Introduction
Esophageal cancer is an invasive and lethal primary
malignant type of cancer and had a worldwide 10% 5-year survival
rate in 2014 (1). Esophageal cancer
has two major histopathologic types: Esophageal squamous cell
carcinoma (ESCC) and esophageal adenocarcinoma (2). ESCC was the major esophageal cancer in
Asia, Africa and South America in 2014 (3). Notably, China alone accounts for ~50%
of new cases worldwide and had multiple areas with incidence rates
of >100 cases per 100,000 individuals in 2013 (4). Although great improvements have been
made in the treatment of ESCC, including surgery, radiotherapy and
chemotherapy, the number of patients who experience recurrence
remains high (5). One of the main
reasons for recurrence in numerous patients with ESCC is high
radioresistance resulting from the disorders of multiple molecular
and signaling pathways, such as ROS, DNA repair and apoptosis
(6). Cancer treatment failure,
particularly radioresistance to the DNA double strand breaks
(DNA-DSBs) repair system, enhancement of the reactive oxygen
species (ROS) scavenging rate, existence of a hypoxic zone and
other disordered molecular expression have been identified in the
tumor microenvironment after radiation (7,8).
Therefore, developing novel radiation sensitizing methods can
facilitate overcoming radioresistance, thus improving the curative
effect of radiotherapy clinically.
Excess ROS integrate with DNA and other molecules
following radiation inside the cells, resulting in genetic defects,
chromatin remodeling and other damages (9). A certain physiological level of ROS is
required for maintaining cell proliferation and signal
transduction; however, ROS accumulation can damage DNA, RNA and
proteins, which leads to increased mutations and altered functions
of numerous enzymes and proteins, as well as activation of oncogene
products and inhibition of cancer suppressor proteins (9). Therefore, radiation induces ROS
accumulation and DNA fragmentation, and elicits cancer cell
apoptosis, which is important in mediating cell death in radiation
therapy (10). Upon DNA damage in
cells, histone H2AX is phosphorylated on serine 139 to generate
γ-H2AX, which is a marker for the cellular response to DNA-DSBs
(11). Therefore, γ-H2AX expression
after ionizing radiation reflects the formation of double stranded
DNA breaks and DNA repair capacity. The MAPK signaling pathway is a
key signaling pathway of malignant biological behaviors in multiple
cancer types, which can stimulate cell proliferation, inhibition of
apoptosis, and resistance to specific chemotherapeutics and
ionizing radiation via DNA damage and accumulation of ROS (12–14).
Furthermore, the MAPK pathway is a signal transduction pathway that
is sensitive to oxidative stress in the majority of cell types,
which could enhance radiation-induced cell proliferation inhibition
by inactivation of ERK and trigger DNA damage by ROS overproduction
(15,16). However, improving the radiation
sensitivity of ESCC remains a great challenge.
High mobility group box 1 (HMGB1) is a highly
conserved DNA-binding protein, which is involved in gene
transcription, chromatin remodeling, and DNA recombination and
repair, as well as stabilizing nucleosome construction (17). Previous studies have demonstrated
that HMGB1 expression is ubiquitously upregulated during the
development and progression of various cancer types, including
ESCC, neuroglioma and breast cancer (18–20).
Endogenous HMGB1, which is involved in the proliferation of cancer
cells and promotes cell invasion, is expressed in both the nucleus
and the cytoplasm (21). In ESCC,
downregulated HMGB1 inhibits vascular endothelial growth factor-C
expression and the proliferation of cancer cells through the
receptor for advanced glycation end-products (RAGE) signaling
pathway, thus functioning as a tumor promoter (22). Furthermore, the non-histone
chromosomal protein HMGB1 serves an important role in enhancing
ligation reactions of DNA-DSBs (23), and downregulation of HMGB1 modulates
telomere homeostasis and sensitizes breast cells to radiotherapy
(24). Furthermore, Zhang et
al (25) reported that the
increase in HMGB1 expression may be associated with radioresistance
in esophageal carcinoma cells. The present study explored the
regulatory role of HMGB1 in radiosensitivity of ESCC cells to
provide a novel target for anticancer strategies.
Materials and methods
Cell culture and irradiation
The human TE-1 ESCC cell line was obtained from the
Shanghai GeneChem Co., Ltd. and was cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(FBS; Biological Industries) and 100 U/l penicillin sodium/100 U/l
streptomycin sulfate (Invitrogen; Thermo Fisher Scientific, Inc.).
The cells were incubated in a 95% air/5% CO2 humidified incubator
at 37°C for 24, 48 and 72 h. Cells were exposed to 6 MV–X-rays
radiation with a linear accelerator source (Varian Medical Systems)
at a cumulative dose of 0–8 Gy at a fixed dose rate of 200 cGy/min
at room temperature. The 0 Gy group was used as the control
group.
MTT assay
An MTT assay was used to assess cell viability.
Cells in the logarithmic growth phase were plated at a density of
4×103 cells per well into 96-well plates in complete
medium and cultured overnight at 37°C. The cells were then
subjected to various treatments (siHMGB1, radiation or both) for
24, 48 and 72 h at 37°C. The remaining medium was discarded from
the wells, 20 µl MTT and 100 µl medium were added to each well, and
cells were incubated at 37 °C for an additional 4 h. Subsequently,
the supernatant was discarded, and the reaction was stopped with
150 µl dimethyl sulfoxide for 10 min to dissolve MTT. Finally, the
absorbance value was quantified at a wavelength of 490 nm on a
microplate reader, and each experiment was repeated independently
at least three times. Cell viability was calculated using the
following formula: Mean optical density of treated cells/mean
optical density of control cells.
siRNA sequences and transfection
HMGB1 and RAGE gene expression specific siRNA
fragments and one scrambled shRNA (negative control) were designed
and synthesized by Shanghai GenePharma Co., Ltd., according to the
manufacturer's protocols. The sequences of the siRNAs were as
follows: si-HMGB1 forward, 5′-GCUCAAGGAGAAUUUGUAATT-3′ and reverse,
5′-UUACAAAUUCUCCUUGAGCTT-3′; si-RAGE forward,
5′-GCCGGAAAUUGUGAAUCCUTT-3′ and reverse,
5′-AGGAUUCACAAUUUCCGCCTT-3′; and scrambled shRNA forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′. A total of TE-1 cells
(4×105) were seeded into 6-well plates for 16 h and then
transfected with 100 pmol siRNA in serum-free medium for 8 h at
37°C. Negative control siRNA (20 µM) and siHMGB1/siRAGE (20 µM)
were transfected into cells using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). After that, the
medium was replaced with serum-supplemented medium for 24 h at
37°C. The time interval between transfection and subsequent
experimentation was 48 h. Negative controls using non-transfected
cells and empty-vector were performed in parallel and then cells
were harvested for analysis of protein expression.
Clonogenic survival assay
A total of 1×103 cells were seeded into
6-well plates in triplicate overnight. The transfection subsection
of cells was performed as previously described. After radiation (0,
2, 4, 6 or 8 Gy), the cells were incubated for 8 days with 5%
CO2 at 37 °C to allow the formation of colonies.
