Introduction
Burkitt's lymphoma (BL) is an aggressive B cell
non-Hodgkin lymphoma (NHL), occurring in three distinct clinical
and epidemiological variants: Sporadic, endemic and
immunodeficiency-associated forms. The hallmark of BL is the
chromosomic translocation t(8;14), which causes the upregulation of
the c-MYC protein transcription factor as well as uncontrolled B
cell proliferation, accounting for the rapid growth rate of BL
tumor cells (1). c-MYC is stabilized
through the phosphorylation function of the human proto-oncogene
proviral integration of the Moloney murine leukemia virus (PIM-1)
serine/threonine kinase, underpinning the critical regulatory role
of PIM-1 in the c-MYC-driven BL tumor cell development (2).
The PIM-1 serine/threonine kinase is a highly
conserved protein. PIM-1 is expressed in normal lymphoid and
myeloid hematopoietic cells and in various human tissues, such as
prostate, testis and oral epithelial cells (3). PIM-1 has a constitutive kinase activity
that critically regulates cell migration, cell cycle, cell
proliferation, cell survival and exerts anti-apoptotic activities
(4). Elevated PIM-1 expression
predicts a poor outcome in hematological malignancies, including
acute myeloid leukemia, B cell chronic lymphocytic leukemia,
diffuse large B cell lymphoma and BL (5). PIM-1-overexpression contributes to
tumorigenesis through three pathways, including inhibiting
apoptosis (i.e., programmed cell death), stimulating cell
proliferation and promoting genomic instability (6). For instance, PIM-1 tumorigenic activity
has a synergizing effect with c-MYC in lymphomagenesis via the
phosphorylation and inactivation of the pro-apoptotic BAD protein,
which inhibits the cleavage of the key pro-apoptotic executioner
caspase-3 (7). Several studies
reported that highly expressed PIM-1 is co-located with c-MYC in
the nucleus of BL B cells, which strongly accelerates c-MYC-driven
lymphomagenesis (8,9).
Current anticancer therapies available for BL are
associated with life-threatening side effects because of tumor
lysis syndrome, especially in older patients with poorer outcomes
compared with younger patients (10). Chemoresistance has also been detected
in tumor cells overexpressing PIM-1, such as BL B cells (11). PIM-1 kinase is important in BL
tumorigenesis and chemoresistance and could be a promising
therapeutic target for patients diagnosed with BL. There is a need
for more specific and less toxic molecular PIM-1-targeted therapy
options for patients with BL. The aim of the present study was to
determine the pro-apoptotic effect of a PIM-1 kinase
pharmacological inhibitor (PIM1-1) in two BL B cell lines, Daudi
and Raji cells, compared with the K562 leukemia cell line that has
high levels of PIM-1.
Materials and methods
Cell culture and treatment
The BL (Raji and Daudi) and leukemia (K562) cell
lines were purchased from the American Type Culture Collection and
authenticated by the supplier. The cells were cultured in complete
medium consisting of RPMI-1640 with 2 mM L-glutamine, 100 µg/ml
streptomycin, 100 IU/ml penicillin and supplemented with 10% fetal
calf serum, (all Gibco; Thermo Fisher Scientific Inc.). The cells
were seeded in T-75 cm2 flasks and maintained in a
humidified incubator under standard conditions (37°C; 5%
CO2).
PIM1-1 (cat. no. 18/144326; 10 mM stock solution;
Tocris Bioscience) was solubilized in DMSO and diluted in
RPMI-1640. The cells were exposed to various concentrations (0.1,
1, 10, 20, 30 and 40 µM) of PIM1-1 and incubated for 48 h. DMSO
0.4% and 1 µM of the protein kinase inhibitor staurosporine (STS)
were used as negative and positive controls, respectively.
Cell viability
B cell lines (Raji and Daudi) and leukemic cell line
(K562) (104) were seeded in 100 µl complete RPMI-1640
medium per well of 96-well plates. The cell treatment was applied
as aforementioned and was performed in triplicate. The number of
viable cells were determined using the CellTiter-Glo®
Luminescent Assay kit (Promega Corporation) and based on the
quantification of ATP generated from metabolic reactions, which was
indicative of active cells. The amount of ATP produced was
proportional to the number of viable cells. The inhibitory
concentration (IC)-50 of PIM1-1 that resulted in a 50% decrease in
the viable cell number was also calculated.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR) analysis
Total RNA was extracted from the untreated and
treated cells (1.5×105/cm2) using the
PureLink™ RNA Mini kit (Ambion; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
High-quality RNA (1 µg) was reverse transcribed to cDNA using an
Applied Biosystems™ High-Capacity cDNA Reverse Transcription kit
(Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. The cDNA was used as the template for the quantitative
PCR reaction. RT-qPCR was performed through real-time monitoring of
the increase in fluorescence of the SYBR® Green dye
(cat. no. 4309155; Qiagen GmbH), using the 7900 Fast Real-Time PCR
system (Thermo Fisher Scientific, Inc.). The primer pair sequences
(Invitrogen; Thermo Fisher Scientific) were used as previously
described (12). The thermocycling
conditions were as follows: Heat denaturing at 95°C for 10 min,
then by 40 cycles of denaturing at 95°C for 10 sec followed by
annealing at 57°C for 20 sec and finally extension at 72°C for 30
sec. The fold-change of PIM-1 RNA expression levels measured
by the cycle threshold (Cq) values were calculated and normalized
to the expression levels of the housekeeping gene GAPDH,
according to 2−ΔΔCq method (13) as follows:
Fold-change=2(−ΔΔCq) with ΔΔCq=ΔCq
(PIM-1treated-GAPDHtreated)-ΔCq
(PIM-1control-GAPDHcontrol).
