Introduction
Pioglitazone is a type 2 anti-diabetic agent
included in the thiazolidinedione (TZD) class and is a ligand for
synthetic peroxisome proliferator-activated receptor (PPARγ). It is
involved in lipid and glucose metabolism and has recently been
reported to be associated with the inhibition of numerous cancer
cells (1). Pioglitazone is reported
to have multiple functions; it is anti-invasive, anti-inflammatory
and prevents angiogenesis (2–4).
Previous studies have shown that pioglitazone shows marked
anti-proliferative and antitumor effects in various types of human
cancers, including cancers of bladder, uterus, thyroid, pancreas
and breast, via inhibiting the signal transducer and activator of
transcription 3 (STAT3), MEK/ERK, p38 mitogen-activated protein
kinase (MAPK) and JAK2/STAT3 signaling pathways, and upregulating
the expression of AIF and death receptors (DRs) such as DR5 and
Fas/CD95 (5–9). Although pioglitazone induces apoptosis
in various cancer cell lines, the detailed molecular mechanism
underlying pioglitazone-induced apoptosis is not understood in Caki
cells derived from human clear cell renal cell carcinoma
(ccRCC).
Cellular FADD-like interleukin-1β-converting enzyme
inhibitory protein (c-FLIP) is an important anti-apoptotic protein
related to cancer cell death. There are three isoforms of c-FLIP,
namely, c-FLIP(L), c-FLIP(S) and
c-FLIP(R) (10). The
c-FLIP(L) shows significant structural similarities with
caspase-8 (10) and is associated
with TNF-related apoptosis-inducing ligand (TRAIL), Fas, TNF-α and
anticancer drug resistance in human malignancies (10–12). The
overexpression of c-FLIP inhibits death ligand-induced apoptosis,
which may impart resistance to anticancer drugs (13). Moreover, c-FLIP is overexpressed in a
wide variety of cancers, including gastric cancer, colorectal
cancer (CRC), bladder urothelial cancer and cervical cancer
(14–17). Therefore, to specifically regulate
the expression and activity of c-FLIP, it is necessary to find a
target molecule that does not interfere with caspases-8 and −10;
moreover, it is necessary to downregulate c-FLIP mRNA expression or
to decrease protein stability via proteasomes.
B-cell lymphoma 2 (Bcl-2) belongs to the Bcl-2
family and can be classified as an anti- or pro-apoptotic protein.
Bcl-2 and Bcl-xL are well-known anti-apoptotic proteins, whereas
Bax and Bak are widely used as pro-apoptotic proteins (18). Anti-apoptotic Bcl-2 family proteins
maintain the mitochondrial membrane, whereas pro-apoptotic Bcl-2
family proteins increase mitochondrial outer membrane
permeabilization (MOMP), which is associated with the induction of
apoptosis (19–21). Bcl-2 plays a critical role in cancer
cell death. Until now, the most effective strategy for targeting
the Bcl-2 family has been to use the BH3 mimetic molecules
(22). Bcl-2 overexpression is an
important mechanism in cancer cells to become resistant to cancer
treatment. Overexpression of Bcl-2 is common in many types of human
cancers, such as gastric cancer and breast cancer (23–25).
Thus, targeting Bcl-2 may be an important strategy to treat
cancers.
In the present study, we found that pioglitazone
induces apoptosis in human ccRCC Caki cells by activating the
caspase-dependent apoptotic signaling pathway via downregulating
c-FLIP(L) and reducing Bcl-2 protein stability.
Materials and methods
Cell culture media and reagents
Human ccRCC Caki cells were obtained from the
American Type Culture Collection (cat. no. HTB-46; ATCC). Caki
cells were maintained in Dulbecco's modified Eagle's medium (DMEM;
cat. no. LM 001-05; Welgene) containing 10% fetal bovine serum
(FBS; cat. no. S001-07; Welgene) and 1% antibiotic antimycotic (AA)
solution (cat. no. LS 203-01, Welgene). Human normal kidney HK-2
cells were purchased from the Korean Cell Line Bank (cat. no.
22190). HK2 cells were maintained in Roswell Park Memorial
Institute (RPMI)-1640 (cat. no. LM 011-01; Welgene) medium
supplemented with 10% FBS and 1% AA solution. Cells were incubated
at 37°C under 5% CO2 environment. The compound z-VAD-fmk
(cat. no. 627610) was purchased from Calbiochem. Pioglitazone (cat.
no. E6910), N-acetylcysteine (NAC; cat. no. A7250) and
cycloheximide (CHX; cat. no. C1988) were purchased from
Sigma-Aldrich.
