Introduction
Progress from G0 to G1 and
through the G1 phase of the mammalian cell cycle is
mediated by the cyclin-dependent kinases 4 and 6 (CDK4, CDK6),
which are activated through binding with their regulatory subunits
D-type cyclins (D1, D2, and D3) (1–4). It is
widely accepted that CDK4 regulates critical aspects of the cell
cycle via phosphorylation of the retinoblastoma (Rb) family of
proteins (5,6). Thus, the so-called CDK4/6-Rb axis is
considered essential to cell-cycle entry and progression. Although
this canonical role of CDK4 as a driver of cell proliferation has
been firmly established, research carried out over the last few
years has suggested cell cycle-independent functions of CDKs and
D-type cyclins (7,8). For example, a novel role for CDK6 in
hematopoietic cells that exceeds its function as a cell-cycle
regulator has been recognized (9).
Increasing evidence suggests that cyclin D1 carries out essential
functions in other processes such as transcription and DNA damage
(7,10,11).
Interestingly, a systematic screen has defined other
potential substrates for CDK4, including the transcription factor
forkhead box protein M1 (FOXM1) (12). Our skin carcinogenesis studies also
suggested that CDK4 plays additional roles unrelated to its
canonical function in the CDK-Rb axis. We reported that transgenic
expression of CDK4 in mouse epidermis favors the malignant
progression of skin tumors (13).
However, forced expression of the other G1-CDKs, such as
CDK6 and CDK2, in mouse keratinocytes resulted in elevated Rb
phosphorylation but did not induce malignant progression such as
observed in CDK4 transgenic mice (14,15).
Moreover, a putative role of CDK4 in chromosome instability, and
consequently in malignant progression, was reported by Adon et
al, in which the absence of CDK4 or CDK2 prevents centrosome
amplification (16).
Herein we report a novel function of CDK4 regulating
the transcriptional expression of genes involved in chromosome
segregation. Chromatin-immunoprecipitation (ChIP) analysis shows
that CDK4 occupies the promoter of genes associated with
chromosomal segregation, such as Aurkb (Aurora-B) and
CENP-P (Centromere Protein P). Gain- and loss-of-function
experiments showed that CDK4 participates in the transcriptional
regulation of AurkB and CENP-P promoters.
Importantly, Aurora-B is a subunit of the chromosome passenger
complex controlling several aspects of chromosome segregation
(17). Thus, deregulation of
Aurora-B through CDK4 expression would result in a malfunction of
the chromosome segregation events and potentially tumorigenesis
(18). Our results suggest that CDK4
may contribute to G2/M regulation in addition to the
prominent role in G0/G1- and
G1/S-transitions. Aurora-B expression peaks during
mitosis have a crucial role in chromosome bi-orientation and the
spindle-assembly checkpoint, whereas CENP-P is required for proper
kinetochore function (19,20), suggests that CDK4 plays a pivotal
role in maintaining chromosomal stability.
Materials and methods
Cell lines and primary mouse
keratinocytes
The 308-cell line was acquired through a previous
research collaboration with Dr. Claudio Conti (MD Anderson Cancer
Center, Texas). This immortalized cell line was derived from
calcium-resistant foci of keratinocytes from adult Balb/c mouse
initiated by 7,12-dimethylbenz[a]anthracene and has been
extensively used as a model of cell proliferative (21–23).
NIH3T3 murine embryo fibroblasts cell line was obtained from the
American Type Culture Collection (Catalog number CRL-1658; ATCC).
Primary keratinocytes were isolated from newborn mice and cultured
in a low Ca2+ medium (EMEM, 06-174 G; Cambrex-bioz) as
described previously (24). Briefly,
four newborns of 48 h of age were washed with ethanol and iodine
solution and put in the refrigerator (4°C) for 30 min to induce
hypothermic anesthesia. After anesthesia by refrigeration, the
newborns were euthanized by decapitation, and skin was removed with
forceps, rinsed, and continue with the cell culture process
(24,25). The generation of mouse primary
keratinocytes and protocols for animal use were approved for the
North Carolina State University Institutional Animal Care and Use
Committee (IACUC) protocol number 18-102-B, as required by federal
regulations.
Cell extraction and immunofluorescence
analysis
To visualize chromatin-bound proteins, unbound
nuclear and cytosolic proteins from keratinocyte cell line 308 and
primary culture of mouse keratinocytes were removed with
cytoskeletal extraction buffer (CSK buffer). 70–80% confluent 308
cells were grown on coverslips coated with poly-L-lysine
(Sigma-Aldrich; Merck KGaA). For CSK extraction, cells were washed
with DPBS (Mediatech Inc.) twice and incubated with CSK buffer [100
mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES (pH.8),
and 0.5% Triton X-100] containing 1 × protease inhibitor cocktail
(Sigma-Aldrich; Merck KGaA) for 2 min on ice. After extraction,
cells were fixed in 10% formalin for 20 min at RT. For DNase
extraction, cells permeabilized with CSK buffer were washed with
DPBS twice and incubated with 10 units of Optizyme Recombinant
DNase I (Thermo Fisher Scientific Inc.) 30 min at 37°C followed by
fixation with 10% formalin.
Immunofluorescence staining cells were blocked with
10% goat serum diluted in 0.01% Triton X-100/PBS solution for 30
min at RT, incubated with primary antibodies against CDK4 (C-22),
and HDAC1 (10E2) (Santa Cruz Biotechnology, Inc.) at 4°C overnight.
Cells were washed with PBS four times and incubated with goat
anti-rabbit-FITC conjugated antibody for CDK4 staining (Thermo
Fisher Scientific Inc.; Pierce Biotechnology Inc.), and goat
anti-mouse-Alexa Fluor 488 (Thermo Fisher Scientific Inc.;
Molecular Probes) for HDAC1 staining. DAPI staining was utilized
for counterstained (Vector Laboratories Inc.). Cells were examined
under a Nikon Eclipse E400 fluorescence microscope (Nikon
Corporation), and images were collected with Qcapture software
(QImaging).
Retroviral infection and generation of
stable cell lines
Murine CDK4 cDNA was subcloned into the pLPCX
retroviral vector (Clontech Laboratories, Inc.) using primers
containing NotI restriction sites. pLPCX-Cdk4 and pLPCX
(empty plasmid) vectors were amplified in DH5α competent E.
coli cells (Invitrogen; Thermo Fisher Scientific Inc.). The
retroviral vector was transfected into the Platinum Retroviral
Packaging Cell line (Plat-E cell) with psPAX2 packing vector
(Addgene) using FuGENE® 6 Transfection Reagent (Promega
Corp.). After 48 h of transfection, the virus-containing medium was
filtered through a 0.45 µm of syringe filter (Corning Inc.).
Harvested CDK4-retroviruses and control-retrovirus were utilized to
infected NIH3T3 and 308 cells with 4 µg/ml of hexadimethrine
bromide (polybrene; Sigma-Aldrich;) and incubated overnight. The
pLPCX-Cdk4 and pLPCX-empty retrovirus infected cells (NIH3T3-CDK4
and 308-CDK4) were selected with 2 µg/ml of puromycin
(Sigma-Aldrich Co. LLC, MO).
Western blot assays
NIH3T3 and 308 cell lines were lysed in RIPA buffer
[150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM
Tris-HCl (pH 8.0)] containing 1× protease inhibitor cocktail
(Sigma-Aldrich; Merck KGaA). Protein concentration was determined
with the DC™ Protein Assay system (Bio-Rad Laboratories), 50 µg of
protein were loaded on 10% SDS-PAGE gel and electrophoretically
transferred onto nitrocellulose membranes. After being blocked with
5% nonfat powdered milk in Dulbecco phosphate-buffered saline, the
membranes were incubated with 1 µg/ml of specific antibodies. The
following antibodies were used: Polyclonal antibodies against CDK4
(C-22), β-actin (I-19) (Santa Cruz Biotechnology, Inc.), CENP-P
(PA5-31186), and Aurora-B (MA5-17226) (Thermo Fisher Scientific
Inc.; Pierce Biotechnology Inc.). Membranes were washed and
incubated with goat anti-rabbit-HRP (sc-2004), donkey
anti-goat-HRP, or goat anti-mouse-HRP secondary antibodies followed
by enhanced chemiluminescence (ECL detection kit; GE Healthcare).