Western blotting
The untreated and treated Daudi, Raji and K562 cells
were lysed in Nonidet P40 (NP40) Cell Lysis Buffer composed of 50
mM Tris (pH 7.4), 250 mM NaCl, 5 mM EDTA, 50 mM NaF, 1 mM
Na3VO4, 1% NP40 and 0.2% NaN3 (all
Invitrogen; Thermo Fisher Scientific, Inc.). The lysates were
separated from the cell debris through centrifugation at 20,000 × g
for 15 min at 4°C and the supernatant was collected. The protein
concentrations were quantified using the Qubit™ Protein Assay kit
(Thermo Fisher Scientific, Inc.). Total proteins (80 µg per lane)
were separated using 12% SDS-PAGE and transferred to a
polyvinylidene difluoride membrane as described previously
(12). The membranes were blocked
for 1 h with 3% BSA at room temperature and probed, overnight at
4°C, with mouse or rabbit primary monoclonal antibodies (1:1,000)
directed against phosphorylated (p)-BAD (Ser112; cat. no. 5284),
total BAD (cat. no. 9239), pro-caspase-3 (cat. no. 14220) and
cleaved caspase-3 (cat. no. 9664) procured from Cell Signaling
Technology, Inc., p-ERK1 (Tyr204 of ERK1, cat. no. sc-7383), total
ERK1 (cat. no. sc-271269), PIM-1 (cat. no. sc-13513) obtained from
Santa Cruz Biotechnology, Inc. and GAPDH (cat. no. ab8245) from
Abcam. The membranes were washed three times in Tris-buffered
saline containing 0.1% Tween-20 (pH 7.4) and incubated with either
goat anti-mouse (1:5,000; cat. no. 170-5047; Bio-Rad Inc.) or
anti-rabbit (1:5,000; cat. no. 170-6515; Bio-Rad Inc.) secondary
antibodies conjugated to horseradish peroxidase or with either
infrared fluorescent IRDye® 680RD-conjugated goat
anti-rabbit (1:10,000; cat. no. 926-68071; Li-COR Biosciences) or
IRDye® 800RD-cojugated goat anti-mouse (1:10,000; cat.
no. 926-32210; Li-COR Biosciences) secondary antibodies, for 1 h at
room temperature. The bands were visualized by incubating the
horseradish peroxidase-stained membranes with enhanced
chemiluminescence reagents (Clarity™ Western ECL Substrate; Bio-Rad
Laboratories, Inc.) and scanned with c-DiGit (LI-COR Biosciences).
The IRDye stained membranes were scanned using a LI-COR
Odyssey® CLx Scanner. The protein expression level was
quantified using ImageJ software v.1.46r (http://rsbweb.nih.gov/ij/index.html).
Statistical analysis
All the data are expressed as mean ± standard
deviation and each experiment was independently repeated three
times. For comparison between the groups, one-way ANOVA with
Tukey's HSD post hoc test was used. SPSS Statistics 64-bit MS
Windows v.22.0.0.0 (IBM, Corp.) was used to analyze the data, and
for gene expression levels, the Applied Biosystems™ QuantStudio™ 6
Flex system (Thermo Fisher Scientific, Inc.) was used. P<0.05
was considered to indicate a statistically significant
difference.
Results
PIM1-1 inhibits Raji, Daudi and K562
cell viability
To study the inhibitory effect of PIM1-1 on Raji,
Daudi and K562 cell viability, the cells were incubated for 48 h
with different concentrations of PIM1-1 (range, 0.1–40 µM), with
0.4% DMSO (negative control) and 1 µM STS (positive control). The
untreated and treated cells were subjected to a luminescent cell
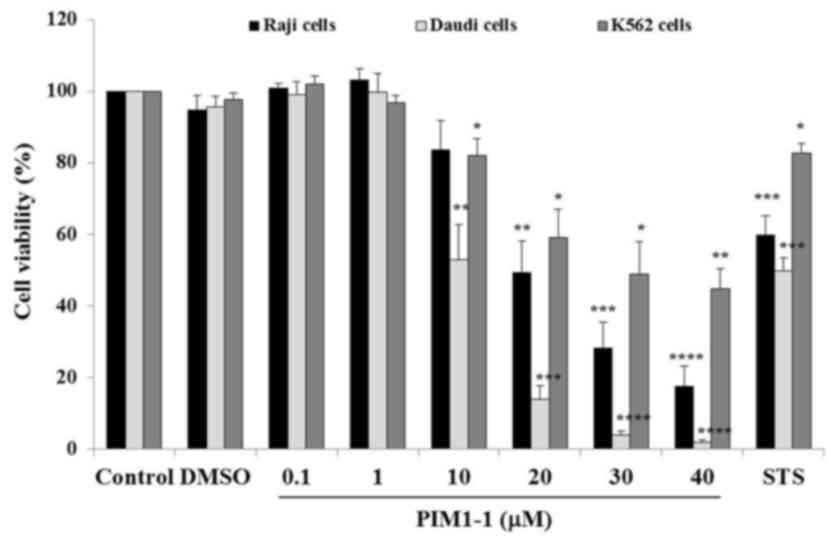
viability assay using CellTiter-Glo™. As shown in Fig. 1, a slight increase of Raji and Daudi
cell viability was observed at low concentrations of PIM1-1 at 0.1
and 1 µM; indicating a slightly higher amount of ATP generated by
treated cells compared with the ATP amount generated by untreated
cells. At higher concentrations, PIM1-1 significantly decreased the
Daudi cell viability by ~50% at 10 µM (P=0.0028) and by 98% at 40
µM (P<0.0001). In the Raji cells, PIM1-1 significantly decreased
cell viability by 50% (P=0.0021) at 20 µM and by ~85% (P<0.0001)
at 40 µM. However, for K562 cells, a 48.8% decrease in viability
was measured at 30 µM PIM1-1 (P=0.015) and 44.8% at 40 µM
(P=0.0051). Increasing concentrations of PIM1-1 decreased the Raji,
Daudi and K562 cell viability in a dose-dependent manner. As
estimated from the sigmoidal dose curve with a variable slope, the
IC50 for the Daudi, Raji and K562 cells were at 10, 20 and 30 µM of
PIM1-1, respectively (data not shown). The PIM-1 kinase
pharmacological inhibitor PIM1-1 inhibited the cell growth of the
BL B cell lines (Raji and Daudi), and the growth of leukemic cell
line K562 was the least affected (Fig.