Cell viability assay
Cell viability assays were performed using a
Welcount Cell Viability Assay Kit (cat. no. TR055-01; WelGene) to
determine cell viability. Caki cells were seeded
(0.25×105 cells/well) in two 96-well plates containing
DMEM supplemented with 10% FBS. The cells were treated with
pioglitazone for 24 h and then incubated with the XTT reagent for 2
h in dark at room temperature. Absorbance was measured at 450 nm
using a microplate spectrophotometer (Thermo Labsystems) at 450/690
nm.
Flow cytometry analysis
Approximately 0.4×106 cells were
suspended in 100 µl cold PBS (cat. no. 70011044; Thermo Fisher
Scientific, Inc.) and 200 µl 95% ethanol (cat. no. 1.00983.1011;
Merck) was added while the sample was being vortexed. The cells
were incubated at 4°C for 2 h, washed with PBS and resuspended in
250 µl of 1.12% sodium citrate buffer (pH 8.4) with 10 mg/ml RNase
A (cat. no. R4875; Sigma-Aldrich). The cells were further incubated
at 37°C for 40 min. Cellular DNA was stained by incubating the
cells with 250 µl propidium iodide (PI) (cat. no. P4170; Sigma) at
37°C for 20 min. The stained cells were analyzed by
fluorescence-activated cell sorting (FACS) using a BD FACSCanto II
flow cytometer (BD Biosciences).
Annexin V-PI staining
Annexin V-FITC (cat. no. 556547; BD Biosciences) and
PI were used for distinguishing cell death mode.
Pioglitazone-treated cells were washed twice in cold PBS and
resuspended in binding buffer at a concentration of
2×106/ml. This suspended cells (100 µl) were stained
with 5 µl of Annexin V-FITC and 10 µl PI. The cells were incubated
for 15 min in the dark at room temperature. After the addition of
400 µl of binding buffer to each tube, the cells were measured by
flow cytometry on a FACSCanto II (BD Biosciences).
Western blot analysis
Whole-cell lysates were prepared by suspending
0.45×106 cells in 30–50 µl lysis buffer consisting of 15
mM ethylene glycol tetraacetic acid (EGTA), 137 mM NaCl, 15 mM
MgCl2, 0.1 mM sodium orthovanadate, 25 mM MOPS, 100 µM
phenylmethanesulfonyl fluoride (PMSF), 0.1% Triton X-100 and 20 µM
leupeptin (pH 7.2). The cells were disrupted by sonication,
followed by protein extraction by incubating the samples at 4°C for
30 min. Total protein in the lysates was quantified using the
bicinchoninic acid (BCA) assay kit (cat. no. 23225; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
proteins (40–60 µg) were separated using 10–12% SDS PAGE gel and
electrotransferred onto nitrocellulose membranes (GE Healthcare).
Target proteins were detected using Immobilon Western
Chemiluminescent HRP Substrate solution (cat. no. WBULS0100,
Millipore). The expressed proteins were visualized using the Image
Quant LAS 4000 imaging system (GE Healthcare). Anti-PARP antibody
(1:1,000; cat. no. 9542) was purchased from Cell Signaling
Technology. Anti-caspase-3 antibody (1:2,000; cat. no. ADI-AAP-113)
and anti-c-FLIP (1:700; cat. no. ALX-804-961-0100) antibody were
purchased from Enzo Life Sciences. Anti-Bcl-2 antibody (1:700; cat.
no. sc-7832), anti-Mcl-1 (1:1,000; cat. no. sc-12756), c-IAP2
(1:1,000; cat. no. sc-517317) and anti-β-actin antibody (1:3,000;
cat. no. sc-47778) were supplied by Santa Cruz Biotechnology, Inc.
and anti-XIAP (1:5,000; cat. no. 610717) antibody was obtained from
BD Biosciences.
RNA isolation and RT-PCR
Bcl-2 mRNA expression was quantified via RT-PCR.
