-mutant AML with higher AXL antigen expression was selected for
AXL-targeted therapy. Furthermore, in AML cell lines and blast
cells, the cytotoxic effects of DAXL-88, DAXL-88-MMAE and R428, and
their molecular mechanisms, were thoroughly explored.
Materials and methods
Cell culture, resistant cell induction
and reagents
The human AML U937 (cat. no. TCHu-159), THP-1 (cat.
no. TCHu-57) and MV4-11 (cat. no. SCSP-5031) cell lines were
obtained from The Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences, where they were characterized by
mycoplasma detection, DNA fingerprinting, isozyme detection and
cell vitality detection. The Adriamycin (ADM)-resistant K562 cell
line (K562/ADM) was kindly provided by Dr Ming Xiong, Central
Laboratory of the People's Liberation Army Navy General Hospital
(Beijing, China). Cell lines were cultured in RPMI-1640 medium
(cat. no. SH30809.01; HyClone; Cytiva) supplemented with 10% FBS
(cat. no. 1997802C; Gibco; Thermo Fisher Scientific, Inc.) and 100
U/ml penicillin-streptomycin at 37°C with 5% CO2. The
AC220-resistant MV4-11 cell line (MV4-11/AC220) was generated by
continuous incubation of the AC220-sensitive MV4-11 cell line with
increasing doses of AC220. The MV4-11 cell line was started in
culture in the presence of 0.1 nM AC220 for ~3 weeks. Between days
22 and 79, the concentration of AC220 was gradually increased from
0.1 to 1 nM. Between days 79 and 96, the concentration of AC220 was
gradually increased from 1 to 5 nM. The cytarabine-resistant U937
cell line (U937/Ara-c) was generated by short-range shock induction
of the cytarabine-sensitive U937 cell line with increasing doses of
cytarabine (cat. no. H20160403; Actavis Italy SpA). The culture
started in the presence of 0.8 µM cytarabine for 24 h.
Subsequently, cytarabine was removed by centrifugation at 45 × g
for 5 min at room temperature, and shock was repeated at double the
dose after cell activity recovery up to a maximum induced dose of
300 µM. DAXL-88, DAXL-88-MMAE and IgG1-MMAE were produced by Dr
Yanting Duan. PKC412 (cat. no. S8064), AC220 (cat. no. S1526), R428
(cat. no. S2841) and MMAE (cat. no. S7721) were purchased from
Selleck Chemicals.
Samples from patients with AML and
clinical data
Cryopreserved leukemic blast cells were obtained
from 57 patients with AML between May 2018 and January 2020,
including 31 males and 26 females, with a mean age ± SD of 50±2.1
years (range, 16–88 years), who provided written consent with the
approval of the Sixth Medical Center of PLA General Hospital Ethics
Committee (research ethics no. HZKY-YJ-2020-1; Beijing, China).
There were a total of 64 blast cell samples, including single
samples from 53 patients, pre- and post-chemotherapy samples from 3
patients, and 5 dynamic samples from 1 patient. After thawing, AML
blast cells were maintained in RPMI-1640 medium containing 20% FBS
(8). The clinical data of patients
with AML included in the present study are summarized in Table I.
 | Table I.AXL receptor tyrosine kinase antigen
expression in 64 AML blast cell samples from 57 patients with
different clinical characteristics. |
Table I.
AXL receptor tyrosine kinase antigen
expression in 64 AML blast cell samples from 57 patients with
different clinical characteristics.
| Patient
characteristics | AML blast cells
(n) | AXL antigen
expression (%), median (range) |
P-valuea |
|---|
| Disease status |
|
| 0.637 |
| De
novo | 29 | 1.98
(0.30–17.76) |
|
|
Relapsed/refractory | 35 | 1.79
(0.27–54.16) |
|
| FLT3-ITD/TKD |
|
| 0.001 |
|
Positive | 26 | 2.70
(1.03–54.16) |
|
|
Negative | 38 | 1.51
(0.27–4.92) |
|
|
FLT3-ITD/TKD-positive |
|
| 0.330 |
| De
novo | 11 | 3.56
(1.18–17.76) |
|
|
Relapsed/refractory | 15 | 2.03
(1.13–54.16) |
|
| ELN2017 genetic
risk stratification |
|
| 0.923b |
|
Favorable | 7 | 2.23
(0.30–4.97) |
|
|
Intermediate | 19 | 1.76
(0.44–6.79) |
|
|
Adverse | 35 | 1.93
(0.27–54.16) |
|
|
Unknown | 3 | 1.56
(1.45–2.15) |
|
Flow cytometry analysis of AXL antigen
expression
AML cells (1×106) were incubated with 2
µl PE-conjugated anti-human AXL antibody (cat. no. FAB154P; R&D
Systems, Inc.). AML blast cells (1×106) were incubated
with 2 µl PE-conjugated anti-human AXL and 3 µl PerCP-conjugated
anti-human CD45 (cat. no. Z6410013; Beijing Quantobio Biotechnology
Co., Ltd.) in PBS for 60 min at room temperature. Cells were washed
after staining and analyzed using a FACSCalibur flow cytometer (BD
Diagnostics; Becton, Dickinson and Company) (15).
Cell Counting Kit-8 (CCK-8) assay of
cytotoxicity and synergistic cytotoxicity
AML cells (2×104) and AML blast cells
(2×105) were seeded in a 96-well tissue culture plates
and exposed to different drug concentrations for 45 or 44 h at 37°C
in a 5% CO2 atmosphere incubator. A total of 10 µl CCK-8
solution (cat. no. CK04; Dojindo Molecular Technologies, Inc.) was
added to each well, followed by incubation for an additional 3 or 4
h. Subsequently, the absorbance was read at 450 nm using a
Multiskan Mk3 microplate reader (16).
Flow cytometry analysis of
apoptosis
Detection of apoptosis was performed using the
Annexin V-FITC Apoptosis Detection Kit (cat. no. 130092052;
Miltenyi Biotec GmbH). Following incubation of the cells with 10 µl
FITC-conjugated Annexin V in the binding buffer for 15 min at room
temperature, cells were washed, incubated with 5 µl PI and analyzed
using a FACSCantoII flow cytometer (BD Diagnostics; Becton,
Dickinson and Company) (15).
Western blot analysis
The anti-p-AXL antibody (Y799; cat. no. AF2228-SP)
was obtained from BD Biosciences. Anti-AXL (cat. no. 8661),
anti-p-FLT3 (cat. no. 3464), anti-FLT3 (cat. no. 3462), anti-AKT
(cat. no. 4691), anti-p-AKT (cat. no. 4060), anti-ERK (cat. no.