1).
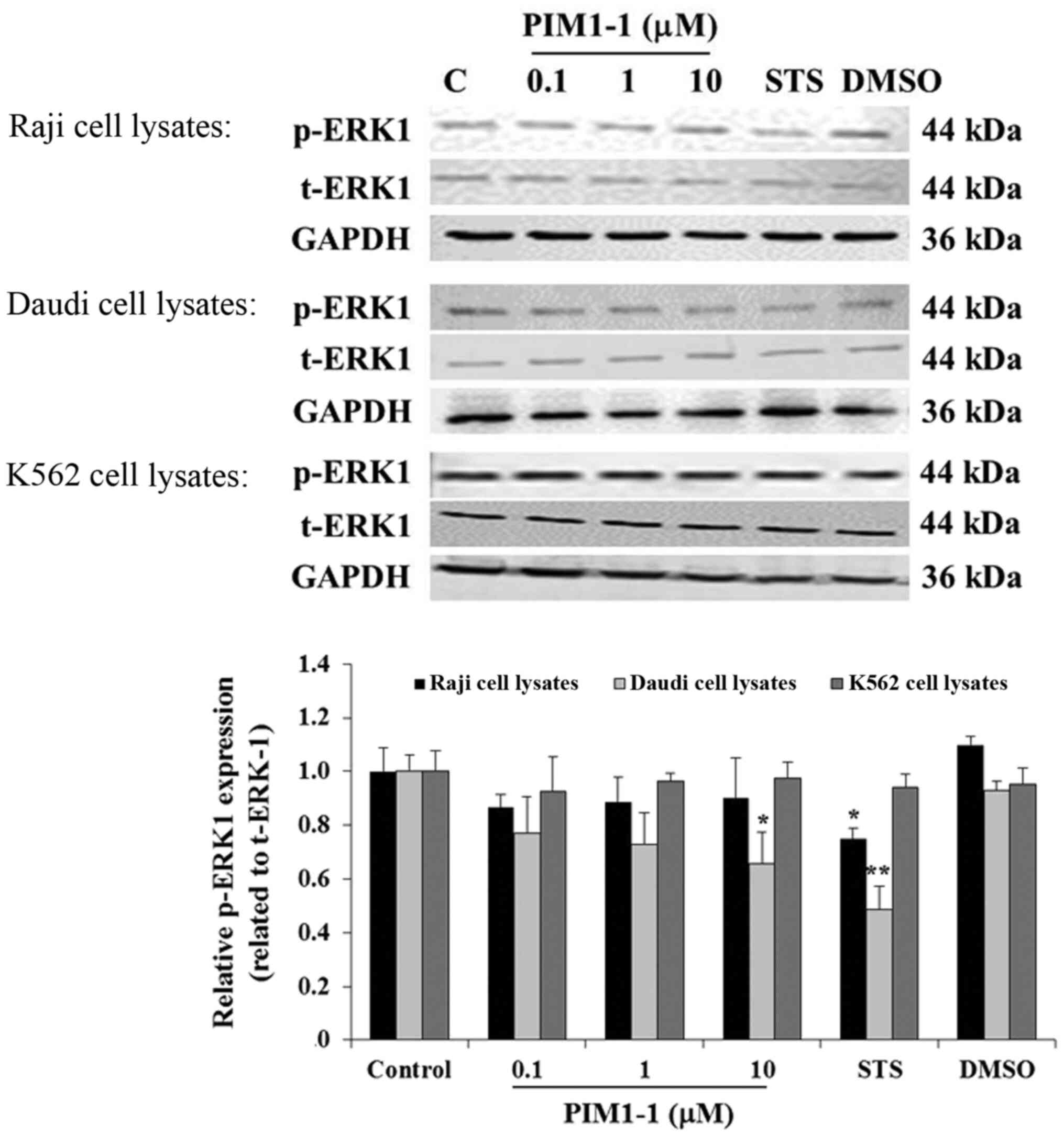
To confirm the decrease of cell proliferation, the
level of p-ERK in the untreated cells and cells treated with DMSO,
STS or (0.1, 1.0 or 10 µM) PIM1-1 were assessed using western
blotting. In the Raji cells, any addition of PIM1-1 slightly
decreased p-ERK1 compared with the untreated cells (Fig. 2). However, a significant decrease in
the level of p-ERK1 was observed in the Daudi cells treated with 10
µM of PIM1-1 compared with the untreated cells. Used as a positive
control, the protein kinase inhibitor STS, when added to Raji and
Daudi cells, resulted in a significant decrease of the level of
p-ERK1 compared with the p-ERK1 expression level detected in the
untreated cells (Fig. 2). No change
was observed in the level of p-ERK1 in the K562 cells in all the
conditions used. Hence, various antiproliferative effects of PIM1-1
on the BL B cell lines Raji and Daudi were confirmed by the
variation of the decrease in the levels of p-ERK1 measured in each
PIM1-1-treated B cell line. At the low concentrations at which the
PIM1-1 was tested (0.1–10 µM), the leukemic cell growth based on
p-ERK1 level remained unchanged.
PIM1-1 induces the downregulation of
PIM-1 and BAD phosphorylation in the Daudi and Raji cell lines
PIM-1 negatively regulates its own expression
(14). PIM1-1 kinase inhibitor
efficiency was assessed based on the PIM-1 kinase expression level.
At the protein level, using western blotting, PIM1-1 tested between
0.1 and 10 µM did not significantly decrease PIM-1 protein
expression in PIM1-1-treated Raji cells (Fig. 3). However, at 1 and 10 µM, PIM1-1
significantly decreased PIM-1 protein expression level by 70%
(P<0.01) in Daudi cells, compared with the expression level
detected in untreated Daudi cells (Fig.
3A). For the K562 cells, PIM1-1 did not change the PIM-1
protein expression level compared with untreated cells (Fig. 3). STS significantly inhibited PIM-1
protein expression level by 85% in both the Raji and Daudi cells,
but not in K562 cells (Fig. 3A). As
expected, DMSO did not affect the PIM-1 expression level in all
types of cells compared with untreated cells (Fig. 3).
