Introduction
Eukaryotic cells have a conserved DNA damage
response (DDR), which comprises cell cycle checkpoints, DNA repair,
apoptosis and senescence, in order to prevent the transfer of
damaged DNA to the next generations. In healthy cells, the DDR not
only acts as a cellular response against DNA damage, but also as an
intrinsic barrier against tumorigenesis (1). Therefore, the disruption of key factors
involved in maintenance of the DDR may result in the initiation of
tumor formation.
The DDR is activated by a large signaling network
cascade controlled by the phosphorylation of ataxia-telangiectasia
mutated (ATM) and ataxia-telangiestasia and Rad3-related (ATR)
kinases, and checkpoint kinase (Chk)1 and Chk2, which are
implicated in the activation of tumor suppressor protein p53. When
activated, p53 induces the activation of p21, which inhibits cell
cycle progression, thus contributing to the temporal arrest of the
cell cycle and the initiation of DNA repair (2,3). The DDR
is terminated by serine/threonine phosphatases, allowing the
recovery of checkpoints and the renewal of cell proliferation
following DNA repair. Wild-type (wt) p53-induced phosphatase 1
(Wip1) is a member of the protein phosphatase 2C family and, as an
important regulator of p53, is responsible for the termination of
the DDR and cellular signals for genotoxic stress (4,5). Wip1
was originally identified as a target of the p53 protein; it is
encoded by the protein phosphatase, Mg2+/Mn2+
dependent 1D (PPM1D) gene and activated upon genotoxic stress
(6). Although Wip1 was initially
identified as a nuclear phosphatase induced by p53 activation,
subsequent studies have demonstrated that other transcription
factors, including estrogen receptor α, c-jun, cAMP response
element-binding protein, E2F and NF-κB, may induce the activation
of Wip1 (7,8). Once activated, Wip1 negatively
regulates p53 activation, either by directly dephosphorylating it
or by activating E3 ubiquitin-protein ligase Mdm2, a p53
antagonist. The targets dephosphorylated by Wip1 also include ATM,
H2A histone family member X (H2AX), Chk1, Chk2 and p38/MAPK, the
key elements of the DDR (9). Wip1 is
now recognized as an oncogenic phosphatase, as it has been found to
be mutated, amplified and overexpressed in several types of human
cancers harboring wt p53 (10–13).
Oncogenic Wip1 essentially leads to the suppression of key elements
of the DDR and prevents the activation of genotoxic stress-induced
cellular responses, such as checkpoint activation, DNA repair,
apoptosis and senescence (14). The
functions of Wip1 have been extensively studied in human solid
tumors harboring functional p53 (15). Thus, Wip1 has emerged as a potential
chemotherapeutic target in solid tumors that may increase
p53-mediated anticancer responses. However, there are limited data
evaluating its potential role in hematological cancers,
particularly in those having an impaired p53 function. Therefore,
in the present study, the role of Wip1 in the regulation of
chemotherapy-induced cellular responses in the p53 mutant (mt)
human ALL Jurkat cell line was investigated.
Materials and methods
Cell culture and drug treatments
The human T-cell acute lymphoblastic leukemia
(T-ALL) cell line (Jurkat; ATCC® TIB-152™), human breast
cancer cell line (MCF-7; ATCC® HTB-22™) and human
foreskin fibroblasts (BJ; ATCC® CRL-2522™) were obtained
from the American Type Culture Collection. The Jurkat cells were
cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal
bovine serum (FBS), 100 IU/ml penicillin and 100 µg/ml streptomycin
(all from Gibco; Thermo Fisher Scientific, Inc.). The MCF-7 and BJ
cells were cultured in high glucose Dulbecco's modified Eagle's
medium (DMEM), supplemented with 10% FBS, 100 U/ml penicillin and
100 g/ml streptomycin (all Gibco; Thermo Fisher Scientific, Inc.).
All cells were maintained at 37°C in a humidified incubator with 5%
CO2. Doxorubicin and etoposide were obtained from Santa
Cruz Biotechnology, Inc. DMSO was used as solvent control in the
relevant experiments.
Genomic DNA isolation and gene copy
number analysis by reverse transcription-quantitative
(RT-q)PCR
Genomic DNA was isolated from Jurkat, MCF-7 and BJ
cells using a GeneJET Genomic DNA Purification kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
TaqMan gene copy number assays, namely PPM1D Gene Copy Number 20X
(Assay ID: Hs05485469_cn) and Ribonuclease P (RNase P) Gene Copy
Number Reference assay 20X (cat. no. 4403326) were from Applied
Biosystems (Thermo Fisher Scientific, Inc.) and were used for qPCR
analysis. RNase P served as the reference gene. The thermocycling
conditions were as follows as: 10-min hot start (95°C), followed by
40 cycles of 2-step qPCR with denaturing for 15 sec at 95°C and
annealing and extension for 1 min at 60°C. The known control sample
(gDNA from BJ fibroblasts carrying 2 alleles) was included in each
reaction plate. All qPCR analyses were performed in triplicate
using gDNA according to the manufacturer's protocol with a StepOne™
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The 2X relative copy number was calculated using StepOne
Software V2.3 (Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the 2−ΔΔCq comparative Cq method (16).
RNA extraction and RT-qPCR
Total RNA was extracted from Jurkat, MCF-7 and BJ
cells using a GeneJet RNA Purification kit (Thermo Fisher
Scientific, Inc.). Complementary DNA (cDNA) was synthesized from
the RNA using a High-Capacity cDNA Reverse Transcription kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.). according to
the manufacturer's instructions. Taqman probes for the PPM1D gene
(Assay ID: Hs01013292_m1) and internal control β-actin (Assay ID:
Hs01060665_g1) were used from commercial assay kits (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: 10 min at 95°C, followed by 40 cycles
at 95°C 15 sec for denaturing and 1 min at 60°C for annealing and
extension. Relative mRNA levels were calculated using StepOne
Software V2.3 according to the 2−ΔΔCq comparative Cq
method (16).
Cell viability assay
Cell viability assays were performed using the water
soluble tetrazolium salt WST-1 (Sigma-Aldrich; Merck KGaA). Jurkat
cells were incubated at 37°C for 24 and 72 h in the absence or
presence of 1 µg/ml doxorubicin or 5 µg/ml etoposide. At the end of
the incubation period, 10 µl WST-1 solution was added to each well
and the cells were incubated for 2 h at 37°C in a humidified
incubator with 5% CO2. The absorbance (A) was read at
450 nm using a Multiskan spectrum microplate reader (Thermo
Labsystems). Cell viability was calculated according to the
following formula: Viability
(%)=[(Asample-Ablank)/(Acontrol-Ablank)]
×100.
Small interfering RNA (siRNA) and
transfection
siRNA oligonucleotides targeting the PPM1D gene
(cat. no. sc-39205) and a control scrambled siRNA oligonucleotide
(cat. no. sc-37007) were purchased from Santa Cruz Biotechnology,
Inc. Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used for the transfection of Jurkat cells. In
brief, 2.5×106 cells/well were cultured overnight at
37°C in 2.5 ml complete RPMI-1640 without antibiotics in 12-well
plates. Then, transfection was conducted at 37°C for 4 h using 40
pmol siRNA/well and Lipofectamine 2000 according to the
manufacturer's protocol. At 16 h post transfection, cells were
treated with appropriate drugs for indicated time points and
subsequently used for Annexin V/7-amino-actinomycin (7AAD),
caspase-3/7 activity, cell cycle and Ser-139 phosphorylated H2AX
(γH2AX) analysis or protein extraction.
Determination of apoptosis using
caspase-3/7 and Annexin V/7AAD assays
Jurkat cells were incubated for 24 or 72 h in the
absence or presence of 1 µg/ml doxorubicin or 5 µg/ml etoposide.
The apoptosis profiles of the cells were determined using
Muse® Annexin V and Dead Cell and Muse Caspase-3/7 kits
(Merck KGaA) according to the manufacturer's instructions.
