Introduction
Advances in chemotherapy and radiation therapy since
the second half of the 20th century have been invaluable in the
effective treatment of a number of types of tumor. However, these
therapeutic approaches have largely reached their potential, and
any improvement in therapeutic outcomes has to involve novel, more
effective therapeutic approaches (1). In recent decades, cancer immunotherapy
has emerged as a promising therapeutic approach for the effective
treatment of various tumors including but not limited to breast
cancer, colorectal cancer and pancreatic cancer (2,3).
Intricately organized, the immune system consists of several
components that are capable of recognizing and reacting to foreign
pathogens, such as lymphoid organs, B and T cells and cytokines,
such as interferons and colony stimulating factors (4). The cells of the immune system are
widely distributed, localizing in the lymph nodes as well as other
tissues and organs (4). However, the
fight against cancer is complex, as cancer cells have been
demonstrated to employ several mechanisms of immune tolerance.
Tumors form immune-suppressive microenvironments through the
presence of T regulatory cells (Tregs) or suppressive myeloid
cells, allowing them to evade the immune system (5). The presence of Tregs and suppressive
myeloid cells in the tumor microenvironment supports the hypothesis
that the immune targeting of cancer is a viable treatment option
(1,5). In addition, the number of available
immunotherapy options is an advantage when it comes to cancer
therapy due to the variations that occur in various tumor types.
One major limitation that cancer immunotherapy presents that
certain treatments, such as cancer vaccines and adoptive cell are
characterized by high inter-patient variability (6). Thus, such treatment may not be
considered universal, and may be effective for only a subgroup of
patients. The present review discusses the following immunotherapy
approaches: The use of cytokines, adoptive cell transfer, immune
checkpoint inhibitors (ICIs), vaccines and monoclonal antibodies.
The different subtypes of checkpoint inhibitors and vaccines have
been tabulated in Tables I and
II, respectively. In addition,
various immunotherapeutic approaches are evaluated, and the
potential use of combination therapy is assessed. The immunotherapy
drugs and techniques available in the market were summarized in
Table III.
 | Table I.Classes of immune checkpoints, their
modes of action, inhibitors, types of cancer they target and the
associated cardiac adverse effects (49). |
Table I.
Classes of immune checkpoints, their
modes of action, inhibitors, types of cancer they target and the
associated cardiac adverse effects (49).
| Inhibitory
receptor | Mode of action | Checkpoint
inhibitors | Types of cancer
treated | Cardiac
effects |
|---|
| CTLA-4 (on T
cells) | Interacts with
HLA-B7-1 and HLA-B7-2 on T cells and delivers an inhibitory signal
to effector T cells while promoting inhibitory function of
regulatory T cells | Ipilimumab | Advanced melanoma,
advanced renal cell cancer | Immune-related
myocarditis, Takotsubo-like syndrome, smoldering myocarditis |
| PD-1 (on T
cells) | PD-L1 is expressed
on the tumor cell surface and through interaction with PD-1 on T
cells causes apoptosis of cytotoxic T cells while inhibiting
apoptosis of regulatory T cells | Nivolumab | Melanoma, Hodgkin
lymphoma, NSCLC, kidney cancers | Acute lymphocytic
myocarditis, heart failure, cancer, head and neck complete
atrioventricular block |
|
|
| Pembrolizumab | Melanoma, Hodgkin
lymphoma, NSCLC, cancers of the urinary tract | Myocarditis,
polymyositis |
| PD-L1 (on cancer
cells) |
| Atezolizumab | Lung cancer, liver
cancer, breast cancer, urothelial cancer | Myositis with
myasthenic syndrome |
|
|
| Avelumab | Merkel cell
carcinoma, cancers of the urinary tract | Immune-related
myocarditis, hypertension |
|
|
| Durvalumab | NSCLC | Immune-related
myocarditis |
| Table II.Types of cancer vaccines. |
Table II.
Types of cancer vaccines.
| Type of
vaccine | Mode of action | Immune
response | Manipulation | Key advantage | Key limitation | Cost |
|---|
| DNA | Bacterial plasmid
encoding tumor antigen coupled to a eukaryotic promoter (89) | Poor (95) | Easy (93) | CpG islands and
post-translational modification in hosT cell (95) | Low uptake rate of
plasmid (94) | Cheap (93) |
| Peptide | Nongenetic
component that induces an immune response (98) | Poor (100) | Easy (99) | Do not overload the
immune system (99) | Inconclusive safety
of vaccine when used with adjuvants (99) | Cheap (99) |
| Dendritic cell | Patient dendritic
cells transfected with the tumor antigen (106,118) | Strong (107) | – | Personalization
(107) | Limitations of
monocyte-derived dendritic cells (109) | Expensive (109) |
| Whole cell | Modified whole
tumor cells (114) | Strong with the
exception of advanced tumors (108) | Easy (114) | Expression of TSAs
and TAAs (115) | Immune evasion
(114) | Expensive (108) |
| Table III.Immunotherapy drugs and techniques
available in the market. |
Table III.
Immunotherapy drugs and techniques
available in the market.
| Drug | Type | Description | Type of cancer | Combination
therapy |
|---|
| IMYGLIC or
T-VEC | Vaccine (133) | Modified HSV1 that
replicates in recurrent lesions causing cell lysis and death
(133) | Melanomas
(133) |
|
| Sipleucel-T or
PROVENGE | Vaccine (112) | Personalized
dendritic cell vaccine for which the patienT cells are harvested
and modified to express tumor antigens in MHC I and II (112) | Metastatic
castration-resistant prostate cancer (112) |
|
| Rituximab | Monoclonal antibody
(125) | Induces tumor cell
death through two mechanisms centered on Fc (125) | CD20+
B-cell non-Hodgkin lymphoma (125) |
|
| Trastuzumab | Monoclonal antibody
(125) | Induces tumor cell
death through two mechanisms centered on Fc (125) | HER2+
breast cancer (125) |
|
| Cetuximab | Monoclonal antibody
(128) | Induces tumor cell
death through two mechanisms centered on Fc (125) | Head and neck
squamous cell carcinoma (128) | Enhanced when
combined with inhibitors of immune checkpoints such as anti PD-1
antibodies and pembrolizumab (128) |
| Pembrolizumab | Monoclonal antibody
(128) | Induces tumor cell
death through two mechanisms centered on Fc (125) | Advanced
HER2+ breast cancer (128) |
|
| Nivolumab | Monoclonal antibody
(130) | Induces tumor cell
death through two mechanisms centered on Fc (125) | Metastatic NSCLC
(130) | Combined with
engineered PEGylated IL-2 (130) |
| Atezolizumab | Monoclonal antibody
(130) | Induces tumor cell
death through two mechanisms centered on Fc (130) | NSCLC (130) | Potential advantage
when combined with PEGylated IL-2 (130) |
Cytokines in cancer immunotherapy
Cytokine immunotherapy is an important area of
cancer immunotherapy that functions by activating the immune system
of patients with cancer (7). The
interleukin-2 (IL-2) family of cytokines, also termed the γ-chain
cytokine family, comprises IL-2, IL-7, IL-15 and IL-21, and is the
most targeted cytokine family in cancer immunotherapy (7). The concept that the immune system may
eradicate cancer through the function of cytokines was first
studied in patients with metastatic melanoma and renal cancer
(7). The IL-2 family of cytokines
activates cytotoxic T cells (CD8+) and natural killer
(NK) cells through the activation of STAT5, which acts as a key
transcription factor in pathways responsible for cellular
differentiation (7). In addition,
IL-2 has been demonstrated to serve an important role not only in
the activation of the immune response, but also in the suppression
of excessive immune reactions such as autoimmunity through the
activation of CD4+ and Foxp3+ regulatory T
cells (8). This part of the review
focuses on the antitumor immune effects of cytokines, specifically
the IL-2 family, and highlights the side effects of using cytokine
immunotherapy.
IL-2 family of cytokines
The IL-2 family of cytokines has been extensively
studied in the field of cancer immunotherapy (9). IL-2 is produced by antigen-activated
CD4+ T cells, CD8+ T cells, NK cells and NKT
cells (9). IL-2 serves roles in the
regulation, proliferation and activation of Tregs, CD4+
and CD8+ T cells, B cells, mature dendritic cells (DCs)
and endothelial cells (9). The main
challenge in using IL-2 in immunotherapy lies in the differential
expression of the IL-2 α receptor (IL-2Rα) on various types of
cells, as IL-2 affects peripheral Tregs (CD25+,
CD4+ and Foxp3+) more efficiently than
effector T cells (CD8+ and NK cells) (10). IL-2Rα and IL-2Rβγ are constitutively
expressed on Tregs at high levels (10). This allows Tregs to respond to low
doses of IL-2 in order to prevent autoimmune pathologies, and thus
suppress the immune response (9). By
contrast, CD4+, CD8+ and NK cells respond
only to high doses of IL-2 due to the transient expression of the
receptor, which is regulated by multiple transcription factors
including nuclear factor of activated T cells, NF-κB and activator
protein 1 (10). The key for using
cytokines in cancer treatment lies in increasing the antitumor
activity of CD8+ T cells and NK cells, while
counteracting the immunosuppressive activity of regulatory T cells
that can hinder the response of the effector cells (10).
IL-2/IL-2Rα fusion protein
High doses of IL-2 have been demonstrated to
increase the activity of low-IL-2 affinity cells (NK and
CD8+) and to initiate an antitumor response (11). However, this response is largely
suppressed and counteracted by the increased activity of Tregs; a
response rate of only 12.5% was observed in patients with
metastatic cancer who received successive high doses of IL-2
(11). On the other hand, low doses
of IL-2 activate the high-affinity receptors present only in Tregs,
thus preventing the activation of the antitumor immune response
(11). As a result, previous studies
have used varying approaches to eliminate the counteraction of
Tregs on the activation of effector T cells and NK cells. One of
the main approaches involves using the ALKS 4230 protein, a fusion
protein of the circularly permutated IL-2 and the extracellular
domain of IL-2Rα (12). This method
targets IL-2 to the intermediate-affinity receptors expressed by NK
and CD8+ cells and not the high-affinity receptors
expressed by immunosuppressive cells (12). A previous study has demonstrated that
ALKS 4230 lowers the frequency of toxicity incidents occurring in
patients undergoing a high-IL-2 dosage regimen and improves the
antitumor efficacy by counteracting the immunosuppressive pathway
(12). ALKS4230 is currently being
considered as a potential cancer immunotherapy treatment (12). Other immunotherapy approaches focus
on using IL-21 rather than IL-2 (12). IL-21 has been demonstrated to
activate NK cells, while poorly activating T cells, including
Tregs; IL-21 does not promote the proliferation and activation of
CD4+, CD25+ and Fox3+ cells nor
the secretion of IL-2 (13). In the
absence of IL-2, IL-21 is unable to compensate for IL-2 by
maintaining the production and activation of Tregs (13). However, IL-21 is capable of promoting
the proliferation of NK and CD2+ T cells (13). Thus, IL-21 indirectly counteracts the
immunosuppressive effect of Tregs, while maintaining the
proliferation of NK and effector T cells (13). IL-21 has been demonstrated to be
relatively safe and tolerated, in contrast to IL-2 treatment, which
leads to Vascular Leak Syndrome (13).
Polyethylene glycol (PEG)-modified
IL-2
The serum half-life of IL-2 is only 7 min; thus, for
effective treatment, patients would have to receive a high dose of
IL-2 every 8 h (14). However, due
to the presence of high-affinity IL-2 receptors on endothelial
cells, consecutive high doses of IL-2 may lead to toxicity
including capillary leak syndrome, which causes a sudden drop in
blood pressure that can potentially lead to organ failure and death
(14). To solve this issue, a number
of studies have examined the impact of the conjugation of IL-2 to
PEG polymers on its serum half-life. PEGylated IL-2 (PEG–IL-2) has
been demonstrated to have the same activity as IL-2, but with a
200% increase in circulating half-life (14). Addition of Polyethylene glycol to
IL-2 increases its retention by protecting it from enzymatic
digestion and renal clearance, thus increasing its serum half-life
to 1 day (14). In a study where
patients treated with two cycles of high dose unconjugated IL-2
were compared with patients receiving a hybrid regimen combining an
initial high dose of IL-2 with successive weekly doses of PEG-IL-2,
the results revealed that the relative response rate to
unconjugated IL-2 was 19%, compared with 17% for the combination of
IL-2 and PEG-IL-2 combination (14).
Thus, the use of high-dose IL-2 followed by PEG-IL-2 is a
well-tolerated regimen that decreases the side effects of constant
high IL-2 doses, but has no significant effects on the antitumor
activity compared with high IL-2 dosages in patients with renal
cell cancer and metastatic melanoma (14).
IL-2 in combination therapy
IL-2 cancer therapy has been studied as a
monotherapy and in combination with other therapeutic approaches
including chemotherapy, peptide vaccines and other combinations of
cytokines (15). IL-2 immunotherapy
has been approved by the US FDA for the treatment of solid tumors
such as metastatic renal cancer and melanoma. In a study where
patients were administered a high dose of IL-2 (600,000-720,000
IU/kg) every 8 h, the overall response rate was 15%, with a
complete response rate of 7% (16).