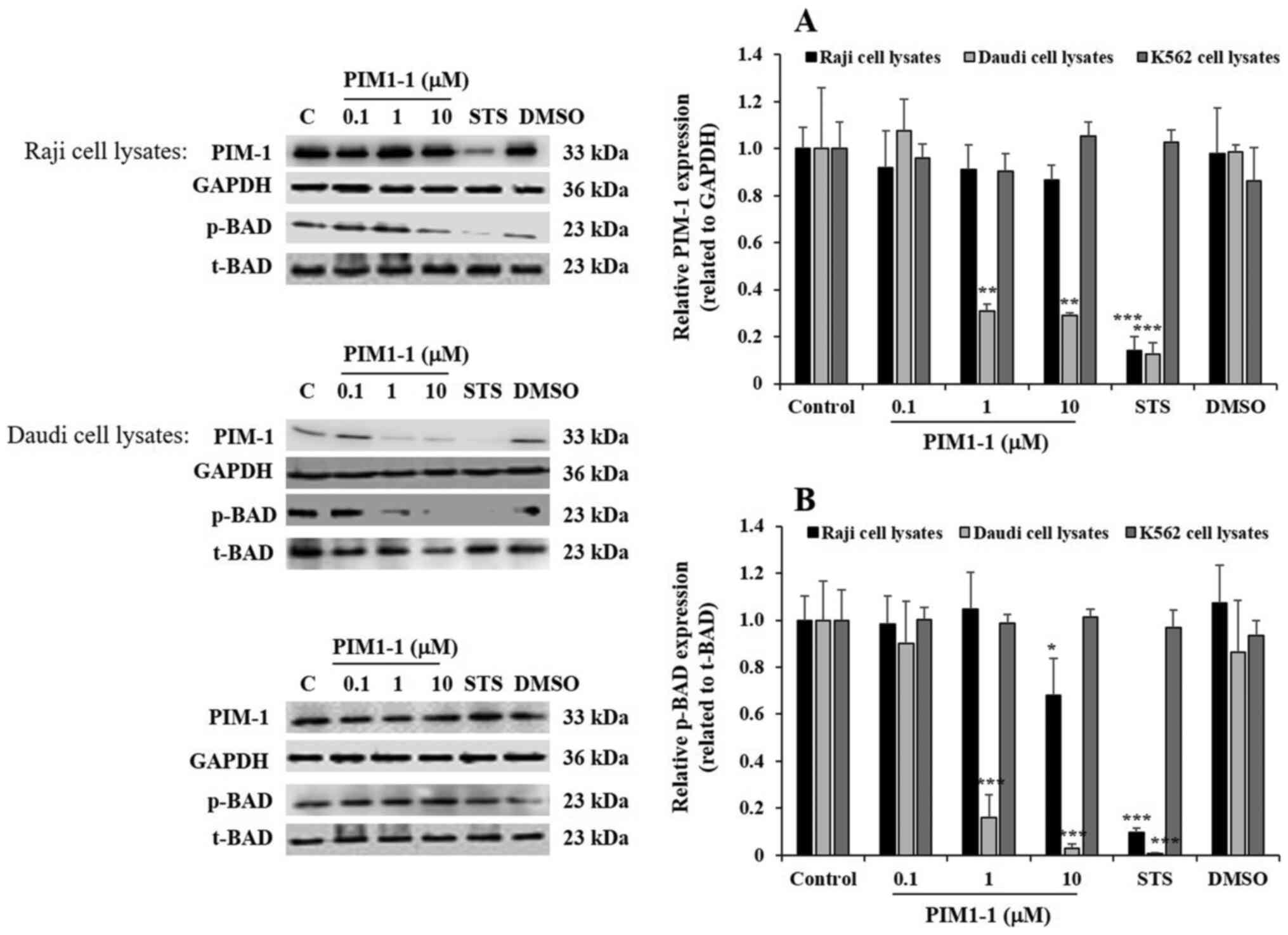 | Figure 3.PIM1-1 induces the downregulation of
PIM-1 protein expression and decreases phosphorylation of BAD
protein in the Raji and Daudi cells. PIM-1 protein expression
levels in the Raji, Daudi and K562 cells following 48 h treatment
with a range of concentrations (range, 0.1–10 µM) of PIM-1.1. Bar
graphs show the relative expression levels of the PIM-1 protein (A)
and of the p-BAD (B) calculated as a ratio of the expression of the
GAPDH and t-BAD loading controls, respectively. *P<0.05,
**P<0.01 and ***P<0.001 compared with the control. PIM-1,
proviral integration of Moloney murine leukemia virus; p-,
phosphorylated-; t-, total-; STS, staurosporine; PIM, proviral
integration of the Moloney virus; PIM1-1, PIM-1 kinase
pharmacological inhibitor. |
PIM-1 kinase phosphorylates the pro-apoptotic
protein BAD and promotes BAD inactivation, which supports cell
survival (7,15). The level of p-BAD was evaluated to
confirm the variation of PIM-1 kinase expression observed in the
PIM1-1-treated cells. Notably, a significant decrease in p-BAD was
observed in the Raji cells treated with 10 µM PIM1-1 compared with
the level detected in the untreated cells (Fig. 3). A significant decrease and
quasi-disappearance of p-BAD was observed in Daudi cells treated
with 1 and 10 µM of PIM1-1 compared with the untreated cells
(Fig. 3). As expected, STS decreased
p-BAD levels in both Raji and Daudi cells, but DMSO did not affect
p-BAD in the three cell lines (Fig.
3). In all the conditions applied, the level of p-BAD in the
K562 cells did not change, compared with the untreated cells
(Fig. 3).