Total RNA was extracted from whole cells using EasyBlue reagent
(cat. no. 17061; Life Technologies). The cDNA was prepared using
M-MLV Reverse Transcriptase (cat. no. 18057018; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Total cellular RNA was reverse-transcribed using random primers and
amplified using PCR. GAPDH was used as an mRNA loading control. The
sequences of primers used for the amplification of Bcl-2 and GAPDH
were as follows: Bcl-2, (forward) 5′-GCCTTCTTTGAGTTCGGTGG-3′ and
(reverse) 5′-ATCTCCCGGTTGACGCTCT-3′; GAPDH: (forward)
5′-AGGTCGGAGTCAACGGATTTG-3′ and (reverse)
5′-GTGATGGCATGGACTGTG-GT-3′ and the PCR cycling conditions were as
follows: for Bcl-2: 95°C for 5 min, followed by 42 cycles of 95°C
for 45 sec, 53°C for 30 sec and 72°C for 30 sec; for GAPDH: 95°C
for 5 min, followed by 25 cycles of 94°C for 30 sec, 57°C for 45
sec and 72°C for 40 sec. The PCR products were analyzed by 1.5%
agarose gel electrophoresis and visualized with 10% ethidium
bromide using a gel system (cat. no. WGD30, Daihan).
Transfection
Caki cells were seeded onto 6-well plates
(0.2×106 cells/well) and incubated overnight at 37°C.
The cells were transfected with pcDNA 3.1 vector and pcDNA 3.1
Bcl-2 plasmid using Lipofectamine 2000 (cat. no. 11668-019;
Invitrogen; Thermo Fisher Scientific, Inc.) in Opti-MEM medium
(cat. no. 31985-070; Invitrogen; Thermo Fisher Scientific, Inc.).
Following transfection, the cells were cultured in DMEM
supplemented with 20% FBS for 18 h. Cells were then treated with
pioglitazone for 24 h and analyzed for Bcl-2 expression by western
blotting.
Statistical analysis
Data were analyzed using one-way ANOVA followed by
post hoc comparisons (Student-Newman-Keuls) using the Statistical
Package for Social Sciences 8.0 (SPSS Inc.). All experiments were
performed in triplicates. The results were expressed as the mean ±
SD and result with P<0.05 were considered statistically
significant.
Results
Pioglitazone mediates apoptosis in
human ccRCC Caki cells
To investigate the anticancer effect of pioglitazone
on Caki cells, a series of experiments were performed. Caki cells
were treated for 24 h with various concentrations of pioglitazone.
As shown in Fig. 1A, treatment with
pioglitazone considerably reduced cell viability. To clarify
pioglitazone-induced cell death mode, we performed Annexin V-PI
double staining using flow cytometry. We confirmed a dose-dependent
increase of apoptotic cells in pioglitazone-treated cells (Fig. 1B). The apoptotic effect of
pioglitazone was also identified. Pioglitazone treatment for 24 h
caused a dose-dependent increase in the sub-G1 cell population
(Fig. 1C). Additionally,
pioglitazone increased the levels of cleaved PARP and
cleaved-caspase-3 in treated cells (Fig.
1D). These results indicate that pioglitazone induces apoptosis
in Caki cells. To examine the underlying molecular mechanism
involved in pioglitazone-mediated apoptosis, the expression levels
of apoptotic-regulatory proteins were confirmed by western
blotting. As shown in Fig. 1E,
c-FLIP(L) and Bcl-2 expression levels moderately or
markedly decreased in pioglitazone-treated Caki cells. However,
Bcl-xL, Mcl-1, XIAP and c-IAP2 protein levels were not affected.
Taken together, these findings indicate that pioglitazone induces
apoptosis and inhibits the expression of c-FLIP(L) and
Bcl-2 in Caki cells.