Another study used IL-2 in combination with lymphokine activated
killer (LAK) cells, and an overall response rate of 25–30% was
observed (16). A subsequent study
focused on combining ex vivo expanded tumor infiltrating
lymphocytes (TILs) with a high-dose IL-2 regimen and
lymphodepletion for the treatment of patients with metastatic
melanoma (17). In this approach,
TILs were rapidly expanded in the presence of anti-CD3, feeder
cells and IL-2, and infused into the patients, and an overall
response of 50% was reported in patients with metastatic melanoma
(17).
Drawbacks of IL2 therapy
The effective use of IL-2 in cancer treatment is
hampered by several drawbacks, the main one being its short serum
half-life, which is mostly due to rapid metabolism and elimination
by the kidneys (18). Thus, IL-2 has
to be administered repeatedly (every 8 h) at high doses, which can
lead to severe toxicity such as hypotension, cardiac problems and
pulmonary edema (18). In addition,
Krieg et al (18) have
reported that the binding of IL-2 to high-affinity
IL-2Ra-expressing endothelial cells causes acute vasodilation
effects that, in turn, lead to the development of vascular leak
syndrome.
Cytokines in combination therapy
Cytokines are a diverse group of molecules, each of
which can be used to target specific cells and activate unique
pathways to obtain an overall therapeutic effect. Cytokines can
also be combined with other therapeutic agents that work
synergistically in order to provide significant advantages over
monotherapy (19). Ovarian cancer is
a type of cancer in which combination immunotherapy has shown
promising results; one of the emerging therapies for ovarian cancer
is targeting its arginine auxotrophy by depriving it of arginine,
using a recombinant human arginase, inducing autophagy and
subsequenT cell death (19). On the
other hand, combination immunotherapy has also been effective in
the treatment of ovarian cancer. A study by Ingersoll et al
has demonstrated that combining cellular therapy using peripheral
blood mononuclear cells with IL-2 and IFN cytokines significantly
increases the survival of an ovarian cancer xenograft mouse model
(20). This is consistent with
another study that has reported that such a combination also
exhibited increased cytotoxic effects on two ovarian cancer cell
lines compared with those of monotherapy (21). Additionally, combining interferon
α-2b and interleukin-2 cytokine treatment with adoptive cell
transfer and cryosurgery has demonstrated promising results in
patients with advanced oral mucosa melanoma (21).
Other interleukins and cytokines in
immunotherapy
IL-2 has been extensively studied in renal cancer
and melanoma; however, other interleukins are also emerging in the
field of cancer immunotherapy (16).
Interleukins such as IL-15, IL-10 and IL-21 are of major importance
in cancer immunotherapeutic studies (7). IL-5 binds to the same receptor as IL-2
and activates STAT5 (7). However,
IL-5 initiates a different downstream signaling pathway, which has
different effects on CDC8+ cell differentiation and
memory cell formation (3).
IL-15 is crucial for the proliferation and
maintenance of memory T cells as well as for promoting the survival
and increasing the cytotoxicity of NK and CD8+ T cells
(22). IL-15 also induces the
release and proliferation of other proinflammatory cytokines such
as IFN-γ (22). Notably, IL-15 has
no affinity for the IL-2Rα chain (CD25) and thus does not activate
Tregs (22). As a result, IL-15 has
been extensively studied as an alternative of IL-2 in cancer
immunotherapy. For example, in a recent clinical trial, patients
with renal cell carcinoma, non-small cell lung cancer (NSCLC) and
melanoma received subcutaneous IL-15 injections, and the results
demonstrated a dose-dependent increase in the number of active NK
cells (23). In addition, a number
of patients exhibited marked disease stabilization, especially
patients with renal cell carcinoma, who presented with >2 years
of increased disease stability (23). However, one study reported that IL-15
has a very short serum half-life of only 2.5 h due to its small
size, resulting in rapid elimination from circulation (24). In addition, high doses of IL-15 have
deleterious effects on the lymphocytes (22). Thus, trans-presentation has been
adopted as a solution for this issue by increasing the specificity
of IL-15 and increasing its half-life (25).
IL-15 binds to the high-affinity IL-15 receptor
IL-15Rα on the cell surface, which stimulates IL-15 signaling in
the neighboring cells (26). This
process is of great importance in activating the cytotoxic immune
response and has been studied as an approach to stimulate direct
cytokine delivery to responsive cells (26).
Several studies have focused on engineering IL-15
variants with a longer half-life (27–29). One
of the promising fusion proteins includes the RNase L inhibitor
protein, where IL-15 is fused to the binding domain of IL-15Rα,
also termed the sushi domain (27).
This fusion protein was shown to increase the survival of mice
bearing multiple myeloma (29).
Another study focused on constructing a triple-fusion protein
termed Sushi-IL15-Apo (28). The
Sushi-IL15-Apo protein comprises the fusion of human IL-15 to the
sushi domain and to apolipoprotein A-I (28). The receptor of the apolipoprotein A-I
is expressed at high levels in tumor cells, and thus IL-15 activity
can be specifically targeted to tumor cells (28).
Another fusion protein of IL-15 is ALT-803, in which
IL-15 is fused to the sushi domain and an IgG1 Fc domain (29). A study in mice has reported that this
fusion protein successfully promotes IL-2 activity (29). In a dose-escalation phase I clinical
study, patients with hematological cancer received ALT-803
following relapse in response to the allotransplantation of bone
marrow (30). Treatment with the
fusion protein was well-tolerated in all patients, and the results
demonstrated increased numbers of NK and CD8+ cells
(30). Another study has reported
that ALT-803 in combination with intravesical bacillus
Calmette-Guérin therapy exhibited sustained and complete responses
in nine patients with localized bladder cancer; as a result, this
combination treatment was approved by the FDA for non-muscle
invasive bladder cancer (31). A
study investigated the use in IL-15 in combination therapies and as
an adjuvant in T- or NK-cell therapies (31).
Another cytokine from the IL-2 family is IL-21,
which exerts poor effects on the activation of Tregs, but is
considered a potent driver of NK cell activation and proliferation
(7). IL-21 has been studied alone
and in combination therapies in cancer trials (7,32). In a
study of the combination therapy of IL-21 with rituximab in
patients with non-Hodgkin lymphoma, eight out of 19 patients
presented with promising clinical responses (32). However, clinical trials with IL-21
are still minimal, and future studies are required that focus on
its use in combination therapy.
Anti-inflammatory cytokines such as IL-10 are also a
major subject of cancer immunotherapy studies. Although IL-10
inhibits the cytokine release in T and NK cells, a previous study
has highlighted that IL-10 inhibits antigen-induced apoptosis of
cytotoxic T cells (CD8+) in cases of chronic infections
and tumors (33). This was
demonstrated in a phase I clinical trial of patients treated with
PEG-conjugated IL-10; the results were promising, with only 15% of
patients presenting with grade 3 immune adverse effects and
PEG-IL-10 being well tolerated in the majority of the patients
(33). The current focus is on the
use of IL-10 in combination therapy with vaccines or other
chemotherapeutic agents (34).
A number of other cytokines, including IFN-α and
TNF-α, have been extensively studied in cancer immunotherapy. In
1986, IFN-α was approved by the FDA for the treatment of patients
with hairy cell leukemia (35).
Since then, it has been a candidate for the treatment of solid
tumors and other hematological malignancies at high doses (35). A major study has demonstrated that
high doses of IFN-α trigger apoptosis and inhibit the proliferation
of cancer cells (35).
Similar to other cytokines, extending the serum
half-life of IFN-α has also been achieved through PEGylation
(36). PEG-IFN-α has been approved
as an adjuvant in the treatment of melanoma (36). A previous study focusing on IFN-α in
combination therapies has reported that IFN-α increases patient
survival rates by enhancing the antitumor responses of cytotoxic
chemotherapies (37). However, the
issue with IFN-α is that high doses induce autoimmune responses due
to its role in potentiating dendritic cells, which induce
cross-priming of apoptotic antigens to cytotoxic T cells (38). One method to overcome the cytotoxic
activity of IFN-α and only provoke the immune-stimulatory activity
involves the fusion of IFN-α to apolipoprotein A-I (39). Apo-IFN-α exhibits an increased
half-life, enhanced immunostimulatory activity and tumor targeting,
as well as decreased cytotoxicity compared with those of IFN-α
(39).
Other approaches to overcome the cytotoxicity of
IFN-α focus on fusing a mutated IFN-α with reduced affinity for its
receptor to a cell-specific targeting domain C-type lectin
domain-containing 9A, which is a molecule expressed on
cross-priming specialized dendritic cells (40). The mutated cytokines termed
activity-on-target cytokines, or AcTakines, exhibit strong
antitumor activity in lymphoma, breast carcinoma and melanoma in
humanized mice with no side effects (40).
Another cytokine that may be used as a potential
cancer therapeutic is the TNF-α. This pro-inflammatory cytokine is
secreted mainly by antigen-presenting cells such as monocytes,
macrophages and DCs, as well as by other cell types such as
endothelial cells, fibroblasts and adipocytes under acute
inflammation and stress conditions (41). TNF-α was the first cytokine studied
for cancer immunotherapy (41).
Numerous studies have focused on the tumor cell apoptosis and
antitumor activity of TNF-α in various malignancies, such as
AIDS-associated Kaposi's sarcoma (41–44).
Phase I clinical trials of TNF-α have revealed dose-limiting
toxicities such as nausea, vomiting, anorexia, hypotension and
thrombocytopenia, with little to no tumor response (42). Chronic TNF-α exposure also induced
the cell death of T lymphocytes and, as a result, promoted tumor
growth (43).
Considering the range of toxicities observed in
animal studies, the antitumor efficacy of TNF-α in humans was not
evident; however, a number of studies described the localized
administration of TNF-α in patients as an approach to avoid the
toxicities associated with its systemic use (44–46). In
a study on localized intratumoral administration of TNF-α to
patients with Kaposi's sarcoma, the results demonstrated a
reduction in the cancer lesions in 15 out of 16 patients (44).
A clinically successful approach of TNF-α therapy is
its use in combination with the alkylating agent melphalan in
isolated limb perfusion protocols (45). This combination therapy approach has
also been adopted for patients with soft tissue sarcomas and
melanomas and has exhibited an 80% complete response rate (45). The high response rates were
associated with increased endothelium permeability, facilitating
the diffusion of chemotherapy into the tumor and the direct killing
of the tumor endothelium (45). In
addition, tumor vascular collapse and hemorrhagic necrosis were
observed upon TNF-α and melphalan administration (46). TNF-α-ILP therapy was approved for
high-grade soft tissue carcinoma treatment in Europe in 1998
(47).
Cytokines are potent yet complex immune mediators
that still require extensive research. Future research should focus
on two main approaches in cytokine therapies: Limiting the effects
of cytokines at the site of tumor to avoid systemic inflammatory
side effects and studying the efficacy of these treatments in
combination therapies.
Immune checkpoint inhibitors
With the increasing understanding of cancer
immunology, researchers have been able to develop novel promising
forms of immunotherapy. For instance, strategies involving the
negative regulation of the immune system have been demonstrated to
exhibit antitumor activity across a wide range of solid tumors,
such as NSCLC and advanced melanoma (48). ICIs are the driving force of this
negative regulation (48).
T cells express receptors to which antigens and
other ligands bind, leading to either their functional activation
or inhibition depending on the type of T-cell receptor targeted,
which is a process based on immune checkpoints (48). Immune checkpoint-related mechanisms
are targeted by cancer cells, allowing them to escape the immune
system through negative feedback mechanisms (48). This is achieved by binding of the
cancer cell to inhibitory receptors on T cells, specifically the
cytotoxic T lymphocyte associated protein 4 (CTLA-4) and programmed
cell death protein 1 (PD-1) inhibitory receptors (48). As the antigen-presenting cancer cells
bind to these receptors, they arrest T-cell activation and
proliferation, thus weakening the immune response against the tumor
(48).
A major turning point in cancer immunotherapy has
been the clinical application of antibodies that block immune
checkpoints (48). These antibodies
are collectively referred to as the ICIs. The main goal of ICIs is
to induce immune cell proliferation and activation against cancer
cells by stimulating the immune system (48). This section of the current review
combines the results of previous studies to provide an overview on
the types of ICIs used in either monotherapy or combination therapy
in various types of cancer. The outcomes and adverse events (AEs)
of ICI therapy as well as the possibility of overcoming these AEs
are also discussed. Table I
summarizes the classes of immune checkpoints, their modes of
action, inhibitors, types of cancers they target and the associated
cardiac adverse effects (49).
ICIs in advanced melanoma
Numerous studies have tested the efficacy and
biological effects of CTLA-4 and PD-1 blockage through either
monotherapy or combined therapy. For instance, to test for the
efficiency of ICIs in advanced melanoma, Wolchok et al
(50) randomly treated previously
untreated patients with either the anti-CTLA-4 human monoclonal
antibody ipilimumab monotherapy, the anti-PD-1 antibody Nivolumab
monotherapy or a combination of ipilimumab and Nivolumab.