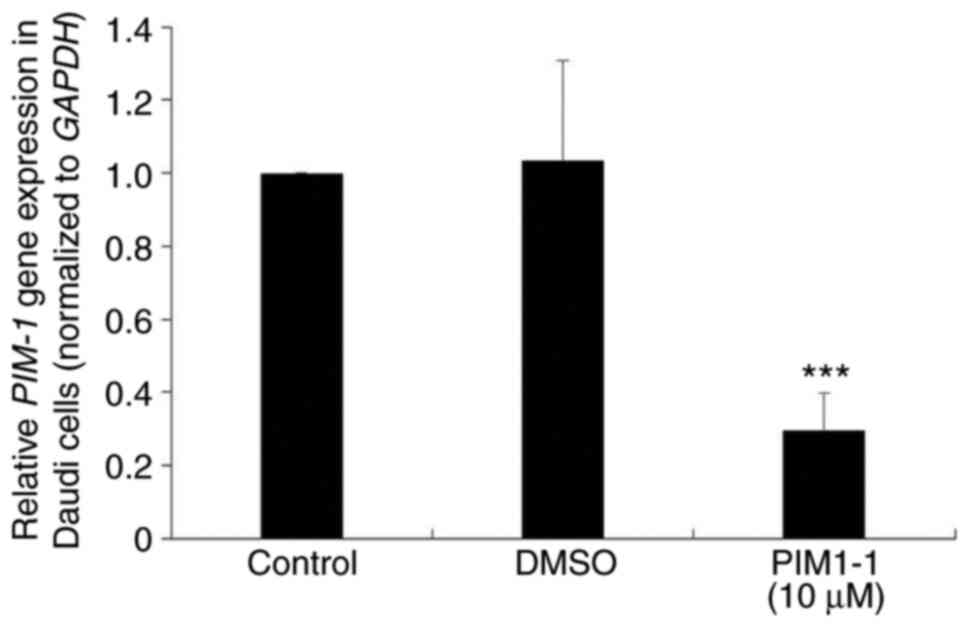
To verify the downregulation of the PIM-1 protein
expression observed in PIM1-1-treated Daudi cells, PIM-1 mRNA
expression level was examined in Daudi cells treated with 10 µM
PIM1-1. A significant decrease (~70%; P=0.0003) of the PIM-1 mRNA
expression level was observed in PIM1-1-treated Daudi cells
compared with the untreated and DMSO-treated cells (Fig. 4).
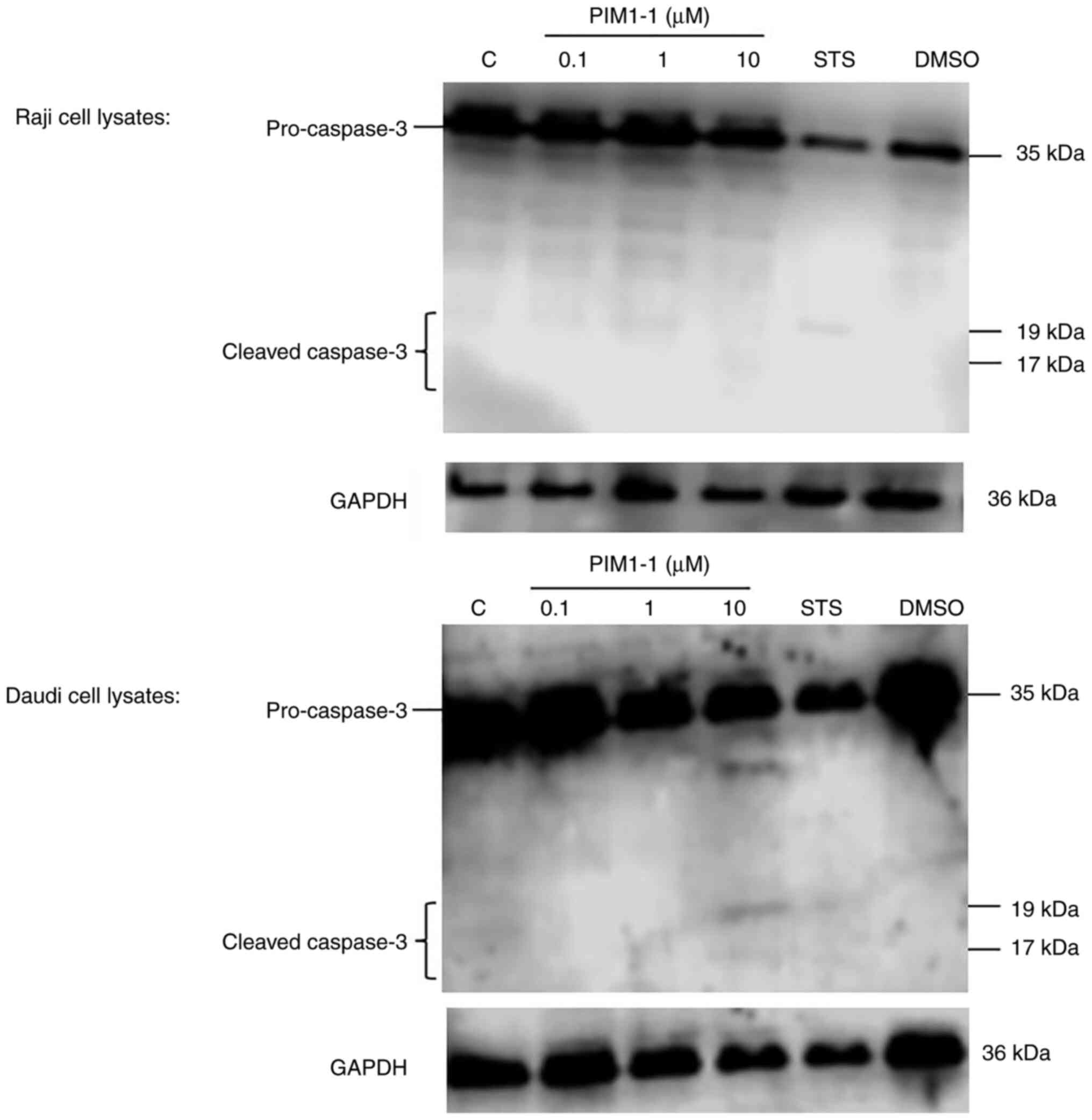
Detection of cleaved caspase-3 in
PIM1-1-treated Raji and Daudi cells
At the functional level, the blockade of PIM-1
kinase activity principally results in the induction of apoptosis
(16). After Raji and Daudi cell
treatment with various concentrations (range, 0.1–10 µM) of PIM1-1,
the protein extracts were subjected to western blotting for the
detection of cleaved caspase-3, a hallmark of apoptosis (17). A weak detection of cleaved caspase-3
was observed in the PIM1-1-treated Raji cells; however, cleaved
caspase-3 was observed in the Daudi cells treated with 10 µM PIM1-1
(Fig. 5). As expected, no cleavage
of pro-caspase-3 was observed in the DMSO-treated cells but some
cleavage occurred in STS-treated cells (Fig. 5).
