Introduction
Colorectal cancer (CRC) is the fourth most commonly
diagnosed malignant disease worldwide (1). Despite surgery, chemotherapy, targeted
therapy and other options, it is estimated that colorectal cancer
ranks as the fourth and fifth most common cause of
cancer-associated death among women and men, respectively, in China
(1). The CRC mortality rate in China
increased annually between 1991 and 2011 (2). By 2012, tobacco smoking, alcohol
consumption, obesity, low levels of physical activity, low fruit
and vegetable intake and high intake of red and processed meat were
responsible for ~46% of CRC cases and associated deaths in China
(3). The development of CRC is a
multistep process that includes the progressive destruction of
epithelial cell proliferation, apoptosis, differentiation and
survival mechanisms (4).
MicroRNAs (miRNAs) are single-stranded RNAs of ~20
nucleotides in length that can degrade and inhibit the translation
of target mRNA (5). Some miRNAs
function as CRC inducers or regulators and can be considered as
biomarkers and therapeutic targets (5). miRNAs are regulated at multiple levels,
including transcription, Drosha and Dicer processing, loading onto
Argonaute proteins and miRNA turnover (6). Each level is mediated by multiple
factors, including transcription factors, RNA-binding proteins,
protein-modifying enzymes, RNA-modifying enzymes, exoribonucleases
and endoribonucleases (6). Intronic
miR-32 is located within intron 14 of the transmembrane protein 245
(TMEM245) gene.
We previously found that miR-32 is upregulated in
CRC tissues and that high miR-32 levels are closely associated with
lymph-node and distant metastases (7). Overall, survival rates are low among
patients with CRC that express abundant miR-32 (7). miR-32-overexpression in CRC cells
results in enhanced proliferation, migration, invasion and reduced
apoptosis through the repression of phosphatase and tensin homolog
(PTEN) expression (8). The
mechanisms of miR-32 upregulation in CRC were explored in more
detail and luciferase reporter assays revealed that miR-32 binds to
the 3′-untranslated region of PTEN (8). The promoter region (~2 kb) of
TMEM245/miR-32 was cut into fragments of different
lengths, cloned into the 5′ end of a luciferase reporter vector and
then transfected into CRC cells. The results of the dual-luciferase
reporter assays suggested that the core promoter region is located
within −320 to −1 bp of the 5′-flanking region. Protein binding to
the core promoter was analyzed using DNA pull-down assays and mass
spectrometry. Transcription factor (TF) analyses identified
forkhead box K1 (FOXK1) as a potentially interactive TF (9).
Forkhead box K1 is involved in tumorigenesis and
cancer development. It promotes ovarian cancer cell proliferation
and metastasis (10), esophageal
cancer cell proliferation and migration, inhibits apoptosis and is
associated with poor differentiation and prognosis (11). Wu et al (12) found that FOXK1 induces the
epithelial-mesenchymal transition and facilitates CRC cell invasion
in vitro and in vivo, and that enhanced FOXK1
expression indicates a poor prognosis in patients with CRC.
The present study aimed to provide novel insights
into the molecular mechanisms underlying CRC by defining FOXK1
expression in CRC tissues and investigating the relationship
between FOXK1 and miR-32 expression. In addition, the study aimed
to clarify the role of FOXK1 in miR-32 regulation and CRC cell
proliferation and apoptosis.
Materials and methods
Patients and specimens
In total, 35 primary CRC and 31 non-cancerous
colonic tissue samples were obtained from 66 patients that had been
treated by surgical resection or colonoscopy at the Affiliated
Hospital of Guangdong Medical University (Guangdong, China) between
July 2017 and September 2019. All the patients were Han Chinese.
The mean age of CRC patients was 59±13 years old (range, 28–84
years; 25 males, 10 females). Patients with newly diagnosed CRC and
>18 years old were included. The patients declined
participation, as well as had received either radiotherapy or
chemotherapy before surgery or colonoscopy were excluded. The study
protocol was approved by The Medical Ethics Committee at the
Affiliated Hospital of Guangdong Medical University and written
informed consent was obtained from all participants. The diagnoses
of the samples were verified by pathologists who were independent
from the present study. Data on clinicopathological
characteristics, including sex, age, tumor diameter,
differentiation, lymphatic and distant metastasis and staging were
collected. Median FOXK1 expression (0.00492) served as the
cut-off for separating the 35 patients with CRC into groups with
high (n=18) and low (n=17) FOXK1 expression. After
collection tissues were immediately frozen in liquid nitrogen and
stored at −80°C until use.
Cell transfection
HCT-116 and HT-29 CRC cells were purchased from The
Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences. The HT-29 cell line was authenticated by STR profiling.
The cells were cultured in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C under a 5% CO2
atmosphere. No antibiotics were used for cell culture. To generate
the FOXK1 expression plasmid, the human FOXK1 coding sequence was
amplified using PCR, as later described. The PCR product was cloned
and inserted into a pcDNA3.1 vector (Promega Corp.). The inserted
fragment was confirmed by sequencing. The FOXK1-overexpressing
plasmid, pcDNA3.1-FOXK1, was synthesized at Guangzhou RiboBio Co.,
Ltd. The pcDNA3.1 empty vector was used as the negative control
(NC) of pcDNA3.1-FOXK1. For plasmid/siRNA/mimic/inhibitor
transfection, cells were seeded at a density of 3×105
cells per well in 6-well plates, or 1×105 cells per well
in 12-well plates, or 5×104 cells per well in 24-well
plates, or 6×103 cells per well in 96-well plates.
Twenty-four hours later, pcDNA3.1-FOXK1 (2.0 µg/ml), pcDNA3.1 (2.0
µg/ml), FOXK1 small interfering (si)RNA (siFOXK1) (150 nM),
siRNA-NC (150 nM), miR-32 mimic (200 nM), mimic-NC (200 nM), miR-32
inhibitor (200 nM) and inhibitor-NC (200 nM) (all RiboBio Co.,
Ltd.) were transfected as described by the manufacturer into
HCT-116 and HT-29 cells using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) in Opti-MEM™ I medium
(Gibco; Thermo Fisher Scientific, Inc.). Cells were harvested 48 h
after transfection for RNA and protein detection, dual-luciferase
reporter assay and apoptosis assay, and 24, 48, 72 and 96 h for
CCK-8 assays, respectively. The siRNA-NC (cat. no. siN0000001-1-5),
mimic-NC (cat. no. miR1N0000001-1-5), inhibitor-NC (cat. no.
miR2N0000001-1-5) were non-targeting and designed and synthesized
by RiboBio Co., Ltd. The following siRNA sequence was used:
siFOXK1, 5′-CCAGTTCACGTCGCTCTAT-3′. The sequences of miR-32 mimic
and inhibitor were as follows: miR-32 mimic, sense:
5′-UAUUGCACAUUACUAAGUUGCA-3′, and antisense:
5′-UGCAACUUAGUAAUGUGCAAUA-3′; mimic-NC, sense:
5′-UUUGUACUACACAAAAGUACUG-3′, and antisense:
5′-CAGUACUUUUGUGUAGUACAAA-3′; miR-32, inhibitor:
5′-UGCAACUUAGUAAUGUGCAAUA-3′ and inhibitor-NC:
5′-CAGUACUUUUGUGUAGUACAAA-3′.
Reverse-transcription quantitative PCR
(RT-qPCR)
Levels of FOXK1, TMEM245 and miR-32
expression in tissues and transfected cells were measured using
RT-qPCR. Total RNA was extracted from tissues and transfected cells
using RNAiso Plus (Takara Bio, Inc.) that was then reverse
transcribed using PrimeScript™ RT Master mix (Perfect Real Time;
Takara Bio, Inc.) for FOXK1 and TMEM245 and Mir-X™
miRNA First-Strand Synthesis kits (Clontech Laboratories, Inc.) for
miR-32 as described by the manufacturers. The endogenous controls
were β-actin mRNA for FOXK1 and TMEM245, and U6
small-nuclear RNA for miR-32. Quantitative PCR was performed using
TB Green® Premix Ex Taq™ II (Tli RNaseH Plus; Takara
Bio, Inc.) and a LightCycler® 480 II system (Roche
Diagnostics) under a cycling program comprising of 30 sec at 95°C
for 1 cycle, then 5 sec at 95°C and 20 sec at 60°C for a total of
40 cycles. Table I lists the qPCR
primers. The mRQ 3′-primer was included in the Mir-X™ miRNA
First-Strand Synthesis kit, but sequence information of this primer
was unavailable. Relative levels of miR-32 and FOXK1 and
TMEM245 mRNA were determined using the 2−∆∆Cq
method (13).
 | Table I.Sequence information for the primers
used in the quantitative PCR assay. |
Table I.