Assessment of the results was based on three variables: i) Overall
survival (OS), the time from randomization of treatments until
death; ii) objective response rate (ORR), the ratio of patients
that exhibited partial or complete response to a specific
treatment; and iii) the rate of progression-free survival (PFS),
the time elapsed between randomization and the first documented
disease or death (50). The results
demonstrated that patients who underwent combined treatment
exhibited the highest ORR (58%), a higher rate of PFS (11.5 months)
and the highest rate of OS among the three groups throughout a
3-year analysis period (50). This
was compared with a 44 and 19% ORR, 6.9 and 2.9 months of PFS, and
52 and 34% OS rate in patients that underwent Nivolumab and
ipilimumab monotherapy, respectively (50). Therefore, for previously untreated
patients with advanced melanoma, more promising results were
observed using the combination therapy of Nivolumab and Ipilimumab
compared with either treatment alone (50).
ICIs in NSCLC
Although NSCLC is a poorly immunogenic malignancy,
ICIs may be encouraging therapeutic agents for this type of tumor,
as they exhibit antitumor activity (50). As previously reported for patients
with advanced melanoma, monotherapy of the anti-CTLA-1 antibody
ipilimumab leads to an improvement in the OS rate (50). However, this is not the case for
patients with NSCLC, unless ipilimumab is combined with other
immunotherapies, such as PD-1 inhibitors (48). When patients with NSCLC were treated
with both ipilimumab and Nivolumab using the same doses used in
previous studies for the treatment of melanoma, the ORR was 16%,
but was accompanied by a high rate of grade 3/4 treatment-related
AEs and treatment-related discontinuations (49 and 35%,
respectively) (48). However, when
the doses were adjusted, increased tolerability and positive
antitumor activities were observed in the patients, with the ORR of
39–47% and a decrease in the rate of treatment-related
discontinuation to 13–14% (48).
ICIs for the treatment of central
nervous system (CNS) metastasis
Systemic cancers are often accompanied by brain
metastasis, a CNS complication which affects ~20% of adults with
systemic malignancies (51). These
most commonly include lung cancer, melanoma and breast cancer
(51). Brain metastasis is becoming
more apparent as awareness about it is increasing and diagnostic
techniques are improving (42).
CNS is considered to be an immune-privileged
environment and, to date, treatments for brain metastasis were
based on stereotactic radiosurgery, whole-brain radiotherapy and
other local therapies. However, increasing knowledge regarding the
lymphatic system of the CNS and the alterations of the blood-brain
barrier by the tumor microenvironment suggests that it is possible
for immune cells to circulate in and out of the CNS (42). Consequently, it is currently
considered possible to use ICIs to trigger peripheral T cells to
exert antitumor effects within the CNS (49). This is supported by the association
between the high density of TILs and OS in both primary tumors and
brain metastasis (42).
The first ICI to exhibit efficacy in the treatment
of brain metastasis was ipilimumab; this was discovered
incidentally when patients treated for metastatic melanoma also
presented with durable CNS responses (51). These results were subsequently
confirmed during treatments of patients with melanoma brain
metastasis using ipilimumab (51).
Clinical data of these studies has demonstrated improvements in
ORR, CNS disease control and response rate and PFS, establishing
the basis for further ICI therapy. Additionally, anti-PD1
monoclonal antibodies nivolumab and pembrolizumab have exhibited
similar efficacy for previously untreated melanoma brain metastasis
(51).
On the other hand, in accordance with what has been
previously observed in the treatment of melanoma and NSCLC,
combined therapy exhibits the most promising CNS response rates to
immunotherapy (51). In one of the
studies aiming to demonstrate the efficacy of combination
immunotherapy for untreated patients with melanoma brain
metastasis, clinical outcomes of monotherapy using nivolumab were
compared with those of combined therapy with Nivolumab and
ipilimumab (51). The CNS response
rates were higher when patients were treated with combined therapy
(46%) compared with those observed following monotherapy (20%),
which included CNS complete response rates of 17 and 12%,
respectively (51). However,
combination therapy was associated with high toxicity, with grade
≥3 AEs observed in 63% of patients receiving Nivolumab and
ipilimumab, compared with 16% of patients receiving nivolumab alone
(51).
Therefore, with promising clinical results for the
treatment of brain metastasis, systemic immunotherapy contradicts
the paradigm of the immune-privileged brain, especially with the
use of combined therapy.
Immune-related (ir)AEs of ICIs
As aforementioned, a number of studies have reported
that combination therapy of anti-CTLA-1 and anti-PD-1 antibodies is
more efficacious compared with the respective monotherapies
(48,50,51).
However, the combination has also been demonstrated to increase the
number and frequency of AEs (52).
The most common AEs are cutaneous, including pruritus, rash and
dermatitis; other AEs are gastrointestinal (mainly diarrhea and
colitis), hepatic, endocrine (thyroid glands), pulmonary (most
commonly pneumonitis), renal, pancreatic, neurological and rarely
cardiac, including autoimmune myocarditis, heart failure,
cardiomyopathy or cardiac fibrosis (52). The cardiac AEs are mostly documented
among patients with history of cardiac pathologies, suggesting that
these AEs represent a deterioration of existing cardiac disorders
(52).
One potential cause of the apparent increase in AEs
compared with each monotherapy may be the assessment of an optimal
dosage regimen (53). In the
treatment of patients with NSCLC, different outcomes were obtained
when the administered doses of ICIs were adjusted (48). AEs may also arise from the
tumor-affected organs, specifically as the disease progresses and
the tumor undergoes biological changes (53). For instance, pulmonary events such as
pneumonitis are more likely occur in patients with NSCLC compared
with those with melanoma, whereas rashes or vitiligo are frequently
observed in patients with melanoma (54,55).
Notably, with the exception of endocrine events which are
considered irreversible and hard to manage (53), most of the other AEs can be well
managed and are reversible, especially with steroid co-treatment
(54). A previous study has reported
that following corticosteroid administration, a patient experienced
a rapid increase in the ejection fraction that had been previously
reduced to 15% due to of autoimmune myocarditis (52).
Rechallenging with ICIs after
irAEs
It remains unclear whether it may be safe to
re-treat patients that presented with grade ≥2 irAEs with ICIs.
Previous studies have reported that re-treating with ICIs is
feasible, but often leads to the recurrence of grade ≥2 irAEs.
Although no deaths were recorded, irAEs occurred on 22 out of 40
patients in one study that evaluated the effectiveness of
rechallenging with ICIs after an irAE (55). Thus, re-administration of ICIs may be
safe if patients are closely monitored, especially if these
patients have previously achieved a complete or partial response
prior to the first occurrence of an irAE in their initial treatment
(56).
Assessing rechallenges remains specific to each case
and to the risk-benefit ratio. For example, re-treatments may only
be considered if the grades of the irAEs decrease to 0 or 1
(56). Treatment decisions also
depend on the potential severity of irAE reoccurrence, especially
if these are cardiac or neurological, as the tools for imaging and
biological monitoring that allow estimations of future recurrence
of irAEs are lacking for neurological irAEs (56).
Potential biomarkers for
immunotherapy
Researchers are currently developing predictive
biomarkers of treatment response, one of which is the tumor cell
expression of PD-1 ligand 1 (PD-L1), which binds to the PD-1
inhibitory receptors on T cells (51,57). The
use of predictive biomarkers may allow improved patient selection
for treatment and may help predict the extent of the response to
treatment using ICIs (57). For
example, in one study, patients with NSCLC with PD-L1+
brain metastasis exhibited a higher intracranial response rate
(29%) compared with no response for patients with PD-L1−
brain metastasis (51). In addition,
the expression of PD-L1 differs with the type of cancer, and has
been reported to be highest among patients with NSCLC and lowest in
melanoma, where PD-L1+ cells were mainly non-tumor cells
that were present in the tumor microenvironment (51). Furthermore, following treatment with
CTLA-4/PD-1 checkpoint inhibitors, PD-L1 expression correlates with
improved PFS and OS rates in NSCLC and renal cell carcinoma
(57). However, PD-L1+
non-tumor cells exhibit a greater degree of response compared with
PD-L1+ tumor cells (57).
One explanation may be that macrophages and activated lymphocytes
that express PD-L1 may interact with PD-1 inhibitory receptors on T
cells, inhibiting their proliferative functions (57).
Therefore, numerous variables and conflicting data,
including tumor type, identity of cells expressing PD-L1, various
reagents and staining methods, as well as the incompatible cut-off
points for determining positivity, complicate the use of PD-L1 as a
predictive biomarker to assess the efficiency of treatment
involving ICIs (57).
Another potential clinical biomarker for the
detection and treatment assessment of brain metastasis is the tumor
mutational burden (TMB). For instance, in one series of
experiments, high TMB in brain metastasis was more common compared
with that in primary tumors and was reported in 39% of the cases
(51).
In conclusion, ICIs are a centerpiece of
immunotherapy, as they exhibit antitumor activity across a range of
tumor types. In addition, combined therapy with anti-CTLA-4 and
anti-PD-1 antibodies results in higher rates of OS and PFS compared
with those following monotherapies with either of the two ICIs.
However, these high response rates are accompanied by a variety of
irAEs, which are, in most types of tumor, reversible and manageable
when steroids are administered. Rechallenging with ICIs following
occurrence of irAEs is considered to be feasible if patients are
closely monitored. Finally, further studies are needed to assess
the predictive role of PD-L1 expression as a biomarker.
Adoptive cell transfer
Adoptive cell transfer (ACT) is an immunotherapy
approach used in the treatment of cancer. The main objective of ACT
is to enhance the function of the immune system (58). ACT is a promising immunotherapy
approach for the treatment of various types of tumor, such as
metastatic cancer, breast cancer, gastric cancer, colon cancer and
pancreatic cancer (58). ACT is
based on the transfer of lymphocytes to the patient following in
vitro expansion and gene modification in order to enhance their
cancer-fighting capabilities (58).
In autologous cell therapy, the cells are extracted from the
patient, cultured in vitro and administered to the same
patient (58). By contrast,
allogeneic therapies involve the transfer of cells from a donor
different from the receiving patient (59,60).
Previous studies have reported that several cell types can be used
in ACT treatment, including T lymphocytes, NK cells, DCs and stem
cells (59,60). In 1955, Mitchison (61) was the first to study ACT for cancer,
and its therapeutic potential was first tested by Fefer and
Rosenberg ~45 years ago (62,63). In
1998, Matsumoto demonstrated that ACT upregulated the expression of
CD3ζ in peripheral lymphocytes and improved the immune response in
patients with gastric and colon cancer (64). Recent advances in cellular and
molecular biology techniques have established ACT as a viable
therapeutic approach for patients who are not responsive to
traditional chemotherapy (59). ACT
takes advantage of the natural abilities of cytotoxic T lymphocytes
against cancer cells by binding to antigens on the surface of these
cells, which has been applied in TIL, engineered T-cell receptor
(TCR), chimeric antigen receptor (CAR) T cell and NK cell therapy
(59). ACT, in combination with
other treatments, is currently being evaluated in several types of
cancer, such as metastatic cancer, breast cancer and melanoma in
clinical trials (60).
Types of ACT
One form of adoptive cell transfer is TIL therapy,
which enhances the function of the naturally occurring T
lymphocytes; T cells infiltrating the tumor are harvested from the
patient, activated and expanded, an infused back into the patient
in order to destroy tumor cells (60).
Engineered TCR therapy is applied to patients whose
T cells do not proliferate sufficiently to challenge the tumor
(60). In this approach, T cells are
harvested, activated, expanded and equipped with receptors that
target specific cancer antigens that are protein, carbohydrate or
glycolipid-based (60).
CAR T-cell therapy has the ability to target cancer
cells even when their antigen is not bound to the surface of major
histocompatibility complex (MHC) by equipping patient T cells with
a synthetic receptor termed CAR (60). However, the range of potential
antigen targets is limited, since CAR T cells only recognize
antigens that naturally occur on cell surfaces (60). CAR T-cell therapy involves genetic
modification of T cells harvested from patients with cancer in
vitro to recognize a cancer-specific antigen (60). The engineering of T cells has
exhibited potential in cancer therapy, especially in overcoming the
limitations of TIL and TCR therapies of being unable to recognize
cancer antigens unless they are expressed by MHC molecules
(65). However, these modified cells
do not undergo normal tolerance mechanisms, such as thymic
education (60). In addition to
their reactivity against target molecules, these cells may react
against antigens present in healthy tissues (59). One potential solution for this
toxicity is to modify the antigen specificity of T cells in a
transient manner (59). Another
strategy uses a drug-inducible suicide gene or a cell-expressed
ligand that is targeted by a depleting antibody in order to allow
the removal of donor T cells (59).
In other approaches, the CAR activity depends on small molecule
drugs, co-expression of two tumor antigens or non-expression of a
non-tumor antigen (59). In
addition, the use of a bi-specific antibody or a biotin-binding
immune receptor in conjunction with a biotinylated molecule
targeting a tumor antigen brings together lymphocytes and tumor
cells, which may aid the selective and conditional antigen
targeting (59).
NK cells have been incorporated in adoptive cell
therapy by equipping NK cells with cancer-targeting CARs (66). NK cells are innate lymphocytes that
recognize and lyse transformed or virally infected cells without
prior activation (67). Adoptive
cancer therapy with NK cells has been tested in clinical trials due
to the ability of NK cells to induce antigen-independent immune
responses against malignancies that are not restricted by the MHC
(68). Allogeneic NK therapy has an
advantage against autologous T cell ACT due to a favorable
therapeutic and safety profile, suggesting that NK cells may be a
suitable alternative for CAR-based therapies (69). Cancer treatment-induced immune
defects associated with the use of patient-derived immune cells may
be avoided by using NK cells obtained from healthy donors (68). In addition, NK cells recognize cancer
cells and trigger tumor lysis by expressing various endogenous
activating receptors (68).