Western blot bands were quantified using UN-SCAN-IT gel version 6.1
software. The experimental data are representative of three
independent repetitions.
Chromatin immunoprecipitation (ChIP)
assay
We utilized the SimpleChIP® Enzymatic
chromatin IP kit (Cell Signaling Technology, Inc., MA) following
the manufacturer's instructions. Briefly, 4×107 NIH3T3
semi-confluent cells were used for ChIP assay. Cell culture media
was replaced with 10 ml of fresh media containing 1% formaldehyde
to crosslink proteins/DNA and incubated for 10 min at RT. The
reaction was stopped by the addition of glycine, a 0.125 M
concentration in cell culture media. Cells were washed with PBS,
scraped, collected into conical tubes, and centrifuged at 1,500
rpm. Chromatin was released by adding lysis buffer containing DTT,
protease inhibitors, and PMSF and further fragmented by partial
digestion with micrococcal nuclease to obtain chromatin fragments
of 1 to 5 nucleosomes in size. Nuclei were pelleted by
centrifugation at 13,000 rpm and resuspended in 1X ChIP buffer
containing SDS, protease inhibitor, and PMSF. Pellets were
sonicated (3 sets of 20-second pulses at setting 2 on a Branson
sonifer 450) and clarified by centrifugation at 10,000 rpm for 10
min. The supernatant was kept at −80°C. 50 µl of chromatin sample
were digested with 2 µl RNase A at 37°C for 30 min and 2 µl
Proteinase K at 65°C for 2 h. DNAs were purified using spin
columns, and 10 µl was used to electrophoresis on 1% agarose gel to
check DNA fragment sizes. DNA concentration was determined with a
NanoDrop 1000 (Thermo Fisher Scientific, Inc.). 10 µg chromatin was
diluted in 500 µl of 1X ChIP buffer containing protease inhibitor
cocktail and used for each immunoprecipitation. 2 µg of rabbit
antibodies against CDK4 (C-22) (Santa Cruz Biotechnology, Inc.),
normal IgG (New England Biolabs) or Histone H3 (D2B12) (New England
Biolabs) were added to DNA samples and incubated overnight at 4°C.
Thirty microliters of ChIP-grade protein G agarose beads (Cell
Signaling Technology, Inc.) were added to each sample and incubated
for 2 h at 4°C, followed by centrifugation 6,000 rpm for 1 min. The
supernatant was transferred to a new tube and processed for
immunoprecipitation 3 times. The agarose pellet was washed with
ChIP buffer three times and ChIP buffer with high salt (350 mM
NaCl). Chromatin was eluted from antibody/protein G bead through
incubations at 65°C, 30 min with gentle vortex, separated by
centrifugation at 6,000 rpm, and transferred to a new tube. RNA and
proteins in Eluted DNA were removed through RNase A and proteinase
K treatment, and DNA was purified through the spin column, as
mentioned above.
Standard PCR was performed using 50 ng DNAs with
specific primers for CENP-P, Aurkb, Ckap2, Zw10, Top2a, and Mlf1ip
genes as previously reported (7)
with KAPA2G Fast PCR kit (KAPA Biosystems). PCR was executed using
34 cycles of amplification: Denaturation at 95°C for 30 sec,
annealing at 50°C (CENP-P, Aurkb), 56.8°C (Mlf1ip) and 62°C (Zw10,
Top2a) for 30 sec, extension at 72°C for 30 sec, and a final
extension at 72°C for 5 min. PCR primers: CENP-P
(CATGGAGATCCGCAGTACC; CATCCCTTCCTCATCGATTT) Aurkb
(CCCAGAGAGTCCTACGGAAG; TGTTCTCAGCCAACTTCTGG) Ckap2
(ATTAAGCGATGGCAGAGTCC; TTTCTTTGTTCTCGGAAGGC) Zw10
(GAAGTGCCAGGATGTGATTG; AGCTTGTGATCAGCATCAGG) Top2A
(ATCACCGACTCGCTCTCATT; GCACATGGACCTTCCTCATT).
Quantitative PCR (qRT-PCR)
Synthesis of cDNA with total RNA was performed using
an iScript cDNA synthesis kit (Bio-Rad Laboratories). Two
micrograms of total RNA and reverse transcriptase H was incubated
in reaction buffer 5 min at 25°C, 30 min at 42°C, and 5 min at
85°C. The iQTM SYBR®-Green Supermix (Bio-Rad
Laboratories) was used for quantitative real-time PCR. The same
primers used for ChIP assays were utilized for this analysis, and
mouse GAPDH as the reference gene. PCR amplification was performed
with a 20 µl reaction mixture containing 2 µl of cDNA, 300 nM of
each primer, and 1× iQTM SYBR®-Green supermix (Bio-Rad
Laboratories). PCR condition as followed: Initial denaturation at
95°C for 3 min, followed by 40 cycles of denaturation at 95°C for
15 sec, annealing, and extension at 60°C for 30 sec. The
transcriptional level of target genes was normalized by the
transcriptional level of GAPDH (CATCACTGCCACCCAGAAGACTG;
ATGCCAGTGAGCTTCCCGTTCAG). For downregulation assays, the
transcriptional levels were compared with control siRNA-treated 308
cells or cell lines, not overexpressing CDK4 (NIH3T3 or 308)
according to the algorithms 2-(ΔΔCt), respectively.
siRNA assay
Cell lines were transfected with CDK4 or control
siRNAs with Lipofectamine® RNAiMAX Reagent (Invitrogen,
Life Technologies) according to the manufacturer's instructions.
308 or 308-CDK4 cells were cultured in 60 mm Petri dishes at 70–80%
of confluence. We utilized commercially available CDK4 specific
siRNA (sc-29262) and control siRNA (sc-37007) (Santa Cruz Biotech).
Briefly, 60 pmol of siRNA was diluted in 300 µl
Opti-MEM® (Invitrogen; Life Technologies), mixed with 18
µl lipofectamine in 300 µl Opti-MEM®, and incubated 5
min at RT. The mixture was added to the cells in 4 ml of culture
media without antibiotics. The media was replaced with fresh media
after 24 h, and cells were harvested 96 h after the transfection.
RNA was isolated using TRIzol® Reagent (Ambion, Life
Technologies).
Statistical analysis
An unpaired Student's t-test was performed using
GraphPad Prism 4 Software (GraphPad Software).
Results
CDK4 loaded onto the chromatin
fraction of mouse keratinocytes
Various studies accomplished in the last decade have
shown that cyclin D1 and CDK6 have additional functions non-related
to their role in the cyclin-cdk-Rb axis (9–11). Based
on these observations, we asked whether CDK4 has similar activities
in transcriptional regulation (26).
Thus, we first examined the CDK4 interaction with the chromatin
fraction of mouse keratinocytes. We performed in situ cell
extraction with the cytoskeleton extraction buffer (CSK), which
removes the soluble proteins from the cytoplasm and nucleoplasm
while leaving the chromatin-bound fraction intact (27–29). We
utilized asynchronous cell cultures of primary mouse keratinocytes
and the keratinocyte cell line 308, a cell line derived from BALB/c
mouse skin treated with 7,12-dimethylbenz[a]anthracene (21–23,30).