Sequence information for the primers
used in the quantitative PCR assay.
| Gene | Forward primer,
5′-3′ | Reverse primer,
5′-3′ |
|---|
| miR-32 |
CGCGCTATTGCACATTACTAAGTTGC | mRQ 3′ primer |
| U6 |
CTCGCTTCGGCAGCACA | mRQ 3′ primer |
| FOXK1 |
TTCCAGGAGCCGCACTTCTA |
GGAAGGTACACTGCTTGGGC |
| TMEM245 |
GACATTCTGGACTGGCAGGA |
AGTGGTGAACAGCAGGCTCA |
| PTEN |
AAAGGGACGAACTGGTGTAATG |
TGGTCCTTACTTCCCCATAGAA |
| β-actin |
GGCGGCAACACCATGTACCCT |
AGGGGCCGGACTCGTCATACT |
Western blotting
Cells transfected with
pcDNA3.1-FOXK1/siFOXK1/pcDNA3.1-FOXK1 + miR-32
inhibitor/siFOXK1+miR-32 mimic were incubated at 37°C for 48 h and
then were digested and total protein was extracted using RIPA lysis
buffer (Beyotime Institute of Biotechnology), and quantified using
BCA protein assay kits (Genstar Technologies Co., Inc.). In total,
30 µg protein was loaded per lane, electrophoresed and separated
using 12% SDS-PAGE and was then transferred to polyvinylidene
difluoride membranes (MilliporeSigma; Merck KGaA). Non-specific
protein binding was blocked by incubating the membranes with
skimmed milk at room temperature for 1 h, then the membranes were
incubated overnight at 4°C with anti-PTEN (1:1,000; cat. no. 9559S;
Cell Signaling Technology, Inc.), anti-FOXK1 (1:1,000; cat. no.
ab18196; Abcam) and anti-GAPDH (1:1,000; cat. no. AG019; Beyotime
Institute of Biotechnology) antibodies. The membranes were then
incubated with horseradish peroxidase-labeled goat anti-rabbit IgG
(1:1,000; cat. no. A0208; Beyotime Institute of Biotechnology) or
horseradish peroxidase-labeled goat anti-mouse IgG (1:1,000; cat.
no. A0216; Beyotime Institute of Biotechnology) secondary antibody
for 1 h at room temperature, washed three times with TBST, then
signals were detected using MilliporeSigma™ Immobilon™ Western
Chemiluminescent HRP Substrate (MilliporeSigma; Merck KGaA). Images
were acquired using an Azure c600 system (Azure Biosystems, Inc.).
Protein bands were quantified using Quantity One software (version
4.6.2; Bio-Rad Laboratories, Inc.). All experiments were repeated
at least three times.
Chromatin
immunoprecipitation-quantitative PCR (ChIP-qPCR)
JASPAR (http://jaspar.genereg.net/) and HOCOMOCO (https://hocomoco11.autosome.ru/) bioinformatics
analysis (performed by Guangzhou RiboBio Co., Ltd.) was used to
identify the binding sites of FOXK1 to the TMEM245/miR-32
promoter. Two putative FOXK1-binding sites were located within −320
to −1 bp of the TMEM245/miR-32 promoter region at −90 to −77
bp (site one) and −166 to −153 (site two) bp. The ChIP assay was
carried out with the Pierce Agarose ChIP Kit (cat. no. 26156;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions. Cells were cross-linked with 1% paraformaldehyde and
genomic DNA was digested to an average size of 100–1,000 bp. Total
sheared chromatin was incubated with FOXK1 antibody (5 µg; cat. no.
ab18196; Abcam) or normal rabbit IgG (2 µl; cat. no.2729S; Cell
Signaling Technology, Inc.) at 4°C overnight. The NC was
non-specific IgG. Immunoprecipitated DNA was analyzed using qPCR on
a LightCycler® 480 II system. The FOXK1-binding sites
one and two in the TMEM245/miR-32 promoter region were
amplified using the respective primers: Site one,
5′-TTCCCCATTTCCCCTTC-3′ and 5′-CGAGATTGTGGGAGTTGTAG-3′; and site
two, 5′-CCTCCAGGAAGATATAGACCC-3′ and 5′-TCCCGAAGGGGAAATG-3′. The
primers were synthesized by Sangon Biotech Co., Ltd. Target
enrichment was expressed as % input according to the formula: Fold
enrichment=2−∆∆Cq (ChIP/IgG) (14).
Dual-luciferase reporter assays
HCT-116 cells were cultured in 24-well plates
(5×104 cells per well), and were transfected with
pGL3-basic (Promega Corporation) luciferase reporter constructs
harboring wild-type (WT) or mutant (MUT) TMEM245/miR-32
promoter target sequences to evaluate the binding potential of
FOXK1 to this promoter. The DNA fragments containing binding sites
one and two were amplified and subcloned into vector pGL3-basic to
create the luciferase pGL3-promoter-WT and the mutated pGL3
reporters: MUT1, MUT2 And MUT1+2, which were mutated at binding
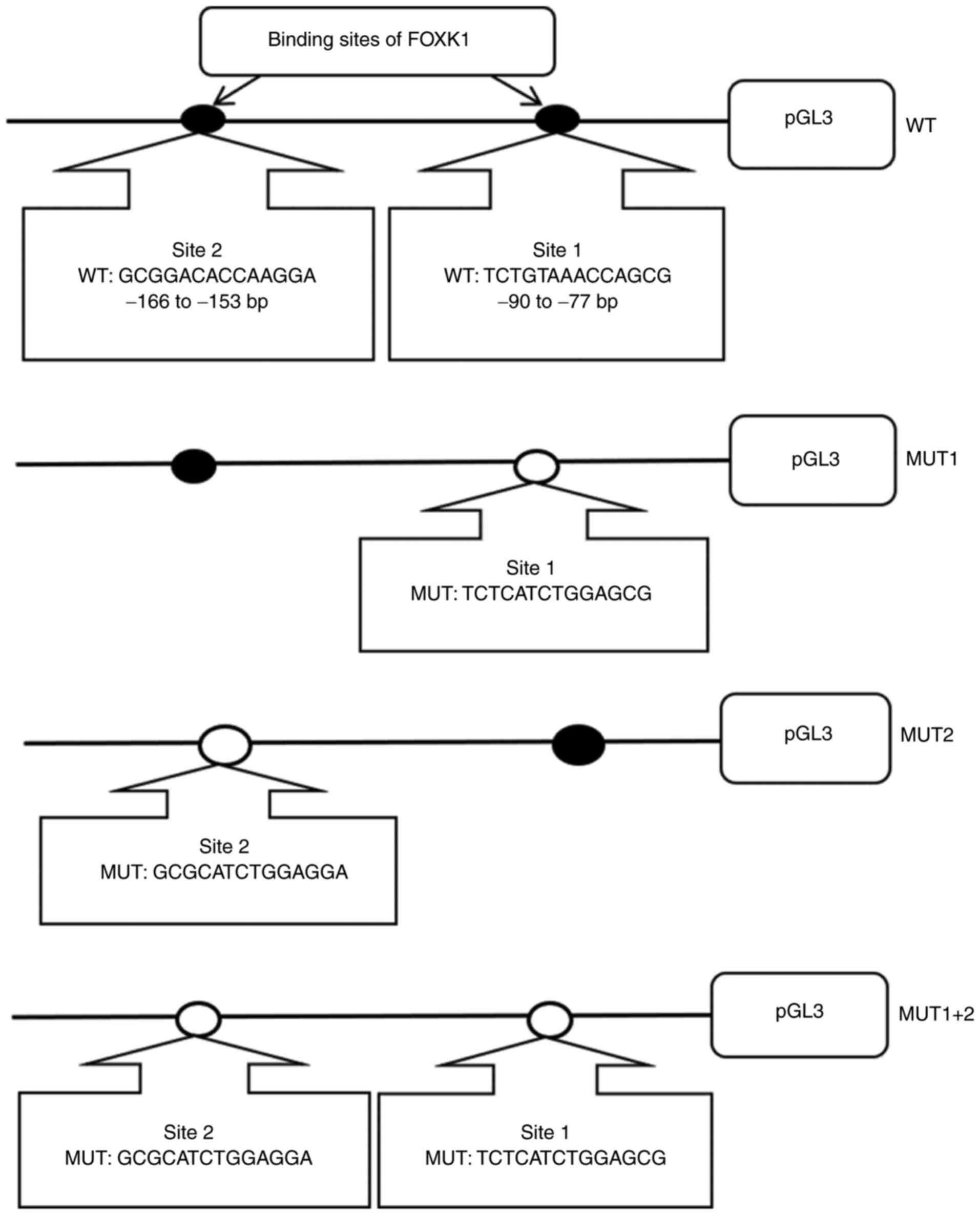
sites one, two or both, respectively (Fig. 1). All WT and MUT plasmids were
synthesized by RiboBio Co, Ltd. The pRL-TK (Promega Corporation)
plasmid was used as an internal control to standardize by Renilla
luciferase activity. Plasmid pGL3-promoter-WT, MUT1, MUT2 or MUT1+2
and pcDNA 3.1-FOXK1 or pcDNA 3.1 vector and pRL-TK were
co-transfected into HCT-116 cells in 24-well plates
(5×104 cells per well) at 37°C for 48 h.