Quantitative analyses of total apoptotic cells or active
caspase-3/7-positive cells were performed using the Muse Cell
Analyzer (Merck KGaA).
Cell cycle analysis
A Muse Cell Cycle Assay kit (Merck KGaA) was used to
analyze of the cell cycle according to the manufacturer's
instructions. In brief, cells were treated with the aforementioned
concentrations of doxorubicin or etoposide for 24 or 72 h in
triplicate. For senescence assays, cells were treated with lower
concentrations (0.2 µg/ml doxorubicin or 1 µg/ml etoposide) for 72
h. At the end of the incubation period, cells were harvested,
washed with 1X PBS and then fixed in 70% ethanol overnight.
Following this, the cells were treated with 200 µl Muse Cell Cycle
reagent and the cell cycle distribution profiles were determined
using the Muse Cell Analyzer.
Quantitative H2AX assay
Cells were treated in triplicate with 0.2 µg/ml
doxorubicin or 1 µg/ml etoposide for 72 h. At the end of the
incubation period, cells were harvested, washed with 1X PBS and
then assayed for the detection of γH2AX. This was conducted using a
Muse H2A.X Activation Dual Detection Assay (Merck KGaA) according
to the manufacturer's instructions. Cells were analyzed using the
Muse Cell Analyzer to quantify activated (γH2AX) and inactivated
(unphosphorylated) H2AX.
Protein extraction and western blot
analysis
After drug treatment and/or transfection, cells were
lysed in RIPA lysis buffer (150 mM NaCl, 5 mM EDTA pH 8.0, 50 mM
Tris pH 8.0, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS)
supplemented with protease inhibitor cocktail (cOmplete™; Roche
Applied Science) and 1 mM Na3VO4. Total
protein concentration was determined using a BCA assay according to
the manufacturer's instructions (Thermo Fisher Scientific, Inc.)
and 100 mg protein/lane was loaded and separated by SDS-PAGE using
4–15% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad
Laboratories, Inc.) and electro-transferred onto PVDF membranes
(Bio-Rad Laboratories, Inc.). The membranes were blocked in 5%
non-fat dried milk dissolved in 1X TBS-Tween for 1 h at room
temperature and then incubated with primary antibodies at 4°C
overnight and with secondary antibodies at room temperature for 1 h
as previously described (17). Mouse
anti-Wip1 (F-10; cat. no. sc-376257; 1:250), mouse anti-Chk1 (G-4;
cat. no. sc-8408; 1:250), mouse anti-Chk2 primary antibodies (cat.
no. sc-17747; 1:250) and horseradish peroxidase-coupled anti-mouse
(cat. no. sc-2357; 1:1,000) or anti-rabbit (cat. no. sc-2004;
1:1,000) secondary antibodies were purchased from Santa Cruz
Biotechnology, Inc. Polyclonal rabbit anti-phosphorylated (p)-Chk1
(S345; cat. no. 2348), polyclonal rabbit anti-p-Chk2 (T68; cat. no.
2197), monoclonal mouse anti-ATM (cat. no. 2873), rabbit anti-p-ATM
(S1981; cat. no. 5883), monoclonal mouse anti-ATR (cat. no. 2790),
polyclonal rabbit anti-p-ATR (S428; cat. no. 2853) antibody and
polyclonal rabbit anti-H2AX (cat. no. 7631) and anti-γH2AX (S139;
cat. no. 9718) were purchased from Cell Signaling Technology, Inc.
(all 1:1,000). Monoclonal mouse anti-GAPDH (1:5,000; cat. no.
60004-1-Ig) was acquired from ProteinTech Group, Inc. Clarity
Western ECL reagent (Bio-Rad Laboratories, Inc.) was used to
visualize the bands, and a ChemiDoc-ItR2 Digital Imager (Analytik
Jena AG) was used to image and analyze the membranes.
Senescence-associated (SA)
β-galactosidase activity assay
Jurkat cells that were either untransfected or 16 h
post transfection with siRNA were incubated in the absence or
presence of 0.2 µg/ml doxorubicin or 1 µg/ml etoposide for at 37°C
72 h, and the staining protocol was performed as described
previously (17). Following staining
photographs were captured with a digital camera under an inverted
microscope (Leica Microsystems GmbH). Cells were counted and the
percentage of SA-β-gal-positive (blue) cells in the total cell
population was calculated.
Statistical analysis
Origin 8.0 software (OriginLab Corporation) was used
to calculate the mean and standard deviations of the results from
at least three independent experiments each with three replicates.
Statistical analysis was performed using one-way ANOVA followed by
Bonferroni's multiple comparisons tests. P≤0.05 was considered to
indicate a statistically significant difference.
Results
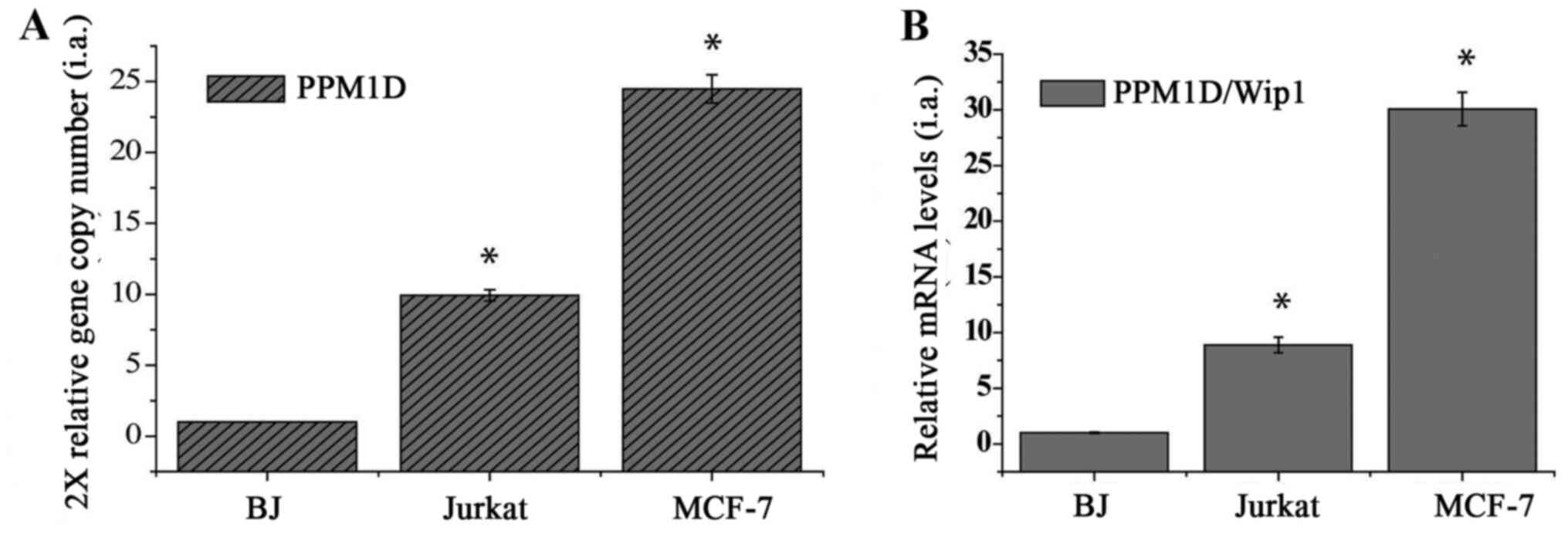
Jurkat cells gain PPM1D amplification
and express increased levels of Wip1 phosphatase
In order to examine the role of Wip1 in
hematological cancers, the Jurkat T-ALL cell line, which harbors mt
p53, was used (18). In human
cancers, oncogenic Wip1 is amplified and overexpressed without
exposure to genotoxic stress (15).