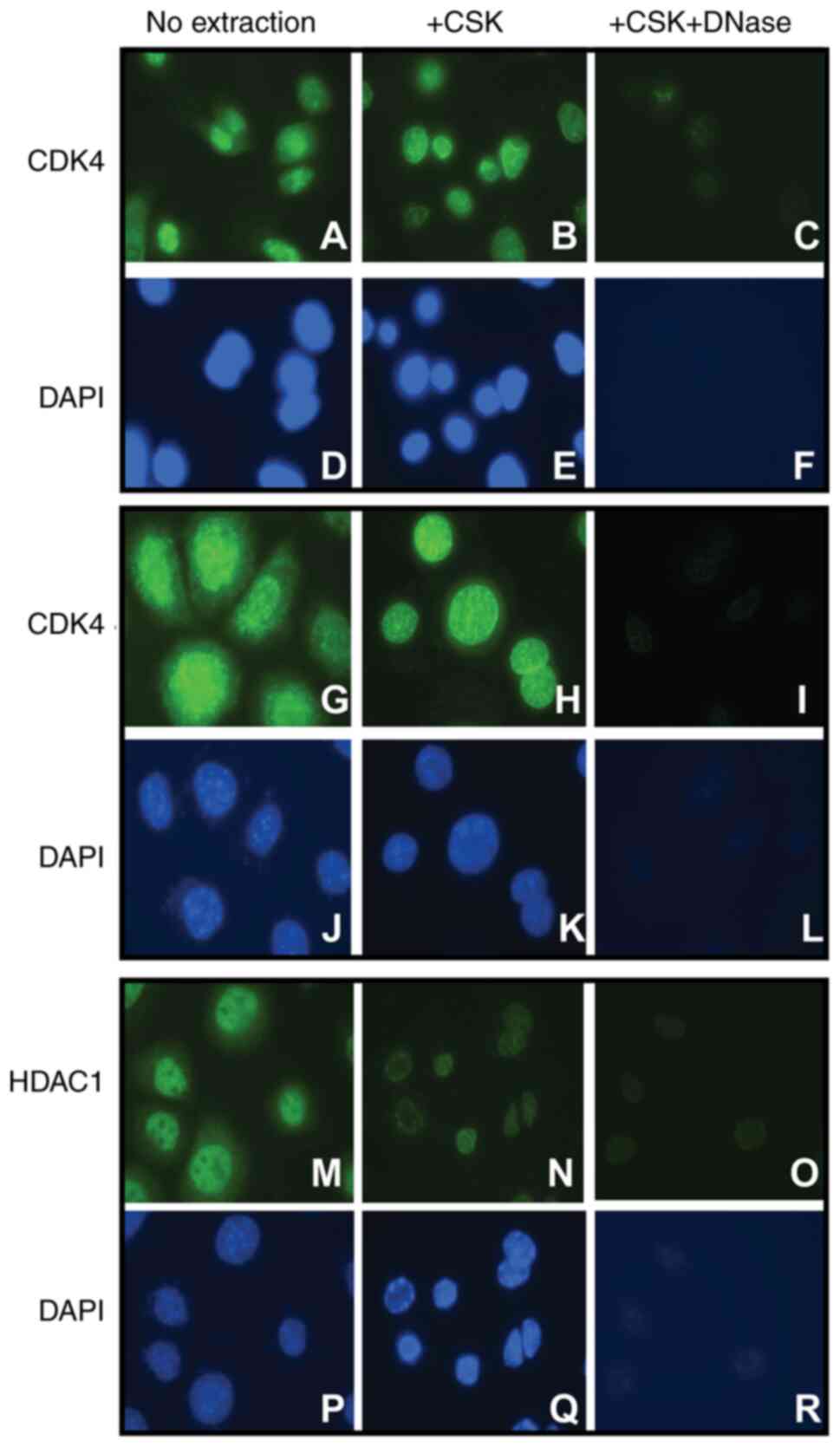
Immunofluorescence analysis of the extracted cells revealed the
presence of CDK4 in the chromatin-bound fraction of both mouse
primary keratinocytes and 308 keratinocyte cell lines (Fig. 1A-L). The CSK extraction buffer
removes the soluble cell fractions, but the DNA and other insoluble
material such as intermediate filament cytoskeleton remain in the
so-called cell ghosts. Therefore, to verifies that CDK4 binds to
DNA, we disrupt the cellular DNA and examine the presence of CDK4
and the positive control chromatin-bound histone deacetylase 1
(HDAC1). DNase treatment released the DNA-bound HDAC1 and CDK4 from
the chromatin fraction (Fig. 1M-R),
confirming that CDK4 is strongly associated with the DNA fraction
of mouse keratinocytes.
CDK4 as a transcriptional regulator on
the Aurkb and CENP-P promoters
In addition to their well-established role in the
cell cycle, cyclin D1 and CDK6 have transcriptional functions
(7,9,10).
Whereas cyclin D1 plays a direct role in transcriptional regulation
of genes governing chromosomal integrity (7), CDK6 is part of a complex that controls
the transcription of p16Ink4 and the angiogenic factor
VEGF-A (9). In view of the presence
of the DNA-bound CDK4, its structural and functional similitudes
with CDK6, and because cyclin D1 is one of the regulatory subunits
of CDK4, we analyzed whether CDK4 may also act as a transcriptional
regulator. We first examined a set of genes transcriptionally
controlled by cyclin D1, which were previously reported that
regulate chromosome segregation (7).
Chromatin immunoprecipitation (ChIP) experiments revealed that CDK4
binds specifically to the Aurkb, CENP-P, and Zw10 promoters, while
no binding to Ckap2, Top2a, and Mlf1ip was detected (Fig. 2A). These findings suggest that CDK4
may behave as a transcriptional regulator or form part of a
transcriptional complex that controls genes associated with
chromosomal segregation.
