Introduction
Endometrial cancer is the most common gynecologic
malignancy in the United States, with >63,000 new cases in 2018
(1). Benefiting from early detection
and advanced therapies, patients with endometrial cancer have shown
improved survival rates during the past few decades (2). However, the cure rate is significantly
low, and the prognosis is poor for patients with advanced
metastatic disease (3). Therefore,
further investigation into the mechanisms underlying the metastasis
of endometrial cancer, with the aim to develop effective treatment
methods and improve patient prognosis, is crucial.
Aerobic glycolysis, the Warburg effect, is a primary
characteristic of cancer cells (4,5). Tumor
cells exhibit increased biosynthesis through glucose uptake and
lactate production via the aerobic glycolysis pathway, thus are
characterized by rapid proliferation, invasiveness and metastatic
potential (6). Emerging evidence has
shown that the deregulation of aerobic glycolysis plays an
important role in cellular proliferation, invasiveness and
angiogenesis of cancers, including cervical cancer (7,8).
Therefore, repression of aerobic glycolysis may be a potential
therapeutic method for preventing cancer progression (9).
Numerous genes and factors have been strongly
implicated in the progression of endometrial cancer, such as p53
and p16 inactivation, PTEN mutations, and the upregulation of
estrogen and/or progesterone receptors (10–12).
Histone deacetylases (HDACs) are important enzymes which induce
deacetylation, resulting in an inactive chromatin structure.
Notably, the genetic alterations in endometrial cancer are strongly
affected by histone-mediated epigenetics, including histone
deacetylation, indicating that HDACs may play an important role in
the progression of endometrial cancer (10–12). For
example, Li et al (13) found
that the administration of romidepsin (FK228), a HDAC inhibitor,
impaired cellular proliferation and accelerated apoptosis by
increasing p53 expression in endometrial cancer Ishikawa and
HEC-1-A cells. De et al (14)
reported that MHY2256, another HDAC inhibitor, induced apoptosis
and autophagy in endometrial cancer cells by elevating p53
acetylation. Furthermore, Zheng et al (15) found that HDAC6 was overexpressed in
endometrial cancer, and that this promoted cellular proliferation,
invasiveness and metastatic potential. HDAC1, together with HDAC2,
3 and 8, are members of the Class I of HDACs family (16). Evidence has demonstrated that HDAC1
exerts an oncogenic role in various human tumor types, including
breast (17,18), lung (19) and ovarian cancer (20). Tang et al (18) demonstrated that HDAC1 was upregulated
in breast cancer cells, where it promoted proliferation and
migration via upregulation of interleukin-8. Liu et al
(20) reported that HDAC1-knockdown
suppressed cellular proliferation, and increased apoptosis and
chemosensitivity in cisplatin-resistant A2780/CDDP ovarian cancer
cells. Moreover, Cao et al (19) found that HDAC1 mRNA and protein
expression was closely associated with the differentiation grade of
lung cancer. Also, the expression level of HDAC1 in
gastrointestinal malignancies, especially in colorectal cancer, was
found to be higher than that in noncancerous tissues, which was
closely associated with advanced tumor stage and poor prognosis
(21). In endometrial cancer,
Weichert et al (22)
suggested that the majority of endometrial cancer cells are
characterized by elevated expression of HDAC1, and that the
expression levels of HDAC1 are associated with cellular
proliferative capacity. However, the role of HDAC1 in the
occurrence and progression of endometrial cancer remains
unknown.
The aim of the present study was to reveal the
effects of HDAC1 on the proliferation, migration, aerobic
glycolysis and tumorigenesis of endometrial cancer by modulating
the level of HDAC1 via knockdown or overexpression.
Materials and methods
Patients
The present study was approved by the ethical
committee of Shanghai Changning Maternity and Infant Health
Hospital (approval. no. CNFBLLKT-2017-011), and adheres to the
Declaration of Helsinki. In total, 64 paired endometrial cancer and
adjacent-normal tissues were obtained from patients diagnosed with
endometrial cancer at the Shanghai Changning Maternity and Infant
Health Hospital (Shanghai, China) between February 2010 and
February 2013. The patient age range was from 34 to 76 years.
Clinicopathological characteristics, including age, parity, body
mass index, International Federation of Gynecology and Obstetrics
[FIGO] stage (23), differentiation
and metastasis status, as well as the survival time after surgery,
were all obtained from electronic medical records. Inclusion
criteria: Patients underwent surgery prior to chemoradiotherapy,
and provided written informed consent. Exclusion criteria: i)
Patients complicated with epithelial ovarian cancer or other
malignant tumors; ii) Patients had received chemoradiotherapy prior
to surgery; and iii) Patients who didn't provide written informed
consent. The relative mRNA level of HDAC1 ≥ the mean was considered
as high HDAC1 expression, and < the average value was considered
as low HDAC1 expression.
Cell culture
Normal human endometrium cells from non-malignant
myoma (KC02-44D hTERT), endometrial cancer cell lines HEC-1-A and
HEC-1-B, and 293T cells were all obtained from the American Type
Culture Collection (ATCC). All cells were maintained in Dulbecco's
modified Eagle's medium, supplemented with 10% fetal bovine serum
(FBS) and 1% penicillin/streptomycin (all Thermo Fisher Scientific,
Inc.) at 37°C with 5% CO2.
Cell transfection and treatment
The lentivirus vectors used to upregulate or
downregulate HDAC1 in human endometrial cancer HEC-1-A cells are
termed overexpression (OE)-HDAC1 (cat. no. RC201745L4V) and short
hairpin (sh)-HDAC1 (cat. no. TL312496V), and were purchased from
OriGene Technologies, Inc. 293T cells were used to generate the
virions through transfection with vectors (20 µg), phelper 1.0 (15
µg) and phelper 2.0 (10 µg) (BioVector NTCC, Inc.) using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C. Viral particles were collected 72 h
post-transfection, then centrifuged at 1,776 × g for 5 min at 4°C,
and filtered with 0.45 µm filter. For cell infection, HEC-1-A cell
culture medium was added with OE-HDAC1, sh-HDAC1, or their negative
control vectors (OE-NC, sh-NC) with a multiplicity of infections of
5, 6, 5 and 6 for 6 h, followed by replacement with fresh medium
and incubation at 37°C for another 24 h. To construct
stably-transfected cell lines, 5 µg/ml puromycin was added to the
culture medium after 24 h of transfection and incubated for 14 days
at 37°C. The shRNA sequences are as follows: Sh-HDAC1-1 Sense,
5′-CACCGAGAAAGACCCAGAGGAGAAGCTCGAGCTTCTCCTCTGGGTCTTTCTC-3′ and
antisense,
5′-AAAAGAGAAAGACCCAGAGGAGAAGCTCGAGCTTCTCCTCTGGGTCTTTCTC-3′;
Sh-HDAC1-2 sense,
5′-CACCGAAGAAAGAAGTCACCGAAGACTCGAGTCTTCGGTGACTTCTTTCTTC-3′ and
antisense,
5′-AAAAGAAGAAAGAAGTCACCGAAGACTCGAGTCTTCGGTGACTTCTTTCTTC-3′.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from tissues and cells using
the RNApure Tissue & Cell Kit (DNase I) according to the
manufacturer's instructions (CWBio). The RNA samples were then
subjected to cDNA synthesis and qPCR using the SuperRT One Step
RT-PCR Kit (CWBio) on a Bio-Rad detection system (Bio-Rad
Laboratories, Inc.). β-actin expression was used to normalize the
mRNA levels of HDAC1, which were calculated using the
2−∆∆Cq method (24). The
qPCR thermocycling conditions were as follows: Initial denaturation
at 95°C for 30 sec, followed by annealing and elongation for 40
cycles of 95°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec, and
a final extension at 72°C for 2 min. Primers targeting HDAC1 and
β-actin were obtained from Invitrogen (Thermo Fisher Scientific,
Inc.), the sequences of which are as follows: HDAC1 forward,
5′-TGCTAAAGTATCACCAGAGGGT-3′ and reverse,
5′-TGGCCTCATAGGACTCGTCA-3′; and β-actin forward,
5′-ACAGAGCCTCGCCTTTGCC-3′ and reverse,
5′-CACACTTGGCGTGTCCTTTG-3′.