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to transfect cells with the plasmids
following the manufacturer's instructions. Luciferase activities
were measured using the Dual-Luciferase Reporter Assay system
(Promega Corp.). Firefly luciferase activity was normalized to that
of Renilla luciferase.
Cell Counting Kit-8 (CCK-8) assay
Cell proliferation was assessed using CCK-8 assays
(Dojindo Molecular Technologies, Inc.). HCT-116 or HT-29 cells
(6×103 cells per well) were seeded into 96-well plates
for 24 h, transfected, then incubated for 24, 48, 72 and 96 h. The
cells were incubated with CCK-8 reagent for 1 h at 37°C, then
absorbance at 450 nm was determined by spectrophotometry.
Apoptosis assays
Apoptosis was assayed using Annexin V- FITC
Apoptosis Detection kit (Dojindo Molecular Technologies, Inc.). At
48 h after transfection, CRC cells were digested, resuspended in
Annexin V Binding Solution, mixed with Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide as described by the
manufacturer instructions, and then analyzed by flow cytometry
using a FACScan (BD Biosciences). The ratios (%) of Annexin
V-FITC-positive cells identified by flow cytometry using FACSDiva
software (version 6.1.3; BD Biosciences) represented the apoptotic
population.
Statistical analysis
All data were statistically analyzed using SPSS
v19.0 software (IBM Corp.). Data are presented as means ± standard
deviation. Two groups were compared using unpaired Student's
t-tests. Associations between clinical and pathological features
with FOXK1 expression was analyzed with Fisher's exact test.
Correlations between TMEM245 and miR-32, FOXK1 and
miR-32 and FOXK1 and TMEM245 expression were
determined using Pearson's correlation coefficients. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of FOXK1, TMEM245 and
miR-32 in CRC tissues
Levels of miR-32, FOXK1 and TMEM245
mRNA expression in CRC and normal colonic tissues were determined
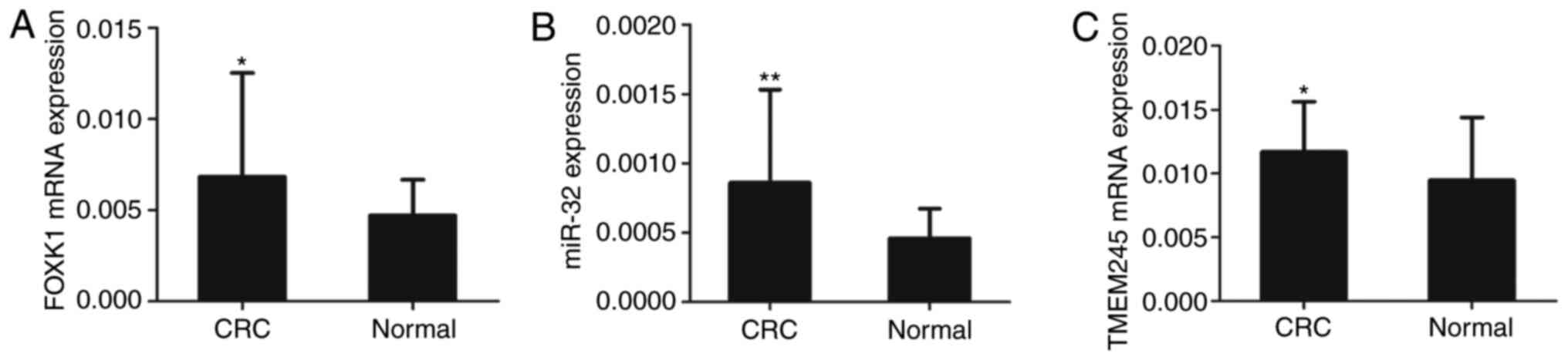
using RT-qPCR. The results showed that the expressions of
FOXK1 (Fig. 2A), miR-32
(Fig. 2B) and TMEM245
(Fig. 2C) were upregulated in CRC
compared with normal tissues (all P<0.05 or P<0.01).
The relationship between increased FOXK1
expression and the clinicopathological features of patients with
CRC was explored. Median FOXK1 expression served as the
cut-off for separating the 35 patients with CRC into groups with
high (n=18) and low (n=17) FOXK1 expression. FOXK1
expression was significantly associated with lymphatic metastasis
and tumor stage (P=0.027 for both; Table II). These results implied a
potential role for FOXK1 in CRC progression.
| Table II.FOXK1 expression and
clinicopathological features of the patients with colorectal
cancer. |
Table II.
FOXK1 expression and
clinicopathological features of the patients with colorectal
cancer.
|
| FOXK1
expression |
|
|---|
|
|
|
|
|---|
| Clinicopathological
features | High, n=18 | Low, n=17 | P-value |
|---|
| Age, years |
|
| 0.315 |
|
<60 | 8 | 11 |
|
|
≥60 | 10 | 6 |
|
| Sex |
|
| 0.471 |
|
Male | 14 | 11 |
|
|
Female | 4 | 6 |
|
| Diameter, cm |
|
| 0.738 |
|
<5 | 9 | 7 |
|
| ≥5 | 9 | 10 |
|
|
Differentiation |
|
| 0.691 |
|
High | 5 | 3 |
|
|
Middle/low | 13 | 14 |
|
| Lymphatic
metastasis |
|
| 0.027a |
|
Positive | 16 | 9 |
|
|
Negative | 2 | 8 |
|
| Distal
metastasis |
|
| >0.999 |
|
Positive | 2 | 2 |
|
|
Negative | 16 | 15 |
|
| Stage |
|
| 0.027a |
|
I–II | 2 | 8 |
|
|
III–IV | 16 | 9 |
|
Correlations between FOXK1, miR-32 and
TMEM245 expression in CRC tissues
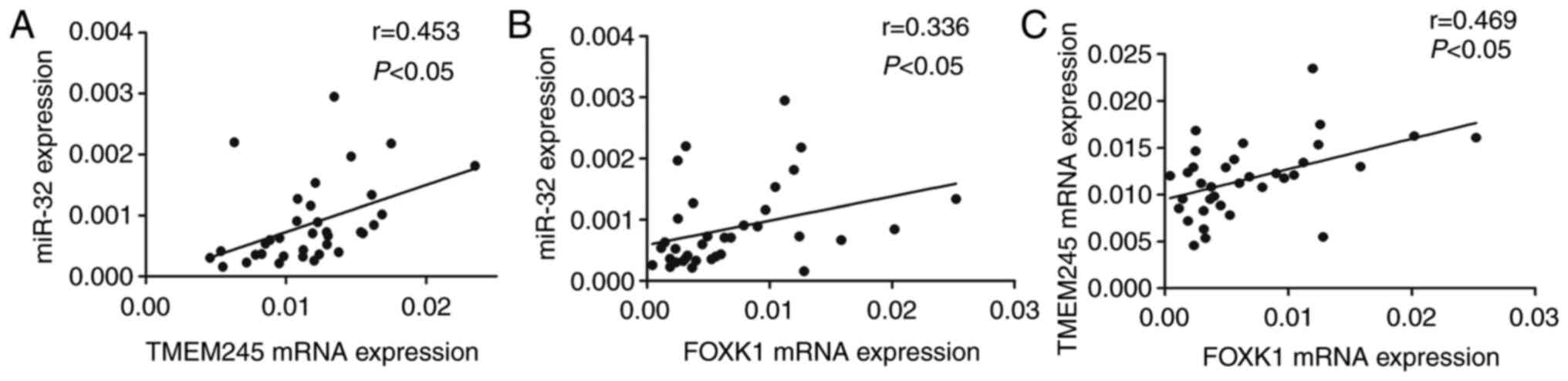
The expression of TMEM245 mRNA and miR-32 was
significantly and positively correlated (r=0.453; P<0.05;
Fig. 3A). Increased FOXK1
mRNA expression was positively correlated with upregulated miR-32
and TMEM245 mRNA (r=0.336 and 0.469, respectively;
P<0.05; Fig. 3B and C). These
results suggest that miR-32 and host gene TMEM245 may be regulated
by FOXK1.