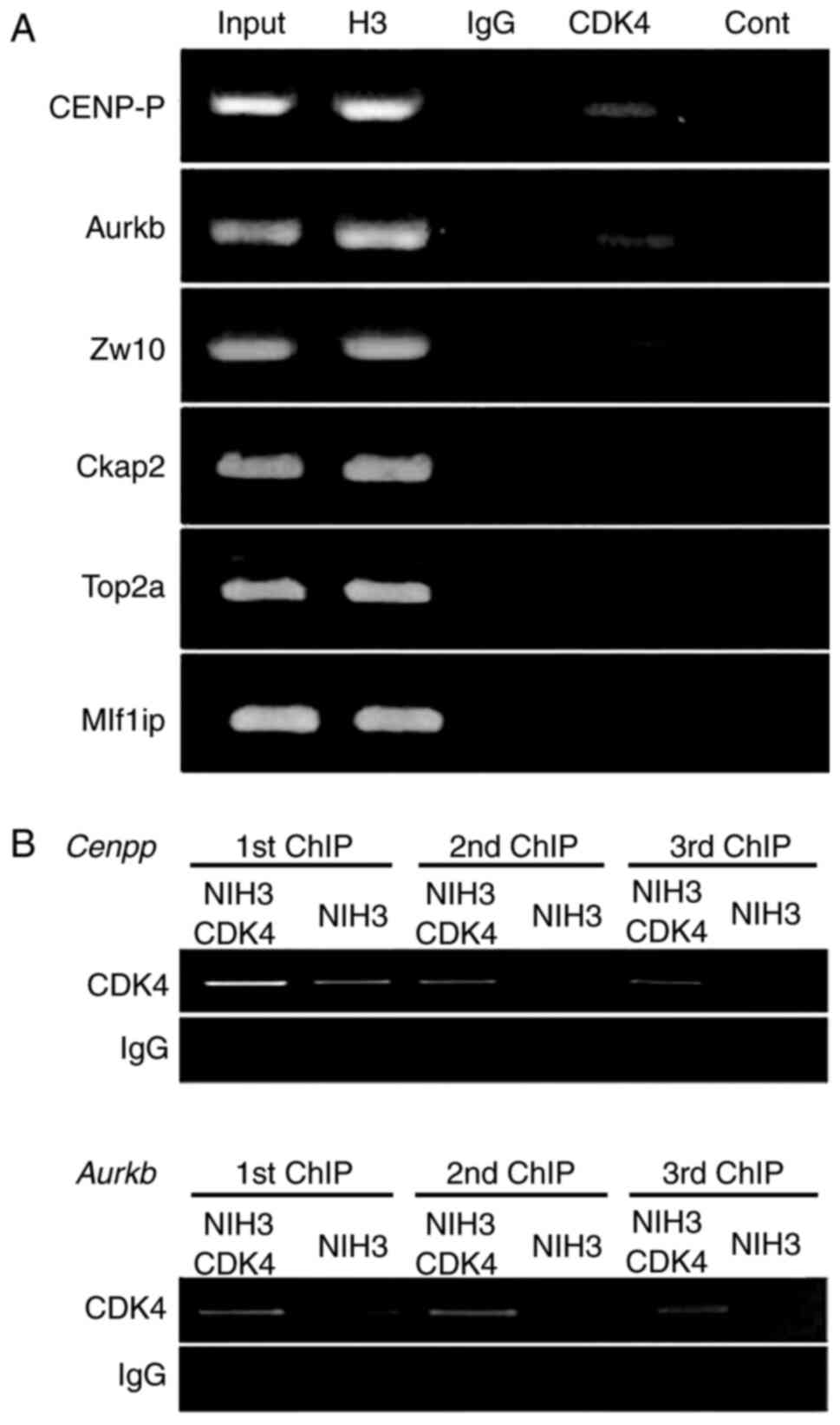 | Figure 2.CDK4 binds to regulatory regions of
genes associated with chromosomal segregation. (A) ChIP analysis of
NIH3T3 cells performed with an anti-CDK4 antibody (CDK4). Chromatin
input and ChIP with anti-H3 antibody were used as positive
controls, and normal IgG was used as a negative control. ‘Cont’
indicated the blank of the PCR reaction. The binding of CDK4 to
specific promoters was analyzed by standard PCR with primers that
amplified the regulatory sequences CENP-P, Aurkb, Ckap2, Zw10,
Top2a and Mlf1ip genes. (B) Increased binding to the regulatory
regions of CENP-P and Aurkb genes upon overexpression of CDK4 in
NIH3T3 cells (NIH3T3-CDK4). ChIP analysis of NIH3T3 and NIH3T3-CDK4
cells was performed in three sequential immunoprecipitations with
an anti-CDK4 antibody. H3, Histone 3; CDK4, cyclin-dependent kinase
4; ChIP, chromatin immunoprecipitation; CENP-P, Centromere Protein
P; Aurkb, Aurora-B; Ckap2, cytoskeleton-associated protein 2; Zw10,
centromere/kinetochore protein zw10 homolog; Top2a, DNA
topoisomerase 2-a; Mlf1ip, MLF1-interacting protein. |
To delineate whether variation in the CDK4 protein
level affects the interaction to the regulatory sites of these
genes, we performed ChIP analysis of NIH3T3 cells overexpressing
murine CDK4 (NIH3T3-CDK4). We carry out ChIP assay in three
sequential immunoprecipitations and quantify the association of
CDK4 with Aurkb and CENP-P promoters in NIH3T3-CDK4 cells and the
parental cell line NIH3T3. CDK4 overexpressing cells showed a 3-,
5- and 9-fold increase binding to the CENP-P promoter in the three
sequential immunoprecipitations, respectively, compared to NIH3T3
cells (Fig. 2B). Similarly,
NIH3T3-CDK4 cells showed 2- and 7-fold increase binding to Aurkb
promoter in the first and second immunoprecipitation, respectively,
compared to NIH3T3 cells (Fig. 2B).
ChIP analysis of the Zw10 promoter showed no differences between
CDK4 overexpressing and parental cell lines. These results support
the specificity of CDK4 binding to Aurkb and CENP-P promoters and
suggest that variation in the CDK4 level might affect the
transcription of Aurkb and CENP-P genes.
To examine whether CDK4 regulates the transcription
of these genes, we quantified the transcription of Aurkb B and
CENP-P genes upon overexpression and downregulation of CDK4. We
performed quantitative PCR (qRT-PCR) of CENP-P, Aurkb, Zw10, Ckap2,
and Top2a on NIH3T3 cells and the keratinocyte cell line 308
overexpressing CDK4 (NIH3T3-CDK4, 308-CDK4) and the parental cells
lines NIH3T3 and 308. The transcriptional levels were normalized to
the Gapdh, and a transcriptional ratio calculated between CDK4
overexpressing and the parental cell lines. NIH3T3-CDK4 cells
showed a 2-fold increase in transcription of Aurkb (P<0.005,
t-test) and 1.5-fold elevate transcription of CENP-P (P<0.05,
t-test) compared to NIH3T3 cells. Similarly, we observed
3-(P<0.05, t-test) and 2-fold (P<0.005, t-test) elevated
transcription of Aurkb and CENP-P, respectively, in the
keratinocyte cell line 308-CDK4 (Fig.
3A). The enhanced expression of CDK4 did not significantly
change the transcriptional levels of Ckap2 gene, although a 2- and
1.3-fold reduction of Zw10 (P<0.05, t-test) and Top2a
(P<0.0005, t-test) genes. Recently reports showed that Top2a and
Zw10 proteins are involved in chromosome segregation and mitotic
checkpoint proteins (31–35); therefore, their potential role
downstream of CDK4 expression in cell proliferation and tumor
development warrants further investigation. We also studied the
protein levels of Aurora-B and CENP-P in both NIH3T3-CDK4 and
308-CDK4 cell lines. Increased protein levels of both CENP-P and
Aurora-B were observed upon overexpression of CDK4 in the 308-cell
line (Fig. 3B). 308-CDK4 cells
showed a 20-fold increase of CENP-P (P<0.05, t-test) and a
4-fold increase of Aurora-B (P<0.05, t-test) compared to 308
cells. Expression of CENP-P protein was elevated 2-fold (P<0.05,
t-test) in NIH3T3-CDK4 cells, although the protein level of
Aurora-B showed a non-statistically significant change compared to
NIH3T3 cells (Fig. 3B). Altogether,
these results demonstrated that the CDK4 protein indeed regulates
the transcription of Aurkb and CENP-P.