4695) and anti-p-ERK (cat. no. 4695) antibodies were obtained from
Cell Signaling Technology, Inc. MV4-11, MV4-11/AC220 and
FLT3-ITD+ AML blast cells were collected and lysed in
ice cold RIPA buffer (Beijing Solarbio Science & Technology
Co., Ltd.) supplemented with protease inhibitor cocktail (Roche
Diagnostics) for 30 min. Protein concentrations were quantified
using a BCA kit (Applygen Technologies, Inc.). Proteins (25
µg/lane) were separated via 8% SDS-PAGE, transferred onto a
nitrocellulose filter membrane (EMD Millipore) and blocked with 5%
TBST skim milk for 1 h at room temperature. The nitrocellulose
membrane was first incubated with primary antibodies against the
aforementioned proteins at a dilution of 1:1,000 (except for
anti-p-AXL which was used at a dilution of 1:200), overnight at
4°C, and then incubated with anti-rabbit secondary antibody
conjugated to horseradish peroxidase (dilution, 1:5,000; cat. no.
AS014; ABclonal Biotech Co., Ltd.) for 1 h at room temperature. An
ImageQuant LAS4000 chemiluminescent imaging analyzer (GE
Healthcare) was used for signal detection. GAPDH (cat. no. AC027;
ABclonal Biotech Co., Ltd.) was used as a reference (13).
Statistical analysis
Differences between mean values of two groups were
evaluated using an independent samples t-test, and that of multiple
groups were evaluated using one-way analysis of variance followed
by Dunnett's or Tukey's post hoc test. Differences between median
values of two groups were evaluated using the Mann-Whitney U test,
and that of multiple groups were evaluated using the Kruskal-Wallis
test. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were performed
using SPSS version 17.0 (SPSS, Inc.), GraphPad Prism version 5.01
(GraphPad Software, Inc.), CalcuSyn version 2.1 (Biosoft) and
ImageJ 1.51j8 (National Institutes of Health).
Results
AXL antigen expression is upregulated
in drug-resistant AML cell lines and FLT3-ITD/tyrosine kinase
domain mutation-positive (TKD)+ AML blast cells
AXL antigen expression in drug-resistant K562/ADM,
U937/Ara-c and MV4-11/AC220 cells was 43.31±1.78, 6.26±0.18 and
30.53±1.14%, respectively, revealing significant upregulation
compared with that in drug-sensitive K562, U937 and MV4-11 cells
(13.03±0.31, 1.12±0.06 and 5.03±0.04%, respectively; P<0.001;
Figs. 1A and S1). AXL antigen expression in 64 blast
cell samples from 57 patients with AML exhibited a skewed
distribution with a median of 1.89% (range, 0.27–54.16%). The
median AXL antigen expression in FLT3-ITD/TKD+ AML blast
cells was 2.70% (range, 1.03–54.16%), which was increased compared
with that in FLT3-ITD/TKD− AML blast cells (1.51%;
range, 0.27–4.92%; P=0.001; Fig.
1B). However, there was no statistical difference in AXL
antigen expression between the de novo and
relapsed/refractory groups, among the favorable, intermediate and
adverse genetic risk groups, and between the
FLT3-ITD/TKD+ AML de novo and relapsed/refractory
groups (Table I).
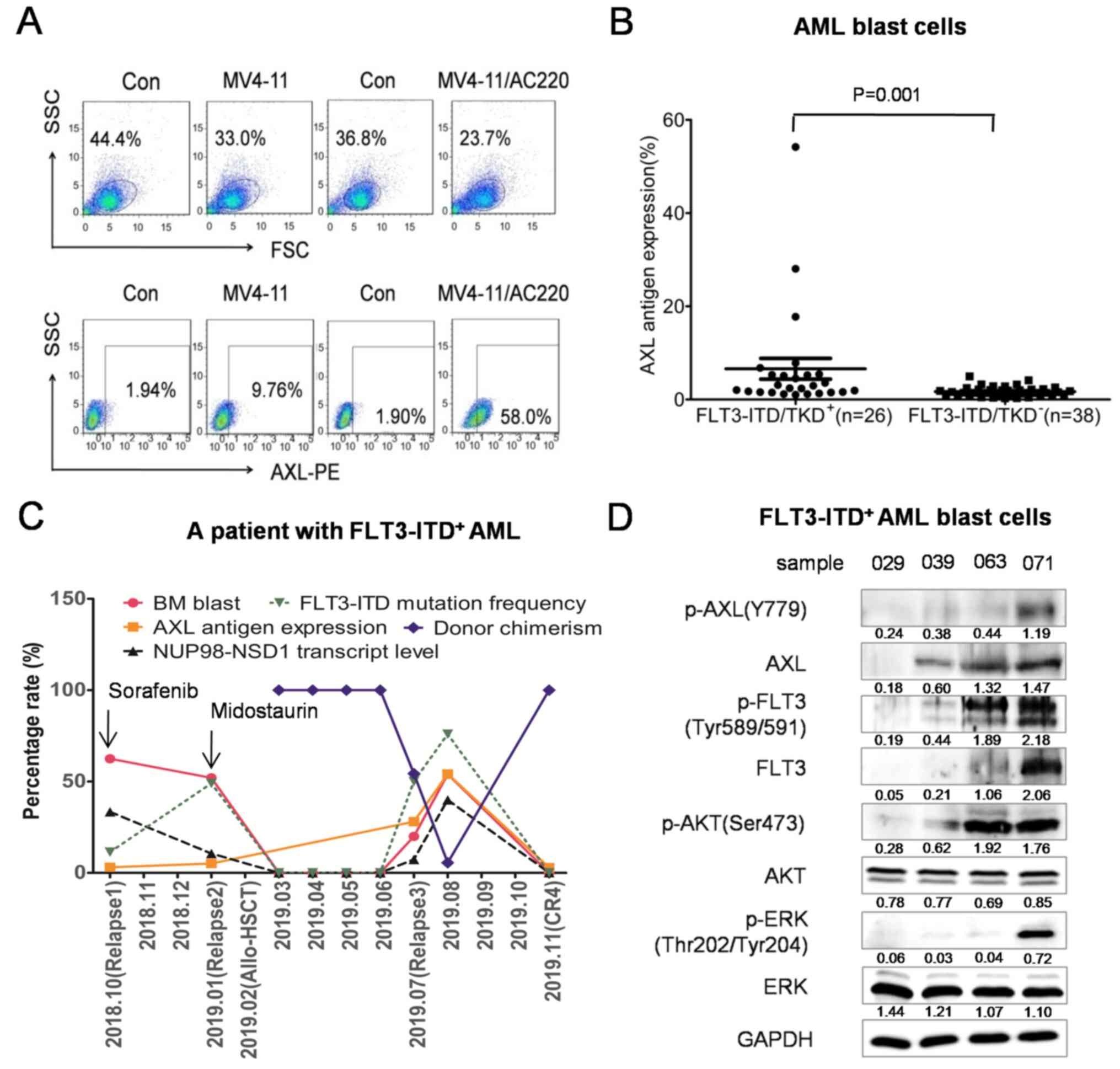 | Figure 1.AXL antigen expression is upregulated
in the MV4-11/AC220 cell line and FLT3-ITD/TKD+ AML
blast cells. (A) AXL antigen expression in the AC220-sensitive
MV4-11 and AC220-resistant MV4-11/AC220 cell lines was detected by
flow cytometry. (B) AXL antigen expression in
FLT3-ITD/TKD+ AML (n=26) and FLT3-ITD/TKD−
AML (n=38) blast cells was detected by flow cytometry. (C) The
dynamic changes of the bone marrow blast percentage, donor
chimerism, AXL antigen expression, FLT3-ITD mutation frequency and
NUP98-NSD1 gene transcript level (transcript copy
number/housekeeping gene Abelson copy number) from a patient with
relapsed/refractory FLT3-ITD+ AML. (D) AML blast cells
(samples 029, 039, 063 and 071) were subjected to western blot
analysis to detect the levels of AXL, p-AXL, FLT3, p-FLT3, ERK,
p-ERK, AKT, p-AKT and GAPDH. The intensity of the bands was
analyzed using ImageJ 1.51j8 and denoted as intensity/GAPDH. BM,
bone marrow; CR, complete remission; allo-HSCT, allogeneic
hematopoietic stem cell transplantation; AXL, AXL receptor tyrosine
kinase; AML, acute myeloid leukemia; FLT3, FMS-like tyrosine kinase
3; ITD, internal tandem duplication; TKD, tyrosine kinase domain
mutations. |
A 19-year-old male patient was diagnosed with
FLT3-ITD+ AML (M4), and experienced three relapses and
four complete remissions during clinical treatment between June
2018 and November 2019 (Fig. S2).