Therefore, the present study initially examined whether the PPM1D
gene is amplified and/or its expression is increased in Jurkat
cells by measuring the gene copy number and mRNA levels using
RT-qPCR. The MCF-7 cell line was used as a positive control,
recognized for PPM1D/Wip1 gene amplification and overexpression
(19). In addition, BJ human normal
diploid fibroblasts were used as a negative control. As shown in
Fig. 1A, the relative gene copy
number of the PPM1D gene in Jurkat cells was significantly
(~10-fold) higher than that in diploid BJ fibroblasts. The MCF-7
cells exhibited a higher copy number and mRNA level compared with
the BJ and Jurkat cells (Fig. 1A).
The relative mRNA level of Wip1 in Jurkat cells was significantly
(~4-fold) higher than that in BJ cells, but lower than that in
MCF-7 cells (Fig. 1B). These results
demonstrate that Jurkat cells harbor PPM1D amplification and
express upregulated levels of Wip1 mRNA without genotoxic
stress.
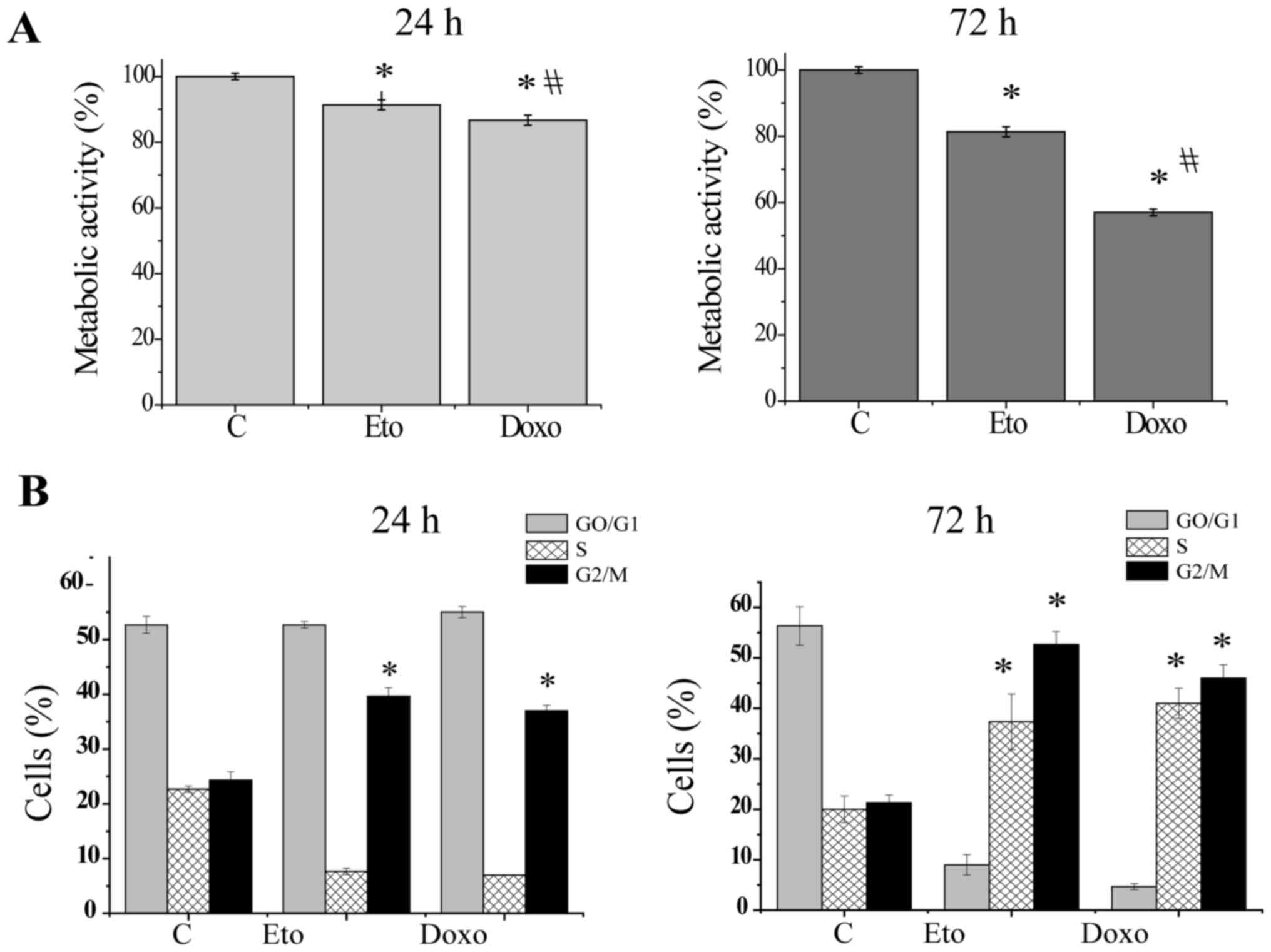
Etoposide and doxorubicin induce cell
cycle arrest at the G2/M phase and decrease the viability of Jurkat
cells
Subsequently, whether increased Wip1 expression
affects the induction of genotoxic stress-mediated cellular
responses in p53 mt Jurkat cells in a similar manner to that in wt
p53 tumors was investigated. The DNA-damaging anticancer agents
etoposide and doxorubicin were used to induce genotoxic stress and
the viability of the Jurkat cells was then examined. As shown in
Fig. 2A, following exposure to
etoposide or doxorubicin for 24 h, a slight but significant
reduction in cell viability was observed, whereas exposure for 72 h
caused a strong and significant reduction in viability (Fig. 2A). In addition, the proportion of
cells in different cell cycle phases after treatment with etoposide
or doxorubicin was examined. As shown in Figs. 2B and S1, 24 h of treatment with etoposide or
doxorubicin induced only a slight increase in the proportion of
cells in the G2 phase, whereas 72 h of treatment strongly induced
the accumulation of cells in the S and G2/M phases. These results
suggest that upregulated Wip1 expression did not prevent cell cycle
arrest or the induction of cell death in Jurkat cells in response
to etoposide or doxorubicin.
Etoposide and doxorubicin induce the
apoptosis of Jurkat cells
Whether the decreased cell viability observed
following treatment with etoposide or doxorubicin was due to the
induction of an apoptotic response was then examined. As determined
by Annexin V/7AAD analysis, treatment with etoposide or doxorubicin
for 24 h induced a modest increase in apoptosis compared with that
in the untreated control (Figs. 3A
and S2A). By contrast, 72 h of
treatment with etoposide or doxorubicin induced a significant
increase in apoptosis to 27±1 and 41±3%, respectively, in Jurkat
cells (Figs. 3A and S2A). Similar results were also obtained
using the caspase-3/7 activity assay. Following 24 h of treatment
of the cells with etoposide or doxorubicin, only a small increase
in the amount of active caspase-3/7 was observed, whereas 72 h of
treatment induced a strong and significant increase (Figs. 3B and S2B). In addition, the phosphorylation
status and total expression levels of known targets of Wip1, namely
ATM, ATR, Chk1, Chk2 and H2AX, were analyzed. Upon treatment with
etoposide or doxorubicin for 72 h, the total protein levels of ATM,
ATR, Chk1 and Chk2 were not markedly altered, and only weak
phosphorylation levels of ATM (S1981), Chk1 (S345) and Chk2 (T68)
were detectable. The phosphorylation of ATR was not detectable
(data not shown). Notably, the phosphorylation of γH2AX (S139) was
increased in response to treatment with etoposide or doxorubicin
(Fig. 3C).