Western blotting
Total protein samples extracted from tissues and
cells were obtained using RIPA lysis buffer containing 50 mM Tris
(pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate,
0.1% sodium dodecyl sulfonate, sodium orthovanadate, sodium
fluoride, EDTA and leupeptin. The proteins were quantified using a
BCA Protein Assay kit (Thermo Fisher Scientific, Inc.).
Subsequently, 20–30 µg protein from each sample was separated by
SDS-PAGE with 10% polyacrylamide gels, and then transferred onto
PVDF membranes (MilliporeSigma). The membranes were blocked with 5%
non-fat milk for 1 h at room temperature, and probed with
anti-HDAC1 (1:2,000; cat. no. #5356; Cell Signaling Technology,
Inc.), anti-E-cadherin (1:1,000; cat. no. ab15148; Abcam),
anti-N-cadherin (1:1,000; cat. no. ab18203; Abcam), anti-cleaved
caspase-3 (1:1,000; cat. no. ab2302; Abcam), anti-hypoxia-inducible
factor 1 (HIF-1) α (1:3,000; cat. no. ab1; Abcam), anti-pyruvate
kinase PKM (1:5,000; cat. no. ab85555; Abcam), anti-L-lactate
dehydrogenase A chain (1:5,000; cat. no. ab101562; Abcam) and
anti-β-actin (1:5,000; cat. no. ab8226; Abcam) antibodies overnight
at 4°C. Then, the membranes were incubated with the corresponding
secondary antibodies, mouse anti-rabbit IgG-HRP (cat. no. sc-2357)
and goat anti-mouse IgG-HRP (cat. no. sc-2005) (both Santa Cruz
Biotechnology, Inc.) for 1 h at room temperature. Protein signaling
was enhanced using ECL reagents (MilliporeSigma) and then examined
using a western blotting imaging and quantitation system (Bio-Rad
Laboratories, Inc.). ImageJ software (v1.8.0; National Institutes
of Health) was used to quantify protein expression.
Cell Counting Kit-8 (CCK-8) assay
To assess cellular proliferation capacity, HEC-1-A
cells were first seeded into 96-well plates at a density of
3×103 cells/well, and cultured at 37°C overnight,
followed by lentiviral infection with sh-HDAC1, sh-NC, OE-HDAC1,
OE-NC. For proliferative assessment, 10 µl CCK-8 reagent (Beyotime
Institute of Biotechnology) and 90 µl fresh medium was added to
each well, and the cells were incubated for another 4 h at 37°C.
The absorbance was recorded at 450 nm every 24 h using a plate
reader (Bio-Rad Laboratories, Inc.).
Flow cytometry
The effects of HDAC1 on apoptosis were assessed by
flow cytometry using the Annexin V (FITC)/Propidium Iodide (PI)
apoptosis detection kit (Nanjing KeyGen Biotech Co., Ltd.)
according to manufacturer's instructions. Then, apoptosis rates
were detected with a CytoFLEX flow cytometer (Beckman Coulter,
Inc.) and analyzed using FlowJo 7.6 software (FlowJo LLC).
Wound-healing assay
HEC-1-A cells (1×105/well) infected with
sh-HDAC1, sh-NC, OE-HDAC1 or OE-NC were seeded into 24-well plate
and incubated at 37°C until confluent. Then, wounds were created
across each monolayer with a 20-µl pipette tip, and the floating
cells were removed. Subsequently, the cells were placed at 37°C and
cultured for another 24 h with serum-free medium (Thermo Fisher
Scientific, Inc.). The wound width was recorded using an inverted
microscope (magnification, ×40) at 0 and 24 h after wound
formation.
Transwell assay
Transwell chambers (8.0-µm; Corning, Inc.) were
precoated with 50 µl Matrigel (at a ratio of 1:1 with culture
medium) for 1 h at 37°C, and then used to assess cellular
invasiveness. Briefly, 1×105 cells in 200 µl FBS-free
culture medium (Thermo Fisher Scientific, Inc.) were placed into
the upper chamber of each well, and 600 µl medium supplemented with
15% FBS was added into the lower chamber. After incubation for 24 h
at 37°C, cells at the upper surface of the membrane were removed
with cotton swabs, while cells on the lower surface were fixed with
4% paraformaldehyde for 15 min at room temperature, followed by
staining with 0.1% crystal violet solution (Beyotime Institute of
Biotechnology) for 10 min at room temperature. Then, the invasive
cells were counted under an inverted microscope with a
magnification of ×200.
Detection of lactate production,
glucose consumption and ATP levels
Lactate production, glucose consumption and ATP
levels were measured according to a previous report (25). A total of 2×105 cells were
seeded into each well of 6-well plates and maintained at 37°C for
48 h, after which cellular lactate production was measured using a
Lactate Colorimetric Assay Kit (cat. no. K627; BioVision, Inc.).
The cells were incubated in cell culture medium without FBS for 1 h
at 37°C, and the supernatant was collected for measurement of
lactate production. The reaction mixture was incubated for 30 min
at room temperature and protected from light, and the lactate
levels were measured at 450 nm using a microplate reader.
Glucose consumption was assessed using the Glucose
Uptake Colorimetric Assay Kit (cat. no. K676; BioVision, Inc.).
Following lentivirus infection, cells were cultured at 37° for 48
h, collected and seeded into 96-well plates with 1×104
cells per well. Then, the cells were glucose-starved by
preincubating with Krebs-Ringer-Phosphate-HEPES buffer (100 µl) for
40 min, and incubated with 10 µl 2-deoxyglucose (10 mM) for 20 min,
followed by Reaction Mix A for 1 h at 37°C. Then, 90 µl extraction
buffer was added to each well, and incubated at 90°C for 40 min,
and then placed in an ice bath for 5 min. Then, Reaction Mix B was
added to each well, followed by centrifugation at 16,000 × g at 4°C
for 2 min. The OD value of the supernatant at 412 nm was measured
using a microplate analyzer.