Subsequently, colonies were washed with PBS, and subsequently fixed
for 20 min with 70% ethanol at room temperature and stained for 20
min with 0.5% crystal violet (Sigma-Aldrich; Merck KGaA) at room
temperature. Colonies containing >50 cells were counted under a
light microscope (×200 magnification; Olympus Corporation). The
surviving fraction was calculated as the ratio of the plating
efficiency of the treated cells to that of control cells. The
sensitization enhancement ratio (SER) was calculated as the mean
inactivation dose in the control group divided by that in the
treated group.
Detection of cell apoptosis by flow
cytometry
Apoptosis was detected using an Annexin
V-phycoerythrin and 7-amino-actinomycin D (Annexin V-PE/7-AAD)
apoptosis kit (Hangzhou Multi Sciences (Lianke) Biotech Co., Ltd.).
Cells (4×105) were plated overnight in 6-well plates,
transfected with or without siHMGB1 for 24 h, and then cultured for
24 h after radiation (4 Gy). A total of 1×106 cells/ml
of treated cells in each group were collected and double-stained
using Annexin V-PE/7-AAD for 15 min at 25°C. Next, cell apoptosis
was quantified by flow cytometry (FACSCalibur; BD Biosciences)
within 1 h, and the results were analyzed using the FlowJo software
(version 7.6.1; Tree Star, Inc.).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA from each group of cultured TE-1 cells was
isolated using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). RNA was dissolved with RNase-free water
and stored at −80°C. RNA concentrations were detected by NanoDrop
spectrophotometry (Thermo Fisher Scientific, Inc.). cDNA synthesis
and its reaction condition were performed with a Prime-Script™ RT
Reagent kit (Takara Biotechnology Co., Ltd.), according to the
manufacturer's protocol. RT-qPCR was carried out using a Stratagene
Mx3000P™ Real-Time PCR System (Agilent Technologies, Inc.) and SYBR
Premix Ex Taq™ (Takara Biotechnology Co., Ltd.) with the following
primers (Generay Biotech Co., Ltd.): NADPH oxidase (NOX)1 forward,
5′-CAAGGCCACTGACATCGT-3′ and reverse, 5′-CAGATTACCGTCCTTATTCCTA-3′;
NOX5 forward, 5′-GATGACCCACCCAATAAGAC-3′ and reverse,
5′-GCCTCTGGTTCCCTCACTT-3′; and GAPDH forward,
5′-TCAACGGATTTGGTCGTATTG-3′ and reverse,
5′-TGGGTGGAATCATATTGGAAC-3′. GAPDH was used as the internal
control. The following thermocycling conditions were used for
amplification: Initial denaturation at 95°C for 2 min, followed by
40 cycles of denaturation at 95°C for 15 sec, annealing at 60°C for
20 sec and extension at 72°C for 15 sec. Data analysis was
performed using the comparative Cq method as previously described
(19).
Detection of intracellular ROS by flow
cytometry
Intracellular ROS levels were detected in living
cells using the 2′,7′-dichlorodihydrofluorescein diacetate
(DCFH-DA) probe (Beyotime Institute of Biotechnology), which is
converted by ROS into the fluorescent product
2′,7′-dichlorofluorescein (10). A
total of 2×106 cells at 37°C were transfected with or
without siHMGB1 for 24 h and subsequently irradiated with 4-Gy
irradiation for 12 h. Subsequently, the cells were isolated and
incubated with 10 µM DCFH-DA diluted in serum-free medium for 25
min at 37°C in a dark humidified incubator, which was added
directly to the cell culture media to a final concentration of 10
µg/ml. After treatment, the cells were washed three times with PBS
at room temperature for a total of 5 min and suspended in
serum-free medium. Subsequently, the ROS levels were measured
immediately via flow cytometry (FACSCalibur; BD Biosciences) with
an excitation wavelength of 488 nm and an emission wavelength of
525 nm. Serum-free medium was used as a negative control.
Fluorescence data were analyzed using the FlowJo software (version
7.6.1; Tree Star, Inc.).
Analysis of DNA-DSBs using flow
cytometry
Cells at 37°C were transfected with siHMGB1 for 24 h
prior to irradiation with 4 Gy X-rays for 1 h. Cells were harvested
with trypsin and washed with ice-cold PBS twice. Subsequently, the
cells were fixed in 4% paraformaldehyde for 30 min at room
temperature, and then permeabilized with Tris-buffered saline
containing 0.25% Triton X-100 on ice for 15 min, followed by final
resuspension containing 5% BSA (Sigma-Aldrich; Merck KGaA) in PBS
for 1 h at 25°C. Next, the cells were incubated with an
anti-phospho-histone H2AX (Ser139) monoclonal antibody (dilution,
1:400 in PBS; cat. no. 9718; Cell Signaling Technology, Inc.) as
the primary antibody overnight at 4°C. The cells were then washed
with PBS three times at room temperature and incubated with
secondary antibody Cy3-conjugated goat anti-rabbit IgG (1:200; cat.
no. GB21303; Wuhan Servicebio Technology Co., Ltd.) for 1 h at 25°C
and then washed with PBS twice. Phospho-γH2AX (p-γH2AX) levels were
measured by flow cytometry (FACSCalibur; BD Biosciences). Untreated
cells were used as the negative control. Data were analyzed using
the FlowJo software (version 7.6.1; Tree Star, Inc.).
Assessment of protein expression by
western blot analysis
Cells were treated as aforementioned with ionizing
radiation, siRNA or a combination of the two treatments, which were
divided into the following groups: TE-1, negative control, 4-Gy,
siHMGB1, siRAGE, 4-Gy+siHMGB1, 4-Gy+siRAGE. Cells were collected
and washed twice with PBS. The cellular protein was extracted using
RIPA lysis buffer (Beijing Solarbio Science & Technology Co.,
Ltd.) at 4°C, according to the manufacturer's protocol, and total
cellular protein concentrations were determined with a BioMate 3S
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocols. Subsequently, 5 µg protein/lane for each sample was
separated by electrophoresis on a 10–15% SDS-polyacrylamide gel,
and then electronically transferred onto a polyvinylidene fluoride
membrane (EMD Millipore). After blocking for 1 h at 37°C with
Tris-buffered saline containing 1% Tween-20 (TBST) or 5% BSA, the
membranes were incubated with the primary antibody at 4°C
overnight. The following primary antibodies were diluted in PBS at
1:500 and purchased from Cell Signaling Technology, Inc.: ERK1/2
(cat. no. 9194S), p-ERK1/2 (cat. no. 4370T), JNK (cat. no. 9252S),
p-JNK (cat. no. 9251S), p38 (cat. no. 9212S), p-p38 (cat. no.
9216S), RAGE (cat. no. 42544S), HMGB1 (cat. no. 3935S), β-actin
(cat. no. 12620S), caspase-3 (cat. no. 14220S) and
cleaved-poly(ADP-ribose) polymerase (PARP; cat. no. 9185S).
Subsequently, the membranes were washed three times with TBST and
incubated for 1 h at 37°C with the anti-rabbit secondary antibody
conjugated to horseradish peroxidase (dilution, 1:5,000 in TBST;
cat. no. 7076; Cell Signaling Technology, Inc.). After washing
three times with TBST, the bands were detected using Pierce ECL
Plus Western Blotting substrate (Thermo Fisher Scientific, Inc.)
and scanned using a FluorChem FC3 imaging system (ProteinSimple).