Expression of miR-32, TMEM245 and PTEN
is modulated by FOXK1 in CRC cells
FOXK1 was overexpressed or knocked down in
CRC cells that were respectively transfected with either
pcDNA3.1-FOXK1 or siFOXK1, to determine whether FOXK1 regulated
miR-32, TMEM245 and PTEN expression using RT-qPCR and
western blotting. The level of FOXK1 was significantly increased in
HCT-116 and HT-29 cells transfected with pcDNA3.1-FOXK1 compared
with control cells transfected with the empty vector pcDNA3.1
(P<0.01; Fig. 4A and B). The
level of FOXK1 was significantly decreased after FOXK1-knockdown
compared with control cells transfected with siRNA-NC (all
P<0.05 or P<0.01; Fig. 4C and
D). Fig. 4E-H shows that
FOXK1-overexpression significantly increased miR-32 and
TMEM245 mRNA expression and suppressed PTEN protein levels
compared with the control empty vector (all P<0.05 or
P<0.01). In contrast, FOXK1-knockdown resulted in
significantly decreased miR-32 and TMEM245 mRNA and
increased PTEN protein expression compared with siRNA-NC
transfected cells (all P<0.05 or P<0.01; Fig. 4I-L) without significant changes in
PTEN mRNA expression. Taken together, these results
suggested that FOXK1 could enhance miR-32 and TMEM245 expression,
and suppress PTEN protein expression.
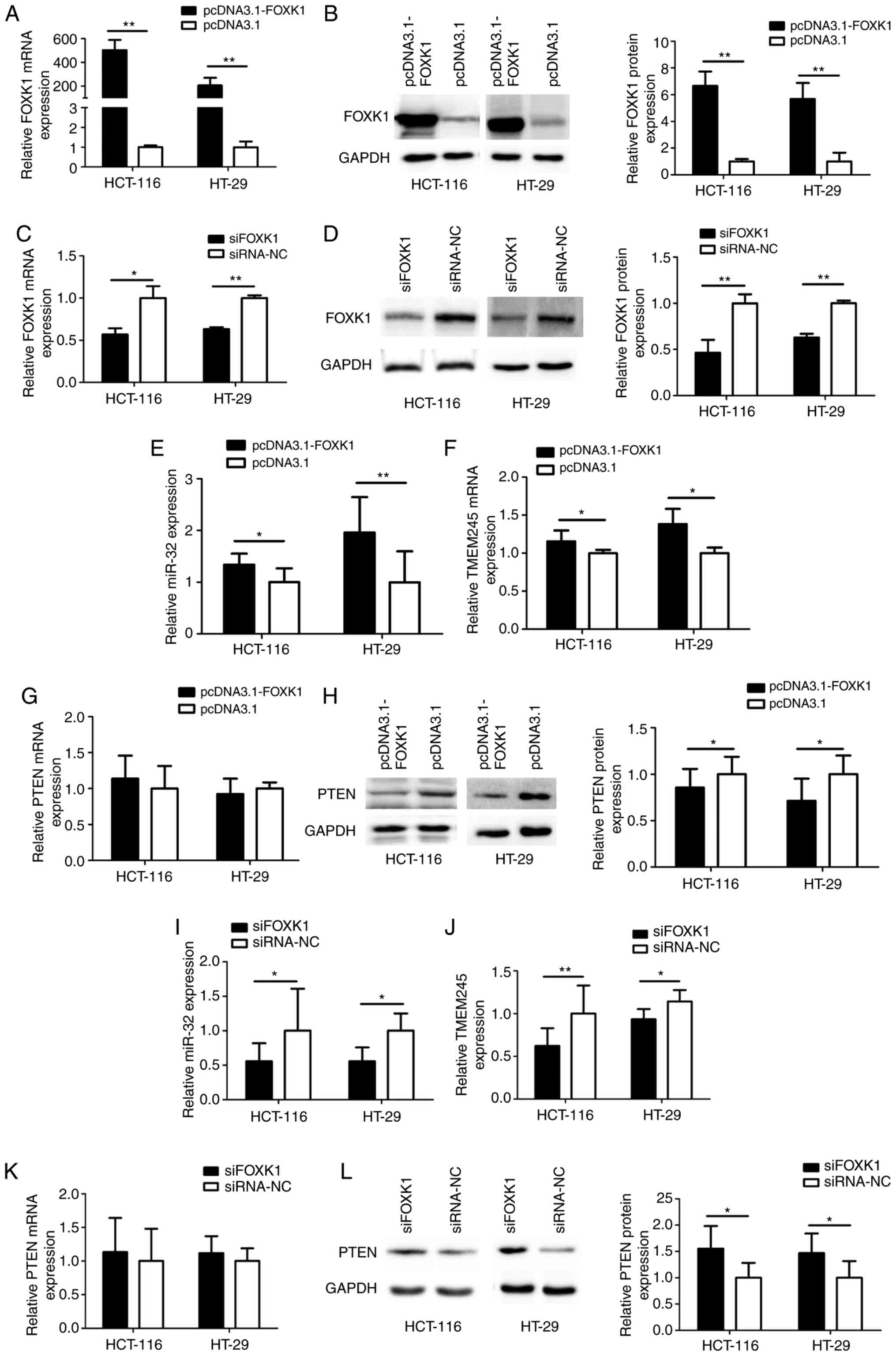 | Figure 4.Expressions of miR-32, TMEM245
and PTEN are modulated by FOXK1 in CRC cells. mRNA and
protein expression was detected using reverse-transcription
quantitative PCR and western blotting, respectively. (A) Expression