An ATP Colorimetric Assay Kit (cat. no. MAK1900;
Sigma-Aldrich; Merck KGaA) was used for ATP level measurement
according to the manufacturer's protocol. Cells (5×105)
were collected and added to 100 µl ATP Assay Buffer. The cells were
centrifuged at 16,000 × g for 5 min at room temperature and the
supernatant was used for ATP measurement. The reaction mixture was
incubated for 30 min at room temperature, protected from light, and
measured at 570 nm in a microplate reader.
Animal experiments
Animal experiments were performed in accordance with
the National Institute of Health Guidelines for the Care and Use of
Laboratory Animals, and were approved by the Animal Care and
Research Committee of Shanghai Changning Maternity and Infant
Health Hospital. Female, 6-week-old BALB/c athymic nude mice
(Beijing Vital River Laboratory Animal Technology) were housed in a
specific pathogen-free animal facility with free access to water
and food, at 22±1°C with 55±2% humidity under a 12 h light/dark
cycle. To construct the tumor-bearing mouse model, 1×106
sh-HDAC1, OE-HDAC1, sh-NC or OE-NC stably transfected HEC-1-A cells
were subcutaneously injected into the nude mice. A total of 12 mice
were used, and three mice were included in each group. The animal
health, behavior and tumor growth were monitored every 3 days; the
tumors were weighed four weeks post cancer cell injection. The mice
were euthanized by cervical dislocation following 28 days of
injection unless the volume reached 1,000 mm3, at which
point the mice were sacrificed early. Mice were considered as dead
when no heartbeat and breathing were observed, and reflexes were
absent. Tumor volume was calculated according to the following
formula: Volume = length × width2/2.
Statistical analysis
With the exception of the animal study, a total of
three independent experiments were performed per assay, and the
data are expressed as mean ± standard deviation. SPSS 22.0 software
(IBM Corp.) was used to performed data analysis. The association
between HDAC1 expression and the clinicopathological feature of
patients with endometrial cancer was assessed using the
χ2 test. Data conformed to Gaussian distribution
(determined by Shapiro-Wilk test), and the t-test and one-way ANOVA
followed by Tukey's test were used for data analysis between 2
groups or multiple groups, respectively, after homogeneity testing
of variance using the F test. Paired Student's t test was applied
for data comparisons between cancer and matched-normal tissues.
P<0.05 was considered to indicate a statistically significant
difference.
Results
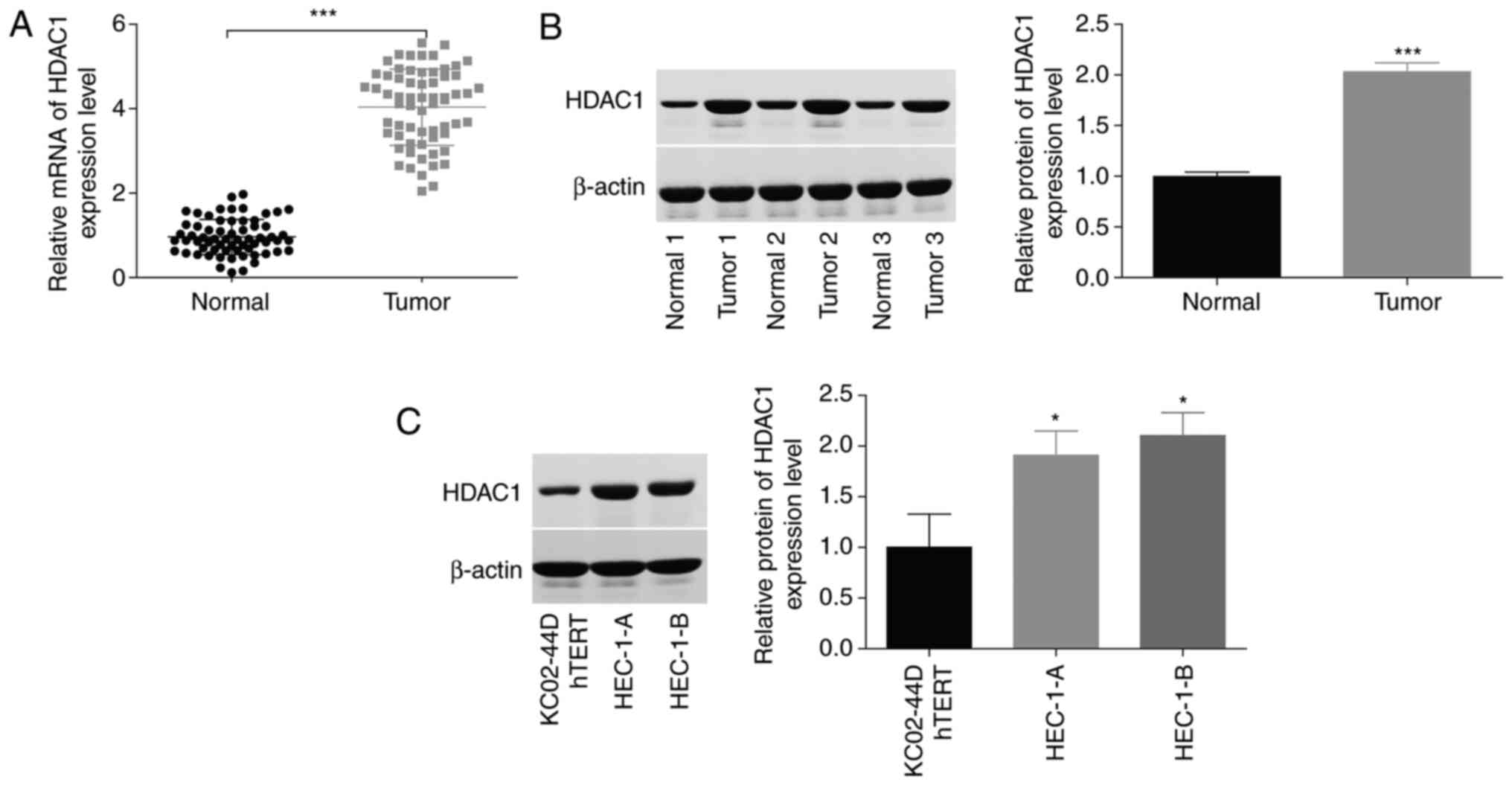
HDAC1 expression is significantly
increased in endometrial cancer tissues and cells
To reveal the function of HDAC1 in the progression
of endometrial cancer, the expression patterns of HDAC1 were
assessed in endometrial cancer tissues and cells. As shown by
RT-qPCR and western blotting, a significant increase (~3-fold) in
HDAC1 expression level was observed in cancer tissues compared with
normal tissue samples (Fig. 1A and
B). In addition, HDAC1 expression levels in human endometrial
cancer cell lines (HEC-1-A and HEC-1-B) were ~2-fold that of those
in KC02-44D hTERT cells (Fig. 1C).
These results indicated that HDAC1 was highly expressed in
endometrial cancer.