The images were semi-quantified with AlphaView software (version
3.4.0; ProteinSimple), normalized to β-actin (standard control) and
expressed as the fold change compared with the control.
Statistical analysis
Data are presented as the mean ± SEM and were
derived from ≥3 independent experiments. Unpaired Student's t-test
was used to compare differences between two groups (Fig. 1B). Multiple group comparisons of the
means were carried out by one-way ANOVA followed by Tukey's post
hoc test. SPSS v20.0 (IBM Corp.) and GraphPad Prism 8.02 (GraphPad
Software, Inc.) were used to perform the analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
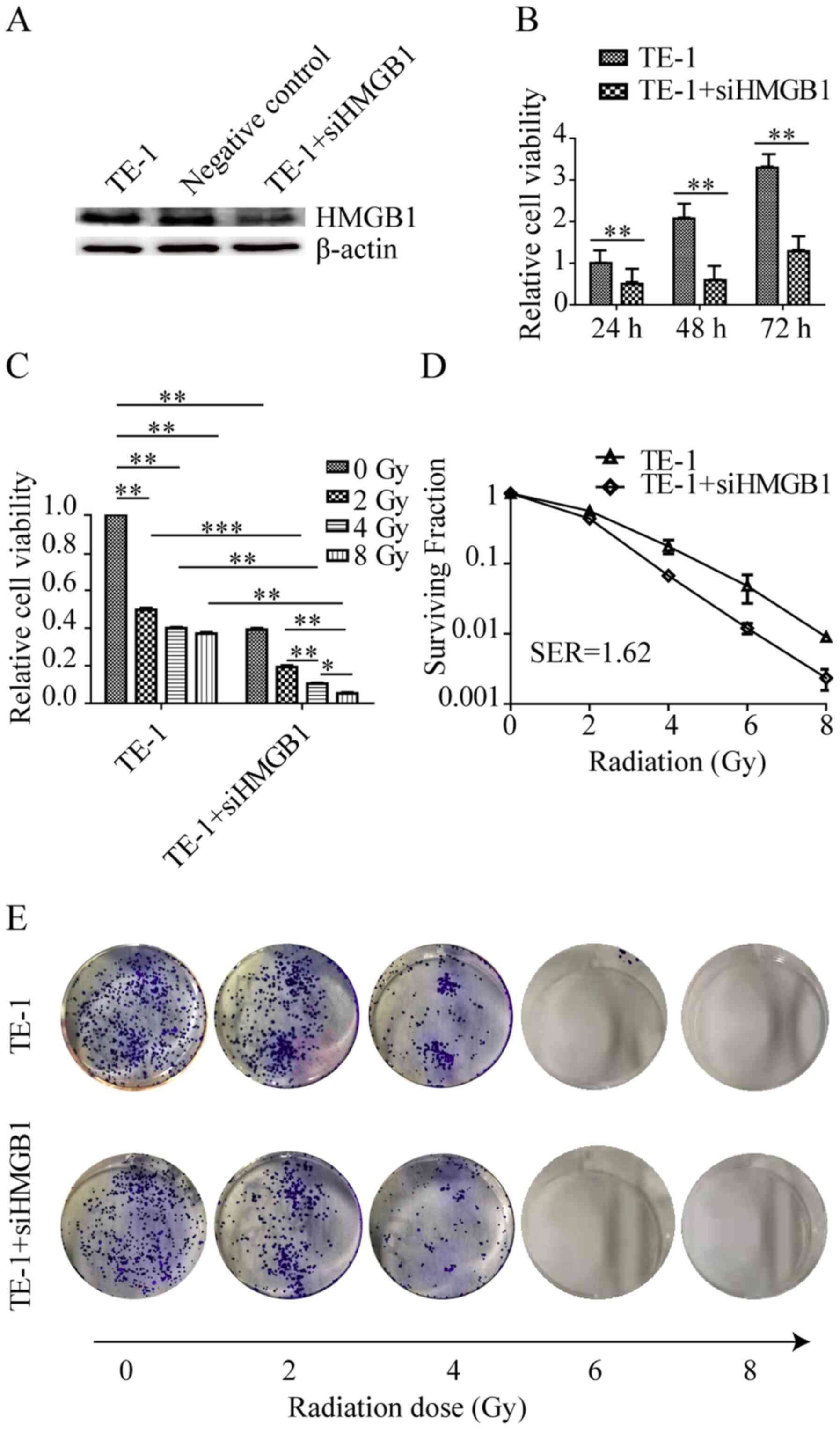
HMGB1 knockdown enhances
radiosensitivity in TE-1 cells
A HMGB1 knockdown cell line was constructed to
investigate the role of HMGB1 in the radiotherapy of ESCC, and the
transfection efficiency was detected using western blot analysis
(Fig. 1A). To investigate the effect
of HMGB1 knockdown on ionizing radiation in TE-1 cells, an MTT
assay was used to detect changes in viability after siHMGB1
transfection at 24, 48 and 72 h in TE-1 cells. As shown in Fig. 1B, siHMGB1 treatment induced a
significant decrease in cell viability compared with that of cells
without transfection at all time points. TE-1 cell viability after
2-Gy radiation significantly decreased compared with the effects of
the non-transfected cells, whereas irradiation at 4 and 8 Gy did
not significantly reduce the number of cells compared with the
effects of 2-Gy irradiation (Fig.
1C). siHMGB1 treatment significantly decreased cell viability
compared with that of cells without transfection after different
doses of radiation (2, 4 and 8-Gy). Cell viability was decreased
after exposure to radiation in a dose-dependent manner in HMGB1
knockdown cells (Fig. 1C).
Furthermore, a clonogenic survival assay was performed to assess
whether HMGB1 knockdown had potential radiosensitization activity.
As shown in Fig. 1D and E, treatmen
of TE-1 cells with siHMGB1 for 24 h prior to radiation led to a
survival curve shift compared with that of untreated TE-1 cells,
with a SER of 1.62. The present results demonstrated that the
viability of TE-1 cells could not be inhibited by increasing the
dose of ionizing radiation (2, 4 and 8-Gy), which was probably due
to radioresistance. However, HMGB1 knockdown could increase the
radiosensitivity of TE-1 cells to radiotherapeutic agents.