of FOXK1 mRNA and (B) FOXK1 protein in CRC cells transfected
with pcDNA3.1. (C) Expression of FOXK1 mRNA and (D) FOXK1
protein in CRC cells transfected with siFOXK1. (E-H) Expression of
miR-32, TMEM245 and PTEN in CRC cells transfected with
pcDNA3.1-FOXK1. (I-L) Expression of miR-32, TMEM245 and PTEN
in CRC cells with FOXK1-knockdown. *P<0.05 and **P<0.01 vs
corresponding control groups. FOXK1, forkhead box K1; TMEM245,
transmembrane protein 245; miR, microRNA; CRC, colorectal cancer;
NC, negative control; si-, short interfering. |
FOXK1 promotes CRC cell
proliferation
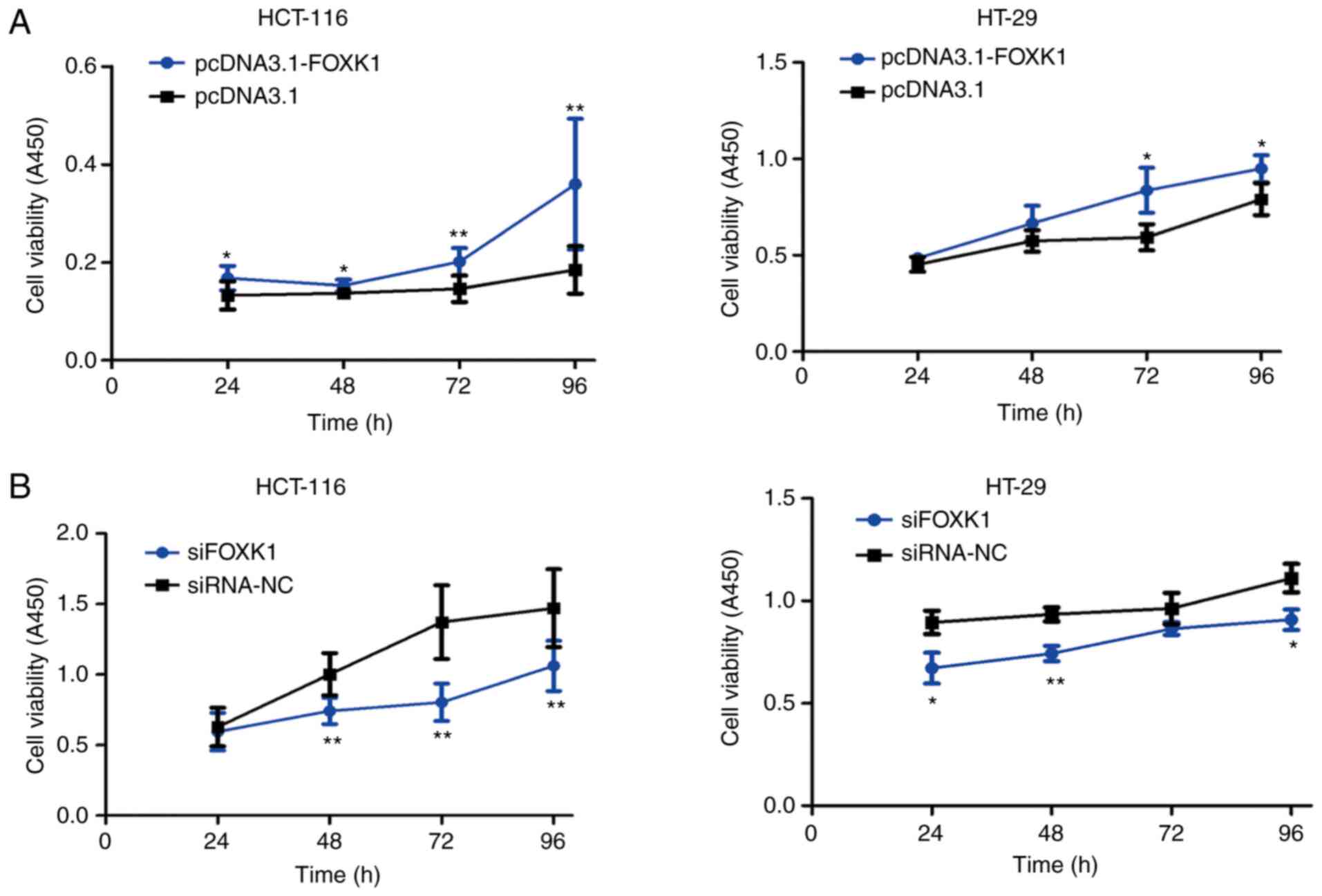
The potential function of FOXK1 in CRC cell
proliferation was investigated using CCK-8 assays. Overexpressed
FOXK1 significantly promoted the proliferation of HCT-116 cells at
24, 48, 72 and 96 h and of HT-29 cells at 72 and 96 h after
transfection (all P<0.05 or P<0.01; Fig. 5A). In contrast, FOXK1
inhibition restricted proliferation of HCT-116 cells at 48, 72 and
96 h and of HT-29 cells at 24, 48 and 96 h after transfection (all
P<0.05 or P<0.01; Fig.
5B).
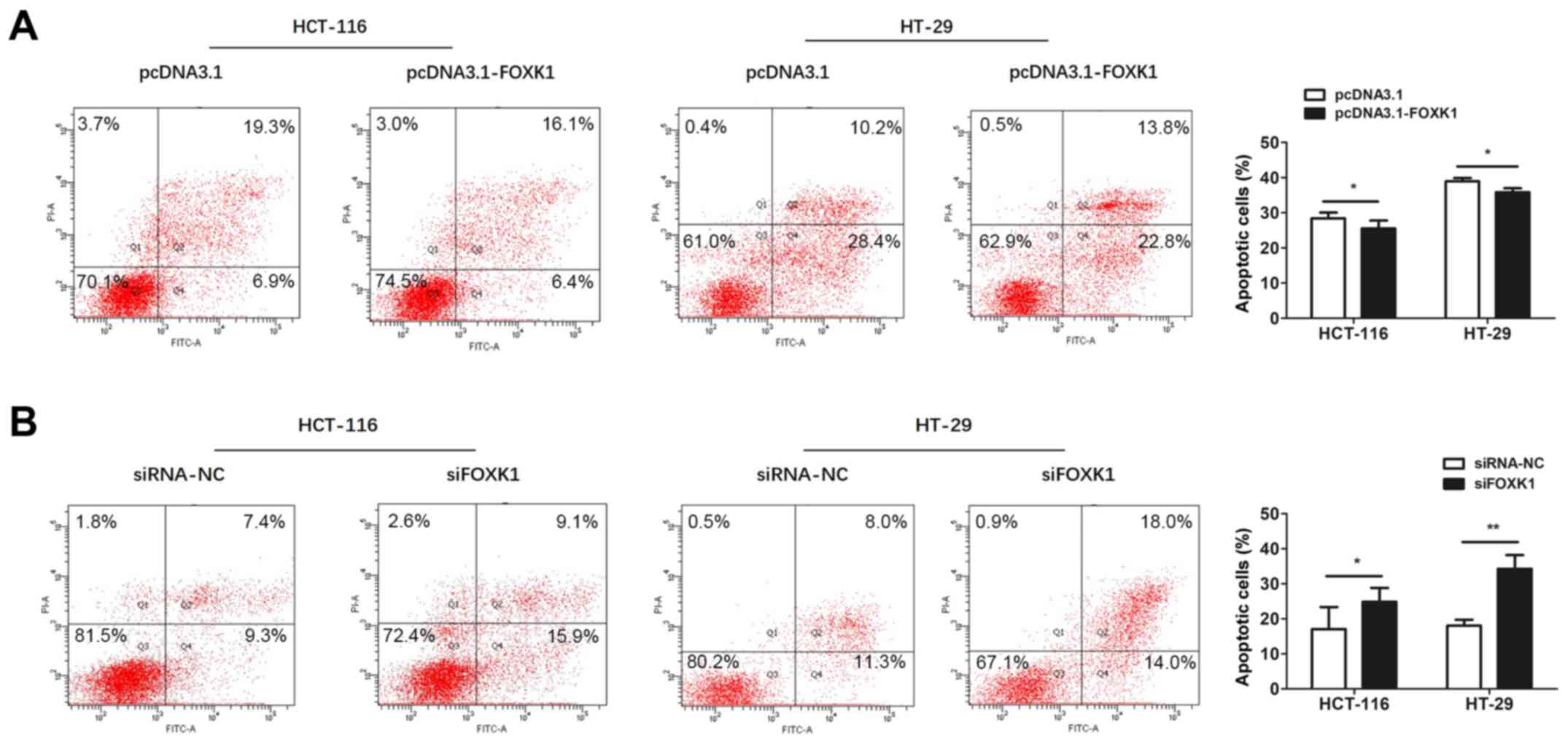
FOXK1 suppresses apoptosis in CRC
cells
Apoptosis was assessed using flow cytometry.
Apoptosis in CRC cells was significantly decreased and increased by
FOXK1-overexpression and knockdown, respectively, compared with
controls (all P<0.05 or P<0.01; Fig. 6). Collectively, these results
suggested that FOXK1 acts as an oncogene in CRC cells.