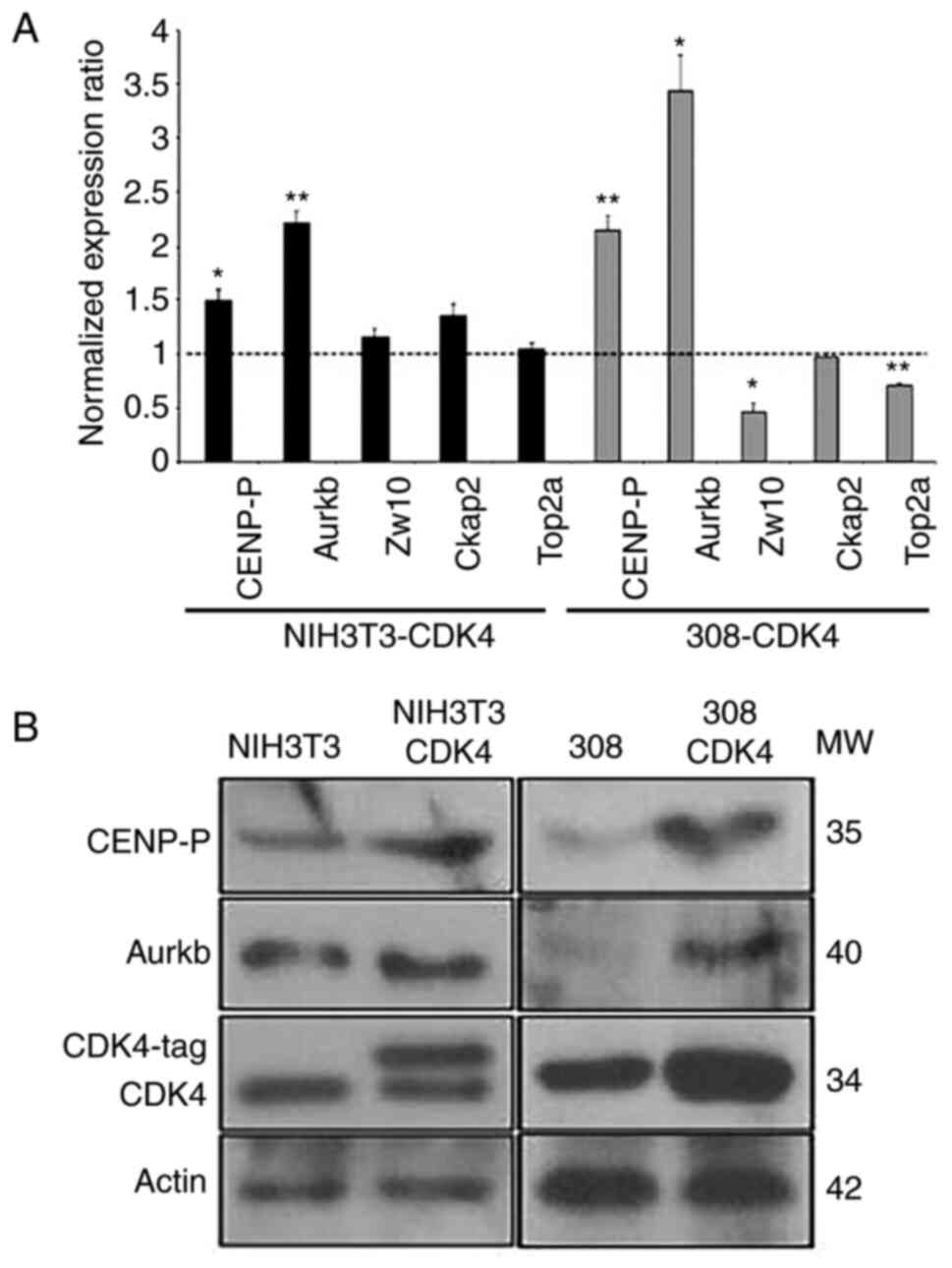 | Figure 3.Transcription and protein expression
levels of CENP-P and Aurkb are increased following overexpression
of CDK4. (A) Transcriptional levels of CENP-P, Aurkb, Zw10, Ckap2
and Top2a were determined by reverse transcription-quantitative PCR
analysis. Shown are normalized expression ratios of cells
overexpressing CDK4 (NIH3T3-CDK4 and 308-CDK4) compared with
parental cells (NIH3T3 and 308). Values >1 denote increased
transcriptional expression in CDK4 overexpressing cells, whereas
values <1 denote reduced or equal transcription levels compared
with parental cell lines. All the results were normalized with
Gapdh expression. n=3 independent experiments, data are presented
as the mean ± SEM. Student's t-test was performed. *P<0.05,
**P<0.005 vs. appropriate parental cell line. (B) Western blot
analysis of CENP-P, Aurkb and CDK4 in cells overexpressing CDK4
(NIH-CDK4, 308-CDK4) and the control cell lines infected with
control retrovirus (NIH3T3, 308). β-Actin was used as a loading
control. CDK4, cyclin-dependent kinase 4; CENP-P, Centromere
Protein P; Aurkb, Aurora-B; Ckap2, cytoskeleton-associated protein
2; Zw10, centromere/kinetochore protein zw10 homolog; Top2a, DNA
topoisomerase 2; MW, molecular weight. |
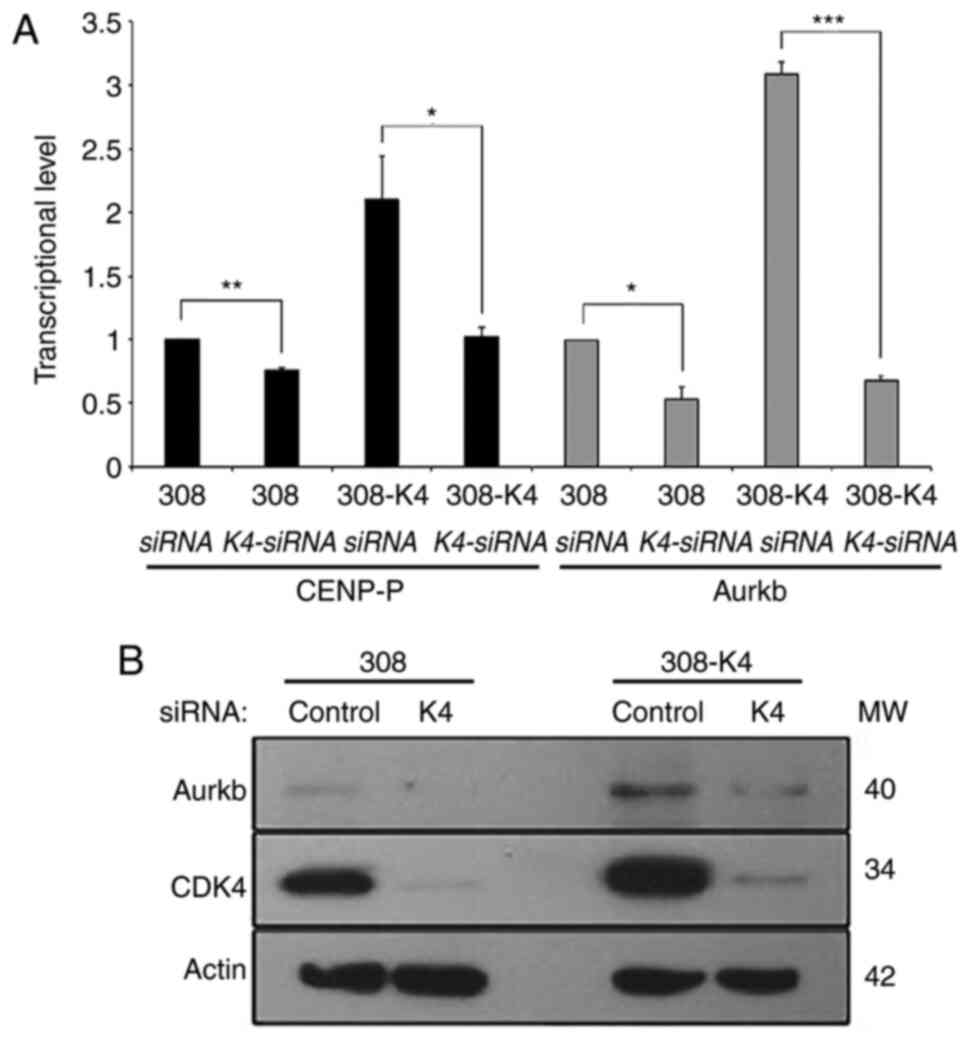
To validate our conclusions, we determined the
effect of the reduction level of CDK4 on Aurkb and CENP-P
transcription. We evaluated the inhibitory effect of CDK4-specific
siRNA on the levels of Aurkb and CENP-P. The keratinocyte cell line
308 overexpressing CDK4 and the parental cell line 308 were
transfected with CDK4-specific siRNA and a control scramble-siRNA.
The transcription levels of Aurkb and CENP-P, normalized to the
transcriptional level of Gapdh, were quantified by qRT-PCR and
compared between CDK4-siRNA and control-siRNA transfected cells. We
observed a reduction in Aurkb and CENP-P RNA levels in both
308-CDK4 and 308 cells 72 h after transfection with CDK4-siRNA
(Fig. 4A). CDK4-siRNA lead to a 24%
(P=0.02) and 51% (P=0.01) decrease of CENP-P RNA expression in 308
and 308-CDK4 cells respectively (Fig.