The dynamic changes of AXL antigen expression in blast cells were
consistent with the clinical resistance to the FLT3 inhibitors
sorafenib and PKC412 (Fig. 1C).
Western blotting was performed on AML blast cells and revealed
that, with the upregulation of p-AXL, the downstream molecules
p-AKT and p-ERK were also upregulated (Fig. 1D). These data suggested that
increased AXL activation by FLT3 inhibitors may mediate resistance
of leukemic cells in patients with FLT3-ITD+ AML.
AXL-targeted agents inhibit the growth
of FLT3-mutant AML cell lines and FLT3-ITD+ AML blast
cells in a dose-dependent manner
AXL antigen expression was upregulated in the
FLT3-ITD+ MV4-11/AC220 resistant cell line,
FLT3-ITD/TKD+ AML blast cells and FLT3
inhibitor-resistant blast cells from a patient with
FLT3-ITD+ AML, suggesting that AXL antigen upregulation
was associated with FLT3-ITD/TKD+ AML, particularly with
FLT3 inhibitor-resistant FLT3-ITD+ AML, and that
targeting AXL may have clinical value. DAXL-88 exerted a
dose-dependent cytotoxic effect on FLT3-wild type (WT)+
THP-1, FLT3-ITD+ MV4-11 and MV4-11/AC220 cells, as well
as FLT3-ITD+ AML blast cells, but had no effect on the
proliferation of FLT3-ITD− U937 and FLT3-ITD−
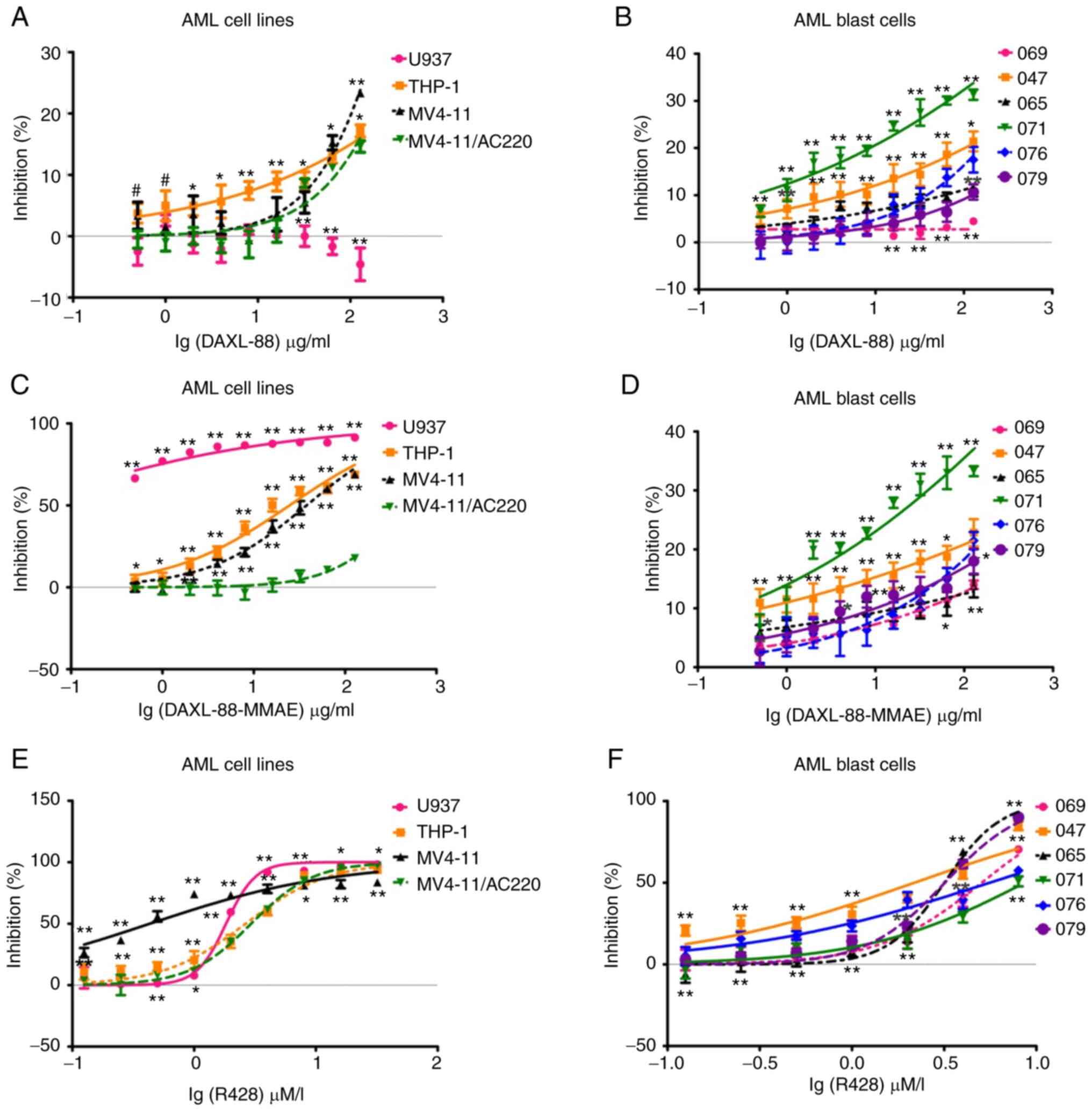
AML blast cells (Fig. 2A and B).