HDAC1 promotes cellular proliferation
and inhibits apoptosis in endometrial cancer
The function of HDAC1 in HEC-1-A cell proliferation
and apoptosis was then investigated. The expression levels of HDAC1
were reduced by 50–70% at the mRNA and protein levels following
infection with sh-HDAC1-1 and sh-HDAC1-2 (Fig. 2A and B). By contrast, HDAC1
expression was increased by 100–250% in HEC-1-A cells infected with
OE-HDAC1 compared with OE-NC (Fig. 2C
and D). Upregulation of HDAC1 induced a 1.5-fold increase in
cellular proliferation (Fig. 2E) and
inhibited apoptosis from 5.8±0.6% to 2.6±0.5% (Fig. 2F-G), whereas downregulation of HDAC1
impaired cellular proliferation and induced cell apoptosis
(Fig. 2E-G) compared with the
corresponding control group. In addition, downregulation of HDAC1
induced an 80% increase in cleaved caspase-3 expression, while
upregulation decreased HDAC1 expression (Fig. 2H). These findings suggested that
HDAC1 facilitated cellular proliferation and inhibited apoptosis in
endometrial cancer.
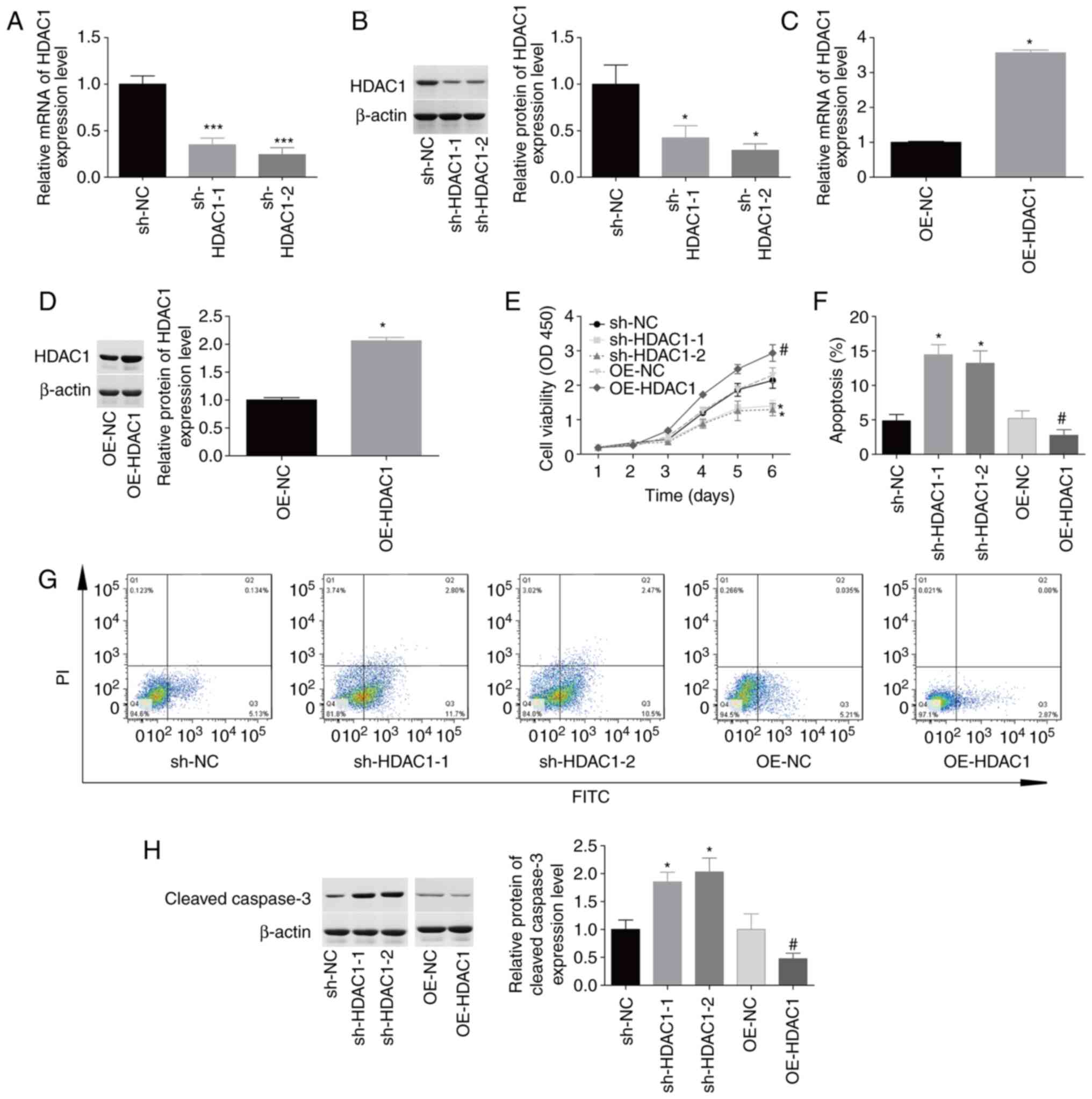 | Figure 2.Evaluation of HDAC1 effects on
cellular proliferation and apoptosis. HEC-1-A cells were infected
with sh-HDAC1-1/-2, sh-NC, OE-HDAC1 or OE-NC, prior to
experimentation. (A-D) Reverse transcription-quantitative PCR and
western blotting were performed to detect HDAC1 mRNA and protein
levels. (E) Cell Counting Kit 8 analysis showed that HDAC1
downregulation inhibited cellular proliferation, which was promoted
by OE-HDAC1. (F and G). Annexin V FITC/PI staining showed that
sh-HDAC1 transfection induced apoptosis and OE-HDAC1 repressed
apoptosis. (H) Expression levels of cleaved caspase-3 were
determined by western blotting. N=3; ANOVA followed by Tukey's
test; *P<0.05, ***P<0.001 sh-HDAC1 vs. sh-NC;
#P<0.05, OE-HDAC1 vs. OE-NC. HDAC1, histone
deacetylase 1; sh, short hairpin; OE, overexpression; PI, propidium
iodide. |
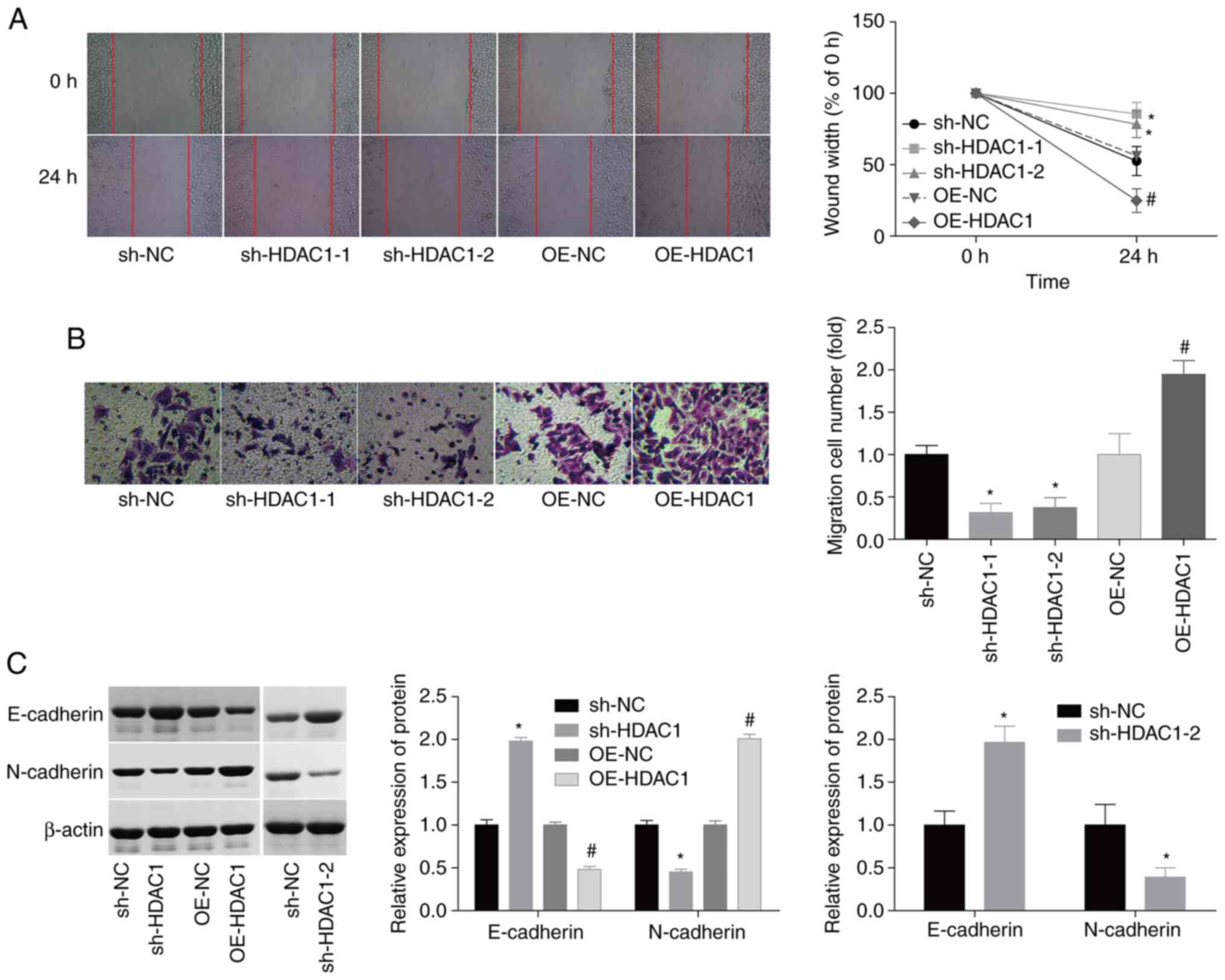
HDAC1 triggers cellular migration and
invasiveness in endometrial cancer
In addition, the effects of HDAC1 on the modulation