Discussion
In hematopoietic cells, PIM-1 is involved in the
development and function of the cells. The overexpression of PIM-1
kinase is found in the majority of hematological malignancies,
including myeloid and lymphoid leukemia and B cell NHL (18). PIM-1-overexpression contributes to
malignant cell proliferation and through dysregulation of the cell
cycle and inhibition of apoptosis (19,20).
Several studies have investigated the effect of
PIM-1-downregulation and its impact on cell survival and apoptosis
(16,21). The results of the present study
established a connection between PIM-1 inhibition using the PIM-1
pharmacological inhibitor, PIM1-1, and the inhibition of cell
survival and increased apoptosis, suggesting that targeting PIM-1
kinase is a potentially promising therapeutic approach in BL. Small
molecule inhibitors of PIM kinases have been investigated in
preclinical and clinical studies to treat hematological and solid
cancer types (21–23). The selective PIM1-1 drug investigated
in the present study belongs to the pyridone-based family of small
molecules inhibitors of PIM-1 kinase which has been shown to exert
an inhibitory effect by binding to the ATP-binding site of the
PIM-1 kinase, suggesting an ATP-competitive inhibitory mechanism
(24). Targeting the kinase action
of PIM-1 is expected to prevent the phosphorylation of the
downstream effectors and to block its capacity to activate or
inactivate proteins involved in cell cycle progression and
apoptosis, such as BAD (7,15). In the present study, the treatment of
the BL B cell lines, Raji and Daudi, with the novel PIM-1 inhibitor
PIM1-1 resulted in decreased cell viability. In addition, a
significant decrease of ERK-1 phosphorylation was detected in
PIM1-1-treated Daudi cells, confirming the PIM1-1 antiproliferative
effect in the BL B cell lines. The current study showed that
inhibiting PIM-1 kinase in Daudi and Raji cells with PIM1-1
resulted in a decrease in BAD phosphorylation and induction of
apoptosis, revealed by caspase-3 cleavage. These data highlighted
the promising therapeutic potential of PIM1-1 against BL B cell
development.
PIM-1 is involved in cell proliferation through the
regulation of cell cycle progression and decreased apoptosis
(19,25). The current study showed that
increasing concentrations of PIM1-1 decreased the Daudi and Raji
cell viability in a dose-dependent manner; however, the leukemia
cell line K562 was less affected. Based on the IC50 determination,
Daudi cells were more sensitive to PIM1-1-inhibition compared with
Raji and K562 cells. This observation can be explained by the
differential PIM-1-expression levels between both the BL B cell
lines and the leukemia cells, with the Daudi cells expressing less
PIM-1 compared with the Raji cells (3). The K562 cells, used as a positive
control for their high-level protein expression of PIM-1 as
previously reported in (3,12), were the least responsive to the
PIM1-1 inhibitor. A previous study has shown that quercetagetin (a
PIM-1 inhibitor) inhibits cell viability and decreases the colony
formation rate of nasopharyngeal carcinoma cells (25). These results are in line with a
previous study in which the inhibition of PIM-1 with K00135
[imidazo (1,2-b) pyridazines] in murine Ba/F3 cells, acute leukemia
cells, and primary blasts from patients with acute myeloid
leukemia, selectively reduces cell survival and suppressed the
colony proliferation of leukemic blasts (26). In the present study, the decrease in
BL B cell line viability caused by PIM1-1 was confirmed by
decreased p-ERK, a key signaling protein involved in cell survival
and proliferation (27,28). A previous study, conducted with
prostate cancer cells, reported that PIM-1-knockdown results in a
decrease of p-ERK (29). It is not
yet clear whether PIM-1 kinase directly or indirectly affects ERK
phosphorylation. However, it would be of interest to investigate
the impact of the PIM1-1 kinase inhibitor on substrates with
similar regulatory survival pathways, such as ERK and
phosphatidylinositol 3-kinase/protein kinase B (PKB also known as
Akt) as well (30,31). In addition, Akt/PKB has been
demonstrated to be activated in two acute myeloid leukemia cell
lines intrinsically resistant to the pan-PIM kinase inhibitor
AZD1208, following to elevation of mitochondrial reactive oxygen
species (ROS) (32). Furthermore,
ROS have been recently reported to be important indicators of drug
resistance (33). An assessment of
Akt/PKB phosphorylation levels and ROS generated in the BL B cells
and leukemic cells could reveal their resistance potential to any
PIM-1 kinase inhibitor, including PIM1-1. Altogether, a combination
of pharmacological inhibitors targeting survival pathway signaling
proteins, ROS production and PIM-1 kinase could enhance the
sensitivity of the BL B cells and leukemic cells to cancer
therapy.
In addition to acting as a survival factor, PIM-1
kinase also plays a role as both a transcription factor and an
activator of several transcription factors, including the signal
transducer activator transcription-3, which stimulates PIM-1 gene
expression (3). In the current
study, 10 µM of the PIM1-1 pharmacological inhibitor reduced the
PIM-1 mRNA and protein expression levels in the Daudi cells, with
no change in the PIM1-1-treated Raji cells compared with the
untreated cells. These findings provide evidence for the
differential expression of PIM-1 in the Daudi and Raji BL B cell
lines, confirming that the Daudi cells are the most responsive to
PIM1-1.