4A). Aurkb RNA levels were reduced by 46% (P=0.01, t-test) and
78% (P=0.0005, t-test) in 308 and 308-CDK4 cells respectively
(Fig. 4A). The effect of CDK4-siRNA
was monitored by Western blot analysis 72 h after transfection
showing a 50- and 10-times reduction of CDK4, correlating with the
10- and 2-fold decreased level of Aurora-B protein in 308 and
308-CDK4 cells, respectively (Fig.
4B). Taken together, these analyses revealed that the binding
of CDK4 to the regulatory site of Aurkb and CENP-P genes leads to
positive transcriptional regulation.
Discussion
The canonical role of CDK4 and D-type cyclins as
drivers of cell proliferation and tumorigenesis via phosphorylation
of the retinoblastoma (Rb) family of proteins has been firmly
established. However, over the last few years, it has been
suggested that CDK4 plays alternative functions in proliferation
and tumorigenesis (8). For example,
CDK4 can also phosphorylate the transcription factor FOXM1, which
in turn induces the transcription of other genes involved in the
G2/M phases (12). Likewise,
additional functions have been identified in different cell cycle
regulators. For instance, the CDK4-related kinase, CDK6, performs
transcriptional functions regulating the expression of VEGF-A and
p16INK4a (9). Cyclin D1,
a regulatory subunit of CDK4 and CDK6, participates in activities
other than cell-cycle regulation, such as interaction with the
androgen and estrogen receptors and DNA repair (8,10,11).
Notably, it was recently reported a transcriptional
role of cyclin D1 regulating chromosome segregation genes such as
Aurkb, CENP-P, Zw10, Ckap2, Top2a and Mlf1ip (7). Our present findings indicate that CDK4
also regulates the transcription of Aurkb and CENP-P, two genes
involved in chromosome segregation. Aurora B expression has a key
role in chromosome bi-orientation and spindle-assembly checkpoint,
whereas Cenpp is required for proper kinetochore function (19,20,36). Our
studies have also established a major difference between the
transcriptional activities of CDK4 and cyclin D1 (7). Both CDK4 and cyclin D1 localizes on the
regulatory sites of Aurkb, CENP-P, and Zw10 genes, but only cyclin
D1 bind to Ckap2, Top2a, and Mlf1ip promoters (7). These results led us to hypothesize that
CDK4 and cyclin D1 may act as a complex at the regulatory sites of
Aurkb and CENP-P genes, whereas cyclin D1 may act independently of
CDK4 in other contexts. Interestingly, the CDK4 binding to the Zw10
promoter does not result in changes in the transcription level of
this gene. It is worth mentioning that the transcriptional role of
cyclin D1 was determined in Ccnd1−/− mouse embryo
fibroblasts (MEF) transfected with an epitope-tagged cyclin D1. In
contrast, we studied the effect of CDK4 gain- and lost-of-function
in keratinocytes and embryo fibroblast cell lines. Therefore,
whether the differences observed in the transcription of Ckap2,
Top2a, Mlf1ip, and Zw10 genes represent cell-specific regulation or
technical discrepancies between these experiments merit further
analysis.
Notably, it has been suggested that CDK4 activity is
necessary for regulation of phase others than G0 and
G1 (8,37). Inhibition of CDK4 activity results in
delayed progression from G2 to mitosis due to a failure
of chromosomes to migrate to the metaphase plate, implying that
CDK4 is necessary for entry into mitosis (38,39).
Consistent with these findings, our studies also showed that the
transcriptional and protein levels of Aurkb and CENP-P correlate
well with CDK4 expression. Notably, Aurora-B mRNA and protein
levels are tightly regulated and peak at the G2-M phases
(40,41), correlating well with the putative
activity of CDK4 in the G2/M phase. Although the
mechanisms regulated by CDK4 in the G2-M phase have not
been clearly defined, it is known that CDK4 inhibition reduces
mitosis's fidelity, implying that CDK4 indeed takes part in the
G2/M phase by regulating Aurora B (38).
Given that CDK4 inhibitors are in active clinical
development (42–45), it is crucial to understand the role
of CDK4 regulating Aurkb and CENP-P in tumor development. In this
regard, we demonstrated that transgenic expression of CDK4 induces
keratinocyte proliferation and accelerates the malignant
progression of mouse skin tumors (13–15). In
contrast, lack of CDK4 expression inhibits skin and oral tumor
development (46,47). We have also determined that CDK4, but
not the related kinases CDK6 and CDK2, induce skin tumor malignant
progression (13–15). Thus, the effect of CDK4 inducing
tumor progression might be related to its role in the
transcriptional activity of Aurkb and CENP-P. Aurora-B is a subunit
of the chromosomal passenger complex (CPC) controlling chromosome
segregation (41,48,49) and
potentially contribute to the Spindle-Assembly-Checkpoint (SAC),
which malfunction leads to aneuploidy and tumorigenesis (18). In fact, long-term overexpression of
Aurora-B in vivo results in defective chromosome
segregation, aneuploidy, and the development of multiple tumors in
mice (50–54). Similarly, CENP-P is a subunit of the
centromeric complex required for proper kinetochore function
contributing to chromosome segregation (20). Thus, the potential effect of CDK4
dysregulation in Aurkb and CENP-P expression leading to CIN and
tumorigenesis warrant further investigation.
Studies demonstrating that inactivation of CDK4 and
D-type cyclins can prevent tumor development in murine models
reinforced the view that CDK4 is suitable for cancer-specific
targets (46,47,55,56).
Based on these results, in the last decade, CDK inhibitors were
designed, which are in clinical development or have already been
approved by the US Food and Drug Administration (42–45,57–59). For
example, the observed preclinical and clinical effects of
palbociclib are consistent with the notion that inhibition of
CDK4/6 is a crucial mechanism underlying tumor growth activity
(60–63). However, some of these drugs have been
met with variable degrees of success in preclinical and clinical
studies. Thus, the CDK4 binding to the promoter regions should be
confirmed with assays in which specific CDK4-inhibitors, such as
Abemaciclib and Palbociclib, are administered to keratinocytes.
Those experiments will be fundamentals to determine the effect of
the CDK4 kinase activity in the transcriptional role of CDK4. If
the function of CDK4 regulating Aurkb and CENP-P levels is not
inhibited by the current drugs, then this new activity might
represent an important therapeutic target to disrupt cell cycle
progression in cancer cells. Such a scenario could help to explain
the reduced efficacy of the existing CDK4 drugs in some cancers and
open new research avenues for future studies directed to provide
new CDK4-related targets for combined therapeutic
interventions.
Acknowledgements
The authors would like to thank the Laboratory
Animal Resources and the Histology Core at the College of
Veterinary Medicine, North Carolina State University (Raleigh, USA)
for helping with the processing and staining of skin samples.
Funding
Research reported in this publication was supported
by grants from the National Cancer Institute (grant no.
RO1CA116328) and the National Institute of Environmental Health
Sciences (grant no. P30ES025128; Center for Human Health and the
Environment).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MLRP was responsible for the design and conception
of the experiments, and was the guarantor of this work and, as
such, takes responsibility for the integrity of the data and the
accuracy of the data analysis. SHL carried out the experiments and
contributed to data analysis and interpretation. LRLR and PLMDM
conceived and designed part of the experiments, and wrote part of
the results section. RM designed the primers utilized in part of
this paper, and performed the immunofluorescence staining and
semi-quantification of the western blots. All authors provided
critical feedback and helped shape the research, analysis and
manuscript. SHL and MLRP wrote the manuscript and confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The generation of mouse primary keratinocytes and
protocols for animal use were approved by the Institutional Animal
Care and Use Committee of North Carolina State University (approval
no. 18-102-B; Raleigh, USA).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CDK4
|
cyclin-dependent kinase 4
|
|
CIN
|
chromosome instability
|
|
ChIP
|
chromatin immunoprecipitation
|
References
|
1
|
Fischer PM, Endicott J and Meijer L:
Cyclin-dependent kinase inhibitors. Prog Cell Cycle Res. 5:235–248.