Considering the non-optimal cytotoxic effect of
DAXL-88, the antibody-drug conjugate DAXL-88-MMAE was further
prepared to improve the cytotoxic effect on these cells.
DAXL-88-MMAE exerted a dose-dependent cytotoxic effect on
AXL-expressing U937, THP-1, MV4-11 and MV4-11/AC220 cells (Fig. 2C), and AML blast cells (Fig. 2D). The cytotoxic effect of
DAXL-88-MMAE was markedly enhanced in U937, THP-1 and MV4-11 cells
compared with that of DAXL-88, and this was independent of the AXL
antigen expression intensity. However, it was associated with the
sensitivity of cell lines to MMAE (Table SI). The cytotoxic effect was not
significantly enhanced in the MV4-11/AC220 cell line (Fig. 2C; Table
SI) and AML blast cells (Fig.
2D; Table SII). In order to
exclude the cytotoxic effect caused by free MMAE from DAXL-88-MMAE,
an IgG1-MMAE antibody was also synthesized as an isotype control,
and this exerted no cytotoxic effect on AML cell lines or blast
cells (data not shown). These data suggested that the cytotoxic
effect of DAXL-88-MMAE was AXL-targeted MMAE cytotoxicity.
R428 also exerted a dose-dependent cytotoxic effect
on AXL-expressing U937, THP-1, MV4-11 and MV4-11/AC220 cells
(Fig. 2E), and AML blast cells
(Fig. 2F). The IC50 at 48
h for FLT3-ITD− AML (sample 069) and relapsed/refractory
FLT3-ITD+ AML (samples 071 and 076) blast cells was
5.59±0.84 µM/l, which was higher compared with that for the de
novo FLT3-ITD+ AML samples (2.84±0.60 µM/l; samples
047, 065 and 079; P=0.012; Table
SII).
AXL-targeted agents in combination
with AC220 synergistically inhibit proliferation and induce
apoptosis of MV4-11/AC220 and FLT3 inhibitor-resistant AML blast
cells
The AXL-targeted agents DAXL-88, DAXL-88-MMAE and
R428 were less effective in killing MV4-11/AC220-resistant cells
and relapsed/refractory FLT3-ITD+ AML blast cells
(Tables SI and SII), suggesting that it is necessary to
target both AXL and FLT3 to overcome drug resistance. DAXL-88,
DAXL-88-MMAE and R428 in combination with AC220 at the indicated
ratio (Fig. 3A and B) exerted
synergistic effects on the MV4-11/AC220 cell line and FLT3
inhibitor-resistant AML blast cells (sample 071).
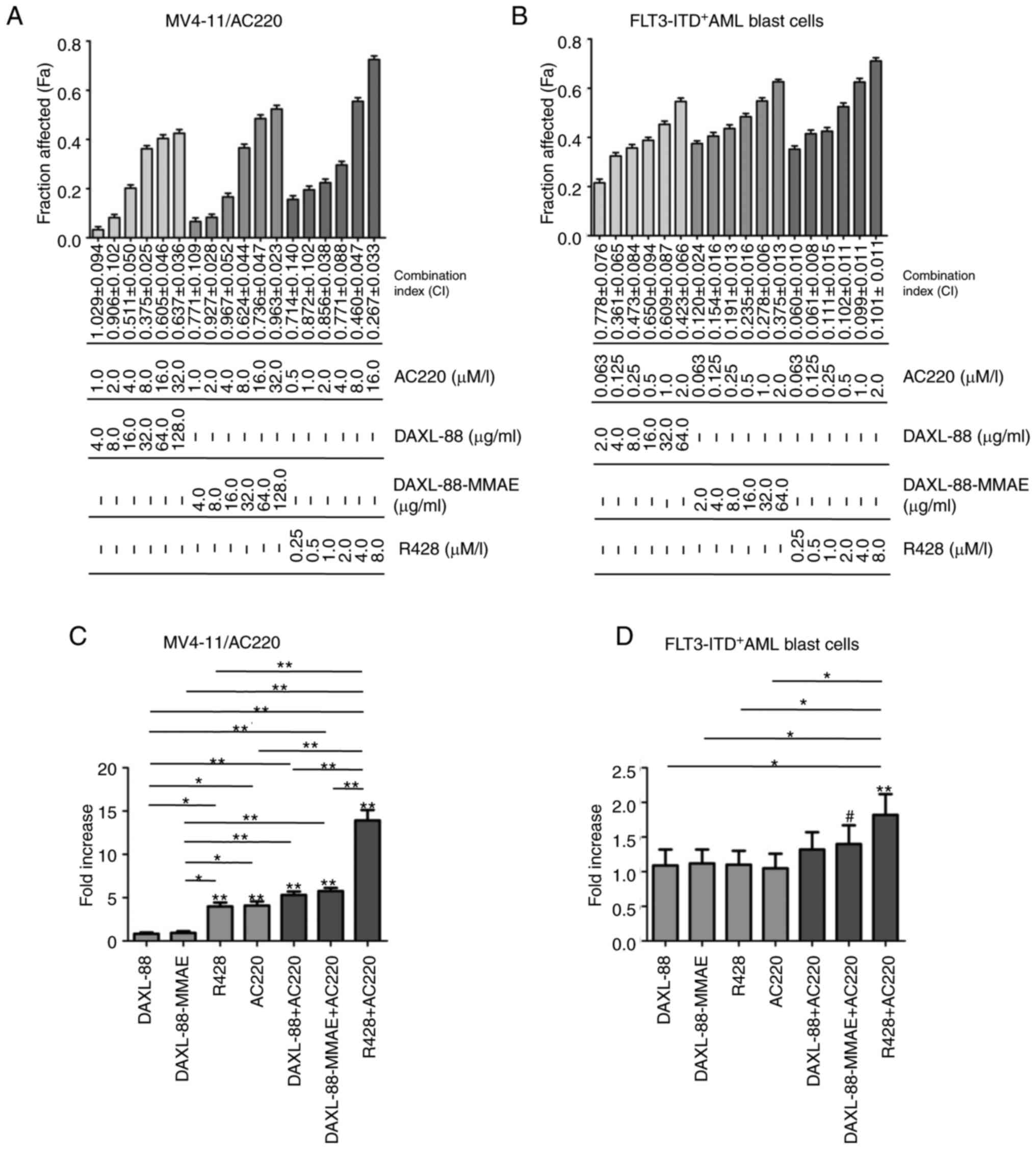 | Figure 3.AXL-targeted agents in combination
with AC220 synergistically inhibit the proliferation and induce the
apoptosis of MV4-11/AC220 cells and FLT3 inhibitor-resistant AML
blast cells. (A) MV4-11/AC220 and (B) AML blast cells (sample 071)
were treated with DAXL-88, DAXL-88-MMAE, R428 and AC220, and their
combinations, at a constant ratio and the indicated concentrations
for 48 h. Cell viability was examined using a Cell Counting Kit-8
assay. The Fa and CI values for each pair of drugs were calculated
using Calcusyn2.1 software. CI<1 indicates a synergistic effect;
CI=1 indicates an additive effect; and CI>1 indicates an
antagonistic effect. (C) MV4-11/AC220 cells treated without
(control culture) or with DAXL-88 (64 µg/ml), DAXL-88-MMAE (64
µg/ml), R428 (3 µM), AC220 (16 µM) and their combination for 48 h.