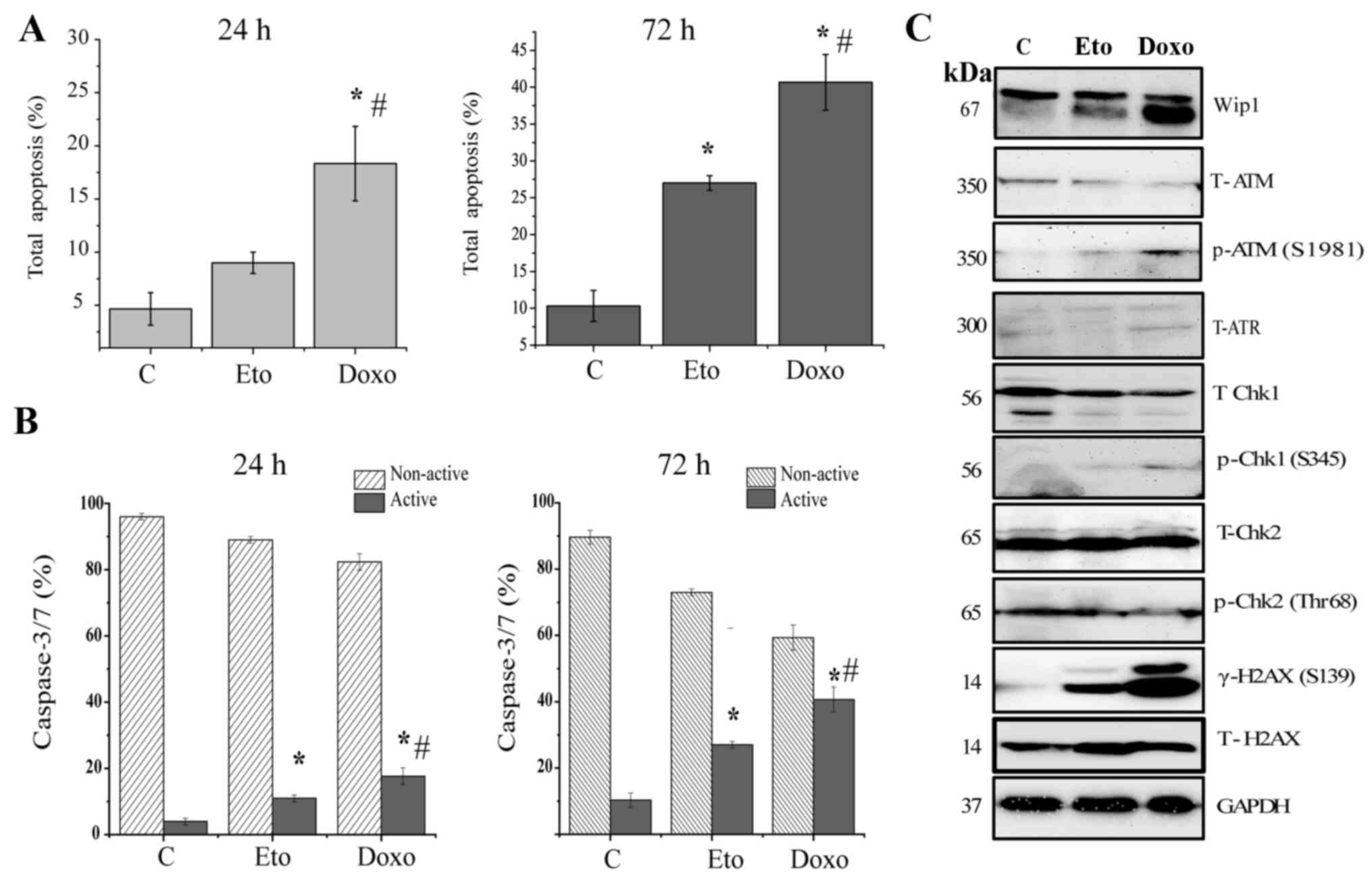 | Figure 3.DNA damage response signaling is
impaired in Jurkat cells, but the induction of apoptosis is
retained. Jurkat cells treated with DMSO, 1 µg/ml Doxo or 5 µg/ml
Eto for 24 h or 72 h were analyzed for (A) apoptosis using Annexin
V/7AAD and (B) caspase-3/7 activity using a Muse Cell Analyzer, and
(C) subjected to the western blot analysis of Wip1, p-ATM, T-ATM,
ATR, p-Chk1, T-Chk1, p-Chk2, T-Chk2, gH2AX and H2AX. GAPDH was used
as the loading control. Data shown are the means ± SD of three
independent experiments. The statistical significance of
differences in the data was analyzed using one-way ANOVA followed
with Bonferroni multiple comparison tests. *P≤0.001 vs. C;
#P≤0.001 vs. Eto. C, DMSO control; Doxo, doxorubicin;
Eto, etoposide; Wip1, p53-induced phosphatase 1; p-, phospho-; T-,
total; ATM, ataxia-telangiectasia mutated; ATR,
ataxia-telangiestasia and Rad3-related; Chk, checkpoint kinase;
H2AX, H2A histone family member X; gH2AX, p-H2AX (S139). |
Etoposide and doxorubicin induce
senescence in Jurkat cells
Since oncogenic Wip1 is known to be involved in the
negative regulation of senescence in response to DNA damage
(15), the present study examined
whether Jurkat cells are capable of undergoing senescence in
response to etoposide or doxorubicin, despite the increased Wip1
activity. In order to avoid the induction of apoptosis, lower
concentrations of etoposide or doxorubicin were used for the
induction of senescence. Accordingly, the effects of the lower
concentrations of etoposide or doxorubicin on cell viability were
examined. As shown in Fig. 4A, no
significant reductions in cell viability occurred following the two
treatments and the majority of cells were viable. One of the
hallmarks of senescence is cell cycle arrest; hence, the cell cycle
distribution of Jurkat cells following 72 h of exposure to the
lower concentrations of etoposide and doxorubicin was examined. As
shown in Fig. 4B, etoposide or
doxorubicin treatment led to the marked and significant arrest of
Jurkat cells in the G2 phase of the cell cycle (Figs. 4B and S3A). In addition, the levels of the
phosphorylated protein γH2AX, which is another senescence marker,
were measured. As shown in Figs. 4C
and S3B, the γH2AX levels were
significantly increased in response to 72 h of etoposide or
doxorubicin treatment. In addition, the etoposide- or
doxorubicin-treated cells were stained for detection of the
senescence marker SA-β-galactosidase. Jurkat cells were positive
for SA-β-galactosidase activity, indicating that both etoposide and
doxorubicin induced senescence (Fig.
4D). Since the induction of senescence is mediated by DDR
signaling, the present study examined whether key elements of the
DDR signaling pathway were phosphorylated. The results were similar
to those for apoptosis; weak levels of p-ATM (S1981), p-Chk1 (S345)
and p-Chk2 (T68) were detectable, whereas the total levels of the
proteins were not altered (Fig.
4E).