2003.PubMed/NCBI
|
|
2
|
Lukasik P, Zaluski M and Gutowska I:
Cyclin-dependent kinases (CDK) and their role in diseases
development-review. Int J Mol Sci. 22:29352021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suryadinata R, Sadowski M and Sarcevic B:
Control of cell cycle progression by phosphorylation of
cyclin-dependent kinase (CDK) substrates. Biosci Rep. 30:243–255.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gitig DM and Koff A: Cdk pathway:
Cyclin-dependent kinases and cyclin-dependent kinase inhibitors.
Methods Mol Biol. 142:109–123. 2000.PubMed/NCBI
|
|
5
|
Weinberg RA: The molecular basis of
carcinogenesis: Understanding the cell cycle clock. Cytokines Mol
Ther. 2:105–110. 1996.PubMed/NCBI
|
|
6
|
Ewen ME, Sluss HK, Sherr CJ, Matsushime H,
Kato J and Livingston DM: Functional interactions of the
retinoblastoma protein with mammalian D-type cyclins. Cell.
73:487–497. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Casimiro MC, Crosariol M, Loro E, Ertel A,
Yu Z, Dampier W, Saria EA, Papanikolaou A, Stanek TJ, Li Z, et al:
ChIP sequencing of cyclin D1 reveals a transcriptional role in
chromosomal instability in mice. J Clin Invest. 122:833–843. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hydbring P, Malumbres M and Sicinski P:
Non-canonical functions of cell cycle cyclins and cyclin-dependent
kinases. Nat Rev Mol Cell Biol. 17:280–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kollmann K, Heller G, Schneckenleithner C,
Warsch W, Scheicher R, Ott RG, Schäfer M, Fajmann S, Schlederer M,
Schiefer AI, et al: A Kinase-Independent function of CDK6 links the
cell cycle to tumor angiogenesis. Cancer Cell. 24:167–181. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bienvenu F, Jirawatnotai S, Elias JE,
Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT,
Odajima J, et al: Transcriptional role of cyclin D1 in development
revealed by a genetic-proteomic screen. Nature. 463:374–378. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jirawatnotai S, Hu Y, Michowski W, Elias
JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB,
et al: A function for cyclin D1 in DNA repair uncovered by protein
interactome analyses in human cancers. Nature. 474:230–234. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anders L, Ke N, Hydbring P, Choi YJ,
Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P and
Sicinski P: A systematic screen for CDK4/6 substrates links FOXM1
phosphorylation to senescence suppression in cancer cells. Cancer
Cell. 20:620–634. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miliani de Marval PL, Macias E, Conti CJ
and Rodriguez-Puebla ML: Enhanced malignant tumorigenesis in Cdk4
transgenic mice. Oncogene. 23:1863–1873. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Macias E, Miliani de Marval PL, De Siervi
A, Conti CJ, Senderowicz AM and Rodriguez-Puebla ML: CDK2
activation in mouse epidermis induces keratinocyte proliferation
but does not affect skin tumor development. Am J Pathol.
173:526–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang X, Sistrunk C and Rodriguez-Puebla
ML: Unexpected reduction of skin tumorigenesis on expression of
cyclin-dependent kinase 6 in mouse epidermis. Am J Pathol.
178:345–354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adon AM, Zeng X, Harrison MK, Sannem S,
Kiyokawa H, Kaldis P and Saavedra HI: Cdk2 and Cdk4 regulate the
centrosome cycle and are critical mediators of centrosome
amplification in p53-null cells. Mol Cell Biol. 30:694–710. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carmena M, Wheelock M, Funabiki H and
Earnshaw WC: The chromosomal passenger complex (CPC): From easy
rider to the godfather of mitosis. Nat Rev Mol Cell Biol.
13:789–803. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Musacchio A and Salmon ED: The
spindle-assembly checkpoint in space and time. Nat Rev Mol Cell
Biol. 8:379–393. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peters AH, Kubicek S, Mechtler K,
O'Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S,
Tachibana M, Shinkai Y, et al: Partitioning and plasticity of
repressive histone methylation states in mammalian chromatin. Mol
Cell. 12:1577–1589. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
D'Avino PP and Capalbo L: New auroras on
the roles of the chromosomal passenger complex in cytokinesis:
Implications for cancer therapies. Front Oncol. 5:2212015.
|
|
21
|
Strickland JE, Greenhalgh DA, Koceva-Chyla
A, Hennings H, Restrepo C, Balaschak M and Yuspa SH: Development of
murine epidermal cell lines which contain an activated rasHa
oncogene and form papillomas in skin grafts on athymic nude mouse
hosts. Cancer Res. 48:165–169. 1988.PubMed/NCBI
|
|
22
|
Hennings H, Michael D, Lichti U and Yuspa
SH: Response of carcinogen-altered mouse epidermal cells to phorbol
ester tumor promoters and calcium. J Invest Dermatol. 88:60–65.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yuspa SH and Morgan DL: Mouse skin cells
resistant to terminal differentiation associated with initiation of
carcinogenesis. Nature. 293:72–74. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lichti U, Anders J and Yuspa SH: Isolation
and short-term culture of primary keratinocytes, hair follicle
populations and dermal cells from newborn mice and keratinocytes
from adult mice for in vitro analysis and for grafting to
immunodeficient mice. Nat Protoc. 3:799–810. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miliani de Marval PL, Kim SH and
Rodriguez-Puebla ML: Isolation and characterization of a stem cell
side-population from mouse hair follicles. Methods Mol Biol.
1195:259–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kitagawa M and Lee SH: The chromosomal
passenger complex (CPC) as a key orchestrator of orderly mitotic
exit and cytokinesis. Front Cell Dev Biol. 3:142015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Madine MA, Swietlik M, Pelizon C,
Romanowski P, Mills AD and Laskey RA: The roles of the MCM, ORC,
and Cdc6 proteins in determining the replication competence of
chromatin in quiescent cells. J Struct Biol. 129:198–210. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martini E, Roche DM, Marheineke K,
Verreault A and Almouzni G: Recruitment of phosphorylated chromatin
assembly factor 1 to chromatin after UV irradiation of human cells.
J Cell Biol. 143:563–575. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geng Y, Whoriskey W, Park MY, Bronson RT,
Medema RH, Li T, Weinberg RA and Sicinski P: Rescue of cyclin D1
deficiency by knockin cyclin E. Cell. 97:767–777. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hennings H, Robinson VA, Michael DM, Petit
GR, Jung R and Yuspa SH: Development of an in vitro analogue of
initiated mouse epidermis to study tumor promoters and
antipromoters. Cancer Res. 50:4794–4800. 1990.PubMed/NCBI
|
|
31
|
Li HN, Zheng WH, Du YY, Wang G, Dong ML,
Yang ZF and Li XR: ZW10 interacting kinetochore protein may serve
as a prognostic biomarker for human breast cancer: An integrated
bioinformatics analysis. Oncol Lett. 19:2163–2174. 2020.PubMed/NCBI
|
|
32
|
Pauleau AL, Bergner A, Kajtez J and
Erhardt S: The checkpoint protein Zw10 connects CAL1-dependent
CENP-A centromeric loading and mitosis duration in
Drosophila cells. PLoS Genet. 15:e10083802019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park Y, Kim JS and Oh JS: Zw10 is a
spindle assembly checkpoint protein that regulates meiotic
maturation in mouse oocytes. Histochem Cell Biol. 152:207–215.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Leonard J, Sen N, Torres R, Sutani T,
Jarmuz A, Shirahige K and Aragón L: Condensin relocalization from
centromeres to chromosome arms promotes Top2 recruitment during
anaphase. Cell Rep. 13:2336–2344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bermejo R, Capra T, Gonzalez-Huici V,
Fachinetti D, Cocito A, Natoli G, Katou Y, Mori H, Kurokawa K,
Shirahige K and Foiani M: Genome-organizing factors Top2 and Hmo1
prevent chromosome fragility at sites of S phase transcription.