CAR-expressing NK cells function even when the CAR target antigens
are downregulated in cancer cells during treatment (68). Additionally, in contrast to T cells,
NK cells do not produce autocrine growth factors such as IL-2,
which limits their lifespan; therefore, NK cells do not remain in
circulation, reducing the risk of long-term AEs and eliminating the
need for introducing suicide genes (68). NK cells have also been reported to
not induce graft-versus-host disease or other types of alloimmune
or autoimmune toxicity, allowing versatile cellular immunotherapy
with allogeneic NK cells from healthy donors (68).
Factors that determine the
effectiveness of ACT
Several factors appear to be important for the
effectiveness of ACT therapy including cell dose, cytolytic
effector function and the long-term persistence of transferred
cells (70). The latter is
associated with a minimal differentiated phenotype, which maintains
the capacity of producing a continuous supply of cytolytic effector
progeny (70). The quality of the
transduced cells that are administered to the patient is also of
great importance, and this area has been extensively studied
(70). Lowering the dose of IL-2
added to the cells and reducing the time that the transduced cells
spend in culture (between 5 and 7 days) has been demonstrated to
provide promising results, as the culture conditions may induce
strong dependence on cytokines (70).
Naïve rather than central memory T cells give rise
to an effector progeny with improved antitumor immunity following
ACT (71). Effector cells derived
from naïve T cells lose the expression of the central memory cell
marker L-selectin more rapidly compared with those derived from
central memory T cells, but do not acquire the expression of killer
cell lectin-like receptor G1, which is a marker for terminal
differentiation and replicative senescence (71). Thus, naïve-derived cells exhibit high
proliferation and cytokine production following ACT (71). These results suggest that superior
efficacy of ACT is obtained by insertion of genes that confer
antitumor specificity into naïve T cells compared with central
memory T cells (71).
The terminal differentiation status of transferred
T cells and their low-affinity binding to tumor-associated antigens
(TAAs) limit the effectiveness of patient-derived tumor-specific T
cell ACT (72). TAAs are expressed
in healthy tissues a low level (72). Transgenic TCRs or CARs recognize TAAs
with high affinity and without MHC restriction, resulting in
cytotoxic T cell activity against the tumor (72). However, a high risk of targeting
healthy self-tissues occurs when central tolerance is overlooked.
In order to achieve the greatest efficiency on tumors with the
lowest risk of off-target and side effects, the choice of the TAA
and of the TCR affinity is crucial (72).
Following transplantation into patients, infused T
cells become functionally fatigued, and consequently their
efficiency decreases (70).
CD8+ T cells lose their antitumor efficacy as they
produce IFN-γ, which activates the innate and adaptive immune
response (73). This limitation may
be resolved by the adoptive transfer of tumor antigen-specific stem
cell memory T (TSCM) cells (73).
TSCM cells are similar to naïve T cells but are also highly
proliferative, exhibit a long lifespan and produce numerous
effector T cells following antigen stimulation (73).
T cell-T cell interactions enhance the function of
T cells within hours of molecular (antigen) stimulation (70). Enhanced cell motility is a short-time
consequence of molecular stimulation, leading to aggregation and
further activation (70). Functional
activation of T cells is halted in the absence of T cell-T cell
interactions even when continuous molecular stimulation is
performed (70). Cryopreservation of
the cellular product to perform lot release testing studies,
followed by direct reinfusion to patients after conditioning
chemotherapy is standard practice of CAR or TCR T cell therapy
(70). An intermediate step of
short-term ex vivo culture with molecular stimulation to
induce T cell-T cell interactions may be favorable and result in
more functional cells reinfused to the patients (70).
ACT in combination therapy
Compared with other immunotherapies, ACT has
certain advantages, including T cell populations having optimal
recognition of autologous tumor antigens that can be preferentially
isolated ex vivo for therapy (74). These selected T cells can be expanded
to large numbers under in vitro conditions that overcome
tolerizing factors within the tumor microenvironment. In addition,
regulatory and immunosuppressive factors can be eliminated by
conditioning the host prior to cell transfer.
Since single-agent monotherapies are continuously
proving futile, combination therapies utilizing two or more types
of ACT or combinations of ACT and cell-based therapies have
exhibited promising potential in the treatment of cancer such as
melanoma (74). Traditional melanoma
treatment includes the use of the chemotherapeutic agent
dacarzabine, which exhibits a modest patient response of 10–20%
with a complete response rate <5% (74). Our previous studies have investigated
the use of anthrax lethal toxin in murine models as a potential
therapeutic agent for this disease and elaborated on the cellular
mechanisms involved in the inhibition of Tregs (75).
In combination therapy, the use of TIL with
high-dose IL2 generated a clinical response rate of 55%, in which T
cell-mediated tumor regression was observed (76). Additionally, T cell-based therapies
that involve the co-delivery of cytokines such as IL-12, IFN-α,
IFN-γ and TNF-α have enhanced the patient response (77). Other types of cancer such as
glioblastoma multiforme (GBM) have been reported to respond to
similar therapies. For example, our previous studies investigated
the use of metformin, a biguanide agent used to treat type 2
diabetes mellitus, as well as human recombinant arginase I
(Co)-PEG5000 [HuArgI (CO)-PRG5000] for the treatment of GBM
(78,79). In the context of immunotherapy, CAR T
cell-based approaches in combination with radiotherapy exert
synergistic efficacy in GBM (77).
Cancer treatments involving ACT
Epstein-Barr virus (EBV)
EBV-induced lymphoma can be effectively treated
with ACT therapy that restores anti-EBV immunity using
viral-antigen-specific T cells (80). Repetitive in vitro stimulation
of peripheral blood lymphocytes (PBLs) with EBV-B lymphoblastic
cell lines has been used to generate lymphocyte cultures highly
enriched for EBV-antigen-specific T cells that are suitable for ACT
therapy and inhibit EBV-induced lymphoproliferation (80).
Non-viral antigen-expressing
tumors
ACT therapies exhibit limited success in treating
tumors that do not express viral antigens due to the difficulty in
generating tumor-antigen-specific cells (80). One review that surveyed clinical
trials involving non-specific activated lymphocytes or non-specific
PBLs activated in vitro with anti-CD3 reported a low
response rate for this therapy, suggesting the necessity of using
tumor antigen-specific lymphocytes in ACT (80).
Limitations
Tumor recurrence is observed when cytotoxic T cell
responses are inhibited by a subset of TGFβ-responsive squamous
cell carcinoma stem cells which evade immunotherapy via the
expression of CD80, a co-stimulatory molecule involved in T-cell
activation and the activity of normal and malignant B cells
(81). Tumor-initiating stem cells
(tSCs) possess self-renewal and differentiation capacities
(81). These cells fuel and sustain
tumor growth by overcoming the immune surveillance barrier
(81). The ability of a regressed
tumor to regrow implies that at least a number of tSCs acquire the
ability to resist the enhanced antitumor immunity (81). One hypothesis is that tSCs exhibit
limited antigen presentation and are therefore undetectable by the
immune system (73). Alternatively,
tSCs may be able to avoid immune attacks due to their
antigen-editing, independent molecular features (82). These tSCs may become invasive in
response to enriched TFGβ around the vessels; they are also more
tumorigenic than tSCs distant from blood vessels, which do not
receive a TGFβ stimulus (82).
TGFβ-responding tSCs survive chemotherapy and induce tumor relapse
(83).
An effective mouse ACT model was established in a
previous study, in which skin squamous cell carcinoma (SCC) tSCs
were tracked, and their susceptibility was directly determined and
compared with antigen-specific antitumor immune responses (81). The results demonstrated that
TGFβ-responding tSCs exhibited resistance to cytotoxic T cell
responses and were responsible for tumor recurrence following ACT
therapy (81). In addition, the
aforementioned study demonstrated that within the tumor, these stem
cells expressed the immune cell ligand CD80, the silencing of which
diminished their resistance to immune surveillance and weakened
tumor relapse (81).
Side effects of ACT
ACT is an effective cancer treatment, but it also
has severe side effects. The variation and severity of these side
effects vary depending on the patient health prior to treatment,
the type and stage of cancer, as well as the type of T-cell
transfer being used (84). The
toxicity is observed as a result of direct T-cell attacks on normal
tissues, i.e., autoimmunity (85).
However, a number of normal tissues tolerate a certain degree of
autoimmunity, and effective targeting of the antigens by these
cells is still possible (85).
In CAR T-cell therapy, cytokine release syndrome is
a side effect defined by the release of large amounts of cytokines
into the blood, which ultimately leads to fever, chills, difficulty
breathing, nausea, tachycardia, fatigue and muscle pain (84). Treatment with the monoclonal antibody
tocilizumab is used to control this side effect (86). In addition, allergic reactions to CAR
T cells have been recorded in a number of patients; the symptoms of
allergic reactions include fever, difficulty breathing and chills
(86). Increased risk of infection
is observed as a side effect of certain types of CAR T-cell therapy
when treating leukemias and lymphomas (86). The increased risk of infection occurs
due to the recognition by CAR T cells of CD19, which is expressed
on the surface of B cells; the CAR T-cells destroy the
CD19+ B cells, leading to a decreased ability to fight
infections (86). Immunoglobulin
therapy is used to treat this side effect by administering
antibodies to help fight infections (86).
TIL therapy causes capillary leak syndrome that is
characterized by the leak of fluid and proteins out of small blood
vessels into the surrounding tissues, leading to severe low blood
pressure, which may lead to multiple organ failure (84). Neurological side effects that affect
the brain have also been observed, and the symptoms include
headaches, altered consciousness, speech changes, seizures and
confusion (86). In addition, severe
rashes leading to vitiligo occur in certain patients (86). A number of patients experience
uveitis with impaired vision and decreased hearing, possibly due to
autoimmunity against pigmented cells in the stria vascularis of the
inner ear (85). These toxicities
respond well to topical corticosteroids (85). In addition, hepatotoxicity has been
observed in certain patients with renal cancer, as well as severe
diarrhea in patients with colorectal cancer (85).
Cancer vaccines: Types and therapeutic
combinations
Various cancer therapies do not evoke long-lasting
immune responses and instead treat the patient immediately, which
leads to the need for improvement of this treatment so that it
induces robust responses that also hinder recurrence. Cancer
vaccines may resolve this issue. Cancer vaccines have an advantage
of being able to use the entire immune system of the individual,
leading to strong and durable responses (87). In addition, mixing vaccines with
other types of treatments, such as ICIs therapy, chemotherapy or
radiotherapy also aims to enhance the immune response and the
potency of treatment (87). Cancer
vaccines may be use used not only used for prophylactic purposes,
but also to ensure that the immune recognition of tumor antigens
occurs more readily (87). A notable
characteristic that should be taken into consideration when
developing cancer vaccines is the classification of antigens into
shared tumor antigens, which are common between most tumors of a
certain histological type, and neoantigens, which are antigens that
are subjected to various mutations that render them distinct from
normal cells (87). These
neoantigens give rise to neoepitopes, which elicit specific immune
cell responses against them (87).
The presence of T cells in the tumor microenvironment is associated
with tumor regression and survival, which indicates the
immunogenicity of most cancers (87). There are various types of DNA
vaccines, allowing physicians and researchers to manipulate certain
criteria of the vaccine for an improved immune response. The
majority of vaccines have minimal AEs and are easily endured by
patients (88). Multiple factors
must be taken into consideration when designing any vaccine,
including the antigen picked, the adjuvant used and the delivery
mode. The various types of cancer vaccines are summarized in
Table II.
DNA vaccines
DNA vaccines are usually bacterial plasmids
containing genes encoding tumor antigens coupled to CMV promoters
(89). These vaccines aim to induce
an innate immune response that augments the adaptive immune
response towards the tumor presenting these antigens (89). Thus, DNA vaccines aim to boost the
production of T cells such as CD8+ and CD4+
cells that specifically target the tumor (5). Another aim of DNA vaccines is breaking
immune tolerance and inducing an immune memory, especially in the
cases where a DNA vaccine is used to prime the T cells against the
antigen to improve the benefits of other therapeutic techniques
(1). In the case of DNA vaccines,
the DNA sequence may be engineered to improve protein expression,
for instance by replacing the leader sequence with other sequences
and adding immunoglobulins as well as performing codon and RNA
optimization (5). In addition, a
novel technique that has emerged recently in DNA vaccines and
provides hope for cancer vaccination is synthetic consensus
vaccines that rely on generating consensus DNA sequences based on
comparing the native sequences between various species; these
synthetic consensus sequences are able to break immune tolerance
due to the increased diversity compared with that of the native
sequences (5,90). DNA vaccines can also be used in
combination with other therapies to overcome certain limitations.
ICIs targeting PD1 and CTLA4 have been demonstrated to enhance the
antitumor activity of a DNA vaccine against mastocytoma by boosting
its antitumor immunity, leading to a ≤90% survival rate (91). A previous clinical study has also
investigated the therapeutic effects of combining DNA vaccines with
cytokines such as IL-2 and IFN-γ (88). In addition, the use of DNA vaccines
is supported by the fact that intrinsic unmethylated CpG islands in
the bacterial plasmid function as an adjuvant, thus stimulating the
immune response to the vaccine (92). To determine the most effective
combination of treatments and DNA vaccines, assessment of the tumor
microenvironment as well as cells involved in the immunosuppressive
behavior in that niche should be performed (93).