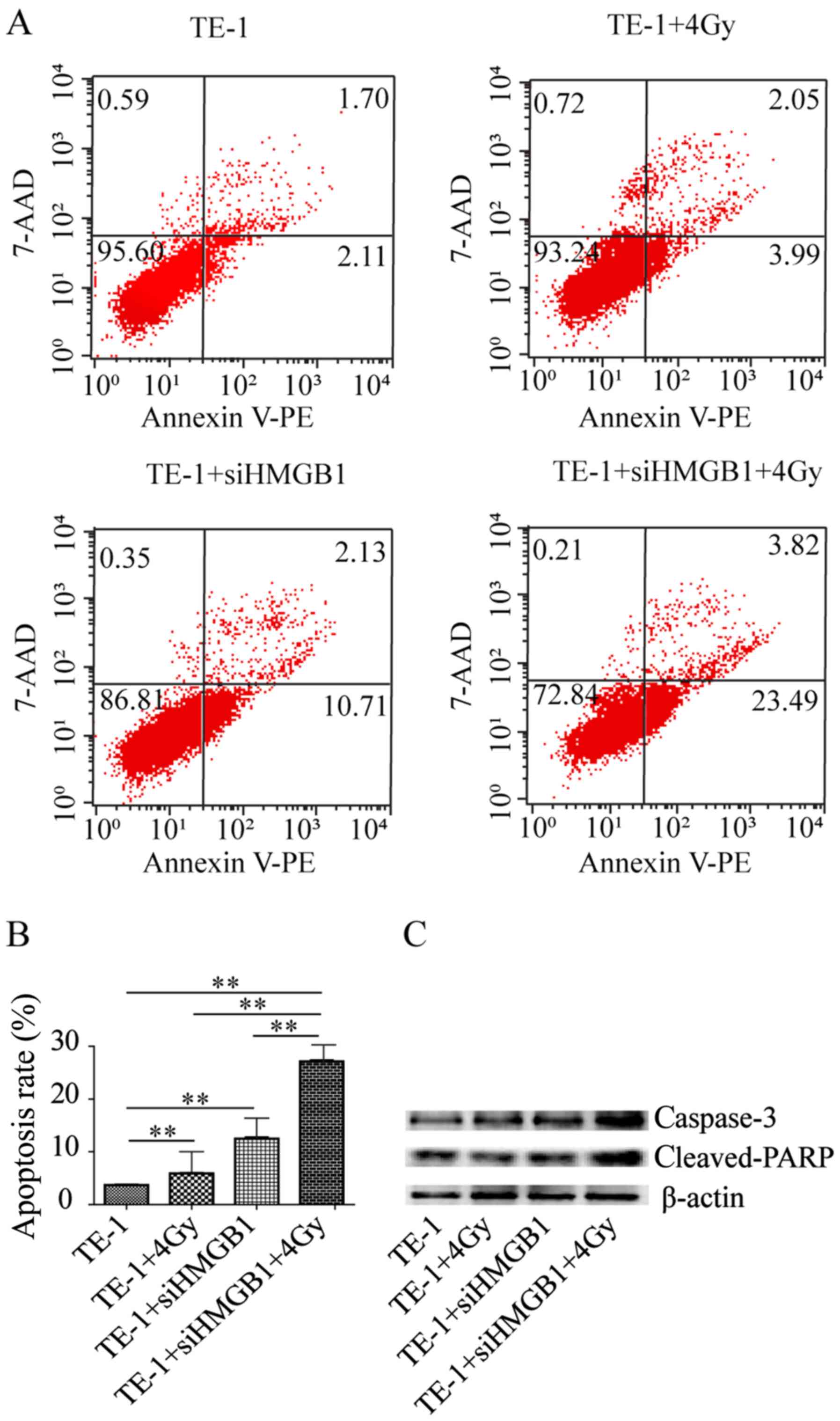
HMGB1 knockdown enhances
radiation-induced apoptosis in TE-1 cells
The levels of apoptosis of TE-1 cells after
radiation were examined using Annexin V-PE and 7-AAD staining to
verify whether HMGB1 knockdown enhanced radiation-induced
inhibition of cell viability through apoptosis. As shown in
Fig. 2A and B, the apoptosis rate of
TE-1 cells after exposure to 4-Gy radiation for 24 h was
significantly increased compared with that of cells that were not
irradiated. However, siHMGB1 treatment significantly increased cell
apoptosis after radiation compared with that of cells without
transfection. Consistent with these findings, a marked increase in
the expression levels of the pro-apoptotic proteins caspase-3 and
cleaved PARP was observed after combined treatment of radiation and
transfection by western blotting (Fig.
2C). These results demonstrated that the radiosensitizing
effect of HMGB1 knockdown may be caused by induction of apoptosis
in TE-1 cells.
HMGB1 knockdown increases NOX-mediated
ROS production in irradiated TE-1 cells
Radiation can induce accumulation of ROS production,
leading to cell damage and apoptosis (26). In the present study, the levels of
ROS production after radiation were detected to examine whether
HMGB1 knockdown could promote radiation-induced ROS production.
Compared with that of TE-1 control cells, ROS production was not
significantly altered after irradiation at 4 Gy, whereas siHMGB1
treatment significantly increased the levels of ROS production
(Fig. 3A and B). However, HMGB1
knockdown combined with radiation resulted in higher ROS production
than either siHMGB1 or radiation alone (Fig. 3A and B). These results demonstrated
that the combination of HMGB1 knockdown and radiation caused the
upregulation of ROS production, which may promote an increase in
apoptosis of TE-1 cells after radiation.
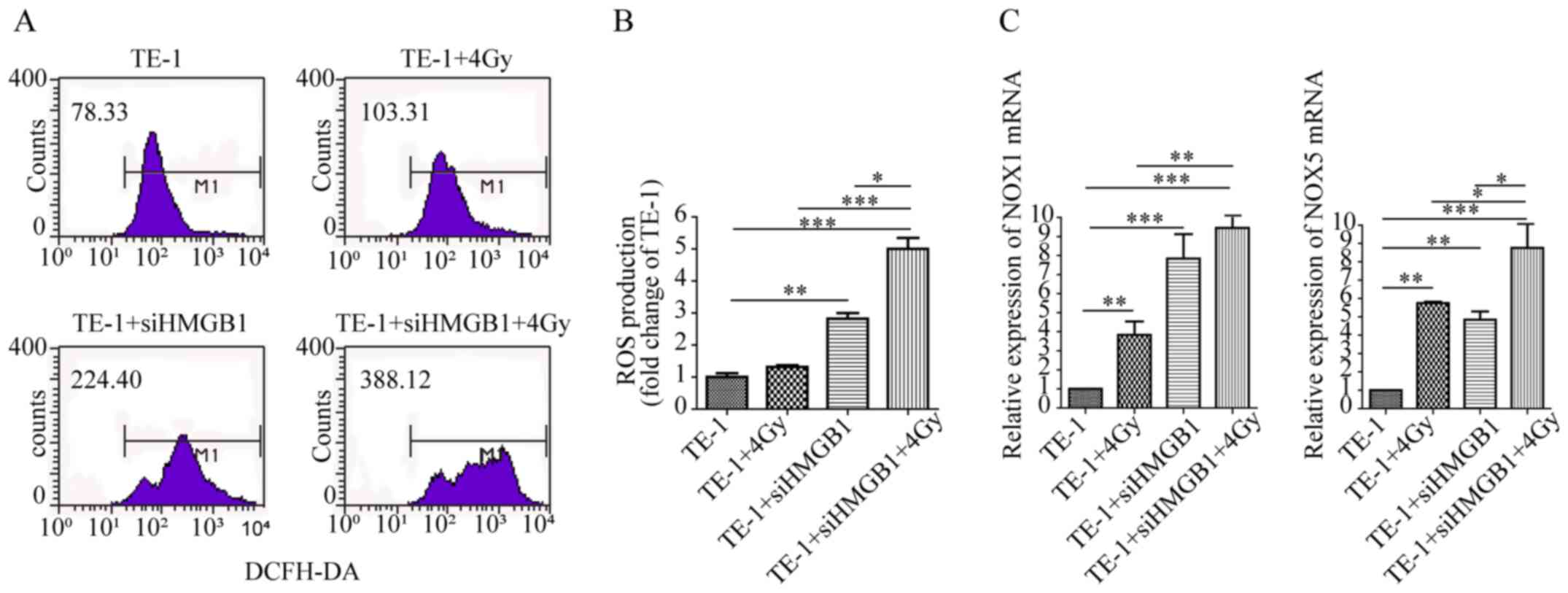 | Figure 3.HMGB1 knockdown increases
NOX-mediated ROS production in irradiated TE-1 cells. (A) ROS
production was assessed via detection of 2′,7′-dichlorofluorescein
expression using flow cytometry in TE-1 cells transfected with or
without siHMGB1 for 24 h and then irradiated with 4-Gy irradiation
for 12 h. (B) Bar graph depicting the fold change in ROS production
in TE-1 cells transfected with siHMGB1, treated with irradiation or
both. All data are presented as the mean ± SEM and were derived
from ≥3 independent experiments. (C) mRNA expression levels of NOX1
and NOX5 in cells transfected with siHMGB1, treated with
irradiation or both are shown as relative levels compared with
those of TE-1 control cells. Data are presented as the mean ± SEM
and were derived from ≥3 independent experiments. *P<0.05,
**P<0.01, ***P<0.001. DCFH-DA,
2′,7′-dichlorodihydrofluorescein diacetate; HMGB1, high mobility
group box 1; NOX, NADPH oxidase; ROS, reactive oxygen species; si,
small interfering RNA. |
The mRNA expression levels of NADPH oxidases were
further detected to examine whether ROS production was induced by
HMGB1 knockdown. It was revealed that the mRNA expression levels of
NOX1 and NOX5 were significantly increased by HMGB1 knockdown or
radiation treatment alone, while HMGB1 knockdown combined with
radiation resulted in higher NOX1 and NOX5 mRNA expression compared
with radiation alone (Fig. 3C).