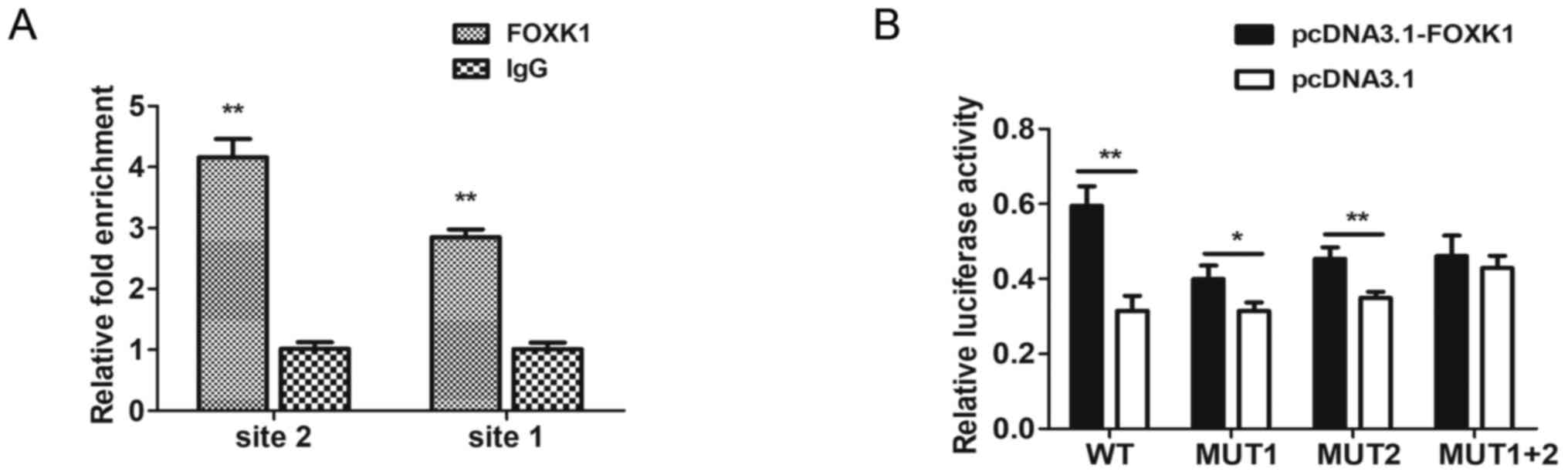
FOXK1 directly binds to the
TMEM245/miR-32 promoter
The findings of the bioinformatics analysis using
JASPAR and HOCOMOCO (RiboBio Co., Ltd.) predicted that FOXK1 could
bind to the core promoter of TMEM245/miR-32. Putative FOXK1-binding
sites one (−90 to −77 bp) and two (−166 to −153 bp) were identified
within the upstream region of the TMEM245/miR-32 gene by
bioinformatics analysis. ChIP-qPCR was used to determine whether
endogenous FOXK1 could directly bind to these sites in CRC cells.
The results revealed that FOXK1 was strongly bound to both sites of
the TMEM245/miR-32 promoter in HCT-116 cells compared with
the IgG control. Fig. 7A shows
substantial enrichment of endogenous FOXK1 at both sites in the
TMEM245/miR-32 promoter, indicating that this promoter was a
direct target of FOXK1.
The relative luciferase activity of
pGL3-promoter-WT, -MUT1 and MUT-2 was significantly increased when
co-transfected with pcDNA3.1-FOXK1 compared with pcDNA3.1 vector
(P<0.05; Fig. 7B). However, there
was no significant difference in relative luciferase activity
between pcDNA3.1-FOXK1 and pcDNA3.1 vector when cotransfected with
pGL3-promoter-MUT1+2 (P>0.05; Fig.
7B). These results indicated that FOXK1 enhanced the
transcriptional activities of the TMEM245/miR-32 promotor
and the involvement of the two binding sites. Collectively, these
data indicated that FOXK1 directly binds to the TMEM245/miR-32
promoter.
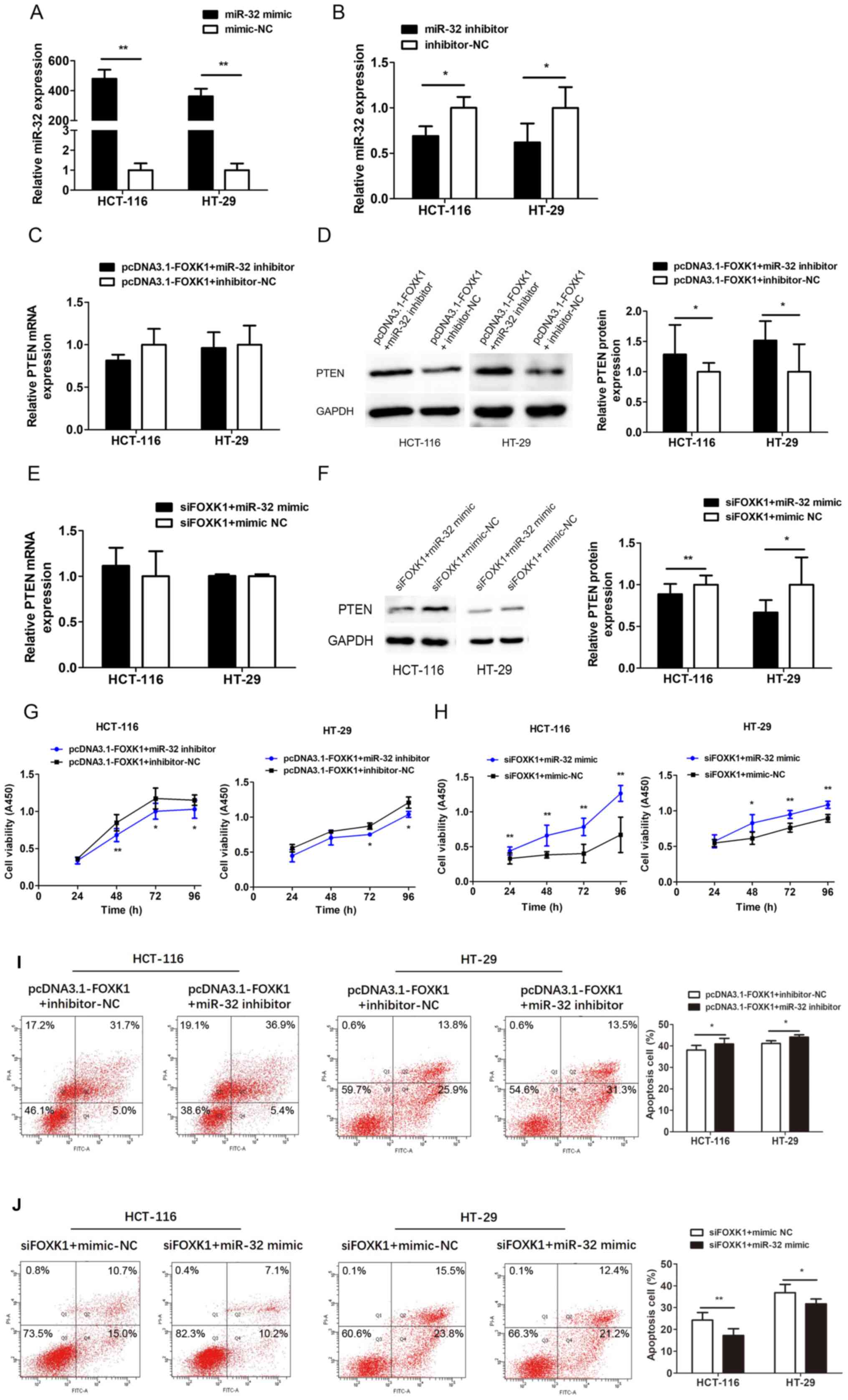
Downregulation of miR-32 reverses the
effects of FOXK1 on PTEN expression and cell proliferation and
apoptosis
miR-32 was explored as a functional target of FOXK1
by co-transfecting HCT-116 and HT-29 cells with pcDNA3.1-FOXK1 and
miR-32 inhibitor, or siFOXK1 and miR-32 mimic. Transfection with
the miR-32 mimic and inhibitor significantly increased and
decreased miR-32 expression, respectively (all P<0.05 or
P<0.01; Fig. 8A and B).
Co-transfection with miR-32 inhibitor partially reversed the
decrease in PTEN expression induced by FOXK1-overexpression
(P<0.05; Fig. 8D), whereas the
miR-32 mimic reversed the PTEN upregulation caused by
FOXK1-knockdown (P<0.05 or P<0.01; Fig. 8F). Levels of PTEN mRNA did not
significantly change (Fig. 8C and
E). Proliferation ability was decreased, whereas apoptosis was
increased after co-transfection with the FOXK1-overexpression
plasmid and miR-32 inhibitor compared with inhibitor-NC (Fig. 8G and I). Proliferation ability was
increased, whereas apoptosis was decreased after co-transfection
with siFOXK1 and miR-32 mimic compared with mimic-NC (Fig. 8H and J). These results indicate that
the promotive effects of FOXK1 on CRC cells are largely mediated by
miR-32 expression and can be reversed by downregulating miR-32.