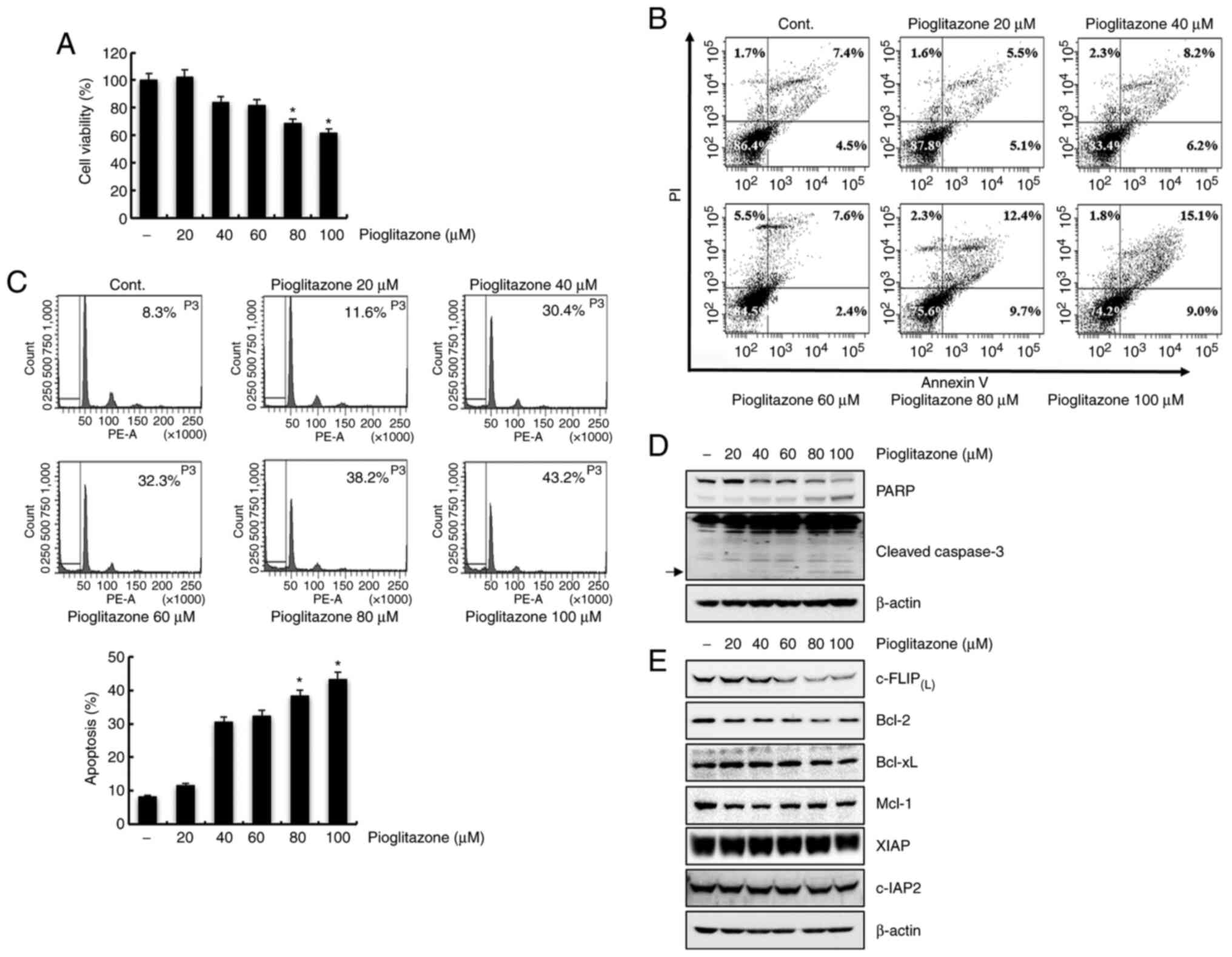 | Figure 1.Pioglitazone mediates apoptosis in
Caki cells. (A) Caki cells were treated with various concentrations
of pioglitazone (0, 20, 40, 60, 80 and 100 µM) for 24 h. Cell
viability was measured using the XTT assay kit. (B) Caki cells were
treated with pioglitazone for 24 h, collected and stained with
Annexin V and PI. Cell death was determined by flow cytometry. Each
value corresponds to the percentage of cells in each quadrant (Q1,
necrotic cells; Q2, late apoptotic cells; Q3, living cells; Q4,
early apoptotic cells). (C) Caki cells were treated with
pioglitazone for 24 h. Apoptosis was analyzed by flow cytometry.