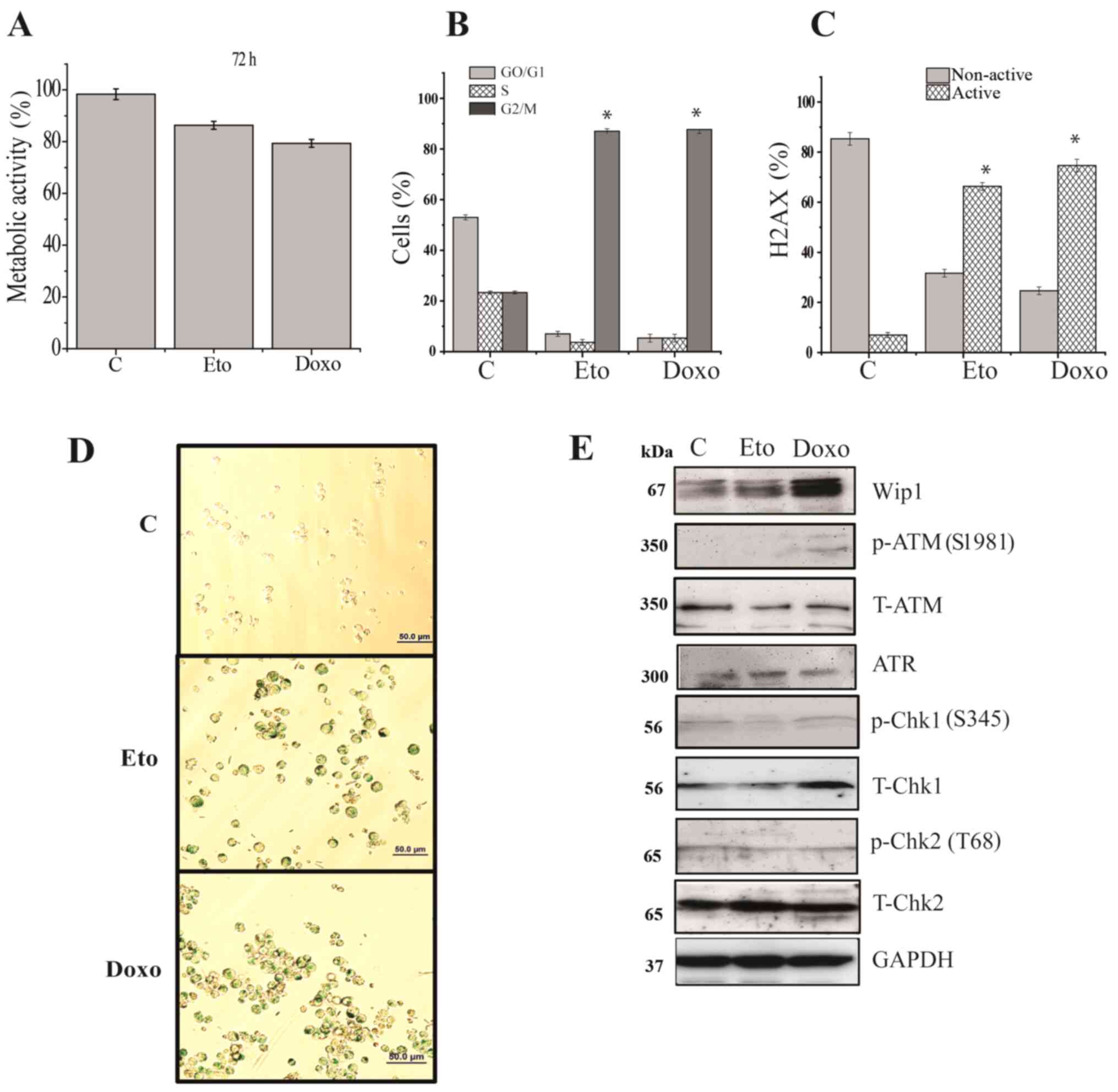 | Figure 4.Induction of senescence is unaffected
by increased Wip1 in Jurkat cells. Jurkat cells were treated with
DMSO, 0.2 µg/ml Doxo or 1 µg/ml Eto for 72 h and the induction of
senescence was assayed by measuring (A) cell viability by WST-1
assay, (B) the cell cycle profile and (C) the amount of active H2AX
[γH2AX; p-H2AX (S139)] and non-active (unphosphorylated) H2AX. (D)
Staining of cells for senescence-associated β-galactosidase
activity. Scale bar, 50 mm. (E) Western blot analysis of Wip1,
p-ATM, T-ATM, ATR, p-Chk1, T-Chk1, p-Chk2 and T-Chk2. GAPDH was
used as the loading control. Data shown are the means ± SD of three
independent experiments. The statistical significance of
differences in the data was analyzed by one-way ANOVA followed by
Bonferroni multiple comparison tests. *P≤0.001 vs. C. C, DMSO
control; Doxo, doxorubicin; Eto, etoposide; H2AX, H2A histone
family member X; Wip1, p53-induced phosphatase 1; p-, phospho-; T-,
total; ATM, ataxia-telangiectasia mutated; ATR,
ataxia-telangiestasia and Rad3-related; Chk, checkpoint kinase. |
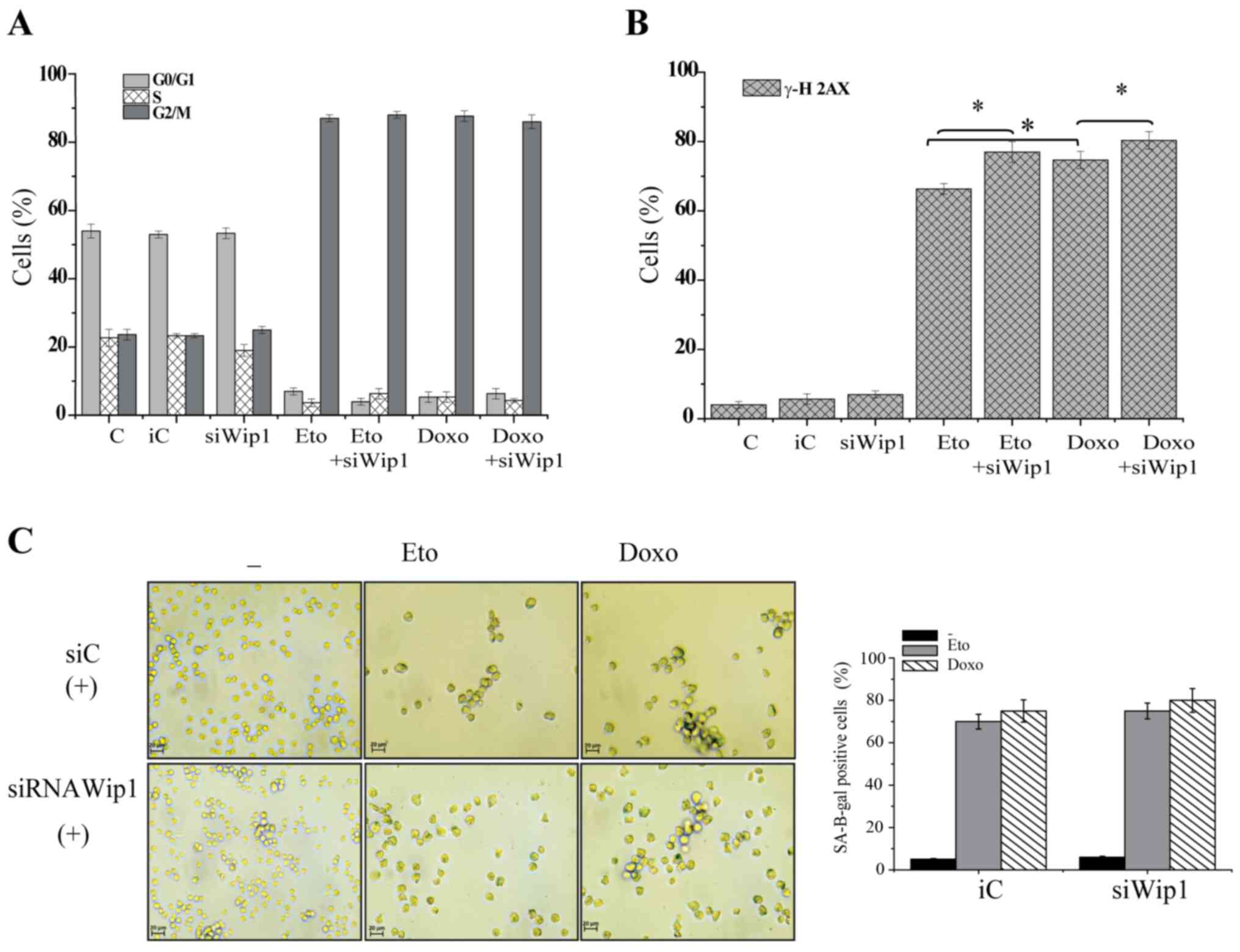
Knockdown of Wip1 restores DDR
signaling, but decreases apoptosis
To gain further insight into the function of Wip1 in
p53 mt Jurkat cells, Wip1 was knocked down and the cellular
responses were further examined. The expression of Wip1 protein was
markedly decreased following the siRNA-mediated knockdown of Wip1
(Fig. 5A). Key elements of DDR
signaling were examined, and western blotting revealed that the
knockdown of Wip1 clearly increased the phosphorylation of ATM
(S1981), Chk1 (S345), Chk2 (T68) and ATR (S428) in the absence of
genotoxic stress. A slight increase in the total protein levels of
ATR, Chk1 and Chk2 was also visible compared with the respective
levels in the cells transfected with control siRNA (Fig. 5A). Whether the knockdown of Wip1
affected the cell cycle status of Jurkat cells in response to
etoposide or doxorubicin treatment was then determined. Notably, no
significant changes were detected in the proportions of cells in
different cell cycle stages when Wip1 was knocked down. Etoposide
or doxorubicin induced G2 cell cycle arrest in a similar manner to
that in the control cells, regardless of whether the cells were
transfected with control siRNA or siRNA targeting Wip1 (Figs. 5B and S4). Subsequently, the effect of the
knockdown of Wip1 on the apoptosis induced by etoposide or
doxorubicin was evaluated. The results demonstrated that the levels
of apoptosis induced by etoposide or doxorubicin were significantly
decreased when Wip1 was knocked down compared with those in the
cells transfected with control siRNA (Figs. 5C and S5A). The amount of total apoptosis was
decreased by ~10% in response to etoposide and doxorubicin
treatment when Wip1 was knocked down. Similar data were obtained in
the caspase-3/7 activity assay. The knockdown of Wip1 significantly
decreased the amount of activated caspase-3/7 induced in response
to etoposide or doxorubicin treatment (Figs. 5D and S5B).