Discussion
Numerous regulatory mechanisms are involved in the
differential processing of miRNA. Mechanisms that regulate miRNA
expression include transcriptional regulation, such as changes in
host gene expression and hypermethylation of host or miRNA gene
promoters, and post-transcriptional regulation, including changes
in miRNA processing and stability (15). The transcription of miRNA can be
regulated by using TFs that bind to specific promoters (16). For example, the TF p65/NFκB can bind
to the miR-224 promoter and acts as a direct transcriptional
regulator of miR-224 expression, and such binding increases in
cells incubated with lipopolysaccharides, tumor necrosis factor-α
or lymphotoxin-α (17). The myogenic
regulatory factor, MyoD, regulates miR-206 expression by directly
binding to its promoter (18). It
can also directly bind to the miR-182 promoter and upregulate
miR-182 expression (19). The basic
leucine zipper TF, C/EBPβ, binds to the let-7f miRNA promoter and
positively modulates let-7f expression (14). It was determined that FOXK1 was a
potentially interactive TF binding to TMEM245/miR-32
promoter in our previous study (9).
Therefore, the present study aimed to clarify the role of FOXK1 in
miR-32 regulation.
miRNAs are classified as intronic or intergenic
according to their genomic location. Some intronic miRNAs can be
transcribed with their host genes, whereas others are not
co-expressed. The co-transcription of intronic miRNA with host
genes might be regulated by the host gene promoter (20). Baskerville and Bartel (21) showed that intronic miRNAs are closely
associated with their host genes. The upstream regions of pre-miRNA
are considered the promoters for intergenic and intronic miRNA that
are independently transcribed (20).
Lerner et al (22) showed
that the putative tumor suppressor gene, deleted in lymphocytic
leukemia 2 (DLEU2), is the host gene of miR-15a/miR-16-1 and
that Myc binding to two alternative DLEU2 promoters reduces
levels of DLEU2 transcription and mature miR-15a/ −16-1. The
host gene of miR-196b-5p, homeobox protein A10 (HOXA10), is
overexpressed in human gastric cancer tissues, and is positively
correlated with miR-196b-5p expression levels. The expression of
HOXA10 and miR-196b-5p in gastric cancer cells increases
when the HOXA10 promoter is demethylated (23).
Intronic miR-32 is encoded by TMEM245. Levels
of TMEM245 and miR-32 transcripts positively correlate in
prostate tumors (24). The present
study found that TMEM245 mRNA expression was positively
correlated with miR-32 levels in CRC tissues. The evolutionarily
conserved FOX proteins comprise of a TF superfamily that is
characterized by a ‘forkhead’ or ‘winged-helix’ DNA-binding domain,
including FOXK1 and FOXK2 (25). The
vital TF FOXK1 protein regulates numerous biological activities,
including cell cycle progression in myogenic progenitor cells
(26), aerobic glycolysis (27), insulin-like growth factor-1
receptor-mediated signaling involved in cell proliferation and
metabolism (28) and diseases such
as cancer (25). This protein also
plays a crucial role in various cancer types by acting as an
oncogene. Li et al (29)
found that FOXK1 expression is significantly increased in human
hepatocellular carcinoma tissues and cell lines, and that
FOXK1-knockdown significantly suppresses hepatocellular
carcinoma cell proliferation, migration and invasion, in part by
inactivating Wnt/β-catenin signaling. Moreover, FOXK1
expression is increased in various malignancies, including CRC,
gastric cancer, glioma and prostate cancer (30–33).
Huang et al (34) reported
that FOXK1 promotes the epithelial-mesenchymal transition, tumor
invasion and metastasis by transactivating cysteine-rich angiogenic
inducer 61 expression in CRC cells.
The present study found that upregulated FOXK1
expression in CRC tissues was positively correlated with miR-32 or
TMEM245 expression. Furthermore, FOXK1 promoted CRC cell
proliferation and reduced apoptosis, thus acting as an oncogene.
However, the proliferation inhibition of siFOXK1 on HT-29 cells at
72 h was not significant, which may be due to the large differences
within the groups. Bioinformatics analysis predicted two binding
sites for FOXK1 in the TMEM245/miR-32 core promotor region.
Gain- and loss-of-function studies revealed that FOXK1 upregulated
the expression of miR-32 and TMEM245 and downregulated PTEN protein
levels. However, there was no significant change in PTEN mRNA
expression. This may be because the main mechanism of
miR-32-induced PTEN suppression is post-transcriptional, which is
consistent with the results in our previous research (8). The binding of FOXK1 to the
TMEM245/miR-32 promoter was identified using ChIP and
dual-luciferase reporter assays. Thus, the present findings
revealed that FOXK1 binds to the TMEM245/miR-32 promotor and
induces miR-32 expression, leading to increased CRC cell
proliferation. In order to determine the mechanism through which
FOXK1 regulates miR-32 functions in cell proliferation and
apoptosis, PTEN expression was assessed and CCK-8 and Annexin
V-FITC apoptosis assays were conducted after simultaneously
interfering with FOXK1 and miR-32 expression. Knockdown of miR-32
in cells overexpressing FOXK1 resulted in decreased cell
proliferation, increased apoptosis and the upregulation of PTEN
protein. Therefore, the promotive effects of FOXK1 on CRC cell
proliferation were mediated, at least in part, by upregulated
miR-32.
However, there were several limitations in the
present study. As a TF, FOXK1 regulates several other genes,
including p21, CCDC43 and Snail (10,35,36).
Moreover, miRNA can be regulated by multiple TFs as well as
non-coding RNAs, such as long non-coding and circular RNA (37,38).
Thus, FOXK1 or miR-32 might affect the onset and development of CRC
via pathways other than the FOXK1-miR-32-PTEN axis. Interactions
between FOXK1 or miR-32 with other genes remain to be clarified.
Animal experiments should be conducted to further evaluate the
mechanisms of FOXK1 in miR-32 regulation.
The findings of the present study suggested that
FOXK1 expression is increased in CRC tissues and positively
correlates with miR-32 levels. FOXK1 was shown to directly bind the
TMEM245/miR-32 promoter to activate miR-32 expression and
downregulate PTEN. Furthermore, miR-32-knockdown mitigated
FOXK1-promoted CRC cell proliferation and FOXK1-inhibited apoptosis
in vitro. Thus, the FOXK1-miR-32-PTEN signaling axis might
play a crucial role in the pathogenesis and development of CRC.
Acknowledgements
Not applicable.
Funding
This study was supported by The Natural Science
Foundation of Guangdong Province (grant no. 2017A030313546).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Author's contributions
WW designed the study. WW, YC, SY, HY and JY carried
out the experiments. SY collected the samples. WW, YC and JQ
analyzed the data. WW and JQ drafted the manuscript. JQ revised the
manuscript. WW, YC, SY, HY, JY and JQ confirm the authenticity of
all raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The Medical Ethics Committees at the Affiliated
Hospital of Guangdong Medical University (Guangdong, China)
approved this study and written informed consent was obtained from
all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Geng F, Wang Z, Yin H, Yu J and Cao B:
Molecular targeted drugs and treatment of colorectal cancer: Recent
progress and future perspectives. Cancer Biother Radiopharm.