These results demonstrated that NADPH oxidase may be crucial in the
radiation-induced generation of ROS following knockdown of
HMGB1.
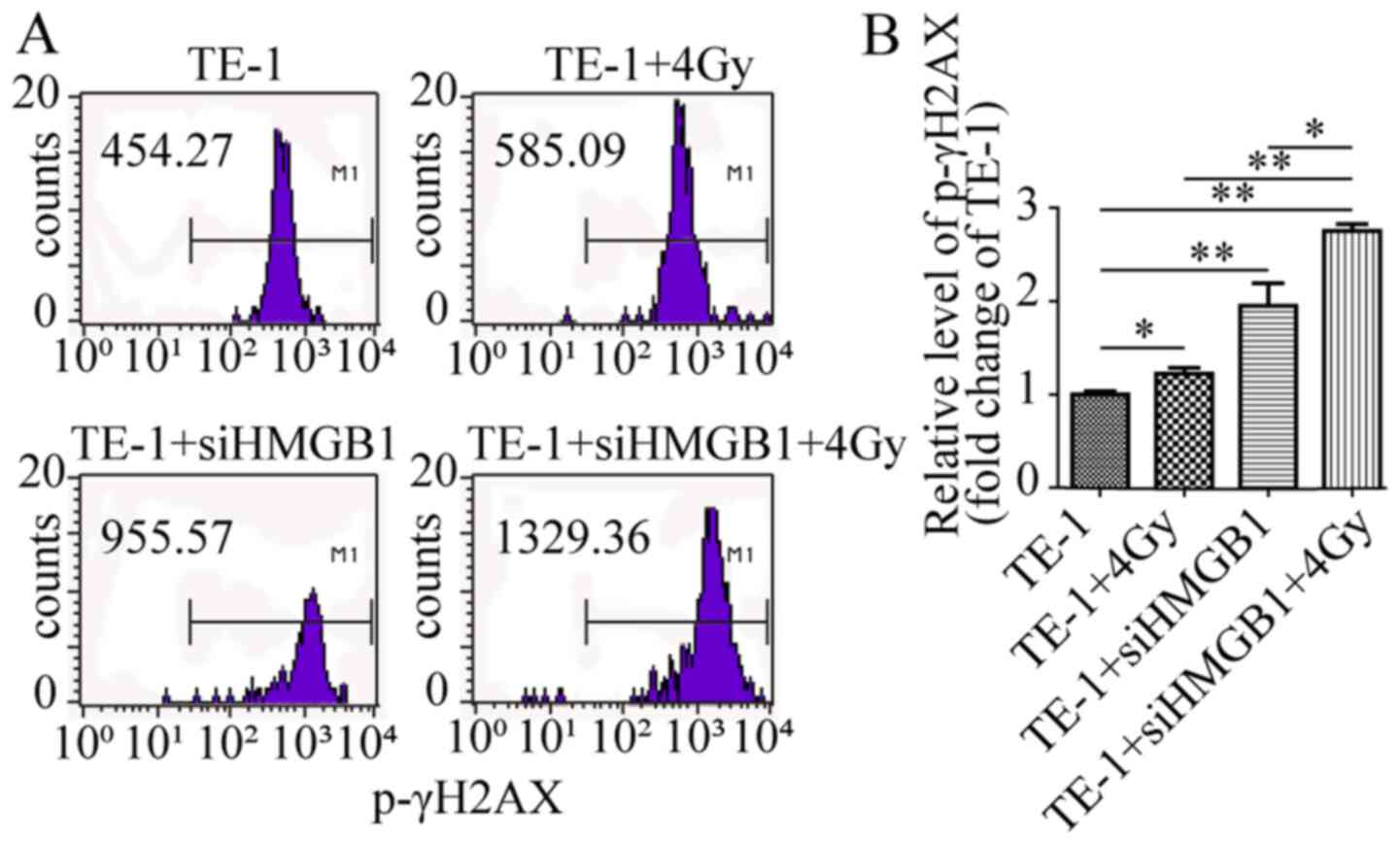
X-rays (4 Gy) enhance HMGB1
knockdown-induced DNA damage in TE-1 cells
ROS accumulation has been demonstrated to cause DNA
damage and trigger radiation-induced cell apoptosis (27). The levels of p-γH2AX after radiation
were examined to investigate whether 4-Gy X-rays could promote
HMGB1 knockdown-induced DNA damage. Compared with that of TE-1
control cells, p-γH2AX expression was significantly altered after
4-Gy radiation or siHMGB1 transfection (Fig. 4). However, HMGB1 knockdown combined
with radiation resulted in higher p-γH2AX production than either
siHMGB1 or radiation alone (Fig. 4).
These results demonstrated that the combination of HMGB1 knockdown
and radiation was associated with increased DNA damage, which may
promote the apoptosis of TE-1 cells after radiation.