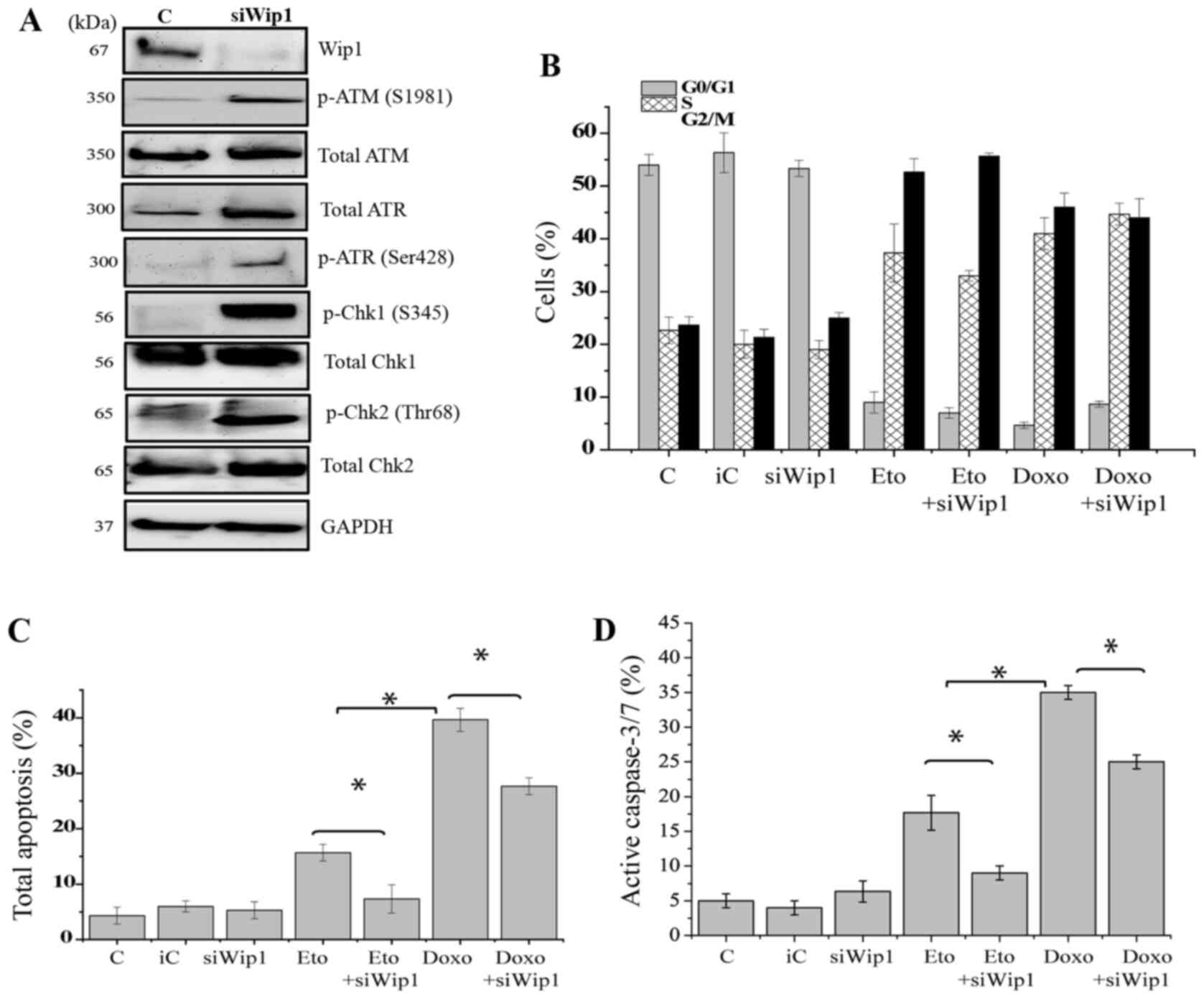 | Figure 5.Knockdown of Wip1 restores DNA damage
response signaling but decreases apoptosis. Jurkat cells were
transfected with siWip1 or siC siRNA. (A) The cells were subjected
to western blot analysis of Wip1, p-ATM, T-ATM, p-ATR, T-ATR,
p-Chk1, T-Chk1, p-Chk2 and T-Chk2. GAPDH was used as the loading
control. siRNA. The transfected cells were treated with DMSO, 1
µg/ml Doxo or 5 µg/ml Eto for 72 h and subjected to (B) cell cycle
analysis (C) Annexin/7AAD staining for apoptosis analysis and (D)
caspase-3/7 activity assay. Data are expressed the as means ± SD of
three independent experiments. The statistical significance of
differences in the data was analyzed using one-way ANOVA followed
by Bonferroni multiple comparison tests. *P≤0.001 as indicated.
Wip1, p53-induced phosphatase 1; siWip1, siRNA targeting Wip1; siC,
inverted siRNA control that does not target any gene; siRNA, small
interfering RNA; C, DMSO control; Doxo, doxorubicin; Eto,
etoposide; p-, phospho-; T-, total; ATM, ataxia-telangiectasia
mutated; ATR, ataxia-telangiestasia and Rad3-related; Chk,
checkpoint kinase. |
Knockdown of Wip1 does not promote the
induction of senescence
The effect of knocking down Wip1 on senescence and
cell cycle arrest in response to etoposide or doxorubicin treatment
in Jurkat cells was investigated. As shown in Figs. 6A and S6A, the knockdown of Wip1 did not alter
the cell cycle status of Jurkat cells in response to etoposide or
doxorubicin. No differences in the proportion of cells in each cell
cycle phase between the Wip1 siRNA- or control siRNA-transfected
cells were observed (Fig. 6A). In
addition, the phosphorylation of γH2AX was measured quantitatively.
Notably, following the knockdown of Wip1, 72 h of exposure to
etoposide or doxorubicin increased the levels of the phosphorylated
protein γH2AX (Figs. 6B and S6B). The SA β-galactosidase activity of
Jurkat cells in response to etoposide or doxorubicin treatment was
also evaluated when Wip1 was knocked down. The results revealed
that the cells in which Wip1 was knocked down were capable of
undergoing senescence, and no significant changes were detected in
the SA β-galactosidase positivity of the cells (Fig. 6C). These results suggest that the
knockdown of Wip1 activity restores DDR signaling and decreases
apoptosis, but does not affect the cell cycle or senescence of p53
mt Jurkat cells.