of cellular migration and invasiveness in endometrial cancer.
Compared with the control group, HEC-1-A cell migration and
invasiveness were enhanced by 55 and 80% following infection with
OE-HDAC1, whereas the migration and invasiveness of HEC-1-A cells
were inhibited by 50 and 55% when infected with sh-HDAC1 (Fig. 3A and B). Due to the crucial role of
epithelial-mesenchymal transition (EMT) in cancer cell migration
(26,27), the potential effects of HDAC1 on EMT
regulation in endometrial cancer were investigated. The expression
levels of N-cadherin, a mesenchymal cell marker, were elevated by
~100%, while the expression of E-cadherin, an epithelial cell
marker, was decreased by 55% when HDAC1 was overexpressed;
sh-HDAC1-1/-2 transfection resulted in the opposite effect
(Fig. 3C). These results suggest
that HDAC1 serves as a promoter of cellular migration and
invasiveness in endometrial cancer.
HDAC1 induces aerobic glycolysis in
endometrial cancer cells
HDAC1 function in the aerobic glycolysis of
endometrial cancer cells was subsequently investigated. The glucose
consumption, lactate secretion and ATP level were all increased by
~1-fold following HEC-1-A cell infection with OE-HDAC1, and
decreased by 40–60% when HDAC1 was downregulated (Fig. 4A-C). In addition, HDAC1
overexpression induced significant increases in HIF-1α, PKM2 and
LDHA levels, while downregulation of HDAC1 decreased the expression
levels of these proteins, compared with the control group (Fig. 4D). These findings suggested that
HDAC1 induced aerobic glycolysis in endometrial cancer.
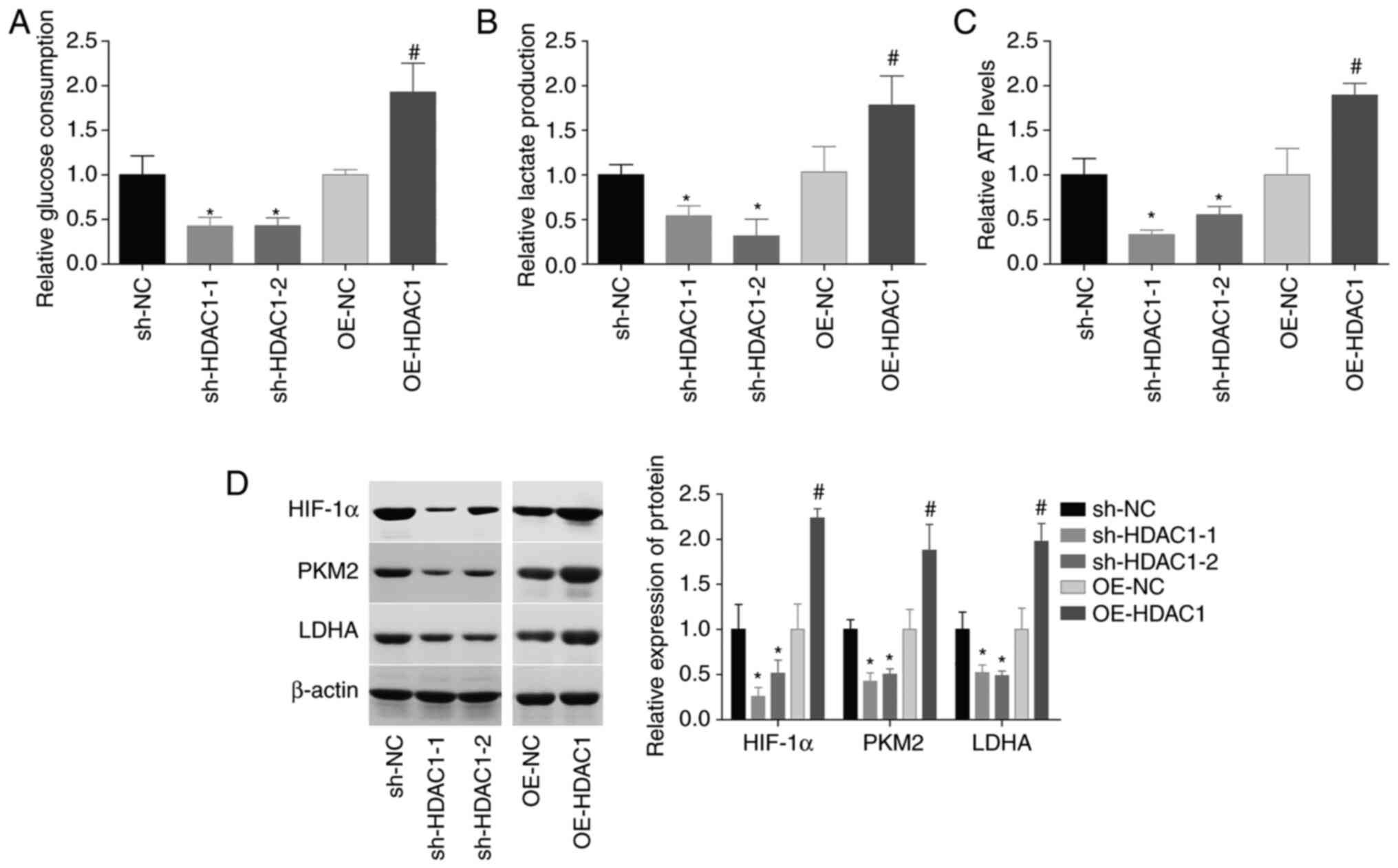 | Figure 4.HDAC1 induces aerobic glycolysis in
endometrial cancer cells. (A) Glucose consumption, (B) lactate
secretion and (C) ATP levels were assessed in HEC-1-A cells
infected with OE-NC, OE-HDAC1, sh-NC or sh-HDAC1-1/-2. (D)
Expression levels of HIF-1α, PKM2 and LDHA were measured by western
blotting. N=3, ANOVA followed by Tukey test; *P<0.05, sh-HDAC1
group vs. sh-NC group; #P<0.05, OE-HDAC1 group vs.