(D) AML blast cells (sample 071) treated without (control culture)
or with DAXL-88 (5 µg/ml), DAXL-88-MMAE (5 µg/ml), R428 (3 µM),
AC220 (1 µM) and their combination for 24 h. Cells were stained
with PI and Annexin V-FITC, and analyzed by flow cytometry. The
fold increase (relative to control untreated cultures of each cell
group) of apoptosis is presented as the mean ± SEM of three
experiments. #P<0.05, *P<0.01 and **P<0.001
(ANOVA and Dunnett test, vs. control untreated cells; ANOVA and
Tukey's test, vs. different treatment groups). CI, combination
index; Fa, fraction affected; MMAE, monomethyl auristatin E; FLT3,
FMS-like tyrosine kinase 3; ITD, internal tandem duplication; AXL,
AXL receptor tyrosine kinase; AML, acute myeloid leukemia. |
DAXL-88 (64 µg/ml) and DAXL-88-MMAE (64 µg/ml) could
not effectively induce apoptosis in MV4-11/AC220 cells; however,
the percentage of apoptotic cells was significantly increased when
combined with AC220 (P<0.001; Fig.
3C). The apoptosis of FLT3 inhibitor-resistant AML blast cells
(sample 071) was not elevated following 24-h sublethal single-dose
treatment with DAXL-88 (5 µg/ml), DAXL-88-MMAE (5 µg/ml), R428 (3
µM) or AC220 (1 µM), but was increased following combination
treatment with DAXL-88-MMAE and AC220 (P<0.05; Fig. 3D), and with R428 and AC220
(P<0.001; Fig. 3D).
DAXL-88 and DAXL-88-MMAE effectively
block AXL, FLT3 and their downstream signaling pathways
To explore the mechanism underlying the antileukemic
effect, the changes in the signal transduction pathways of MV4-11,
MV4-11/AC220 and FLT3 inhibitor-resistant AML blast cells (sample
071) following treatment with DAXL-88, DAXL-88-MMAE, R428, AC220
and their combinations were analyzed by western blotting. In the
MV4-11 cell line, the levels of p-AXL and p-FLT3, and those of
their downstream molecules p-AKT and p-ERK, were downregulated by
DAXL-88 and DAXL-88-MMAE. Furthermore, the expression levels of AXL
and FLT3 were also downregulated by DAXL-88-MMAE. AXL expression
was upregulated, and the levels of p-AXL, p-AKT and p-ERK were
downregulated by R428 (Fig. 4A). In
FLT3 inhibitor-resistant AML blast cells (sample 071), the levels
of AXL, FLT3 and its downstream target p-AKT were also
downregulated by treatment with DAXL-88 and DAXL-88-MMAE (Fig. 4C). Compared with those in MV4-11
cells, the AXL and p-AXL levels in MV4-11/AC220 cells were
increased (data not shown), and the expression of FLT3 and p-FLT3
was almost completely inhibited (Fig.
4B). p-AXL and p-AKT levels were not downregulated by DAXL-88
(64 µg/ml), DAXL-88-MMAE (64 µg/ml) or R428 (3 µM). When DAXL-88,
DAXL-88-MMAE and R428 were combined with AC220, the p-AXL, AXL and
p-AKT levels were decreased compared with those in the single-agent
groups (Fig. 4B).
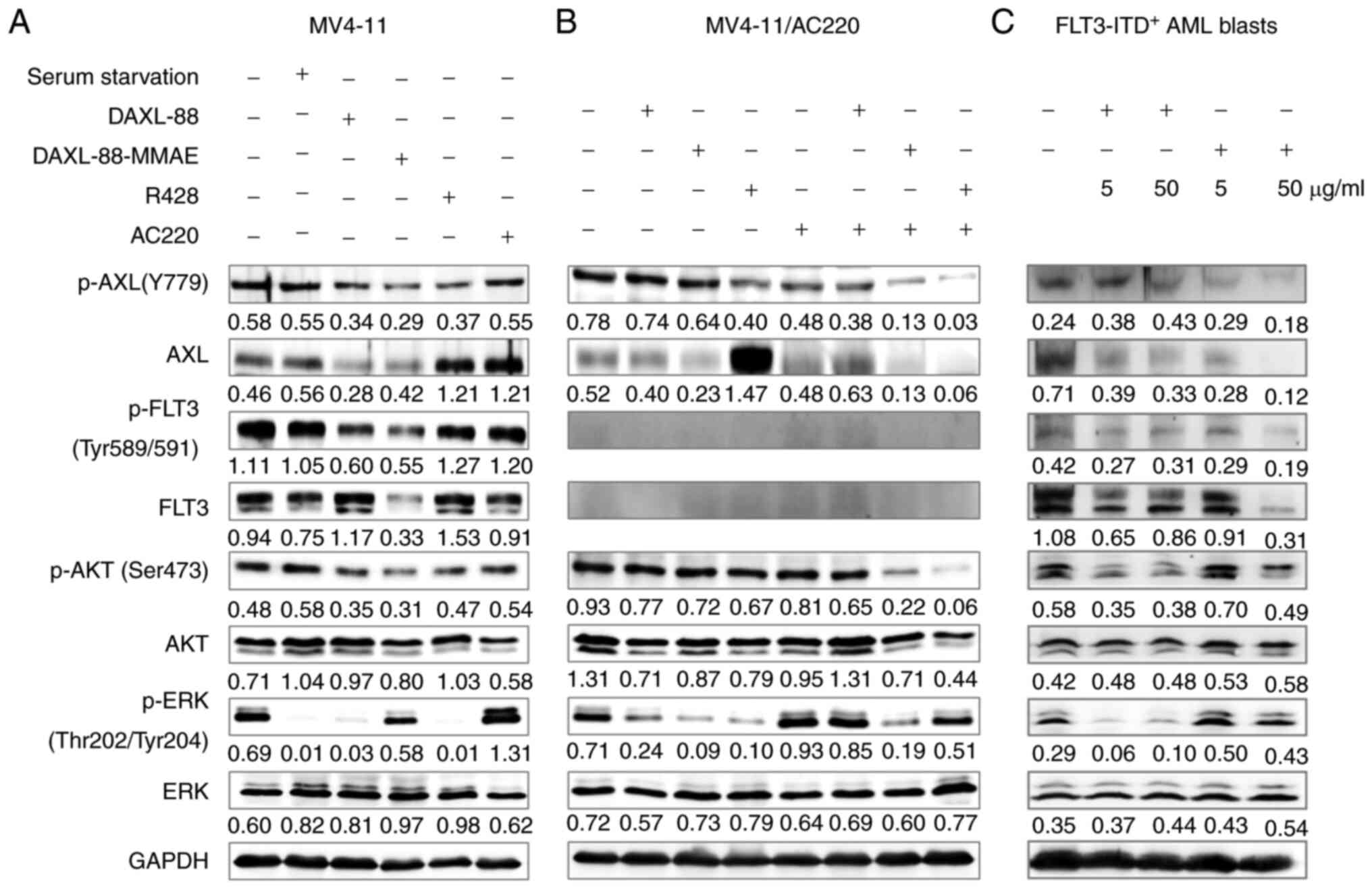 | Figure 4.DAXL-88 and DAXL-88-MMAE effectively
block AXL, FLT3 and their downstream signaling pathways. (A) MV4-11
cells treated without (control culture) or with serum starvation
(2% FBS), DAXL-88 (64 µg/ml), DAXL-88-MMAE (64 µg/ml), R428 (0.4
µM) or AC220 (2 nM) for 48 h. (B) MV4-11/AC220 cells treated
without (control culture) or with DAXL-88 (64 µg/ml), DAXL-88-MMAE
(64 µg/ml), R428 (3 µM), AC220 (16 µM) and their combinations for
48 h. (C) AML blast cells (sample 071) treated without (control
culture) or with DAXL-88 (5 or 50 µg/ml) and DAXL-88-MMAE (5 or 50
µg/ml) for 48 h. Cells were subjected to western blot analysis to
detect the levels of AXL, p-AXL, FLT3, p-FLT3, ERK, p-ERK, AKT,
p-AKT and GAPDH. The intensity of the bands was analyzed using
ImageJ 1.51j8 and denoted as intensity/GAPDH. AXL, AXL receptor
tyrosine kinase; AML, acute myeloid leukemia; FLT3, FMS-like
tyrosine kinase 3; ITD, internal tandem duplication. |
Discussion
The present study selected daunorubicin, ADM and
cytarabine to induce the resistance of AML cell lines with
reference to the clinical AML standard ‘3+7’ induction chemotherapy