The mode of delivery should be taken into
consideration when discussing the cellular mechanisms of DNA
vaccines. DNA vaccines can be delivered through a variety of
different routes including intramuscular, subcutaneous, mucosal or
transdermal delivery (94). In
addition, it is important to ensure that the plasmid transfects the
cells, for which electroporation may be used (94). Furthermore, when the vaccine infects
the cell, the plasmid is replicated in the cell, which is driven by
the eukaryotic CMV promoter to produce that target antigens;
following production of the antigens, they are then expressed in
MHC class I and II, which are recognized by various immune cells
and are capable of producing humoral and cellular immune responses
(5).
The advantages of using DNA vaccines include their
low cost, ability to elicit antigen-specific immune responses, easy
sequence alteration, large-scale production and storage (93). In addition, DNA vaccines are not
infectious and do not cause diseases or elicit immune responses in
individuals of a specific MHC class (93). Therefore, in contrast to personalized
vaccines, DNA vaccines may be considered versatile (93). Finally, as these antigens are
expressed in the cells, they undergo post-translational
modifications that form proteins similar to the host proteins,
which ensures appropriate immune presentation (94).
On the other hand, certain disadvantages associated
with DNA vaccines have been reported. DNA vaccines are negatively
charged due to their DNA base and have a low molecular weight,
which may cause difficulty in cellular uptake and poor antigenic
production, leading to poor immune responses (92). Additionally, challenges occur in
selecting the appropriate tumor antigen and combating the problems
of poor immunogenicity and immunosuppressive microenvironments,
which may lead to weak results in this field (95).
The only DNA cancer vaccine that has reached a
phase III clinical trial is VGX-3100, which is a DNA vaccine
targeting precancerous lesions that may progress to cervical
cancer, termed CIN2/3 (96). This
vaccine appears to elicit durable responses, but has not yet been
approved by the FDA (96). In
addition, DNA vaccines have been studied for breast, prostate,
cervical, ovarian and pancreatic cancer, glioblastoma, melanoma,
renal cell and urothelial carcinomas, but no significant progress
has been reported beyond phase II clinical trials or even
preclinical animal models (97).
Peptide vaccines
Peptide vaccines consist of nongenetic components
of the disease to be treated, such a tumor or an infectious agent,
in order to elicit an immune response (98). Peptide vaccines do not unnecessarily
overload the immune system, as only the epitope of interest is
delivered to the immune system of the individual (99). These vaccines are also cheap and can
be easily produced (99). However,
they usually fail to elicit an immune response due to the presence
of immunosuppressive molecules in the tumor microenvironment
(100), and the safety of these
vaccines when combined with adjuvants remains unclear; thus, the
safety of usage of adjuvants should be assessed and eliminated in
order to ensure the efficacy of peptide vaccines (99). Therefore, current research aims to
improve peptide vaccines. For example, peptide vaccines may be
potentiated by combination with other molecules, such as avasimibe,
a molecule that aids in cholesterol metabolism and increases the
potentiation of a Kras vaccine targeting lung tumors (101), indomecathin, which reduces the rate
of proliferation in tumor cells and increases the apoptotic rate
when used in conjunction with a MUC1 peptide vaccine (100), or by adding adjuvants as the
antigens by themselves provide low immunogenicity and MHC
restriction (102). Future research
is required to enhance the immune response in other manners such as
utilizing protein nanoparticles that surround the peptides, which
is an approach that offers numerous merits including a symmetrical
structure, biodegradability and an appropriate size for the
delivery of vaccines (103).
One FDA-approved vaccine that is not strictly a
peptide vaccine, but may be considered to be one, is IMLYGIC or
T-VEC, which is a modified herpes simplex virus 1 that replicates
in the lesions of patients with recurrent melanoma, causing cell
lysis and death (104). The reason
why it may be considered a peptide vaccine is that this virus is
engineered to express the granulocyte-macrophage colony-stimulating
factor, which promotes the antitumor response and priming of T
cells (104).
DC vaccines
DCs were first described by Robert Steinman in 1973
due to their unique shape and high MHC expression (105). DC vaccines are vaccines produced by
harvesting patient DCs and transfecting them with the tumor antigen
ex vivo; these cells are then re-introduced into the
patient's body which can usually occur via many routes including
intradermal, intravenous, intranodal and intralymphatic routes, to
destroy the tumor cells that they readily recognize (106). DCs possess an ability to modulate
adaptive immune responses, which renders them as potent stimulatory
tools for memory and immune cells (91,106).
In addition, DCs stimulate naïve T cells (106). However, DCs are low in number in
the peripheral blood and tissues, and thus, their population needs
to be enhanced in vivo or ex vivo prior to vaccine
administration (107). The
advantages of DC vaccines include their personalized nature, the
feasibility of producing an immune response since dendritic cells
are major players in the activation of adaptive responses, their
ability to specify the response towards one antigen or a group of
antigens and the relatively safety in individuals as they do not
cause AEs (108). By contrast,
DC-based vaccines, similar to other vaccines, face certain hurdles,
such as tumor immunosuppression and the limited functions of
monocyte-derived dendritic cells, which are the cells used to
generate this type of vaccines (109). In addition, since they are
personalized, DC vaccines tend to be expensive (109). As with other types of vaccines, DC
vaccines suppress tumor growth and increase the number of T cells
infiltrating the tumor microenvironment, which can be more
efficient compared with other types of vaccines in certain cases,
such as in murine lung carcinoma (LL2), the study was done in
vitro and the DC vaccine was able to exhibit superior
immunogenicity compared with an antigen-adjuvant vaccine (102). Furthermore, DC cancer vaccines may
also be used with other treatments for enhanced results, including
ICIs such as anti-PD-1 and adjuvants (110). Using a mathematical model, Lai and
Friedman (111) have demonstrated a
synergistic effect of the combination of a DC cancer vaccine and
anti-PD1 in cancer therapy. In laboratory preparation of DC
vaccines, observing and controlling culture conditions is crucial
since DCs are highly sensitive to their environment (110). Particular care should be taken in
identifying the molecules that allow efficient delivery of DC cells
to the lymph nodes for maturation (110).
One example of an FDA-approved DC cancer vaccine is
Sipuleucel-T or PROVENGE, which is used in the treatment of
metastatic castration-resistant prostate cancer with metastasis
beyond the prostate gland that cannot be treated by hormone therapy
(112). DC vaccines for other types
of cancer have not been approved; however, current efforts are
aimed towards metastatic melanoma, breast and prostate cancer
(113).
Whole-cell vaccines
Whole-cell cancer vaccines contain whole tumor
cells that have been modified ex vivo (114). Despite the lack of strong results
in cancer vaccination, whole-cell vaccines offer numerous
advantages. One advantage is that whole-cell vaccines express both
tumor-specific and tumor-associated antigens (114). Additionally, whole-cell vaccines
elicit strong cellular as well as humoral immune responses
(114). However, tumor cells
possess immune evasion strategies, which must be considered when
modifying tumor cells to achieve adequate immunogenicity (115). In addition, whole-cell vaccines
have not elicited strong immune responses in clinical trials,
possibly because advanced tumors build immune tolerance that
hinders immune recognition (114).
Whole-cell vaccines are also time consuming, laborious and costly
due to the numerous procedures needed to acquire, maintain,
manipulate and reintroduce tumor cells to the individual (108). Carbohydrate antigens in the context
of whole-cell vaccines are also important as they are expressed on
the surface of cells in the form glycoproteins, proteoglycans and
glycolipids, and are necessary for various biological processes
including aberrant processes implicated in carcinogenesis (116). Xia et al (116) have developed a glycol-antigen
microarray to study antibody responses to carbohydrates induced by
whole-cell vaccines and reported high IgG and IgM responses induced
by the GVAX pancreatic cancer vaccine, indicating the importance of
other types of antigens in addition to peptide antigens (116). In terms of combination therapy, one
study by Chen et al (117)
has reported that combining a whole-cell breast cancer vaccine with
the standard chemotherapeutic alkylating agent cyclophosphamide
increases the clinical benefits of this agent in treating
Her2+ metastatic breast cancer.
Cancer vaccination has been a potent field of
research for the treatment of cancer in the past 30 years (88). Cancer vaccines can be used in two
different manners: A prophylactic application that aims to prevent
disease or as vaccine-mediated postoperative immunotherapy for
recurrent and metastatic tumors. One major challenge for cancer
vaccines is the interpatient variability of cellular responses,
which causes a robust immune response in certain individuals, but
no response in others (88). Another
challenge is that cancer vaccines by themselves may not be
efficient and require prior priming or co-priming of the immune
system using other substances that potentiate the vaccine (101,102,118).
In addition, limited knowledge is currently available on the
intracellular processing and tracking in humans as opposed to
animals due to ethical concerns (88). However, despite these drawbacks,
cancer vaccines may have positive inclinations in the future,
especially when combined with other types of treatment (88).
Monoclonal antibodies in immunotherapy
(IT)
George Köhler and Cesar Milstein defined their
attempt at circumventing the deficiencies of the prior methods that
were considered permanent cultures of Myeloma cells that lacked ‘a
satisfactory source of monoclonal antibodies of predefined
specificity’ as capable of being ‘valuable for medical use’
(119). In order to benefit from
the boundaries of antibody yield and lifespan, George Köhler and
Cesar Milstein fused a myeloma cell line P3-X63-Ag8 with spleen
cells from a mouse immunized with sheep red blood cells to generate
hybridoma cells that release antibodies with homogeneous
specificity towards one antigen: Monoclonal antibodies (mAbs)
(119).
The antibody modes of action
Antibodies are proteins engineered to target an
epitope present on cancer cells (when used for immunotherapeutic
purposes) and are produced, collected and administered to a patient
and they circulate throughout the body until they meet the
aforementioned antigen. They recruit other immune system agents to
eliminate the antigen via three main mechanisms: Activating
antibody-dependenT cellular cytotoxicity (ADCC), blocking growth
factor (GF) receptors, and activating complement-dependent
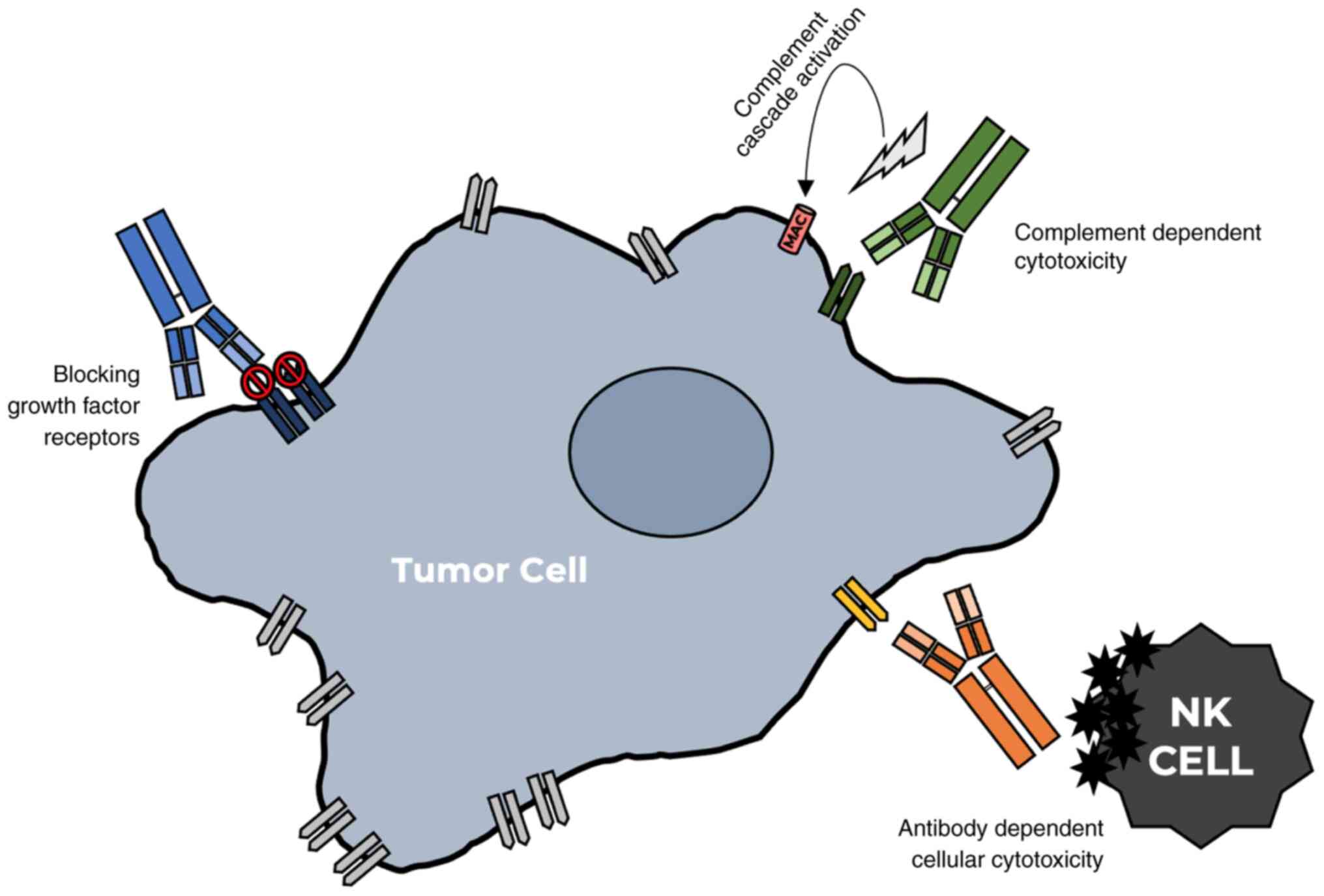
cytotoxicity (CDC) by complement activation (Fig. 1) (120). Understanding the mechanisms by
which mAbs work must precede their application to ensure
efficacy.