OE-NC group). HDAC1, histone deacetylase 1; sh, short hairpin; OE,
overexpression; HIF-1α, hypoxia-inducible factor 1; PKM2, pyruvate
kinase PKM; LDHA, L-lactate dehydrogenase A chain. |
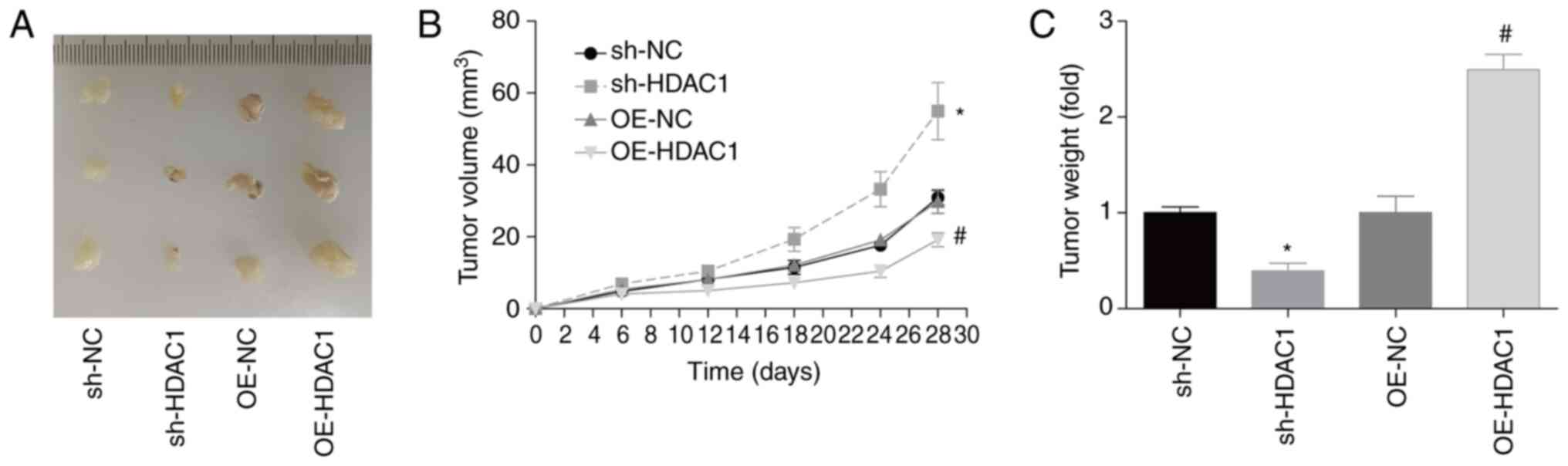
HDAC1 promotes endometrial cancer
growth in vivo
In addition, the role of HDAC1 in endometrial cancer
growth in vivo was investigated. Compared with the control
group, tumor volume and weight 28 days post-cell injection were
increased by 90 and 120% when HDAC1 was overexpressed in HEC-1-A
cells, and decreased by 40 and 60% when HDAC1 was downregulated,
respectively (Fig. 5A-C). This assay
confirmed that HDAC1 promoted endometrial cancer growth in
vivo.
High expression of HDAC1 is closely
associated with the advanced clinicopathological features of
patients with endometrial cancer
The clinical value of HDAC1 in endometrial cancer
was then evaluated. As shown in Table
I, it was observed that high HDAC1 expression level was
significantly associated with poor differentiation (P=0.005),
advanced FIGO stage (P=0.019), higher incidence rate of lymphatic
metastasis (P=0.009) and deeper muscular wall invasion depth
(P=0.043), indicating the potential of HDAC1 as a diagnosed marker
for endometrial cancer.
 | Table I.Evaluation of the clinical value of
histone deacetylase 1 in endometrial cancer. |
Table I.
Evaluation of the clinical value of
histone deacetylase 1 in endometrial cancer.
| Index | High expression
(n=37) | Low expression
(n=27) | P-value |
|---|
| Age, years | 35-75 | 34-76 | 0.449 |
|
≤60 | 22 | 13 |
|
|
>60 | 15 | 14 |
|
| Degree of
differentiation |
|
| 0.005 |
|
Well | 14 | 20 |
|
|
Moderate-poor | 23 | 7 |
|
| FIGO stage |
|
| 0.019 |
|
I–II | 17 | 21 |
|
|
III–IV | 20 | 6 |
|
| Lymphatic
metastasis |
|
| 0.009 |
|
Negative | 18 | 22 |
|
|
Positive | 19 | 5 |
|
| Muscular wall
invasion depth |
|
| 0.043 |
|
<1/2 | 16 | 19 |
|
|
≥1/2 | 21 | 8 |
|
Discussion
Through the regulation of a wider spectrum of
substrate proteins, HDACs have been identified to be closely
involved in a range of cellular processes, such as viability,
survival, apoptosis and differentiation (28,29), and
have been identified as potential targets for the treatment of
multiple cancer types, (30,31), including endometrial cancer (32). In the present study, the role HDAC1
in endometrial cancer progression was investigated. Consistent with
the work of Weichert et al (22), HDAC1 expression was found to be
significantly elevated in endometrial cancer tissues and cells,
compared with adjacent-normal tissues and control cells. In
addition, the high expression level of HDAC1 was closely linked to
a malignant clinical phenotype in patients with endometrial cancer.