(1). Additionally, the FLT3
inhibitors PKC412 and AC220 were selected as the
resistance-inducing medications for the following reasons: i)
20–30% of patients with AML harbor the FLT3-ITD/TKD mutation; ii)
FLT3-ITD and FLT3-TKD are high-risk AML biomarkers (17); and iii) FLT3-targeted therapies have
been widely used in the clinical treatment of
FLT3-ITD/TKD+ AML, and resistance to FLT3 inhibitors is
a recent problem. Only three stable drug-resistant AML cell lines
(K562/ADM, U937/Ara-c and MV4-11/AC220) were obtained, and AXL
antigen expression in these cells was markedly upregulated compared
with that in drug-sensitive cell lines, indicating that
upregulation of AXL antigen expression may be associated with
resistance in leukemic cells (Fig.
1A and Fig. S1). In particular,
K562/ADM and MV4-11/AC220 cells may be transformed into
semi-adherent cells due to the upregulation of the adhesion
molecule AXL (18).
Compared with that in FLT3-ITD/TKD− AML,
AXL antigen expression level in FLT3-ITD/TKD+ AML blast
cells (7 cases of FLT3-ITD+ and 2 cases of
FLT3-TKD+) was increased (Fig. 1B). The dynamic upregulation of AXL
antigen expression (Fig. 1C) and
p-AXL protein levels (Fig. 1D) in
blast cells from a typical patient with relapsed/refractory
FLT3-ITD+ AML was consistent with clinical resistance to
FLT3 inhibitors, which is characterized by increased FLT3 mutation
frequency and new inserted fragments (Fig. S2). These data suggested that AXL
antigen upregulation was associated with FLT3-ITD/TKD+
AML, particularly with drug-resistant FLT3-ITD+ AML.
Therefore, targeting AXL has clinical value in FLT3-mutant AML. The
present study further compared AXL antigen expression in de
novo FLT3-ITD/TKD+ AML blast cells with that in
relapsed/refractory FLT3-ITD/TKD+ AML, and no
significant difference was observed (P=0.330; Table I). This may be associated with the
insufficient sample size and the insufficient cases using FLT3
inhibitors. Additional clinical specimens will be collected in the
future to confirm higher AXL antigen expression in
relapsed/refractory FLT3-ITD/TKD+ AML.
DAXL-88 only exerted a dose-dependent cytotoxic
effect on FLT3-WT+ THP-1, FLT3-ITD+ MV4-11
and MV4-11/AC220 cells, as well as on FLT3-ITD+ AML
blast cells, and this was independent of AXL antigen expression
intensity (Fig. 2A and B; Tables SI and SII). Duan et al (14) constructed a three-dimensional model
of AXL and DAXL-88 Fv fragments, and identified the interaction
sites as Q122-E129 and
H201-G205. The interaction sites are not the
key binding sites for GAS6, suggesting that DAXL-88 may rely on
steric hindrance to block the binding of GAS6 to AXL. DAXL-88
exerted no cytotoxic effect on the SKOV3 human ovarian cancer cell
line, the A549 non-small cell lung cancer cell line, the MDA-MB-231
breast cancer cell line with high AXL antigen expression (data not
shown) or the U937 AML cell line with low AXL antigen expression,
indicating that the cytotoxic mechanism of DAXL-88 is not mediated
via blocking of the GAS6/AXL signaling pathway or the AXL
self-activation signaling pathway (19). Park et al (7) demonstrated that, in the
FLT3-ITD+ MV4-11 cell line, there is a physical
interaction between AXL and FLT3, and AXL can regulate FLT3
phosphorylation by affecting this interaction. In the present
study, DAXL-88 may have blocked the physical interaction between
AXL and FLT3 in FLT3-mutant AML cells by spatial steric hindrance,
blocked the formation of AXL heterodimer, and inhibited the
phosphorylation of AXL, FLT3 and their downstream molecules AKT and
ERK, thus inducing cell apoptosis and inhibiting cell
proliferation. DAXL-88-MMAE exerted stronger growth inhibitory and
apoptosis-inducing effects on the FLT3-ITD+ MV4-11 cell
line (Fig. 2C; Table SI), which was associated with the
downregulation of the levels of p-AXL, AXL, p-FLT3, FLT3 and their
downstream molecules p-AKT and p-ERK (Fig. 4A).