For activating ADCC, a specifically designed mAb
attaches to a previously specified antigen and allows NK cells to
recognize and eliminate it by releasing cytotoxic agents, which
kills the cell by apoptosis (121,122).
Targeting GF receptors is achieved by using several mAbs to
competitively inhibit these receptors (123). For instance, targeting epidermal
growth factor receptor blocks its use, causing the internalization
and downregulation of the receptor (123). In CDC, a mAb binds to antigens on
the targeT cell surface and triggers multiple pathways in the
complement cascade, where complements (various soluble plasma
proteins and membrane proteins) bind to the mAbs, induce an attack
and lead to the lysis of the targeted cell (124).
Types of mAbs used in IT
There are three types of mAbs that are used in
cancer treatment: Naked, conjugated and bi-specific. Naked mAbs are
used without any drugs (125). For
example, rituximab induces tumor cell death through the dependent
and independent mechanisms centered on Fc (125). The dependent mechanism contains the
aforementioned ADCC mechanism mediated by NK cells and macrophages,
antibody-dependenT cellular phagocytosis mediated by macrophages
and CDC (125). Apoptosis is
directly induced by the independent Fc mechanisms by the binding of
a mAb to its receptor or by its blocking of receptor-ligand
interactions, as is the case with the HER2 receptor-mediated GF
signaling on cancer cells (126).
Conjugated mAbs are tools through which chemotherapeutic agents and
radioactive molecules are led directly to targeted cells (125). The conjugation of mAbs with
radioactive substances, such as iodine-131 or yttrium-90, generates
cytotoxic molecules, such as monomethyl auristatin E or emtansine,
improves efficacy compared with that of naked mAbs and generates
antibody-drug conjugates (125).
This facilitates the delivery of cytotoxic agents to tumor cells,
increasing their efficiency (126).
Bispecific (or bifunctional) mAbs are a type of mAbs in which two
IgG chains of different specificity are amalgamated; they can be
administered to effectively bind two different surface antigens,
which eases the burden of the patient significantly by decreasing
the frequency with which this procedure is administered (127).
mAbs and combination therapy
mAb monotherapy has been successful in treating
certain patients with cancer, but current studies are shifting
towards combining mAb with other antitumor therapies for a more
effective treatment: mAbs and ICIs (128,129).
The effect of the mAb cetuximab in treating head and neck squamous
cell carcinoma appears to be enhanced when combined with inhibitors
of the immune checkpoint PD-1/PD-1L such as anti-PD-1 antibodies
and pembrolizumab (128). In
addition, resistance to the monotherapy of the mAb trastuzumab in
advanced HER2+ breast cancer may be overcome with the
use of pembrolizumab (129).
In addition to immune checkpoint blockage, cytokine
immunotherapy is also synergistic with monoclonal antibody therapy.
An engineered PEGylated IL-2 is currently undergoing clinical
trials in which it is combined with the mAb Nivolumab for the
treatment of advanced solid tumors (130). PEGylated IL-2 has also exhibited
potential therapeutic advantage when combined with the mAb
Atezolizumab in treating NSCLC (131).
The downsides of mAbs in IT
There are several side effects for mAb treatments
against cancer. In one case, upon treating 496 patients with
metastatic melanoma with anti-PD1 antibodies, 242 side effects were
described in 138 patients, including skin, gastrointestinal and
renal system side effects, and, in certain cases, rare side effects
including diabetes mellitus and pancreatitis (23). In addition, in patients with melanoma
treated with Nivolumab or pembrolizumab, side effects were observed
in the respiratory tract, musculoskeletal system, nervous system,
eyes, heart and blood (132).
The future of mAbs in IT
As far as the future of monoclonal antibodies is
concerned, this technology may improve the current shortcomings in
cancer therapeutics. mAbs possess specificity for certain
conformations to ensure improved selectivity and fulfillment of
therapeutic goals. However, the most prominent limitation currently
is that the majority of mAbs are unable to recognize 3D
configurations. Thus, 3D-recognizing antibodies may be valuable to
offer an extra level of neutralization.
Conclusions
The present review of the various approaches of
cancer immunotherapy aimed to comprehensively describe the modes of
action and limitations of the current approaches to cancer
treatment via immunotherapy. The use of cytokines in cancer
immunotherapy is among the most promising approaches. This review
focused on one of the most extensively studied cytokines, IL-2, and
highlighted other promising cytokines reported in literature.
Current research is focusing on using multiple cytokines together
in therapy, combined therapies and fusion proteins that provide
improved specificity and longer half-lives. ICIs have been reported
to have a positive effect on the treatment of various types of
cancer, especially if applied in combined therapy. These promising
findings may revolutionize immunological cancer treatments. ACT is
an effective type of immunotherapy in which T cells obtained from
the patient or donors are administered to patients to help fight
diseases such as cancer. The present review discussed several types
of ACT therapy in addition to the factors that determine the
efficacy of this treatment. In addition, the advantages and side
effects of ACT therapy were described. Although several limitations
hinder the efficiency of ACT, research and clinical trials are
focusing on the development and future of this technique. In the
cancer vaccines section, the types of therapeutic cancer vaccines
were introduced, with an emphasis on their advantages and
disadvantages as well as their function in combination therapy. The
review on cancer vaccines also included specific examples of
approved vaccines and those undergoing clinical trials. Finally,
the current review discussed mAbs, their modes of action,
advantages and limitations of using them as well as their future
and applications in combination therapy. In addition, specific
downsides of immunotherapy were discussed in this review. Notably,
the information presented here does not constitute the entirety of
the knowledge on this topic; the present review highlights basic
knowledge pertaining to cytokines, ICIs, ACT, cancer vaccines and
mAbs in cancer immunotherapy. Finally, this review does not seek to
give the impression that immunotherapy is the only method of
treating cancer, as there are other methods used in a complementary
manner to immunotherapy or by themselves to treat cancer.
Acknowledgements
The authors would like to thank Dr Ralph Abi-Habib
(Lebanese American University, Beirut, Lebanon) for his critical
reading of the manuscript.
Funding
This work was supported by the Department of
Natural Sciences at the School of Arts and Science at the Lebanese
American University.
Availability of data and materials
Not applicable.
Authors' contributions
MH, ZO, RR, FW, AZ and MF all wrote different
sections of the manuscript. EAK and MES wrote the final draft and
edited it for important intellectual content. Data authentication
is not applicable. All the authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Koo SL, Wang WW and Toh HC: Cancer
Immunotherapy-The target is precisely on the cancer and also not.
Ann Acad Med Singap. 47:381–387. 2018.PubMed/NCBI
|
|
2
|
Meng J, Zhou Y, Lu X, Bian Z, Chen Y, Zhou
J, Zhang L, Hao Z, Zhang M and Liang C: Immune response drives
outcomes in prostate cancer: Implications for immunotherapy. Mol
Oncol. 15:1358–1375. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Balachandran VP, Beatty GL and Dougan SK:
Broadening the impact of immunotherapy to pancreatic cancer:
Challenges and opportunities. Gastroenterology. 156:2056–2072.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parkin J and Cohen B: An overview of the
immune system. Lancet. 357:1777–1789. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Perales-Puchalt A, Wojtak K, Duperret EK,
Yang X, Slager AM, Yan J, Muthumani K, Montaner LJ and Weiner DB:
Engineered DNA vaccination against follicle-stimulating hormone
receptor delays ovarian cancer progression in animal models. Mol
Ther. 27:314–325. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pedersen M, Westergaard MCW, Milne K,
Nielsen M, Borch TH, Poulsen LG, Hendel HW, Kennedy M, Briggs G,
Ledoux S, et al: Adoptive cell therapy with tumor-infiltrating
lymphocytes in patients with metastatic ovarian cancer: A pilot
study. OncoImmunology. 7:e15029052018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mitchell DM, Ravkov EV and Williams MA:
Distinct roles for IL-2 and IL-15 in the differentiation and
survival of CD8+ effector and memory T cells. J Immunol.
184:6719–6730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaeckel E, Kretschmer K, Apostolou I and
von Boehmer H: Instruction of Treg commitment in peripheral T cells
is suited to reverse autoimmunity. Semin Immunol. 18:89–92. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brisslert M, Bokarewa M, Larsson P, Wing
K, Collins LV and Tarkowski A: Phenotypic and functional
characterization of human CD25+ B cells. Immunology. 117:548–557.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim HP, Imbert J and Leonard WJ: Both
integrated and differential regulation of components of the
IL-2/IL-2 receptor system. Cytokine Growth Factor Rev. 17:349–366.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Smith FO, Downey SG, Klapper JA, Yang JC,
Sherry RM, Royal RE, Kammula US, Hughes MS, Restifo NP, Levy CL, et
al: Treatment of metastatic melanoma using interleukin-2 alone or
in conjunction with vaccines. Clin Cancer Res. 14:5610–5618. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lopes JE, Fisher JL, Flick HL, Wang C, Sun
L, Ernstoff MS, Alvarez JC and Losey HC: ALKS 4230: A novel
engineered IL-2 fusion protein with an improved cellular
selectivity profile for cancer immunotherapy. J Immunother Cancer.
8:e0006732020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Attridge K, Wang CJ, Wardzinski L,
Kenefeck R, Chamberlain JL, Manzotti C, Kopf M and Walker LS: IL-21
inhibits T cell IL-2 production and impairs Treg homeostasis.
Blood. 119:4656–4664. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zimmerman RJ, Aukerman SL, Katre NV,
Winkelhake JL and Young JD: Schedule dependency of the antitumor
activity and toxicity of polyethylene glycol-modified interleukin 2
in murine tumor models. Cancer Res. 49:6521–6528. 1989.PubMed/NCBI
|
|
15
|
Rosenberg SA: IL-2: The first effective
immunotherapy for human cancer. J Immunol. 192:5451–5458. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grimm EA, Mazumder A, Zhang HZ and
Rosenberg SA: Lymphokine-activated killer cell phenomenon. Lysis of
natural killer-resistant fresh solid tumor cells by interleukin
2-activated autologous human peripheral blood lymphocytes. J Exp
Med. 155:1823–1841. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dudley ME, Wunderlich JR, Yang JC, Sherry
RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE,
Mavroukakis SA, et al: Adoptive cell transfer therapy following
non-myeloablative but lymphodepleting chemotherapy for the
treatment of patients with refractory metastatic melanoma. J Clin
Oncol. 23:2346–2357. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krieg C, Létourneau S, Pantaleo G and
Boyman O: Improved IL-2 immunotherapy by selective stimulation of
IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad
Sci USA. 107:11906–11911. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nasreddine G, El-Sibai M and Abi-Habib RJ:
Cytotoxicity of [HuArgI (co)-PEG5000]-induced arginine deprivation
to ovarian cancer cells is autophagy dependent. Invest New Drugs.
38:10–19. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ingersoll SB, Ahmad S, McGann HC, Banks
RK, Stavitzski NM, Srivastava M, Ali G, Finkler NJ, Edwards JR and
Holloway RW: Cellular therapy in combination with cytokines
improves survival in a xenograft mouse model of ovarian cancer. Mol
Cell Biochem. 407:281–287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ingersoll SB, Patel S, Caballero L, Ahmad
S, Edwards D, Holloway RW and Edwards JR: Synergistic cytotoxicity
of interferonalpha-2b and interleukin-2 in combination with PBMC
against ovarian cancer: Development of an experimental model for
cellular therapy. Gynecol Oncol. 112:192–198. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Di Scala M, Gil-Fariña I, Olagüe C, Vales
A, Sobrevals L, Fortes P, Corbacho D and González-Aseguinolaza G:
Identification of IFN-γ-producing T cells as the main mediators of
the side effects associated to mouse interleukin-15 sustained
exposure. Oncotarget. 7:49008–49026. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miller JS, Morishima C, McNeel DG, Patel
MR, Kohrt HEK, Thompson JA, Sondel PM, Wakelee HA, Disis ML, Kaiser
JC, et al: A First-in-Human Phase I Study of subcutaneous
outpatient recombinant human IL15 (rhIL15) in adults with advanced
solid tumors. Clin Cancer Res. 24:1525–1535. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Conlon KC, Lugli E, Welles HC, Rosenberg
SA, Fojo AT, Morris JC, Fleisher TA, Dubois SP, Perera LP, Stewart
DM, et al: Redistribution, hyperproliferation, activation of
natural killer cells and CD8 T cells, and cytokine production
during First-in-Human clinical trial of recombinant human
Interleukin-15 in patients with cancer. J Clin Oncol. 33:74–82.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rubinstein MP, Kovar M, Purton JF, Cho JH,
Boyman O, Surh CD and Sprent J: Converting IL-15 to a superagonist
by binding to soluble IL-15R{alpha}. Proc Natl Acad Sci USA.