MAPK signaling pathway is involved in
the HMGB1-mediated promotion of radioresistance
The MAPK signaling pathway is an oxidative
stress-sensitive signal transduction pathway that is involved in
radiation-induced cell apoptosis (12–14). The
present study evaluated whether the MAPK signaling pathway also
mediated the effects of apoptosis of HMGB1 knockdown during
radiation of TE-1 cells. It was revealed that the levels of
p-p38/p-38 and p-ERK1/2/ERK1/2 decreased, while p-JNK/JNK increased
by HMGB1 knockdown along with radiation compared with the effects
produced by either siHMGB1 or radiation alone (Fig. 5A). To determine whether HMGB1
promoted MAPK signaling-mediated radioresistance via RAGE, which is
an HMGB1 receptor, TE-1 cells were transfected with siRAGE
(Fig. 5B) and treated with radiation
alone or transfected with siRAGE and treated with radiation. As
shown in Fig. 5C, RAGE knockdown
combined with radiation increased p-JNK levels, and decreased p-p38
and p-ERK1/2 levels. Therefore, these results indicated that the
MAPK signaling pathway is involved in the process of
HMGB1-promoting radioresistance via RAGE in TE-1 cells.
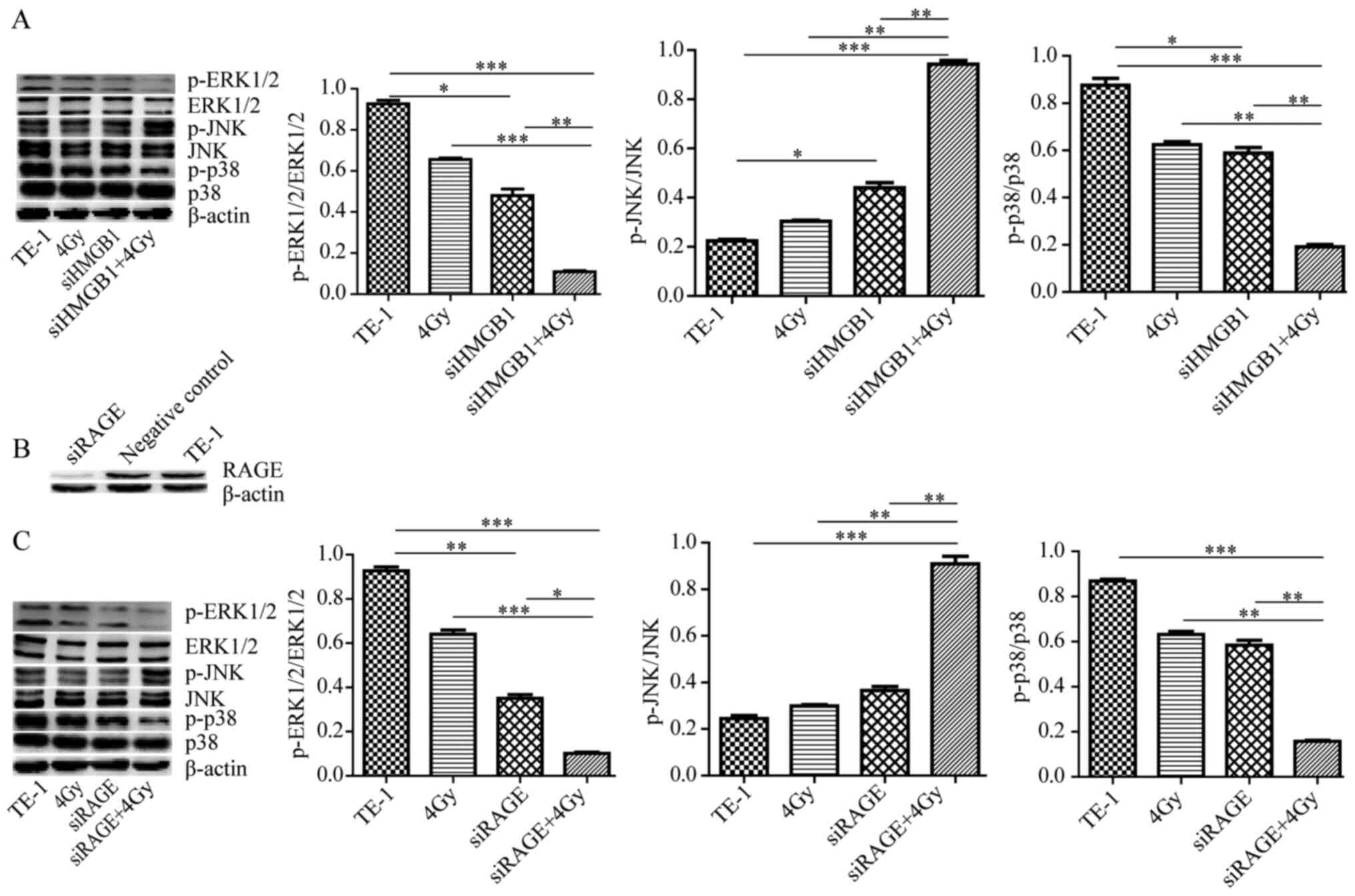 | Figure 5.MAPK signaling pathway is involved in
the HMGB1-mediated promoting radioresistance. (A) Western blotting
was performed to detect the expression of β-actin, p-ERK1/2,
ERK1/2, p-JNK, JNK, p-38 and p-p38 after transfection with or
without siHMGB1 for 24 h prior to radiation for 24 h. (B) Western
blotting was performed to validate the transfection efficiency with
siRAGE or negative control siRNA for 48 h. (C) Western blotting was
performed to detect the expression of β-actin, p-ERK1/2, ERK1/2,
p-JNK, JNK, p-38 and p-p38 after transfection with or without
siRAGE for 24 h prior to radiation for 24 h. The cells were lysed
for western blot analysis. Experiments were repeated at least three
times with similar results, and only one representative result is
shown. *P<0.05, **P<0.01, ***P<0.001. HMGB1, high mobility
group box 1; si, small interfering RNA; RAGE, receptor for advanced
glycation end production; p-, phosphorylated. |
Discussion
HMGB1 expression is frequently upregulated in human
tumors (18–20); however, to the best of our knowledge,
its relevance in cancer radiotherapy is unknown. HMGB1-deficient
tumors have an impaired ability to recruit innate immune cells into
chemotherapy-treated tumor tissues, indicating that HMGB1 serves an
important role in antitumor immunity (28). In the majority of epithelial cancer
types, including ESCC, HMGB1 knockdown can induce apoptosis
(22). Activated caspase-3 and
cleaved PARP are well-documented measurements of apoptosis
(29,30). The present study revealed that HMGB1
knockdown combined with radiation reduced cancer cell viability and
increased apoptosis, indicating that HMGB1 knockdown-induced cell
apoptosis is a major mechanism for HMGB1 knockdown-mediated
radiosensitivity.