Similarly, HDAC1 expression was closely associated with the
differentiation grade of lung cancer (19). Also, the expression level of HDAC1 in
gastrointestinal malignancies, especially in colorectal cancer, was
higher than that in noncancerous tissues, which closely was
associated with advanced tumor stage and poor prognosis (21).
To date, the role of HDAC1 has been revealed in
various types of cancer (33). For
instance, HDAC1 was upregulated in breast cancer, and HDAC1
overexpression induced cellular proliferation and migration, while
HDAC1 silencing inhibited proliferation (17,18). Liu
et al (20) reported that
HDAC1-knockdown suppressed cellular proliferation, and increased
apoptosis and chemosensitivity in cisplatin-resistant ovarian
cancer A2780/CDDP cells. Downregulation of HDAC1 also induced
apoptosis in advanced thyroid cancer cells (34), as well as inhibiting invasiveness and
inducing apoptosis in non-small cell lung cancer cells (35). These aforementioned studies have
demonstrated that HDAC1 serves as an oncogene in multiple cancer
types. Similarly, in the current study, the role of HDAC1 in
modulating cellular function was investigated in an endometrial
cancer setting. Consistent with other cancer types, the results
showed that the upregulation of HDAC1 significantly promoted
proliferation, tumorigenesis, migration and invasiveness abilities,
and repressed apoptosis in HEC-1-A cells, indicating that HDAC1
exerts an oncogenic role in endometrial cancer.
EMT is a key process in cancer metastasis (26,27), and
studies have shown that HDAC inhibitors play different roles in EMT
in various cancers. HDAC inhibitors induced EMT in colon carcinoma
(36,37), but have also been shown to revert EMT
in non-small cell lung (38),
biliary tract (39), prostate
(40) and head and neck cancers
(41). These findings suggest that
the regulatory role of HDACs during EMT is associated with the
cancer type. In the present study, the role of HDAC1 in endometrial
cancer cell EMT was investigated. The results demonstrated that the
expression of N-cadherin was significantly elevated, while that of
E-cadherin was decreased when HEC-1-A cells were infected with
OE-HDAC1; sh-HDAC1 caused an opposing effect, indicating that HDAC1
accelerated EMT, which then contributed to cancer cell
migration.
To further elucidate the mechanisms underlying the
effects of HDAC1 in endometrial cancer, its effect on glycolysis
[through which cancer cells obtain additional energy to maintain
rapid growth and migration (8,42)] were
assessed. The results showed that HDAC1 overexpression
significantly increased the glucose consumption, lactate secretion
and ATP level of HEC-1-A cells, suggesting that HDAC1 induces
aerobic glycolysis in endometrial cancer. Aerobic glycolysis may be
the mechanism by which HDAC1 accelerates endometrial cancer
progression. Chen et al (43)
reported that HDAC1 increased the expression of HIF-1α in
colorectal cancer. HIF-1α is a well-known master regulator of
glycolysis and can enhance the expression of glycolytic genes
(25). Consistently, the effects of
HDAC1 on HIF-1α expression were assessed in the present study, and
revealed that HDAC1 overexpression promoted HIF-1α expression,
indicating that HDAC1 potentially facilitated aerobic glycolysis in
endometrial cancer through HIF-1α. To this end, rescue experiments
should be carried out in the future.
In conclusion, to the best of our knowledge, the
present study was the first to reveal that HDAC1 facilitates
aerobic glycolysis and growth in endometrial cancer, which may
provide a potential target for endometrial cancer treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Fund Project
of the Changning District Science and Technology Commission (grant.
no. CNKW2017Y16).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CM conceived the project and revised the manuscript.
QW, WZ and YL conducted the experiments. QW wrote the paper. QW, YH
and HW analyzed the data. CM and QW confirmed the authenticity of
all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethical
committee of Shanghai Changning Maternity and Infant Health
Hospital (approval. no. CNFBLLKT-2017-011). Written consent was
obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morice P, Leary A, Creutzberg C,
Abu-Rustum N and Darai E: Endometrial cancer. Lancet.
387:1094–1108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ma J, Li D, Kong FF, Yang D, Yang H and Ma
XX: miR-302a-5p/367-3p-HMGA2 axis regulates malignant processes
during endometrial cancer development. J Exp Clin Cancer Res.
37:192018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen G, Zhang Y, Liang J, Li W, Zhu Y,
Zhang M, Wang C and Hou J: Deregulation of hexokinase II is
associated with glycolysis, autophagy, and the
epithelial-mesenchymal transition in tongue squamous cell carcinoma
under hypoxia. Biomed Res Int. 2018:84807622018.PubMed/NCBI
|
|
5
|
Chen G, Liu H, Zhang Y, Liang J, Zhu Y,
Zhang M, Yu D, Wang C and Hou J: Silencing PFKP inhibits
starvation-induced autophagy, glycolysis, and epithelial
mesenchymal transition in oral squamous cell carcinoma. Exp Cell
Res. 370:46–57. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee M and Yoon JH: Metabolic interplay
between glycolysis and mitochondrial oxidation: The reverse Warburg
effect and its therapeutic implication. World J Biol Chem.
6:148–161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han J, Zhang L, Guo H, Wysham WZ, Roque
DR, Willson AK, Sheng X, Zhou C and Bae-Jump VL: Glucose promotes
cell proliferation, glucose uptake and invasion in endometrial
cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol.
138:668–675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han X, Ren C, Yang T, Qiao P, Wang L,
Jiang A, Meng Y, Liu Z, Du Y and Yu Z: Negative regulation of
AMPKalpha1 by PIM2 promotes aerobic glycolysis and tumorigenesis in
endometrial cancer. Oncogene. 38:6537–6549. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ganapathy-Kanniappan S: Molecular
intricacies of aerobic glycolysis in cancer: Current insights into
the classic metabolic phenotype. Crit Rev Biochemi Mol Biol.
53:667–682. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mackay HJ, Eisenhauer EA, Kamel-Reid S,
Tsao M, Clarke B, Karakasis K, Werner HM, Trovik J, Akslen LA,
Salvesen HB, et al: Molecular determinants of outcome with
mammalian target of rapamycin inhibition in endometrial cancer.
Cancer. 120:603–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gehrig PA, Le LV, Olatidoye B and Geradts
J: Estrogen receptor status, determined by immunohistochemistry, as
a predictor of the recurrence of stage I endometrial carcinoma.