103:9166–9171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ochoa MC, Fioravanti J, Rodriguez I,
Hervas-Stubbs S, Azpilikueta A, Mazzolini G, Gúrpide A, Prieto J,
Pardo J, Berraondo P and Melero I: Antitumor immunotherapeutic and
toxic properties of an HDL-Conjugated Chimeric IL-15 fusion
protein. Cancer Res. 73:139–149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mortier E, Quéméner A, Vusio P, Lorenzen
I, Boublik Y, Grötzinger J, Plet A and Jacques Y: Soluble
interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective
and potent agonist of IL-15 action through IL-15R beta/gamma.
Hyperagonist IL-15 × IL-15R alpha fusion proteins. J Biol Chem.
281:1612–1619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ochoa MC, Minute L, López A, Pérez-Ruiz E,
Gomar C, Vasquez M, Inoges S, Etxeberria I, Rodriguez I, Garasa S,
et al: Enhancement of antibody-dependenT cellular cytotoxicity of
cetuximab by a chimeric protein encompassing interleukin-15.
Oncoimmunology. 7:e13935972017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rhode PR, Egan JO, Xu W, Hong H, Webb GM,
Chen X, Liu B, Zhu X, Wen J, You L, et al: Comparison of the
superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics
in animal models. Cancer Immunol Res. 4:49–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Romee R, Cooley S, Berrien-Elliott MM,
Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun
C, DeFor TE, et al: First-in-human phase 1 clinical study of the
IL-15 superagonist complex ALT-803 to treat relapse after
transplantation. Blood. 131:2515–2527. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rosser CJ, Nix L, Ferguson L, Hernandez L
and Wong HC: Phase Ib trial of ALT-803, an IL-15 superagonist, plus
BCG for the treatment of BCG-naïve patients with
non-muscle-invasive bladder cancer. J Clin Oncol. 36 (Suppl
6):5102021. View Article : Google Scholar
|
|
32
|
Timmerman JM, Byrd JC, Andorsky DJ, Yamada
RE, Kramer J, Muthusamy N, Hunder N and Pagel JM: A phase I
dose-finding trial of recombinant interleukin-21 and rituximab in
relapsed and refractory low grade B-cell lymphoproliferative
disorders. Clin Cancer Res. 18:5752–5760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fioravanti J, Di Lucia P, Magini D, Moalli
F, Boni C, Benechet AP, Fumagalli V, Inverso D, Vecchi A, Fiocchi
A, et al: Effector CD8+ T cell-derived interleukin-10 enhances
acute liver immunopathology. J Hepatol. 67:543–548. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koski A, Kangasniemi L, Escutenaire S,
Pesonen S, Cerullo V, Diaconu I, Nokisalmi P, Raki M, Rajecki M,
Guse K, et al: Treatment of cancer patients with a serotype 5/3
chimeric oncolytic adenovirus expressing GMCSF. Mol Ther.
18:1874–1884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Spaapen RM, Leung MY, Fuertes MB, Kline
JP, Zhang L, Zheng Y, Fu YX, Luo X, Cohen KS and Gajewski TF:
Therapeutic activity of High-Dose Intratumoral IFN-β requires
direct effect on the tumor vasculature. J Immunol. 193:4254–4260.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Herndon TM, Demko SG, Jiang X, He K,
Gootenberg JE, Cohen MH, Keegan P and Pazdur R: U.S. Food and Drug
Administration Approval: Peginterferon-alfa-2b for the adjuvant
treatment of patients with melanoma. Oncologist. 17:1323–1328.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bellobuono A, Mondazzi L, Tempini S,
Silini E, Vicari F and Idéo; G. Ribavirin and interferon-alpha
combination therapy vs interferon-alpha alone in the retreatment of
chronic hepatitis C, : A randomized clinical trial. J Viral Hepat.
4:185–191. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gogas H, Ioannovich J, Dafni U,
Stavropoulou-Giokas C, Frangia K, Tsoutsos D, Panagiotou P, Polyzos
A, Papadopoulos O, Stratigos A, et al: Prognostic significance of
autoimmunity during treatment of melanoma with interferon. N Engl J
Med. 354:709–718. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fioravanti J, González I, Medina-Echeverz
J, Larrea E, Ardaiz N, González-Aseguinolaza G, Prieto J and
Berraondo P: Anchoring interferon alpha to apolipoprotein A-I
reduces hematological toxicity while enhancing immunostimulatory
properties. Hepatology. 53:1864–1873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cauwels A, Van Lint S, Paul F, Garcin G,
De Koker S, Van Parys A, Wueest T, Gerlo S, Van der Heyden J,
Bordat Y, et al: Delivering Type I interferon to dendritic cells
empowers tumor eradication and immune combination treatments.
Cancer Res. 78:463–474. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Palladino MA, Bahjat FR, Theodorakis EA
and Moldawer LL: Anti-TNF-alpha therapies: The next generation. Nat
Rev Drug Discov. 2:736–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Creaven PJ, Plager JE, Dupere S, Huben RP,
Takita H, Mittelman A and Proefrock A: Phase I clinical trial of
recombinant human tumor necrosis factor. Cancer Chemother
Pharmacol. 20:137–144. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zheng L, Fisher G, Miller RE, Peschon J,
Lynch DH and Lenardo MJ: Induction of apoptosis in mature T cells
by tumour necrosis factor. Nature. 377:348–351. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kahn JO, Kaplan LD, Volberding PA, Ziegler
JL, Crowe S, Saks SR and Abrams DI: Intralesional recombinant tumor
necrosis factor-alpha for AIDS-associated Kaposi's sarcoma: A
randomized, double-blind trial. J Acquir Immune Defic Syndr.
2:217–223. 1989.PubMed/NCBI
|
|
45
|
Manusama ER, Nooijen PT, Stavast J,
Durante NM, Marquet RL and Eggermont AM: Synergistic antitumour
effect of recombinant human tumour necrosis factor alpha with
melphalan in isolated limb perfusion in the rat. Br J Surg.
83:551–555. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lejeune FJ, Liénard D, Matter M and Rüegg
C: Efficiency of recombinant human TNF in human cancer therapy.
Cancer Immun. 6:62006.PubMed/NCBI
|
|
47
|
van Horssen R, Ten Hagen TL and Eggermont
AM: TNF-alpha in cancer treatment: Molecular insights, antitumor
effects, and clinical utility. Oncologist. 11:397–408. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Herzberg B, Campo MJ and Gainor JF: Immune
checkpoint inhibitors in non-small cell lung cancer. Oncologist.
22:81–88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Delgobo M and Frantz S: Heart failure in
cancer: Role of checkpoint inhibitors. J Thorac Dis. 10 (Suppl
35):S4323–S4334. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wolchok JD, Chiarion-Sileni V, Gonzalez R,
Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D,
Ferrucci PF, et al: Overall survival with combined nivolumab and
ipilimumab in advanced melanoma. N Engl J Med. 377:1345–1356. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kamath SD and Kumthekar PU: Immune
checkpoint inhibitors for the treatment of central nervous system
(CNS) metastatic disease. Front Oncol. 8:4142018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Heinzerling L, Ott PA, Hodi FS, Husain AN,
Tajmir-Riahi A, Tawbi H, Pauschinger M, Gajewski TF, Lipson EJ and
Luke JJ: Cardiotoxicity associated with CTLA4 and PD1 blocking
immunotherapy. J Immunother Cancer. 4:502016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sznol M, Postow MA, Davies MJ, Pavlick AC,
Plimack ER, Shaheen M, Veloski C and Robert C: Endocrine-related
adverse events associated with immune checkpoint blockade and
expert insights on their management. Cancer Treat Rev. 58:70–76.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hassel JC, Heinzerling L, Aberle J, Bähr
O, Eigentler TK, Grimm MO, Grünwald V, Leipe J, Reinmuth N, Tietze
JK, et al: Combined immune checkpoint blockade
(anti-PD-1/anti-CTLA-4): Evaluation and management of adverse drug
reactions. Cancer Treat Rev. 57:36–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Simonaggio A, Michot JM, Voisin AL, Le
Pavec J, Collins M, Lallart A, Cengizalp G, Vozy A, Laparra A,
Varga A, et al: Evaluation of readministration of immune checkpoint
inhibitors after immune-related adverse events in patients with
cancer. JAMA Oncol. 5:1310–1317. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Santini FC, Rizvi H, Plodkowski AJ, Ni A,
Lacouture ME, Gambarin-Gelwan M, Wilkins O, Panora E, Halpenny DF,
Long NM, et al: Safety and efficacy of re-treating with
immunotherapy after immune-related adverse events in patients with
NSCLC. Cancer Immunol Res. 6:1093–1099. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kluger HM, Zito CR, Turcu G, Baine MK,
Zhang H, Adeniran A, Sznol M, Rimm DL, Kluger Y, Chen L, et al:
PD-L1 studies across tumor types, its differential expression and
predictive value in patients treated with immune checkpoint
inhibitors. Clin Cancer Res. 23:4270–4279. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fan J, Shang D, Han B, Song J, Chen H and
Yang JM: Adoptive cell transfer: Is it a promising immunotherapy
for colorectal cancer? Theranostics. 8:5784–5800. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wrangle J, Paulos CM, Smith TW, Nishimura
MI and Rubinstein MP: Inducible enhancement of T cell function and
anti-tumor activity after adoptive transfer. Mol Ther.
25:1995–1996. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rohaan MW, Wilgenhof S and Haanen JBAG:
Adoptive cellular therapies: The current landscape. Virchows Arch.
474:449–461. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mitchison NA: Studies on the immunological
response to foreign tumor transplants in the mouse. I. The role of
lymph node cells in conferring immunity by adoptive transfer. J Exp
Med. 102:157–177. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Fefer A: Immunotherapy and chemotherapy of
Moloney sarcoma virus-induced tumors in mice. Cancer Res.
29:2177–2183. 1969.PubMed/NCBI
|
|
63
|
Rosenberg SA and Terry WD: Passive
immunotherapy of cancer in animals and man. Adv Cancer Res.
25:323–388. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kono K, Ichihara F, Iizuka H, Sekikawa T
and Matsumoto Y: Expression of signal transducing T-cell receptor
zeta molecules after adoptive immunotherapy in patients with
gastric and colon cancer. Int J Cancer. 78:301–305. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lu TL, Pugach O, Somerville R, Rosenberg
SA, Kochenderfer JN, Better M and Feldman SA: A Rapid cell
expansion process for production of engineered autologous CAR-T
cell therapies. Hum Gene Ther Methods. 27:209–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xiao L, Cen D, Gan H, Sun Y, Huang N,
Xiong H, Jin Q, Su L, Liu X, Wang K, et al: Adoptive transfer of
NKG2D CAR mRNA-Engineered natural killer cells in colorectal cancer
patients. Mol Ther. 27:1114–1125. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Vivier E, Raulet DH, Moretta A, Caligiuri
MA, Zitvogel L, Lanier LL, Yokoyama WM and Ugolini S: Innate or
adaptive immunity? The example of natural killer cells. Science.
331:44–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Morvan MG and Lanier LL: NK cells and
cancer: You can teach innate cells new tricks. Nat Rev Cancer.
16:7–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Basar R, Daher M and Rezvani K:
Next-generation cell therapies: The emerging role of CAR-NK cells.
Blood Adv. 4:5868–5876. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhou J, Bethune MT, Malkova N, Sutherland
AM, Comin-Anduix B, Su Y, Baltimore D, Ribas A and Heath JR: A
kinetic investigation of interacting, stimulated T cells identifies
conditions for rapid functional enhancement, minimal phenotype
differentiation, and improved adoptive cell transfer tumor
eradication. PLoS One. 13:e01916342018. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hinrichs CS, Borman ZA, Cassard L,
Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, Muranski P, Kern SJ,
Logun C, et al: Adoptively transferred effector cells derived from
naive rather than central memory CD8+ T cells mediate superior
antitumor immunity. Proc Natl Acad Sci USA. 106:17469–17474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
De Sanctis F, Trovato R and Ugel S:
Anti-telomerase T cells adoptive transfer. Aging (Albany NY).
9:2239–2240. 2017. View Article : Google Scholar
|
|
73
|
Kondo T, Imura Y, Chikuma S, Hibino S,
Omata-Mise S, Ando M, Akanuma T, Iizuka M, Sakai R, Morita R and
Yoshimura A: Generation and application of human induced-stem cell
memory T cells for adoptive immunotherapy. Cancer Sci.
109:2130–2140. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Foley KC, Nishimura MI and Moore TV:
Combination immunotherapies implementing adoptive T-cell transfer
for advanced-stage melanoma. Melanoma Res. 28:171–184. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Abi-Habib RJ, Singh R, Leppla SH, Greene
JJ, Ding Y, Berghuis B, Duesbery NS and Frankel AE: Systemic
anthrax lethal toxin therapy produces regressions of subcutaneous
human melanoma tumors in athymic nude mice. Clin Cancer Res.