However, the exact mechanism by which HMGB1
knockdown increases radiation-induced apoptosis is unknown. ROS may
be a risk factor in radiation-induced DNA damage (24). There are numerous types of ROS,
including superoxide, hydrogen peroxide and highly toxic hydroxyl
radicals (31). NOX is the main
source of ROS, and is mainly composed of six subunits: NOX1, NOX3,
NOX4, NOX5, dual oxidase (DUOX)1 and DUOX2 (32). Consistent with previously reported
observations that HMGB1 knockdown combined with cordycepin
treatment had anti-proliferative and pro-apoptotic effects via
increasing the ROS levels in the K562 human chronic myeloid
leukemia cell line (33), siHMGB1
treatment enhanced the mRNA expression levels of NOX1 and NOX5, and
increased intracellular ROS levels in irradiated TE-1 cells in the
present study. It has been reported that diabetes and intravitreal
injection of HMGB1 in normal rats induces upregulation of ROS and
NOX2 in the retina (34), and that
the rate of generation of ROS decreases upon exposure to HMGB1
inhibition in rat tubulo-epithelial cells (35). The role of HMGB1 in ROS production
remains to be investigated in future studies. Therefore, it was
concluded that HMGB1 knockdown promoted radiation-induced NOX1 and
NOX5-derived ROS generation in ESCC. In addition, ROS accumulation
was involved in the regulation of DNA damage, triggering apoptosis
signal transduction pathways and resulting in the promotion of
radiation sensitivity in TE-1 cells.
Ionizing radiation induces various types of damage
in cellular DNA, which is considered to be the target of biological
effects of radiation, by both direct energy deposition on DNA and
reactions with diffusible water radicals (36). Radioresistance decreases the
therapeutic effect of cervical cancer, and one of the main reasons
for this is the influence of apoptosis and DSB repair (37). Increased p-γH2AX is a sensitive and
precise hallmark for chromatin-induced DNA-DSBs by ionizing
radiation or oxidative damage (11).
It was revealed that HMGB1 knockdown or its combination with
radiation significantly increased the levels of p-γH2AX compared
with radiation treatment alone. Therefore, HMGB1 knockdown markedly
increased DNA-DSBs in irradiated cells, and sensitized TE-1 cells
to radiation-induced apoptosis by increasing DNA damage.
Endogenous HMGB1 binds to RAGE on the cell surface
and to Toll-like receptor (TLR)2, TLR4 and TLR9 in the cytoplasm,
which activates MAPK, thus promoting angiogenesis, unlimited
replicative potential, tissue invasion and metastasis (38,39).
Elevated RAGE expression promotes cell viability in ESCC (22). RAGE knockdown increases cell
apoptosis and diminishes cell survival by ROS-induced oxidative
injury in pancreatic tumor cells (40). Furthermore, the MAPK signaling
pathway is involved in the regulation of cell apoptosis (15,16).
Pro-survival ERK signaling is a critical effector downstream of
epidermal growth factor receptor signaling, which enhances DNA-DSB
repair in human glioma cells (16).
JNK, a stress-activated protein kinase (41), can be activated by ionizing radiation
or ROS (41,42). Activation of JNK by radiation is
associated with apoptotic cell death-mediated Bcl-2 downregulation
(43). Furthermore, previous studies
have demonstrated that pretreatment with JNK inhibitor can prevent
the activation of caspase-3 and cleavage PARP triggered by
radiation (29,30), indicating that ERK downregulation and
JNK upregulation are the main mechanisms promoting cell death in
cancer in response to radiation (14,16).
Consistent with a previous study (24), the present results demonstrated that
HMGB1 or RAGE knockdown induces upregulation of p-JNK in TE-1 cells
after radiation, whereas p-ERK levels decrease, suggesting that the
MAPK signaling pathway is involved in regulating DNA damage and ROS
generation, increasing apoptosis when HMGB1 knockdown is combined
with radiation.
In contrast to eosinophils from patients with asthma
and cutaneous squamous cell carcinoma cells, p38 inhibition reduces
Bcl-2 expression and increases apoptosis or augments cutaneous
squamous cell carcinoma tumorigenesis via NOX2-driven ROS
generation (44,45). The present study revealed that
enhancement of apoptosis after radiation combined with HMGB1
knockdown significantly induced downregulation of p38. This finding
suggested that the enhancement of radiation-induced apoptosis by
HMGB1 knockdown may be p38-dependent. Whether these differences are
due to different cell lines, different disease models or different
interventions remains to be investigated in future studies.
The present study focused on the effect of HMGB1 on
radiosensitivity and it preliminarily examined the mechanism. The
exact mechanism by which HMGB1 promotes radioresistance by using
agonists/inhibitors/siRNA of key molecules in the MAPK signaling
pathway via RAGE needs further verification as well as evaluation
of apoptosis and cell viability in future studies.
In summary, HMGB1 knockdown promoted cell apoptosis
and inhibited cell viability. HMGB1 knockdown-enhanced and
radiation-induced apoptosis may be involved in the regulation of
DNA damage and ROS generation via the MAPK signaling pathway.
Therefore, targeting HMGB1 is an attractive strategy to increase
the efficacy of radiation therapy for ESCC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GH, SL and LY designed the experiments. GH, SL and
RL performed the experiments. GH, XW and CS analyzed the data and
prepared the figures. XW, SL, GH and YZ drafted the manuscript and
revised it for academic content. YZ made substantial contributions
to the design and conception of the present study. GH, RL, CS, XW,
YZ, LY and SL confirm the authenticity of the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Song Y, Li L, Ou Y, Gao Z, Li E, Li X,
Zhang W, Wang J, Xu L, Zhou Y, et al: Identification of genomic
alterations in oesophageal squamous cell cancer. Nature. 509:91–95.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu J, Chen Y, Zhang R, Song Y, Cao J, Bi
N, Wang J, He J, Bai J, Dong L, et al: Global and targeted
metabolomics of esophageal squamous cell carcinoma discovers
potential diagnostic and therapeutic biomarkers. Mol Cell
Proteomics. 12:1306–1318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rustgi AK and El-Serag HB: Esophageal
carcinoma. N Engl J Med. 371:2499–2509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin Y, Totsuka Y, He Y, Kikuchi S, Qiao Y,
Ueda J, Wei W, Inoue M and Tanaka H: Epidemiology of esophageal
cancer in Japan and China. J Epidemiol. 23:233–242. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Y: Epidemiology of esophageal
cancer. World J Gastroenterol. 19:5598–5606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang G, Liu L, Sharma S, Liu H, Yang W,
Sun X and Dong Q: Bmi-1 confers adaptive radioresistance to
KYSE-150R esophageal carcinoma cells. Biochem Biophys Res Commun.
425:309–314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hosseinimehr SJ: The protective effects of
trace elements against side effects induced by ionizing radiation.
Radiat Oncol J. 33:66–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luo D, Wang Z and Wu J, Jiang C and Wu J:
The role of hypoxia inducible factor-1 in hepatocellular carcinoma.
BioMed Res Int. 2014:4092722014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maraldi T: Natural compounds as modulators
of NADPH oxidases. Oxid Med Cell Longev. 2013:2716022013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Endres L, Begley U, Clark R, Gu C,
Dziergowska A, Małkiewicz A, Melendez JA, Dedon PC and Begley TJ:
Alkbh8 regulates selenocysteine-protein expression to protect
against reactive oxygen species damage. PLoS One. 10:e01313352015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Probin V, Wang Y and Zhou D:
Busulfan-induced senescence is dependent on ROS production upstream
of the MAPK pathway. Free Radic Biol Med. 42:1858–1865. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chung EJ, Urick ME, Kurshan N, Shield W
III, Asano H, Smith PD, Scroggins BS, Burkeen J and Citrin DE:
MEK1/2 inhibition enhances the radiosensitivity of cancer cells by
downregulating survival and growth signals mediated by EGFR
ligands. Int J Oncol. 42:2028–2036. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ameziane-El-Hassani R, Talbot M, de Souza
Dos Santos MC, Al Ghuzlan A, Hartl D, Bidart JM, De Deken X, Miot
F, Diallo I, de Vathaire F, et al: NADPH oxidase DUOX1 promotes
long-term persistence of oxidative stress after an exposure to
irradiation. Proc Natl Acad Sci USA. 112:5051–5056. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Golding SE, Morgan RN, Adams BR, Hawkins
AJ, Povirk LF and Valerie K: Pro-survival AKT and ERK signaling
from EGFR and mutant EGFRvIII enhances DNA double-strand break
repair in human glioma cells. Cancer Biol Ther. 8:730–738. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lotze MT and Tracey KJ: High-mobility
group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal.