32:149–160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fang JY, Dong HL, Sang XJ, Xie B, Wu KS,
Du PL, Xu ZX, Jia XY and Lin K: Colorectal cancer mortality
characteristics and predictions in China, 1991–2011. Asian Pac J
Cancer Prev. 16:7991–7995. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gu MJ, Huang QC, Bao CZ, Li YJ, Li XQ, Ye
D, Ye ZH, Chen K and Wang JB: Attributable causes of colorectal
cancer in China. BMC Cancer. 18:382018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun L, Xue H, Jiang C, Zhou H, Gu L, Liu
Y, Xu C and Xu Q: LncRNA DQ786243 contributes to proliferation and
metastasis of colorectal cancer both in vitro and in vivo. Biosci
Rep. 36:e003282016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Strubberg AM and Madison BB: MicroRNAs in
the etiology of colorectal cancer: Pathways and clinical
implications. Dis Model Mech. 10:197–214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu W, Yang P, Feng X, Wang H, Qiu Y, Tian
T, He Y, Yu C, Yang J, Ye S and Zhou Y: The relationship between
and clinical signifcance of microRNA-32 and phosphatase and tensin
homologue expression in colorectal cancer. Genes Chromosomes
Cancer. 52:1130–1140. 2013. View Article : Google Scholar
|
|
8
|
Wu W, Yang J, Feng X, Wang H, Ye S, Yang
P, Tan W, Wei G and Zhou Y: MicroRNA-32 (miR-32) regulates
phosphatase and tensin homologue (PTEN) expression and promotes
growth, migration, and invasion in colorectal carcinoma cells. Mol
Cancer. 12:302013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu W, Tan W, Ye S, Zhou Y and Quan J:
Analysis of the promoter region of the human miR-32 gene in
colorectal cancer. Oncol Lett. 17:3743–3750. 2019.PubMed/NCBI
|
|
10
|
Li L, Gong M, Zhao Y, Zhao X and Li Q:
FOXK1 facilitates cell proliferation through regulating the
expression of p21, and promotes metastasis in ovarian cancer.
Oncotarget. 8:70441–70451. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen D, Wang K, Li X, Jiang M, Ni L, Xu B,
Chu Y, Wang W, Wang H, Kang H, et al: FOXK1 plays an oncogenic role
in the development of esophageal cancer. Biochem Biophys Res
Commun. 494:88–94. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu Y, Peng Y, Wu M, Zhang W, Zhang M, Xie
R, Zhang P, Bai Y, Zhao J, Li A, et al: Oncogene FOXK1 enhances
invasion of colorectal carcinoma by inducing epithelial-mesenchymal
transition. Oncotarget. 7:51150–51162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ayyar K and Reddy KVR: Transcription
factor CCAAT/enhancer-binding protein-β upregulates microRNA,
let-7f-1 in human endocervical cells. Am J Reprod Immunol. 78:Epub
2017. Sep 16–2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gulyaeva LF and Kushlinskiy NE: Regulatory
mechanisms of microRNA expression. J Transl Med. 14:1432016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Treiber T, Treiber N, Plessmann U,
Harlander S, Daiß JL, Eichner N, Lehmann G, Schall K, Urlaub H and
Meister G: A compendium of RNA-binding proteins that regulate
microRNA biogenesis. Mol Cell. 66:270–284.e13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Scisciani C, Vossio S, Guerrieri F,
Schinzari V, De Iaco R, D'Onorio de Meo P, Cervello M, Montalto G,
Pollicino T, Raimondo G, et al: Transcriptional regulation of
miR-224 upregulated in human HCCs by NFκB inflammatory pathways. J
Hepatol. 56:855–861. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Koutalianos D, Koutsoulidou A,
Mastroyiannopoulos NP, Furling D and Phylactou LA: MyoD
transcription factor induces myogenesis by inhibiting Twist-1
through miR-206. J Cell Sci. 128:3631–3645. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dodd RD, Sachdeva M, Mito JK, Eward WC,
Brigman BE, Ma Y, Dodd L, Kim Y, Lev D and Kirsch DG: Myogenic
transcription factors regulate pro-metastatic miR-182. Oncogene.
35:1868–1875. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang HM, Kuang S, Xiong X, Gao T, Liu C
and Guo AY: Transcription factor and microRNA co-regulatory loops:
Important regulatory motifs in biological processes and diseases.
Brief Bioinform. 16:45–58. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baskerville S and Bartel DP: Microarray
profiling of microRNAs reveals frequent coexpression with
neighboring miRNAs and host genes. RNA. 11:241–247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lerner M, Harada M, Lovén J, Castro J,
Davis Z, Oscier D, Henriksson M, Sangfelt O, Grandér D and Corcoran
MM: DLEU2, frequently deleted in malignancy, functions as a
critical host gene of the cell cycle inhibitory microRNAs miR-15a
and miR-16-1. Exp Cell Re. 315:2941–2952. 2009. View Article : Google Scholar
|
|
23
|
Shao L, Chen Z, Peng D, Soutto M, Zhu S,
Bates A, Zhang S and El-Rifai W: Methylation of the HOXA10 promoter
directs miR-196b-5p-dependent cell proliferation and invasion of
gastric cancer cells. Mol Cancer Res. 16:696–706. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ambs S, Prueitt RL, Yi M, Hudson RS, Howe
TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, et al:
Genomic profiling of microRNA and messenger RNA reveals deregulated
microRNA expression in prostate cancer. Cancer Res. 68:6162–6170.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y, Ding W, Ge H, Ponnusamy M, Wang Q,
Hao X, Wu W, Zhang Y, Yu W, Ao X and Wang J: FOXK transcription
factors: Regulation and critical role in cancer. Cancer Lett.
458:1–12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi X and Garry DJ: Sin3 interacts with
Foxk1 and regulates myogenic progenitors. Mol Cell Biochem.
366:251–258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sukonina V, Ma H, Zhang W, Bartesaghi S,
Subhash S, Heglind M, Foyn H, Betz MJ, Nilsson D, Lidell ME, et al:
FOXK1 and FOXK2 regulate aerobic glycolysis. Nature. 566:279–283.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sakaguchi M, Cai W, Wang CH, Cederquist
CT, Damasio M, Homan EP, Batista T, Ramirez AK, Gupta MK, Steger M,
et al: FoxK1 and FoxK2 in insulin regulation of cellular and
mitochondrial metabolism. Nat Commun. 10:15822019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li P, Yu Z, He L, Zhou D, Xie S, Hou H and
Geng X: Knockdown of FOXK1 inhibited the proliferation, migration
and invasion in hepatocellular carcinoma cells. Biomed
Pharmacother. 92:270–276. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu M, Wang J, Tang W, Zhan X, Li Y, Peng
Y, Huang X, Bai Y, Zhao J, Li A, et al: FOXK1 interaction with FHL2
promotes proliferation, invasion and metastasis in colorectal
cancer. Oncogenesis. 5:e2712016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang H, Wu X, Xiao Y, Wu L, Peng Y, Tang
W, Liu G, Sun Y, Wang J, Zhu H, et al: Coexpression of FOXK1 and
vimentin promotes EMT, migration, and invasion in gastric cancer
cells. J Mol Med (Berl). 97:163–176. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ji ZG, Jiang HT and Zhang PS: FOXK1
promotes cell growth through activating wnt/β-catenin pathway and
emerges as a novel target of miR-137 in glioma. Am J Transl Res.
10:1784–1792. 2018.PubMed/NCBI
|
|
33
|
Chen F, Xiong W, Dou K and Ran Q:
Knockdown of FOXK1 suppresses proliferation, migration, and
invasion in prostate cancer cells. Oncol Res. 25:1261–1267. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang X, Xiang L, Li Y, Zhao Y, Zhu H,
Xiao Y, Liu M, Wu X, Wang Z, Jiang P, et al: Snail/FOXK1/Cyr61
signaling axis regulates the epithelial-mesenchymal transition and
metastasis in colorectal cancer. Cell Physiol Biochem. 47:590–603.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Liu G, Liu M, Xiang L, Xiao Y, Zhu
H, Wu X, Peng Y, Zhang W, Jiang P, et al: The FOXK1-CCDC43 axis
promotes the invasion and metastasis of colorectal cancer cells.
Cell Physiol Biochem. 51:2547–2563. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu H, Huang S, Zhu X, Zhang W and Zhang X:
FOXK1 promotes glioblastoma proliferation and metastasis through
activation of Snail transcription. Exp Ther Med. 15:3108–3116.
2018.PubMed/NCBI
|
|
37
|
Zhang Z, Li M and Zhang Z: LncRNA MALAT1
modulates oxaliplatin resistance of gastric cancer via sponging
miR-22-3p. Onco Targets Ther. 13:1343–1354. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang L and Ding F: Hsa_circ_0008945
promoted breast cancer progression by targeting miR-338-3p. Onco
Targets Ther. 12:6577–6589. 2019. View Article : Google Scholar : PubMed/NCBI
|