The cytotoxic effect of DAXL-88-MMAE was not
significantly enhanced in the MV4-11/AC220 cell line (Fig. 2C; Table
SI) and AML blast cells (Fig.
2D; Table SII) compared with
that of DAXL-88 (Fig. 2A and B;
Tables SI and SII), which differed from U937, THP-1 and
MV4-11 cell lines (Fig. 2A and C;
Table SI). The sensitivity of cells
to free MMAE is an important factor (12); however, there are other potential
factors involved in the difference in cytotoxic effects, such as
antigen expression (20), somatic
mutations (21) and p-glycoprotein
(p-gp)-related multidrug resistance. Gemtuzumab ozogamicin
(CMA-676) (22,23), an anti-CD33 antibody conjugate, is
actively pumped out by resistant leukemic cells and blast cells
expressing p-gp, thereby reducing its intracellular accumulation
and cytotoxic effect. The drug resistance mechanism of MV4-11/AC220
cells and the optimized modification of DAXL-88-MMAE will be
explored in future studies.
R428 could effectively inhibit the proliferation
(Fig. 2E and F; Tables SI and SII) and induce the apoptosis of MV4-11 and
MV4-11/AC220 cells (Fig. 3C),
revealing upregulation of AXL and slight downregulation of p-AXL
(Fig. 4A and B). Mild inhibition of
p-AXL was insufficient to explain the effective cytotoxicity of
R428, suggesting that there may be other mechanisms independent of
AXL to be explored, such as blocking of lysosomal acidification and
recycling (24).
In addition to its role in driving drug resistance,
recent studies have revealed that AXL serves an important role in
the regulation of leukemic stem cells and the bone marrow
hematopoietic niche. Leukemic stem cells, which are responsible for
leukemia initiation, progression and relapse, are considered to be
a key factor in eliminating leukemia. Jin et al (25) identified that the GAS6/AXL paracrine
loop is a critical regulator of the self-renewal capacity of
chronic myelogenous leukemic stem cells conferring imatinib
resistance. Wang et al (26)
reported that alkB homolog 5 RNA demethylase (ALKBH5) is
specifically required to maintain the function of AML stem cells,
and regulates AXL stability in leukemic cells in an
N6-methyladenosine-dependent manner. These findings
indicate that targeting AXL/GAS6 or AXL upstream molecule ALKBH5
could eliminate leukemic stem cells. Dumas et al (27) demonstrated that the bone marrow
hematopoietic niche enhances AXL expression through canonical
ligand GAS6, STAT5-activating soluble factors and the local hypoxic
environment, thus providing protection for FLT3-ITD+ AML
cells against AC220. The study suggested that dual inhibition of
AXL and FLT3 not only diminished the AML burden, but also prevented
AXL expression from alleviating protection against the
leukemia-initiating cells provided by the hematopoietic niche.
DAXL-88, DAXL-88-MMAE and R428 in combination with
AC220 exerted synergistic cytotoxic effects in the MV4-11/AC220
cell line and FLT3 inhibitor-resistant AML blast cells (sample 071;
Fig. 3A and B). Flow cytometry
revealed that their combination was more effective in inducing
apoptosis than treatment with a single agent (Fig. 3C and D). Furthermore, western
blotting demonstrated that the levels of p-AXL, AXL and p-AKT were
downregulated by combination treatment compared with single-agent
treatment (Fig. 4B). Therefore,
AXL-targeted agents could overcome the resistance of MV4-11/AC220
and FLT3 inhibitor-resistant AML blast cells to AC220. The efficacy
of the AXL-targeted agents in combination with FLT3 inhibitors
would be expected in FLT3-ITD+ AML mouse
xenotransplantation models, considering the possible triple
inhibition of leukemic cells, leukemic stem cells and the bone
marrow hematopoietic niche.
In conclusion, upregulation of AXL antigen
expression was associated with FLT3-ITD/TKD+ AML,
particularly drug-resistant FLT3-ITD+ AML. Therefore,
targeting AXL has clinical value in FLT3-mutant AML. The
AXL-targeted agents DAXL-88, DAXL-88-MMAE and R428 could
effectively inhibit the growth of FLT3-mutant AML cells and
FLT3-ITD+ AML blast cells, and overcome resistance in
the AC220-resistant MV4-11/AC220 cell line and FLT3
inhibitor-resistant AML blast cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was partially supported by the
National Key Clinical Specialized Military Construction Project
(Clinical Medicine).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and JW conceptualized the project, performed the
experiments, analyzed the data and wrote the article. JL and WM
performed the experiments. YD produced the anti-AXL antibody
DAXL-88 and DAXL-88-MMAE. DL conceptualized the project, oversaw
the experiments and edited the article. YL, JW and DL confirm the
authenticity of the raw data. All the authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Sixth Medical Center of PLA General Hospital. The
patients provided written informed consent for the use of
peripheral blood or bone marrow samples.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AXL
|
AXL receptor tyrosine kinase
|
|
AML
|
acute myeloid leukemia
|
|
K562/ADM
|
Adriamycin-resistant K562 cell
line
|
|
U937/Ara-c
|
cytarabine-resistant U937 cell
line
|
|
MV4-11/AC220
|
AC220-resistant MV4-11 cell line
|
|
FLT3
|
FMS-like tyrosine kinase 3
|
|
WT
|
wild-type
|
|
ITD
|
internal tandem duplication
|
|
TKD
|
tyrosine kinase domain mutations
|
|
MMAE
|
monomethyl auristatin E
|
References
|
1
|
Döhner H, Estey E, Grimwade D, Amadori S,
Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA,
et al: Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood.
129:424–447. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kell J: Considerations and challenges for
patients with refractory and relapsed acute myeloid leukaemia. Leuk
Res. 47:149–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Varnum BC, Young C, Elliott G, Garcia A,
Bartley TD, Fridell YW, Hunt RW, Trail G, Clogston C, Toso RJ, et
al: Axl receptor tyrosine kinase stimulated by the vitamin
K-dependent protein encoded by growth-arrest-specific gene 6.
Nature. 373:623–626. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paccez JD, Vogelsang M, Parker MI and
Zerbini LF: The receptor tyrosine kinase Axl in cancer: Biological
functions and therapeutic implications. Int J Cancer.