Nat Rev Immunol. 5:331–342. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chuangui C, Peng T and Zhentao Y: The
expression of high mobility group box 1 is associated with lymph
node metastasis and poor prognosis in esophageal squamous cell
carcinoma. Pathol Oncol Res. 18:1021–1027. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiao Y, Wang HC and Fan SJ: Growth
suppression and radiosensitivity increase by HMGB1 in breast
cancer. Acta Pharmacol Sin. 28:1957–1967. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ellerman JE, Brown CK, de Vera M, Zeh HJ,
Billiar T, Rubartelli A and Lotze MT: Masquerader: High mobility
group box-1 and cancer. Clin Cancer Res. 13:2836–2848. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen CG, Tang P and Yu ZT: Effect of HMGB1
on the VEGF-C expression and proliferation of esophageal squamous
cancer cells. Zhonghua Zhong Liu Za Zhi. 34:566–570. 2012.(In
Chinese). PubMed/NCBI
|
|
23
|
Yamanaka S, Katayama E, Yoshioka K, Nagaki
S, Yoshida M and Teraoka H: Nucleosome linker proteins HMGB1 and
histone H1 differentially enhance DNA ligation reactions. Biochem
Biophys Res Commun. 292:268–273. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ke S, Zhou F, Yang H, Wei Y, Gong J, Mei
Z, Wu L, Yu H and Zhou Y: Downregulation of high mobility group box
1 modulates telomere homeostasis and increases the radiosensitivity
of human breast cancer cells. Int J Oncol. 46:1051–1058. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang H, Gao XS, Zhao J, Xiong W, Zhang M,
Li HZ, Zhou DM, Jin X and Zhang DS: Differential gene expression
profiles of DNA repair genes in esophageal cancer cells after X-ray
irradiation. Chin J Cancer. 29:865–872. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie Q, Zhou Y, Lan G, Yang L, Zheng W,
Liang Y and Chen T: Sensitization of cancer cells to radiation by
selenadiazole derivatives by regulation of ROS-mediated DNA damage
and ERK and AKT pathways. Biochem Biophys Res Commun. 449:88–93.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lam RK, Fung YK, Han W and Yu KN: Rescue
effects: Irradiated cells helped by unirradiated bystander cells.
Int J Mol Sci. 16:2591–2609. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guerriero JL, Ditsworth D, Catanzaro JM,
Sabino G, Furie MB, Kew RR, Crawford HC and Zong WX: DNA alkylating
therapy induces tumor regression through an HMGB1-mediated
activation of innate immunity. J Immunol. 186:3517–3526. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nicholson DW, Ali A, Thornberry NA,
Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle
M, Lazebnik YA, et al: Identification and inhibition of the
ICE/CED-3 protease necessary for mammalian apoptosis. Nature.
376:37–43. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly(ADP-ribose) polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roehlecke C, Schumann U, Ader M, Brunssen
C, Bramke S, Morawietz H and Funk RH: Stress reaction in outer
segments of photoreceptors after blue light irradiation. PLoS One.
8:e715702013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: Physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen X, Wang Y, Liu J, Xu P, Zhang XM,
Tian YY, Xue YM, Gao XY, Liu Y and Wang JH: Synergistic effect of
HMGB1 knockdown and cordycepin in the K562 human chronic myeloid
leukemia cell line. Mol Med Rep. 12:4462–4468. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mohammad G, Alam K, Nawaz MI, Siddiquei
MM, Mousa A and Abu El-Asrar AM: Mutual enhancement between
high-mobility group box-1 and NADPH oxidase-derived reactive oxygen
species mediates diabetes-induced upregulation of retinal apoptotic
markers. J Physiol Biochem. 71:359–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nair AR, Ebenezer PJ, Saini Y and Francis
J: Angiotensin II-induced hypertensive renal inflammation is
mediated through HMGB1-TLR4 signaling in rat tubulo-epithelial
cells. Exp Cell Res. 335:238–247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kehrer JP: Free radicals as mediators of
tissue injury and disease. Crit Rev Toxicol. 23:21–48. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ye C, Sun NX, Ma Y, Zhao Q, Zhang Q, Xu C,
Wang SB, Sun SH, Wang F and Li W: MicroRNA-145 contributes to
enhancing radiosensitivity of cervical cancer cells. FEBS Lett.
589:702–709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weng H, Deng Y, Xie Y, Liu H and Gong F:
Expression and significance of HMGB1, TLR4 and NF-κB p65 in human
epidermal tumors. BMC Cancer. 13:3112013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Moser B, Janik S, Schiefer AI, Müllauer L,
Bekos C, Scharrer A, Mildner M, Rényi-Vámos F, Klepetko W and
Ankersmit HJ: Expression of RAGE and HMGB1 in thymic epithelial
tumors, thymic hyperplasia and regular thymic morphology. PLoS One.
9:e941182014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kang R, Tang D, Livesey KM, Schapiro NE,
Lotze MT and Zeh HJ III: The Receptor for Advanced Glycation
End-products (RAGE) protects pancreatic tumor cells against
oxidative injury. Antioxid Redox Signal. 15:2175–2184. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen YR, Meyer CF and Tan TH: Persistent
activation of c-Jun N-terminal kinase 1 (JNK1) in gamma
radiation-induced apoptosis. J Biol Chem. 271:631–634. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shao C, Lyng FM, Folkard M and Prise KM:
Calcium fluxes modulate the radiation-induced bystander responses
in targeted glioma and fibroblast cells. Radiat Res. 166:479–487.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Faqihi F, Neshastehriz A, Soleymanifard S,
Shabani R and Eivazzadeh N: Radiation-induced bystander effect in
non-irradiated glioblastoma spheroid cells. J Radiat Res (Tokyo).
56:777–783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Maa SH, Wang CH, Liu CY, Lin HC, Huang KH
and Kuo HP: Endogenous nitric oxide downregulates the Bcl-2
expression of eosinophils through mitogen-activated protein kinase
in bronchial asthma. J Allergy Clin Immunol. 112:761–767. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu L, Rezvani HR, Back JH, Hosseini M,
Tang X, Zhu Y, Mahfouf W, Raad H, Ragi G, Athar M, et al:
Inhibition of p38 MAPK signaling augments skin tumorigenesis via
NOX2 driven ROS generation. PLoS One. 9:e972452014. View Article : Google Scholar : PubMed/NCBI
|