PIM1-1 deactivates the pro-apoptotic BAD protein by
phosphorylation of Ser112, and changes in the level of this
phosphorylation indicates variation of PIM-1 kinase activity
(15). BAD is a pro-apoptotic member
of a Bcl-2 group that assists with cell death. The phosphorylation
of BAD enhances the binding of BAD to 14-3-3 proteins to block the
associations between BAD with the anti-apoptotic proteins Bcl2 and
Bcl-xL (34). It has been reported
that PIM-1 may play a critical role in the control of survival
signaling through the inhibition of mitochondrial pro-apoptotic
members of the Bcl-2 family, such as BAD (15,35). In
the present study, administration of the PIM1-1 inhibitor caused a
significant reduction in the BAD phosphorylation level in the Daudi
cells from 1 µM of PIM1-1, while the Raji cells showed a reduction
on the p-BAD expression level at 10 µM of PIM1-1. These findings
are similar to those reported by Chen et al (16) in 2011 who demonstrated the
downregulation of p-BAD in acute myeloid leukemia cells treated
with the pan-PIM inhibitor SGI-1776. Forshell et al
(9) also reported in 2011 that
treating Myc-induced B cell lymphoma with the pan-PIM kinase
inhibitor (named as Pim1) caused the dephosphorylation of BAD and
the induction of apoptosis. The reduction in PIM-1 expression
observed in the current study may have contributed to the decrease
in BAD phosphorylation and to the induction of apoptosis, revealed
by caspase-3 cleavage. Although executioner caspases, including
caspase-3, function in natural cancer cell death (36), PIM-1 phosphorylates the endogenous
apoptosis signaling kinase (ASK)1, which suppresses the activation
of pro-caspase-3 and maintains cancer cell survival (37). In the present study, inhibition of
PIM-1 kinase allowed caspase-3-activation in BL B cell lines, which
resulted in strong cleaved caspase-3 expression in Daudi cells and
weak expression in the Raji cells after treatment with 1–10 µM
PIM1-1. An assessment of ASK1 phosphorylation level using western
blotting, expected to decrease following BL B cell treatment with
PIM1-1, could confirm apoptosis induction through caspase
activation.
In conclusion, the present study demonstrated that
the novel PIM1-1 pharmacological inhibitor effectively
downregulated PIM-1 expression, markedly decreased cell viability
and induced apoptosis, which was revealed by caspase-3 cleavage, in
BL B cell lines. These findings provide new evidence for PIM-1
kinase inhibition as a potential therapeutic target for BL
treatment. Due to the limited number of cell lines used in the
current study, it would be interesting to evaluate the effects of
PIM1-1 using other B cell lymphoma cell lines or animal models.
Studying the effect of PIM-1-knockdown on other genes involved in
cell proliferation, survival, homing, cell signaling, apoptosis and
migration is also required. Future studies should examine the
effect of PIM1-1 on primary BL cells obtained from patients and to
analyze the chromosomal translocation frequency and c-MYC
expression levels, which are the main genetic hallmarks of BL
(38). In addition, the synergistic
effect between PIM1-1 and current chemotherapies should be
evaluated to reduce resistance to chemotherapy and to improve the
response to the available therapies, which could provide an
effective treatment and improve the survival rate for patients
diagnosed with BL.
Acknowledgements
Not applicable.
Funding
This project was fully funded by King Abdullah
International Medical Research Center (grant no. RC12/092).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contribution
IA and AhA conceived the study. IA, SMN and AbA
conducted the study. MoA, HAE, SA, KA and MaA conducted the
experiments, collected the data, analyzed the data and reviewed the
manuscript. Authenticity of all raw data were confirmed by SMN and
AbA. MoA, SMN, AbA, AhA and IA wrote the manuscript. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kalisz K, Alessandrino F, Beck R, Smith D,
Kikano E, Ramaiya NH and Tirumani SH: An update on burkitt
lymphoma: A review of pathogenesis and multimodality imaging
assessment of disease presentation, treatment response, and
recurrence. Insights Imaging. 10:562019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tursynbay Y, Zhang J, Li Z, Tokay T,
Zhumadilov Z, Wu D and Xie Y: Pim-1 kinase as cancer drug target:
An update. Biomed Rep. 4:140–146. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bachmann M and Möröy T: The
serine/threonine kinase Pim-1. Int J Biochem Cell Biol. 37:726–730.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mondello P, Cuzzocrea S and Mian M: Pim
kinases in hematological malignancies: Where are we now and where
are we going? J Hematol Oncol. 7:952014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Panchal NK and Sabina EP: A
serine/threonine protein PIM kinase as a biomarker of cancer and a
target for anti-tumor therapy. Life Sci. 255:1178662020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Magnuson NS, Wang Z, Ding G and Reeves R:
Why target PIM1 for cancer diagnosis and treatment? Future Oncol.
6:1461–1478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Macdonald A, Campbell DG, Toth R,
McLauchlan H, Hastie CJ and Arthur JS: Pim kinases phosphorylate
multiple sites on Bad and promote 14-3-3 binding and dissociation
from Bcl-XL. BMC Cell Biol. 7:12006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ionov Y, Le X, Tunquist BJ, Sweetenham J,
Sachs T, Ryder J, Johnson T, Lilly MB and Kraft AS: Pim-1 protein
kinase is nuclear in Burkitt's lymphoma: Nuclear localization is
necessary for its biologic effects. Anticancer Res. 23:167–178.
2003.PubMed/NCBI
|
|
9
|
Forshell LP, Li Y, Forshell TZ, Rudelius
M, Nilsson L, Keller U and Nilsson J: The direct Myc target Pim3
cooperates with other Pim kinases in supporting viability of
Myc-induced B-cell lymphomas. Oncotarget. 2:448–460. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McBride A, Trifilio S, Baxter N, Gregory
TK and Howard SC: Managing tumor lysis syndrome in the era of novel
cancer therapies. J Adv Pract Oncol. 8:705–720. 2017.PubMed/NCBI
|
|
11
|
Zemskova M, Sahakian E, Bashkirova S and
Lilly M: The PIM1 kinase is a critical component of a survival
pathway activated by docetaxel and promotes survival of
docetaxel-treated prostate cancer cells. J Biol Chem.