134:1024–1033. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ben-Batalla I, Schultze A, Wroblewski M,
Erdmann R, Heuser M, Waizenegger JS, Riecken K, Binder M, Schewe D,
Sawall S, et al: Axl, a prognostic and therapeutic target in acute
myeloid leukemia mediates paracrine crosstalk of leukemia cells
with bone marrow stroma. Blood. 122:2443–2452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hong CC, Lay JD, Huang JS, Cheng AL, Tang
JL, Lin MT, Lai GM and Chuang SE: Receptor tyrosine kinase AXL is
induced by chemotherapy drugs and overexpression of AXL confers
drug resistance in acute myeloid leukemia. Cancer Lett.
268:314–324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park IK, Mishra A, Chandler J, Whitman SP,
Marcucci G and Caligiuri MA: Inhibition of the receptor tyrosine
kinase Axl impedes activation of the FLT3 internal tandem
duplication in human acute myeloid leukemia: Implications for Axl
as a potential therapeutic target. Blood. 121:2064–2073. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park IK, Mundy-Bosse B, Whitman SP, Zhang
X, Warner SL, Bearss DJ, Blum W, Marcucci G and Caligiuri MA:
Receptor tyrosine kinase Axl is required for resistance of leukemic
cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia.
29:2382–2389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kariolis MS, Miao YR, Diep A, Nash SE,
Olcina MM, Jiang D, Jones DS II, Kapur S, Mathews II, Koong AC, et
al: Inhibition of the GAS6/AXL pathway augments the efficacy of
chemotherapies. J Clin Invest. 127:183–198. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kariolis MS, Miao YR, Jones DS II, Kapur
S, Mathews II, Giaccia AJ and Cochran JR: An engineered Axl ‘decoy
receptor’ effectively silences the Gas6-Axl signaling axis. Nat
Chem Biol. 10:977–983. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ye X, Li Y, Stawicki S, Couto S,
Eastham-Anderson J, Kallop D, Weimer R, Wu Y and Pei L: An anti-Axl
monoclonal antibody attenuates xenograft tumor growth and enhances
the effect of multiple anticancer therapies. Oncogene.
29:5254–5264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boshuizen J, Koopman LA, Krijgsman O,
Shahrabi A, van den Heuvel EG, Ligtenberg MA, Vredevoogd DW, Kemper
K, Kuilman T, Song JY, et al: Cooperative targeting of melanoma
heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK
inhibitors. Nat Med. 24:203–212. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duan Y, Hu B, Qiao C, Luo L, Li X, Wang J,
Liu H, Zhou T, Shen B, Lv M, et al: Engineered AXL-ECD-Fc variants
that abolish the AXL/Gas6 interaction suppress tumor cell
migration. Oncol Lett. 17:5784–5792. 2019.PubMed/NCBI
|
|
14
|
Duan Y, Luo L, Qiao C, Li X, Wang J, Liu
H, Zhou T, Shen B, Lv M and Feng J: A novel human anti-AXL
monoclonal antibody attenuates tumour cell migration. Scand J
Immunol. 90:e127772019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cossarizza A, Chang HD, Radbruch A, Acs A,
Adam D, Adam-Klages S, Agace WW, Aghaeepour N, Akdis M, Allez M, et
al: Guidelines for the use of flow cytometry and cell sorting in
immunological studies(second edition). Eur J Immunol. 49:1457–1973.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zou Y, Huang Y and Ma X: Phenylhexyl
isothiocyanate suppresses cell proliferation and promotes apoptosis
via repairing mutant P53 in human myeloid leukemia M2 cells. Oncol
Lett. 18:3358–3366. 2019.PubMed/NCBI
|
|
17
|
Zhao J, Song Y and Liu D: Gilteritinib: A
novel FLT3 inhibitor for acute myeloid leukemia. Biomark Res.
7:192019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Neubauer A, Fiebeler A, Graham DK, O'Bryan
JP, Schmidt CA, Barckow P, Serke S, Siegert W, Snodgrass HR and
Huhn D: Expression of axl, a transforming receptor tyrosine kinase,
in normal and malignant hematopoiesis. Blood. 84:1931–1941. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shen Y, Chen X, He J, Liao D and Zu X: Axl
inhibitors as novel cancer therapeutic agents. Life Sci.
198:99–111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Francisco JA, Cerveny CG, Meyer DL, Mixan
BJ, Klussman K, Chace DF, Rejniak SX, Gordon KA, DeBlanc R, Toki
BE, et al: cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E
conjugate with potent and selective antitumor activity. Blood.
102:1458–1465. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dornan D, Bennett F, Chen Y, Dennis M,
Eaton D, Elkins K, French D, Go MA, Jack A, Junutula JR, et al:
Therapeutic potential of an anti-CD79b antibody-drug conjugate,
anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma.
Blood. 114:2721–2729. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Naito K, Takeshita A, Shigeno K, Nakamura
S, Fujisawa S, Shinjo K, Yoshida H, Ohnishi K, Mori M, Terakawa S,
et al: Calicheamicin-conjugated humanized anti-CD33 monoclonal
antibody (gemtuzumab zogamicin, CMA-676) shows cytocidal effect on
CD33-positive leukemia cell lines, but is inactive on
P-glycoprotein-expressing sublines. Leukemia. 14:1436–1443. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matsui H, Takeshita A, Naito K, Shinjo K,
Shigeno K, Maekawa M, Yamakawa Y, Tanimoto M, Kobayashi M, Ohnishi
K, et al: Reduced effect of gemtuzumab ozogamicin (CMA-676) on
P-glycoprotein and/or CD34-positive leukemia cells and its
restoration by multidrug resistance modifiers. Leukemia.
16:813–819. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen F, Song Q and Yu Q: Axl inhibitor
R428 induces apoptosis of cancer cells by blocking lysosomal
acidification and recycling independent of Axl inhibition. Am J
Cancer Res. 8:1466–1482. 2018.PubMed/NCBI
|
|
25
|
Jin Y, Nie D, Li J, Du X, Lu Y, Li Y, Liu
C, Zhou J and Pan J: Gas6/AXL signaling regulates self-renewal of
chronic myelogenous leukemia stem cells by stabilizing β-catenin.
Clin Cancer Res. 23:2842–2855. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang J, Li Y, Wang P, Han G, Zhang T,
Chang J, Yin R, Shan Y, Wen J, Xie X, et al: Leukemogenic Chromatin
Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL
Signaling Axis. Cell Stem Cell. 27:81–97.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dumas PY, Naudin C, Martin-Lannerée S,
Izac B, Casetti L, Mansier O, Rousseau B, Artus A, Dufossée M,
Giese A, et al: Hematopoietic niche drives FLT3-ITD acute myeloid
leukemia resistance to quizartinib via STAT5-and hypoxia-dependent
upregulation of AXL. Haematologica. 104:2017–2027. 2019. View Article : Google Scholar : PubMed/NCBI
|