Cancer. 86:2083–2089. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Myers AP: New strategies in endometrial
cancer: Targeting the PI3K/mTOR pathway-the devil is in the
details. Clin Cancer Res. 19:5264–5274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li LH, Zhang PR, Cai PY and Li ZC: Histone
deacetylase inhibitor, romidepsin (FK228) inhibits endometrial
cancer cell growth through augmentation of p53-p21 pathway. Biomed
Pharmacother. 82:161–166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De U, Son JY, Sachan R, Park YJ, Kang D,
Yoon K, Lee BM, Kim IS, Moon HR and Kim HS: A new synthetic histone
deacetylase inhibitor, MHY2256, induces apoptosis and autophagy
cell death in endometrial cancer cells via p53 acetylation. Int J
Mol Sci. 19:27432018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zheng Y and Yang X, Wang C, Zhang S, Wang
Z, Li M, Wang Y, Wang X and Yang X: HDAC6, modulated by miR-206,
promotes endometrial cancer progression through the PTEN/AKT/mTOR
pathway. Sci Rep. 10:35762020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weichert W: HDAC expression and clinical
prognosis in human malignancies. Cancer Lett. 280:168–176. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Senese S, Zaragoza K, Minardi S, Muradore
I, Ronzoni S, Passafaro A, Bernard L, Draetta GF, Alcalay M, Seiser
C and Chiocca S: Role for histone deacetylase 1 in human tumor cell
proliferation. Mol Cell Biol. 27:4784–4795. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang Z, Ding S, Huang H, Luo P, Qing B,
Zhang S and Tang R: HDAC1 triggers the proliferation and migration
of breast cancer cells via upregulation of interleukin-8. Biol
Chem. 398:1347–1356. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao LL, Song X, Pei L, Liu L, Wang H and
Jia M: Histone deacetylase HDAC1 expression correlates with the
progression and prognosis of lung cancer: A meta-analysis. Medicine
(Baltimore). 96:e76632017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Yu Y, Zhang J, Lu C, Wang L, Liu P
and Song H: HDAC1 silencing in ovarian cancer enhances the
chemotherapy response. Cell Physiol Biochem. 48:1505–1518. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao LL, Yue Z, Liu L, Pei L, Yin Y, Qin L,
Zhao J, Liu H, Wang H and Jia M: The expression of histone
deacetylase HDAC1 correlates with the progression and prognosis of
gastrointestinal malignancy. Oncotarget. 8:39241–39253. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weichert W, Roske A, Niesporek S, Noske A,
Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T and Denkert
C: Class I histone deacetylase expression has independent
prognostic impact in human colorectal cancer: Specific role of
class I histone deacetylases in vitro and in vivo. Clin Cancer Res.
14:1669–1677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prat J: FIGO staging for uterine sarcomas.
Int J Gynaecol Obstet. 104:177–178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li L, Liang Y, Kang L, Liu Y, Gao S, Chen
S, Li Y, You W, Dong Q, Hong T, et al: Transcriptional regulation
of the Warburg effect in cancer by SIX1. Cancer Cell. 33:368–385.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu F, Gu LN, Shan BE, Geng CZ and Sang
MX: Biomarkers for EMT and MET in breast cancer: An update. Oncol
Lett. 12:4869–4876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nantajit D, Lin D and Li JJ: The network
of epithelial-mesenchymal transition: Potential new targets for
tumor resistance. J Cancer Res Clin Oncol. 141:1697–1713. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang XJ and Seto E: HATs and HDACs: From
structure, function and regulation to novel strategies for therapy
and prevention. Oncogene. 26:5310–5318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Villagra A, Sotomayor EM and Seto E:
Histone deacetylases and the immunological network: Implications in
cancer and inflammation. Oncogene. 29:157–173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Glozak MA and Seto E: Histone deacetylases
and cancer. Oncogene. 26:5420–5432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stazi G, Fioravanti R, Mai A, Mattevi A
and Valente S: Histone deacetylases as an epigenetic pillar for the
development of hybrid inhibitors in cancer. Curr Opin Chem Biol.
50:89–100. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garmpis N, Damaskos C, Garmpi A, Spartalis
E, Kalampokas E, Kalampokas T, Margonis GA, Schizas D, Andreatos N,
Angelou A, et al: Targeting histone deacetylases in endometrial
cancer: A paradigm-shifting therapeutic strategy? Eur Rev Med
Pharmacol Sci. 22:950–960. 2018.PubMed/NCBI
|
|
33
|
Khabele D, Son DS, Parl AK, Goldberg GL,
Augenlicht LH, Mariadason JM and Rice VM: Drug-induced inactivation
or gene silencing of class I histone deacetylases suppresses
ovarian cancer cell growth: Implications for therapy. Cancer Biol
Ther. 6:795–801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin CL, Tsai ML, Lin CY, Hsu KW, Hsieh WS,
Chi WM, Huang LC and Lee CH: HDAC1 and HDAC2 double knockout
triggers cell apoptosis in advanced thyroid cancer. Int J Mol Sci.
20:4542019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang L, Bu L, Hu J, Xu Z, Ruan L, Fang Y
and Wang P: HDAC1 knockdown inhibits invasion and induces apoptosis
in non-small cell lung cancer cells. Biol Chem. 399:603–610. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji M, Lee EJ, Kim KB, Kim Y, Sung R, Lee
SJ, Kim DS and Park SM: HDAC inhibitors induce
epithelial-mesenchymal transition in colon carcinoma cells. Oncol
Rep. 33:2299–2308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Feng J, Cen J, Li J, Zhao R, Zhu C, Wang
Z, Xie J and Tang W: Histone deacetylase inhibitor valproic acid
(VPA) promotes the epithelial mesenchymal transition of colorectal
cancer cells via up regulation of snail. Cell Adh Migr. 9:495–501.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jakobsen KR, Demuth C, Sorensen BS and
Nielsen AL: The role of epithelial to mesenchymal transition in
resistance to epidermal growth factor receptor tyrosine kinase
inhibitors in non-small cell lung cancer. Transl Lung Cancer Res.
5:172–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sakamoto T, Kobayashi S, Yamada D, Nagano
H, Tomokuni A, Tomimaru Y, Noda T, Gotoh K, Asaoka T, Wada H, et
al: A histone deacetylase inhibitor suppresses
epithelial-mesenchymal transition and attenuates chemoresistance in
biliary tract cancer. PLoS One. 11:e01459852016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ruscetti M, Dadashian EL, Guo W, Quach B,
Mulholland DJ, Park JW, Tran LM, Kobayashi N, Bianchi-Frias D, Xing
Y, et al: HDAC inhibition impedes epithelial-mesenchymal plasticity
and suppresses metastatic, castration-resistant prostate cancer.
Oncogene. 35:3781–3795. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bruzzese F, Leone A, Rocco M, Carbone C,
Piro G, Caraglia M, Gennaro ED and Budillon A: HDAC inhibitor
vorinostat enhances the antitumor effect of gefitinib in squamous
cell carcinoma of head and neck by modulating ErbB receptor
expression and reverting EMT. J Cell Physiol. 226:2378–2390. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lunt SY and Heiden MG: Aerobic glycolysis:
Meeting the metabolic requirements of cell proliferation. Ann Rev
Cell Dev Biol. 27:441–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen C, Wei M, Wang C, Sun D, Liu P, Zhong
X, He Q and Yu W: The histone deacetylase HDAC1 activates
HIF1α/VEGFA signal pathway in colorectal cancer. Gene.
754:1448512020. View Article : Google Scholar : PubMed/NCBI
|