Discussion
The function of the p53 tumor suppressor gene is
impaired in more than half of human cancers (20), whereas in cancers harboring wt p53,
mechanisms have developed to bypass the tumor suppressor function
of p53 (21). The increased activity
of serine threonine phosphatase Wip1 is recognized as one of the
mechanisms by which the functions of p53 associated with genotoxic
stress responses are bypassed (22).
In healthy cells, upon genotoxic stress, Wip1 is activated and
provides feedback inhibition to p53 that terminates DDR signaling.
By contrast, Wip1 is amplified in cancer cells; it is upregulated
and acts as an oncogene, suppressing DDR and p53 activation.
Oncogenic Wip1 has been extensively studied and is accepted as a
therapeutic target in human solid tumors harboring functional p53
(15). However, its function in
p53-impaired tumors has rarely been evaluated (23). Thus, the present study aimed to
evaluate the role of Wip1 in the regulation of chemotherapy-induced
cellular responses in the p53 mt human ALL cell line Jurkat
(18). To the best of our knowledge,
the present study is the first to demonstrate that p53 mt Jurkat
cells exhibit PPM1D gene amplification and increased expression of
Wip1 phosphatase. The increased Wip1 expression enhances the
apoptosis and senescence sensitivity of Jurkat cells by attenuating
DDR signaling and dephosphorylating ATM, ATR, Chk1 and Chk2. By
contrast, the knockdown of Wip1 restores DDR signaling, but
decreases the sensitivity of Jurkat cells to chemotherapeutic
agents.
This conclusion was confirmed by several lines of
evidence. First, evidence was provided by gene copy number and gene
expression analysis, which demonstrated that Jurkat cells exhibit
PPM1D amplification and express high levels of Wip1 mRNA in the
absence of genotoxic stress. Second, cell cycle analysis
demonstrated that etoposide and doxorubicin each caused a
significant increase in the accumulation of cells in the S and G2/M
phases. More importantly, it demonstrated that the cell cycle
status of Jurkat cells in response to etoposide or doxorubicin
treatment was not altered by the knockdown of Wip1. In a previous
study on neural progenitor cells, it was observed that Wip1 did not
influence the cell cycle status of p53 knockout neural progenitor
cells, but it did affect it in p53 wt cells (24). Furthermore, another study reported
that Wip1 did not affect the cell cycle distribution of p53- or
p21-knockout-MCF7 cells, suggesting that the effects of Wip1 on
cell cycle distribution depend mainly on p53 and p21 activity
(13). Hence, the findings of the
present study are in line with previous research.
An important finding of the present study is that
upregulated expression of Wip1 enhances the apoptotic sensitivity
of Jurkat cells to etoposide and doxorubicin treatment. This was
confirmed by Annexin V/7AAD and caspase-3/7 activity measurements,
which showed that the knockdown of Wip1 significantly decreased the
levels of apoptosis in response to etoposide or doxorubicin.
Previous studies have demonstrated that targeting Wip1 enhances
sensitivity to chemotherapy and promotes apoptosis in human cancers
harboring wt p53. However, contradictory data have been reported
for p53-deficient tumors (23,25). For
example, the study conducted by Goloudina et al (23) demonstrated that the inhibition of
Wip1 increased the sensitivity of wt p53 HCT116 colon cancer cells
to cisplatin-induced apoptosis, but did not have such an effect on
p53−/− HCT116 or Saos-2 cells. The study also
demonstrated that in response to anticancer drugs, Wip1
overexpression promoted apoptosis via the induction of Bax through
activation of the transcription factor RUNX2 in cells with inactive
p53. These data suggest that Wip1 functions as a sensitization
factor to anticancer drugs in p53 mt Jurkat cells.
The present study provides evidence that the
activation of DDR signaling is attenuated in Jurkat cells in
response to etoposide or doxorubicin. Therefore, it is suggested
that the increased expression of Wip1 phosphatase in Jurkat cells
may be involved in the suppression of DDR signaling. This
hypothesis was confirmed by the results demonstrating that the
knockdown of Wip1 increased the phosphorylation of ATM, ATR, Chk1
and Chk2, even in the control cells without any genotoxic
stress.
Another noteworthy finding of the present study is
that γH2AX levels were increased, although the phosphorylation
status of other targets of Wip1, namely ATM, ATR, Chk1 and Chk2,
was not altered in response to etoposide or doxorubicin treatment.
Previous studies have demonstrated that Wip1 targets and
dephosphorylates key elements of the DDR, including ATM, ATR, Chk1
and Chk2 kinases, as well as γH2AX. The phosphorylation of H2AX on
serine 139 is induced in response to DNA damage and is designated
as γH2AX. The phosphorylation of H2AX to form γH2AX has been
specifically recognized as a marker for the generation of DNA
double-strand breaks (26).
Generally, ATM kinase is considered to be the main physiological
mediator of the phosphorylation of H2AX in response to DNA damage.
However, studies have suggested that H2AX can also be
phosphorylated by other phosphoinositide 3-kinase-associated
protein kinases, including ATR and/or DNA-dependent protein kinase
(DNA-PKc) (27,28). Thus, since the present study did not
detect any marked phosphorylation of ATM or ATR, it is possible
that other kinases, such as DNA-PKc, may be responsible for the
increased phosphorylation of γH2AX in response to etoposide or
doxorubicin treatment. The data demonstrating that the knockdown of
Wip1 significantly increased the phosphorylation of ATM and ATR in
control cells, but not the phosphorylation of H2AX in a similar
proportion, support the hypothesis.
In healthy cells, the DDR maintains the integrity of
the genome and defects in the DDR result in damaged DNA being
unrepaired, which ultimately leads to the accumulation of
mutations, genomic instability and cancer initiation. Indeed,
previous research has indicated that deficiencies in the DDR
frequently occur in human cancers. However, DDR defects also
provide targetable susceptibilities that are relatively specific to
cancer cells, which may be exploited for clinical benefit with the
use of DDR inhibitors (29). In line
with previous findings, the present study demonstrated that the
knockdown of Wip1 increased DDR activity, but decreased apoptotic
sensitivity, also suggesting that apoptotic resistance may be due
to increased DDR signaling and DNA repair, as previously reported
(29). However, based on the current
findings that the knockdown of Wip1 increased the phosphorylation
of ATM, ATR, Chk1 and Chk2 without any genotoxic stress, it may be
deduced that the increased expression of Wip1 is an early event
occurring during the tumorigenesis of Jurkat cells in order to
bypass the DDR.
DNA damage-induced senescence is known as another
important cellular response, besides apoptosis, that is activated
by chemotherapeutic agents (30). In
particular, treatment with sublethal concentrations of conventional
DNA-damaging anticancer agents readily induces premature senescence
in cancer cells (23,30). Previous research has demonstrated the
negative effects of oncogenic Wip1 on the induction of senescence
by chemotherapeutic agents in wt p53 cell lines (13). However, other research has provided
contrasting data, suggesting that Wip1 phosphatase is downregulated
during persistent DNA damage and p53-dependent senescence. The
inhibitory effects of Wip1 on the induction of senescence are
mostly dependent on p53 and p21 activity (31). The present study provides evidence
that, despite the increased expression of Wip1, etoposide and
doxorubicin induced senescence in p53 mt Jurkat cells. Furthermore,
the induction of senescence was not affected by the knockdown of
Wip1, and Wip1 was not downregulated during senescence. In Jurkat
cells, the induction of senescence was independent of p53 and,
thus, Wip1 was ineffective in this response.