Cell. 138:870–884. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gruneberg U, Neef R, Honda R, Nigg EA and
Barr FA: Relocation of Aurora B from centromeres to the central
spindle at the metaphase to anaphase transition requires MKlp2. J
Cell Biol. 166:167–172. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Neuman E, Ladha MH, Lin N, Upton TM,
Miller SJ, DiRenzo J, Pestell RG, Hinds PW, Dowdy SF, Brown M and
Ewen ME: Cyclin D1 stimulation of estrogen receptor transcriptional
activity independent of cdk4. Mol Cell Biol. 17:5338–5347. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Burgess A, Wigan M, Giles N, Depinto W,
Gillespie P, Stevens F and Gabrielli B: Inhibition of S/G2 phase
CDK4 reduces mitotic fidelity. J Biol Chem. 281:9987–9995. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gabrielli BG, Sarcevic B, Sinnamon J,
Walker G, Castellano M, Wang XQ and Ellem KA: A cyclin D-Cdk4
activity required for G2 phase cell cycle progression is inhibited
in ultraviolet radiation-induced G2 phase delay. J Biol Chem.
274:13961–13969. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kimura M, Uchida C, Takano Y, Kitagawa M
and Okano Y: Cell cycle-dependent regulation of the human aurora B
promoter. Biochem Biophys Res Commun. 316:930–936. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kimura M, Kotani S, Hattori T, Sumi N,
Yoshioka T, Todokoro K and kano Y: Cell cycle-dependent expression
and spindle pole localization of a novel human protein kinase, Aik,
related to Aurora of Drosophila and yeast Ipl1. J Biol Chem.
272:13766–13771. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fry DW, Harvey PJ, Keller PR, Elliott WL,
Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK and
Toogood PL: Specific inhibition of cyclin-dependent kinase 4/6 by
PD 0332991 and associated antitumor activity in human tumor
xenografts. Mol Cancer Ther. 3:1427–1438. 2004.PubMed/NCBI
|
|
43
|
Toogood PL, Harvey PJ, Repine JT, Sheehan
DJ, VanderWel SN, Zhou H, Keller PR, McNamara DJ, Sherry D, Zhu T,
et al: Discovery of a potent and selective inhibitor of
cyclin-dependent kinase 4/6. J Med Chem. 48:2388–2406. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Infante JR, Cassier PA, Gerecitano JF,
Witteveen PO, Chugh R, Ribrag V, Chakraborty A, Matano A, Dobson
JR, Crystal AS, et al: A Phase I study of the cyclin-dependent
kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced
solid tumors and lymphomas. Clin Cancer Res. 22:5696–5705. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Patnaik A, Rosen LS, Tolaney SM, Tolcher
AW, Goldman JW, Gandhi L, Papadopoulos KP, Beeram M, Rasco DW,
Hilton JF, et al: Efficacy and safety of abemaciclib, an inhibitor
of CDK4 and CDK6, for patients with breast cancer, non-small cell
lung cancer, and other solid tumors. Cancer Discov. 6:740–753.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rodriguez-Puebla ML, Miliani de Marval PL,
LaCava M, Moons DS, Kiyokawa H and Conti CJ: Cdk4 deficiency
inhibits skin tumor development but does not affect normal
keratinocyte proliferation. Am J Pathol. 161:405–411. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Miliani de Marval PL, Macias E, Rounbehler
R, Sicinski P, Kiyokawa H, Johnson DG, Conti CJ and
Rodriguez-Puebla ML: Lack of cyclin-dependent kinase 4 inhibits
c-myc tumorigenic activities in epithelial tissues. Mol Cell Biol.
24:7538–7547. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Glover DM, Leibowitz MH, McLean DA and
Parry H: Mutations in aurora prevent centrosome separation leading
to the formation of monopolar spindles. Cell. 81:95–105. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gopalan G, Chan CS and Donovan PJ: A novel
mammalian, mitotic spindle-associated kinase is related to yeast
and fly chromosome segregation regulators. J Cell Biol.
138:643–656. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gonzalez-Loyola A, Fernandez-Miranda G,
Trakala M, Partida D, Samejima K, Ogawa H, Cañamero M, de Martino
A, Martínez-Ramírez Á, de Cárcer G, et al: Aurora B overexpression
causes aneuploidy and p21Cip1 repression during tumor development.
Mol Cell Biol. 35:3566–3578. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nguyen HG, Makitalo M, Yang D, Chinnappan
D, St Hilaire C and Ravid K: Deregulated Aurora-B induced
tetraploidy promotes tumorigenesis. FASEB J. 23:2741–2748. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nguyen HG and Ravid K:
Tetraploidy/aneuploidy and stem cells in cancer promotion: The role
of chromosome passenger proteins. J Cell Physiol. 208:12–22. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Giet R, Petretti C and Prigent C: Aurora
kinases, aneuploidy and cancer, a coincidence or a real link?
Trends Cell Biol. 15:241–250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Katayama H, Brinkley WR and Sen S: The
Aurora kinases: Role in cell transformation and tumorigenesis.
Cancer Metastasis Rev. 22:451–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu Q, Geng Y and Sicinski P: Specific
protection against breast cancers by cyclin D1 ablation. Nature.
411:1017–1021. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yu Q, Sicinska E, Geng Y, Ahnström M,
Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stål O and
Sicinski P: Requirement for CDK4 kinase function in breast cancer.
Cancer Cell. 9:23–32. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Finn RS, Martin M, Rugo HS, Jones S, Im
SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al:
Palbociclib and letrozole in advanced breast cancer. N Engl J Med.
375:1925–1936. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Long F, He Y, Fu H, Li Y, Bao X, Wang Q,
Wang Y, Xie C and Lou L: Preclinical characterization of SHR6390, a
novel CDK 4/6 inhibitor, in vitro and in human tumor xenograft
models. Cancer Sci. 110:1420–1430. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bisi JE, Sorrentino JA, Jordan JL, Darr
DD, Roberts PJ, Tavares FX and Strum JC: Preclinical development of
G1T38: A novel, potent and selective inhibitor of cyclin dependent
kinases 4/6 for use as an oral antineoplastic in patients with
CDK4/6 sensitive tumors. Oncotarget. 8:42343–42358. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dean JL, Thangavel C, McClendon AK, Reed
CA and Knudsen ES: Therapeutic CDK4/6 inhibition in breast cancer:
Key mechanisms of response and failure. Oncogene. 29:4018–4032.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dean JL, McClendon AK, Hickey TE, Butler
LM, Tilley WD, Witkiewicz AK and Knudsen ES: Therapeutic response
to CDK4/6 inhibition in breast cancer defined by ex vivo analyses
of human tumors. Cell Cycle. 11:2756–2761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Schwartz GK, LoRusso PM, Dickson MA,
Randolph SS, Shaik MN, Wilner KD, Courtney R and O'Dwyer PJ: Phase
I study of PD 0332991, a cyclin-dependent kinase inhibitor,
administered in 3-week cycles (Schedule 2/1). Br J Cancer.
104:1862–1868. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang YX, Sicinska E, Czaplinski JT,
Remillard SP, Moss S, Wang Y, Brain C, Loo A, Snyder EL, Demetri
GD, et al: Antiproliferative effects of CDK4/6 inhibition in
CDK4-amplified human liposarcoma in vitro and in vivo. Mol Cancer
Ther. 13:2184–2193. 2014. View Article : Google Scholar : PubMed/NCBI
|