12:7437–7443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kong LY, Abou-Ghazal MK, Wei J,
Chakraborty A, Sun W, Qiao W, Fuller GN, Fokt I, Grimm EA,
Schmittling RJ, et al: A novel inhibitor of signal transducers and
activators of transcription 3 activation is efficacious against
established central nervous system melanoma and inhibits regulatory
T cells. Clin Cancer Res. 14:5759–5768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Weiss T, Weller M, Guckenberger M, Sentman
CL and Roth P: NKG2D-Based CAR T cells and radiotherapy exert
synergistic efficacy in glioblastoma. Cancer Res. 78:1031–1043.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Al Hassan M, Fakhoury I, El Masri Z,
Ghazale N, Dennaoui R, El Atat O, Kanaan A and El-Sibai M:
Metformin treatment inhibits motility and invasion of glioblastoma
cancer cells. Anal Cell Pathol (Amst). 2018:59174702018.
|
|
79
|
Khoury O, Ghazale N, Stone E, El-Sibai M,
Frankel AE and Abi-Habib RJ: Human recombinant arginase I
(Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced arginine depletion is
selectively cytotoxic to human glioblastoma cells. J Neurooncol.
122:75–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Dudley ME and Rosenberg SA:
Adoptive-cell-transfer therapy for the treatment of patients with
cancer. Nat Rev Cancer. 3:666–675. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Miao Y, Yang H, Levorse J, Yuan S, Polak
L, Sribour M, Singh B, Rosenblum MD and Fuchs E: Adaptive immune
resistance emerges from tumor-initiating stem cells. Cell.
177:1172–1186.e14. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Agudo J, Park ES, Rose SA, Alibo E,
Sweeney R, Dhainaut M, Kobayashi KS, Sachidanandam R, Baccarini A,
Merad M and Brown BD: Quiescent tissue stem cells evade immune
surveillance. Immunity. 48:271–285.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Brown JA, Yonekubo Y, Hanson N,
Sastre-Perona A, Basin A, Rytlewski JA, Dolgalev I, Meehan S,
Tsirigos A, Beronja S and Schober M: TGF-β-induced quiescence
mediates chemoresistance of tumor-propagating cells in squamous
cell carcinoma. Cell Stem Cell. 21:650–664.e8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tey SK: Adoptive T-cell therapy: Adverse
events and safety switches. Clin Transl Immunology. 3:e172014.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yang JC: Toxicities associated with
adoptive T-cell transfer for cancer. Cancer. 21:506–509. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Miliotou AN and Papadopoulou LC: CAR
T-cell therapy: A new era in cancer immunotherapy. Curr Pharm
Biotechnol. 19:5–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Maeng HM and Berzofsky JA: Strategies for
developing and optimizing cancer vaccines. F1000Res 8: F1000
Faculty Rev-654. 2019. View Article : Google Scholar
|
|
88
|
Gatti-Mays ME, Redman JM, Collins JM and
Bilusic M: Cancer vaccines: Enhanced immunogenic modulation through
therapeutic combinations. Hum Vaccines Immunother. 13:2561–2574.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Manthorpe M, Cornefert-Jensen F, Hartikka
J, Felgner J, Rundell A, Margalith M and Dwarki V: Gene therapy by
intramuscular injection of plasmid DNA: Studies on firefly
luciferase gene expression in mice. Hum Gene Ther. 4:419–431. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Walters JN, Ferraro B, Duperret EK,
Kraynyak KA, Chu J, Saint-Fleur A, Yan J, Levitsky H, Khan AS,
Sardesai NY and Weiner DB: A Novel DNA vaccine platform enhances
neo-antigen-like T cell responses against WT1 to break tolerance
and induce anti-tumor immunity. Mol Ther. 25:976–988. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lopes A, Vanvarenberg K, Kos Š, Lucas S,
Colau D, Van den Eynde B, Préat V and Vandermeulen G: Combination
of immune checkpoint blockade with DNA cancer vaccine induces
potent antitumor immunity against P815 mastocytoma. Sci Rep.
8:157322018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Paston SJ, Brentville VA, Symonds P and
Durrant LG: Cancer vaccines, adjuvants, and delivery systems. Front
Immunol. 12:6279322021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Gamat-Huber M, Jeon D, Johnson LE, Moseman
JE, Muralidhar A, Potluri HK, Rastogi I, Wargowski E, Zahm CD and
McNeel DG: Treatment combinations with DNA vaccines for the
treatment of Metastatic Castration-Resistant Prostate Cancer
(mCRPC). Cancers (Basel). 12:28312020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Li L and Petrovsky N: Molecular mechanisms
for enhanced DNA vaccine immunogenicity. Expert Rev Vaccines.
15:313–329. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Jahanafrooz Z, Baradaran B, Mosafer J,
Hashemzaei M, Rezaei T, Mokhtarzadeh A and Hamblin MR: Comparison
of DNA and mRNA vaccines against cancer. Drug Discov Today.
25:552–560. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bhuyan PK, Dallas M, Kraynyak K, Herring
T, Morrow M, Boyer J, Duff S, Kim J and Weiner DB: Durability of
response to VGX-3100 treatment of HPV16/18 positive cervical HSIL.
Hum Vaccin Immunother. 17:1288–1293. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lopes A, Vandermeulen G and Préat V:
Cancer DNA vaccines: Current preclinical and clinical developments
and future perspectives. J Exp Clin Cancer Res. 38:1462019.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Malonis RJ, Lai JR and Vergnolle O:
Peptide-based vaccines: Current progress and future challenges.
Chem Rev. 120:3210–3229. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Li W, Joshi MD, Singhania S, Ramsey KH and
Murthy AK: Peptide vaccine: Progress and challenges. Vaccines
(Basel). 2:515–536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Curry JM, Besmer DM, Erick TK, Steuerwald
N, Das Roy L, Grover P, Rao S, Nath S, Ferrier JW, Reid RW and
Mukherjee P: Indomethacin enhances anti-tumor efficacy of a MUC1
peptide vaccine against breast cancer in MUC1 transgenic mice. PLoS
One. 14:e02243092019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Pan J, Zhang Q, Palen K, Wang L, Qiao L,
Johnson B, Sei S, Shoemaker RH, Lubet RA, Wang Y and You M:
Potentiation of Kras peptide cancer vaccine by avasimibe, a
cholesterol modulator. EBioMedicine. 49:72–81. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zhang R, Yuan F, Shu Y, Tian Y, Zhou B, Yi
L, Zhang X, Ding Z, Xu H and Yang L: Personalized neoantigen-pulsed
dendritic cell vaccines show superior immunogenicity to
neoantigen-adjuvant vaccines in mouse tumor models. Cancer Immunol
Immunother. 69:135–145. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Neek M, Kim TI and Wang SW: Protein-based
nanoparticles in cancer vaccine development. Nanomedicine.
15:164–174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Rousseau RF, Hirschmann-Jax C, Takahashi S
and Brenner MK: Cancer vaccines. Hematol Oncol Clin North Am.
15:741–773. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Steinman RM and Cohn ZA: Identification of
a novel cell type in peripheral lymphoid organs of mice. I.
Morphology, quantitation, tissue distribution. J Exp Med.
137:1142–1162. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Santos PM and Butterfield LH: Dendritic
cell-based cancer vaccines. J Immunol. 200:443–449. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Alvarez-Dominguez C, Calderón-Gonzalez R,
Terán-Navarro H, Salcines-Cuevas D, Garcia-Castaño A, Freire J,
Gomez-Roman J and Rivera F: Dendritic cell therapy in melanoma. Ann
Transl Med. 5:3862017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
de Gruijl TD, van den Eertwegh AJM, Pinedo
HM and Scheper RJ: Whole-cell cancer vaccination: From autologous
to allogeneic tumor- and dendritic cell-based vaccines. Cancer
Immunol Immunother. 57:1569–1577. 2008. View Article : Google Scholar
|
|
109
|
Fu C, Zhou L, Mi QS and Jiang A: DC-Based
vaccines for cancer immunotherapy. Vaccines (Basel). 8:7062020.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wculek SK, Amores-Iniesta J, Conde-Garrosa
R, Khouili SC, Melero I and Sancho D: Effective cancer
immunotherapy by natural mouse conventional type-1 dendritic cells
bearing dead tumor antigen. J Immunother Cancer. 7:1002019.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lai X and Friedman A: Combination therapy
of cancer with cancer vaccine and immune checkpoint inhibitors: A
mathematical model. PLoS One. 12:e01784792017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Anassi E and Ndefo UA: Sipuleucel-T
(provenge) injection: The first immunotherapy agent (vaccine) for
hormone-refractory prostate cancer. P T. 36:197–202.
2011.PubMed/NCBI
|
|
113
|
Ayoub NM, Al-Shami KM and Yaghan RJ:
Immunotherapy for HER2-positive breast cancer: Recent advances and
combination therapeutic approaches. Breast Cancer (Dove Med Press).
11:53–69. 2019.PubMed/NCBI
|
|
114
|
Han Q, Wang Y, Pang M and Zhang J:
STAT3-blocked whole-cell hepatoma vaccine induces cellular and
humoral immune response against HCC. J Exp Clin Cancer Res.
36:1562017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sheikhi A, Jafarzadeh A, Kokhaei P and
Hojjat-Farsangi M: Whole tumor cell vaccine adjuvants: Comparing
IL-12 to IL-2 and IL-15. Iran J Immunol. 13:148–166.
2016.PubMed/NCBI
|
|
116
|
Xia L, Schrump DS and Gildersleeve JC:
Whole-Cell cancer vaccines induce large antibody responses to
carbohydrates and glycoproteins. Cell Chem Biol. 23:1515–1525.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Chen G, Gupta R, Petrik S, Laiko M,
Leatherman JM, Asquith JM, Daphtary MM, Garrett-Mayer E, Davidson
NE, Hirt K, et al: A feasibility study of cyclophosphamide,
trastuzumab, and an allogeneic GM-CSF-secreting breast tumor
vaccine for HER2+ Metastatic breast cancer. Cancer Immunol Res.
2:949–961. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Constantino J, Gomes C, Falcão A, Cruz MT
and Neves BM: Antitumor dendritic cell-based vaccines: Lessons from
20 years of clinical trials and future perspectives. Transl Res.
168:74–95. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Köhler G and Milstein C: Continuous
cultures of fused cells secreting antibody of predefined
specificity. Nature. 256:495–497. 1975. View Article : Google Scholar
|
|
120
|
Chung S, Lin YL, Reed C, Ng C, Cheng ZJ,
Malavasi F, Yang J, Quarmby V and Song A: Characterization of in
vitro antibody-dependenT cell-mediated cytotoxicity activity of
therapeutic antibodies-impact of effector cells. J Immunol Methods.
407:63–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Wang W, Erbe AK, Hank JA, Morris ZS and
Sondel PM: NK Cell-Mediated Antibody-DependenT cellular
cytotoxicity in cancer immunotherapy. Front Immunol. 6:3682015.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Harris TJ and Drake CG: Primer on tumor
immunology and cancer immunotherapy. J Immunother Cancer. 1:122013.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Mayor M, Yang N, Sterman D, Jones DR and
Adusumilli PS: Immunotherapy for non-small cell lung cancer:
Current concepts and clinical trials. Eur J Cardiothorac Surg.
49:1324–1333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Kimiz-Gebologlu I, Gulce-Iz S and
Biray-Avci C: Monoclonal antibodies in cancer immunotherapy. Mol
Biol Rep. 45:2935–2940. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Karlitepe A, Ozalp O and Avci CB: New
approaches for cancer immunotherapy. Tumour Biol. 36:4075–4078.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Sathyanarayanan V and Neelapu SS: Cancer
immunotherapy: Strategies for personalization and combinatorial
approaches. Mol Oncol. 9:2043–2053. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Posner J, Barrington P, Brier T and
Datta-Mannan A: Monoclonal antibodies: Past, present and future.
Handb Exp Pharmacol. 260:81–141. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zahavi D and Weiner L: Monoclonal
Antibodies in Cancer Therapy. Antibodies (Basel). 9:342020.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Loi S, Giobbie-Hurder A, Gombos A,
Bachelot T, Hui R, Curigliano G, Campone M, Biganzoli L, Bonnefoi
H, Jerusalem G, et al: Pembrolizumab plus trastuzumab in
trastuzumab-resistant, advanced, HER2-positive breast cancer
(PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet
Oncol. 20:371–382. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
ClinicalTrials.gov, . A Dose Escalation
and Cohort Expansion Study of NKTR-214 in Combination With
Nivolumab and Other Anti-Cancer Therapies in Patients With Select
Advanced Solid Tumors (PIVOT-02). linicalTrials.gov Identifier:
NCT02983045. U.S.National Library of Medicine; Bethesda, MD: 2016,
https://clinicaltrials.gov/ct2/show/NCT02983045December
6–2016
|
|
131
|
ClinicalTrials.gov, . Bempegaldesleukin
and Pembrolizumab With or Without Chemotherapy in Locally Advanced
or Metastatic Solid Tumors (PROPEL). ClinicalTrials.gov Identifier:
NCT03138889. U.S.National Library of Medicine; Bethesda, MD: 2017,
https://clinicaltrials.gov/ct2/show/NCT03138889May
3–2017
|
|
132
|
Zimmer L, Goldinger SM, Hofmann L, Loquai
C, Ugurel S, Thomas I, Schmidgen MI, Gutzmer R, Utikal JS, Göppner
D, et al: Neurological, respiratory, musculoskeletal, cardiac and
ocular side-effects of anti-PD-1 therapy. Eur J Cancer. 60:210–225.
2016. View Article : Google Scholar : PubMed/NCBI
|