In conclusion, the present study demonstrated that
p53 mt Jurkat cells exhibit amplification of the PPM1D gene and the
upregulated expression of Wip1 phosphatase. The increased
expression of Wip1 attenuates DDR signaling by dephosphorylating
ATM, ATR, Chk1 and Chk2; however, the ability of chemotherapeutic
agents to induce apoptosis and senescence is retained. Thus, the
present study, to the best of our knowledge, is the first to
demonstrate that increased Wip1 expression enhances the sensitivity
of the p53 mt ALL cell line to chemotherapy-induced apoptosis,
unlike the effect observed in solid tumors with wt p53. The present
study highlights the importance of careful consideration when
devising future treatment strategies that aim to manipulate or
target Wip1, as targeting Wip1 in human cancers lacking p53 may not
yield the same results as in tumors harboring wt p53.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Aydın Adnan
Menderes University, Scientific Research Projects Foundation (grant
no. TPF1-16029) and The Scientific and Technological Research
Council of Turkey (TUBITAK; grant no. 214S200).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MKE designed the study, performed the experiments,
analyzed the data, wrote the manuscript, provided funding and
obtained the grants. NBK and HP performed the experiments and
analyzed the data. NBK and MKE confirm the authenticity of all the
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
The ORCIDs of the authors are as follows: Mehtap
Kilic Eren, https://orcid.org/0000-0003-3811-9819; Nur Betül
Kartal, https://orcid.org/0000-0003-1750-1672; Hatice
Pilevneli, https://orcid.org//0000-0003-1455-7283.
References
|
1
|
Bartek J, Bartkova J and Lukas J: DNA
damage signalling guards against activated oncogenes and tumour
progression. Oncogene. 26:7773–7779. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Halazonetis TD, Gorgoulis VG and Bartek J:
An oncogene-induced DNA damage model for cancer development.
Science. 319:1352–1355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shimada M and Nakanishi M: Response to DNA
damage: Why do we need to focus on protein phosphatases? Front
Oncol. 3:82013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang ZP, Tian Y and Lin J: Role of
wild-type p53-induced phosphatase 1 in cancer. Oncol Lett.
14:3893–3898. 2107. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fiscella M, Zhang HL, Fan S, Sakaguchi K,
Shen S, Mercer EW, Vande Woude GF, O'Connor MP and Appella E: Wip1,
a novel human protein phosphatase that is induced in response to
ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci
USA. 94:6048–6053. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chew J, Biswas S, Shreeram S, Humaidi M,
Wong ET, Dhillion MK, Teo H, Hazra A, Fang CC, López-Collazo E, et
al: WIP1 phosphatase is a negative regulator of NF-kappaB
signalling. Nat Cell Biol. 11:659–666. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Le Guezennec X and Bulavin DV: WIP1
phosphatase at the crossroads of cancer and aging. Trends Biochem
Sci. 35:109–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lowe J, Cha H, Lee MO, Mazur SJ, Appella E
and Fornace AJ Jr: Regulation of the Wip1 phosphatase and its
effects on the stress response. Front Biosci (Landmark Ed).
17:1480–1498. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao M, Zhang H, Zhu G, Liang J, Chen N,
Yang Y, Liang X, Cai H and Liu W: Association between
overexpression of Wip1 and prognosis of patients with non-small
cell lung cancer. Oncol Lett. 11:2365–2370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goloudina AR, Kochetkova EY, Pospelova TV
and Demidov ON: Wip1 phosphatase: Between p53 and MAPK kinases
pathways. Oncotarget. 7:31563–31571. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yin S, Wang P, Yang L, Liu Y, Wang Y, Liu
M, Qi Z, Meng J, Shi TY, Yang G and Zang R: Wip1 suppresses ovarian
cancer metastasis through the ATM/AKT/Snail mediated signaling.
Oncotarget. 7:29359–29370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pechackova S, Burdova K, Benada J,
Kleiblova P, Jenikova G and Macurek L: Inhibition of WIP1
phosphatase sensitizes breast cancer cells to genotoxic stress and
to MDM2 antagonist nutlin-3. Oncotarget. 7:14458–14475. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Macurek L, Benada J, Müllers E, Halim VA,
Krejčíková K, Burdová K, Pecháčková S, Hodný Z, Lindqvist A, Medema
RH and Bartek J: Downregulation of Wip1 phosphatase modulates the
cellular threshold of DNA damage signaling in mitosis. Cell Cycle.
12:251–262. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pecháčková S, Burdová K and Macurek L:
WIP1 phosphatase as pharmacological target in cancer therapy. J Mol
Med (Berl). 95:589–599. 2017. View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eren MK and Tabor V: The role of hypoxia
inducible factor-1 alpha in bypassing oncogene-induced senescence.
PLoS One. 9:e1010642014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Drexler HG, Fombonne S, Matsuo Y, Hu ZB,
Hamaguchi H and Uphoff CC: p53 alterations in human
leukemia-lymphoma cell lines: In vitro artifact or prerequisite for
cell immortalization? Leukemia. 14:198–206. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bulavin DV, Demidov ON, Saito S,
Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G,
Nebreda AR, Anderson CW, et al: Amplification of PPM1D in human
tumors abrogates p53 tumor-suppressor activity. Nat Genet.
31:210–215. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martin AC, Facchiano AM, Cuff AL,
Hernandez-Boussard T, Olivier M, Hainaut P and Thornton JM:
Integrating mutation data and structural analysis of the TP53
tumor-suppressor protein. Human Mutat. 19:149–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bond GL, Hu W and Levine AJ: MDM2 is a
central node in the p53 pathway: 12 years and counting. Curr Cancer
Drug Targets. 5:3–8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu X, Ma O, Nguyen TA, Jones SN, Oren M
and Donehower LA: The Wip1 Phosphatase acts as a gatekeeper in the
p53-Mdm2 autoregulatory loop. Cancer Cell. 12:342–54. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goloudina AR, Tanoue K, Hammann A,
Fourmaux E, Le Guezennec X, Bulavin DV, Mazur SJ, Appella E,
Garrido C and Demidov ON: Wip1 promotes RUNX2-dependent apoptosis
in p53-negative tumors and protects normal tissues during treatment
with anticancer agents. Proc Natl Acad Sci USA. 109:E68–E75. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu YH, Zhang CW, Lu L, Demidov ON, Sun L,
Yang L, Bulavin DV and Xiao ZC: Wip1 regulates the generation of
new neural cells in the adult olfactory bulb through p53-dependent
cell cycle control. Stem Cells. 27:1433–1442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia ZS, Wu D, Zhong W, Lu XJ, Yu T and
Chen QK: Wip1 gene silencing enhances the chemosensitivity of human
colon cancer cells. Oncol Lett. 14:1875–1883. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Burma S, Chen BP, Murphy M, Kurimasa A and
Chen DJ: ATM phosphorylates histone H2AX in response to DNA
double-strand breaks. J Biol Chem. 276:42462–42467. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mukherjee B, Kessinger C, Kobayashi J,
Chen BP, Chen DJ, Chatterjee A and Burma S: DNA-PK phosphorylates
histone H2AX during apoptotic DNA fragmentation in mammalian cells.
DNA Repair. 5:575–590. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang H, Wang M, Wang H, Böcker W and
Iliakis G: Complex H2AX phosphorylation patterns by multiple
kinases including ATM and DNA-PK in human cells exposed to ionizing
radiation and treated with kinase inhibitors. J Cell Physiol.
202:492–502. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pilié PG, Tang C, Mills GB and Yap TA:
State-of-the-art strategies for targeting the DNA damage response
in cancer. Nat Rev Clin Oncol. 16:81–104. 2019. View Article : Google Scholar
|
|
30
|
Kilic M and Schmitt CA: Exploiting drug
induced senescence in transgenic mouse models. Beyond Apoptosis:
Cellular Outcomes of Cancer Therapy. Roninson IB, Brown JM and
Bredesen DE: Informa Health Care Publication; pp. 273–295. 2008,
View Article : Google Scholar
|
|
31
|
Crescenzi E, Raia Z, Pacifico F, Mellone
S, Moscato F, Palumbo G and Leonardi A: Down-regulation of
wild-type p53-induced phosphatase 1 (Wip1) plays a critical role in
regulating several p53-dependent functions in premature senescent
tumor cells. J Biol Chem. 288:16212–24. 2013. View Article : Google Scholar : PubMed/NCBI
|