283:20635–20644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matou-Nasri S, Rhaban Z, Al-Baijan H,
Al-Eidi H, Yahya WB, Al Abdulrahman A, Almobadel N, Alsubeai M, Al
Ghamdi S, Alaskar A, et al: CD95-mediated apoptosis in Burkitt's
lymphoma B-cells is associated with Pim-1 down-regulation. Biochim
Biophys Acta Mol Basis Dis. 1863:239–252. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peltola KJ, Paukku K, Aho TL, Ruuska M,
Silvennoinen O and Koskinen PJ: Pim-1 kinase inhibits
STAT5-dependent transcription via its interactions with SOCS1 and
SOCS3. Blood. 103:3744–3750. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aho TL, Sandholm J, Peltola KJ, Mankonen
HP, Lilly M and Koskinen PJ: Pim-1 kinase promotes inactivation of
the pro-apoptotic Bad protein by phosphorylating it on the Ser112
gatekeeper site. FEBS Lett. 571:43–49. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen LS, Redkar S, Taverna P, Cortes JE
and Gandhi V: Mechanisms of cytotoxicity to Pim kinase inhibitor,
SGI-1776, in acute myeloid leukemia. Blood. 118:693–702. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shah N, Pang B, Yeoh KG, Thorn S, Chen CS,
Lilly MB and Salto-Tellez M: Potential roles for the PIM1 kinase in
human cancer-a molecular and therapeutic appraisal. Eur J Cancer.
44:2144–2151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morishita D, Katayama R, Sekimizu K,
Tsuruo T and Fujita N: Pim kinases promote cell cycle progression
by phosphorylating and down-regulating p27Kip1 at the
transcriptional and posttranscriptional levels. Cancer Res.
68:5076–5085. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ouhtit A, Gupta I, Muzumdar S,
Shanmuganathan S and Tamimi Y: Understanding the functional
discrepancy of Pim-1 in cancer. Front Biosci (Elite Ed). 7:208–214.
2015.PubMed/NCBI
|
|
21
|
Luszczak S, Kumar C, Sathyadevan VK,
Simpson BS, Gately KA, Whitaker HC and Heavey S: PIM kinase
inhibition: Co-targeted therapeutic approaches in prostate cancer.
Signal Transduct Target Ther. 5:72020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blanco-Aparicio C and Carnero A: Pim
kinases in cancer: Diagnostic, prognostic and treatment
opportunities. Biochem Pharmacol. 85:629–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang X, Song M, Kundu JK, Lee MH and Liu
ZZ: PIM kinase as an executional target in cancer. J Cancer Prev.
23:109–116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheney IW, Yan S, Appleby T, Walker H, Vo
T, Yao N, Hamatake R, Hong Z and Wu JZ: Identification and
structure-activity relationships of substituted pyridones as
inhibitors of Pim-1 kinase. Bioorg Med Chem Lett. 17:1679–1683.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jie W, He QY, Luo BT, Zheng SJ, Kong YQ,
Jiang HG, Li RJ, Guo JL and Shen ZH: Inhibition of Pim-1 attenuates
the proliferation and migration in nasopharyngeal carcinoma cells.
Asian Pac J Trop Med. 5:645–650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pogacic V, Bullock AN, Fedorov O,
Filippakopoulos P, Gasser C, Biondi A, Meyer-Monard S, Knapp S and
Schwaller J: Structural analysis identifies
imidazo[1,2-b]pyridazines as PIM kinase inhibitors with in vitro
antileukemic activity. Cancer Res. 67:6916–6924. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu Z and Xu S: ERK1/2 MAP kinases in cell
survival and apoptosis. IUBMB Life. 58:621–631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee S, Rauch J and Kolch W: Targeting MAPK
signaling in cancer: Drug resistance and sensitivity. Int J Mol
Sci. 21:11022020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang J, Anderson PD, Luo W, Gius D, Roh M
and Abdulkadir SA: Pim1 kinase is required to maintain
tumorigenicity in MYC-expressing prostate cancer cells. Oncogene.
31:1794–1803. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aziz AR, Farid S, Qin K, Wang H and Liu B:
PIM kinases and their relevance to the PI3K/Akt/mTOR pathway in the
regulation of ovarian cancer. Biomolecules. 8:72018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Warfel NA and Kraft AS: PIM kinase (and
Akt) biology and signaling in tumors. Pharmacol Ther. 151:41–49.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brunen D, García-Barchino MJ, Malani D,
Basheer NJ, Lieftink C, Beijersbergen RL, Murumägi A, Porkka K,
Wolf M, Zwaan CM, et al: Intrinsic resistance to PIM kinase
inhibition in AML through p38α-mediated feedback activation of mTOR
signaling. Oncotarget. 7:37407–37419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bhardwaj V and He J: Reactive oxygen
species, metabolic plasticity, and drug resistance in cancer. Int J
Mol Sci. 21:34122020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zha J, Harada H, Yang E, Jockel J and
Korsmeyer SJ: Serine phosphorylation of death agonist BAD in
response to survival factor results in binding to 14-3-3 Not
BCL-X(L). Cell. 87:619–628. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lilly M, Sandholm J, Cooper JJ, Koskinen
PJ and Kraft A: The PIM-1 serine kinase prolongs survival and
inhibits apoptosis-related mitochondrial dysfunction in part
through a bcl-2-dependent pathway. Oncogene. 18:4022–4031. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 5:a0086562013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gu JJ, Wang Z, Reeves R and Magnuson NS:
PIM1 phosphorylates and negatively regulates ASK1-mediated
apoptosis. Oncogene. 28:4261–4271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nguyen L, Papenhausen P and Shao H: The
role of c-MYC in B-cell lymphomas: Diagnostic and molecular
aspects. Genes (Basel). 8:1162017. View Article : Google Scholar : PubMed/NCBI
|