Representative FACS histograms are presented in the upper panel and
cumulative data in the lower panel. (D) The cells were cultured
with the indicated concentrations of pioglitazone. PARP,
cleaved-caspase-3 and β-actin protein expression levels were
determined using western blotting. (E) Caki cells were treated with
pioglitazone for 24 h. The expression levels of
c-FLIP(L), Bcl-2, Bcl-xL, Mcl-1, XIAP, c-IAP2 and
β-actin proteins were detected by western blot analysis. β-actin
was used as a control for protein loading. Arrows indicate cleaved
forms of caspase-3. The data were obtained from three independent
experiments. Data are expressed as the mean ± SD (n=3). *P<0.05
vs. non-treated cells. FACS, Fluorescence-activated cell sorting;
PI, propidium iodide; PARP, poly (ADP-ribose) polymerase; c-FLIP,
cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory
protein; Bcl-2, B cell lymphoma 2; Bcl-xL, B cell lymphoma-extra
large; Mcl-1, myeloid cell leukemia-1; XIAP, X-linked inhibitor of
apoptosis protein; c-IAP, cellular inhibitor of apoptosis
protein. |
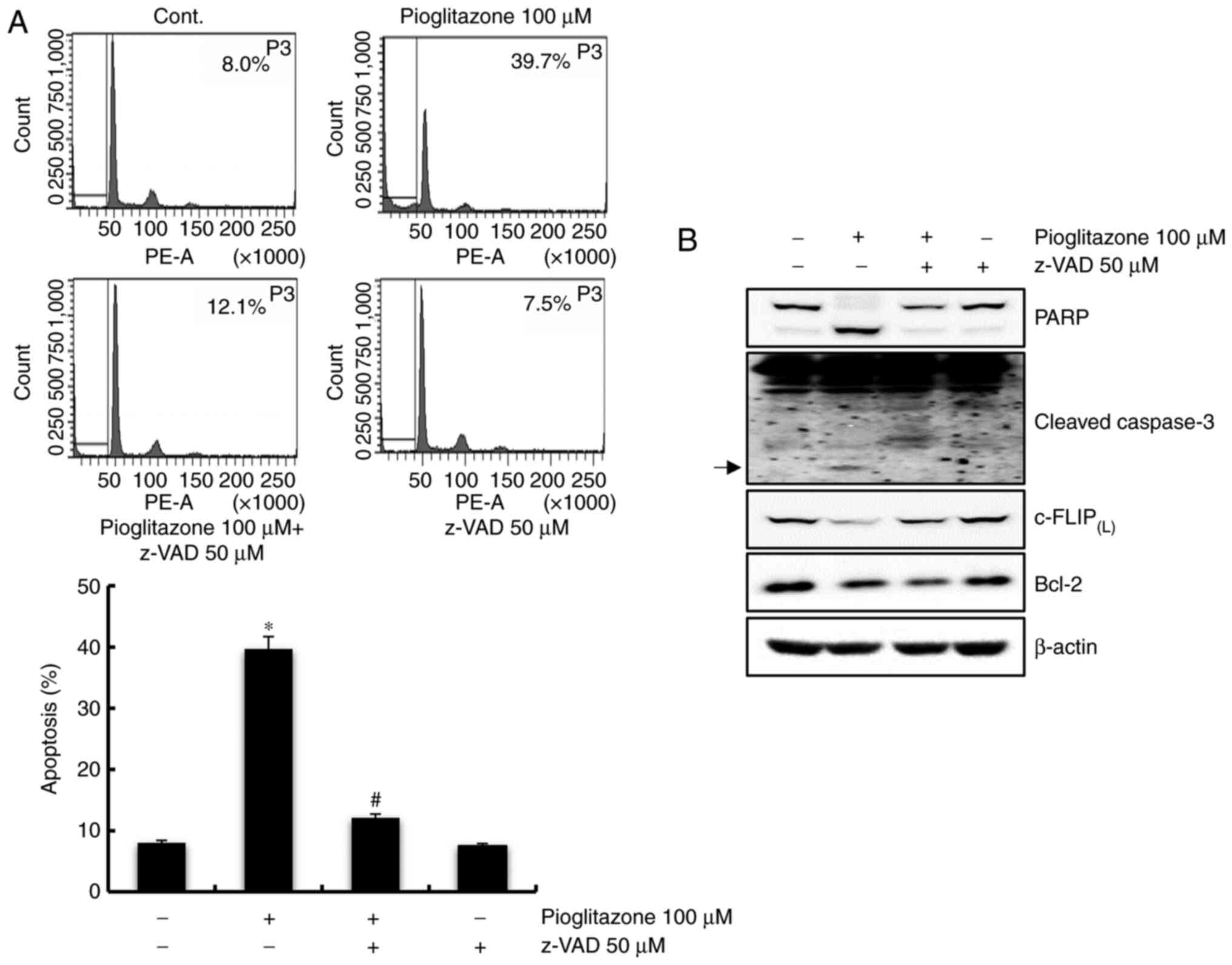
Pioglitazone-induced apoptosis is
markedly blocked by a caspase-dependent pathway in Caki cells
To determine whether the caspase-dependent pathway
plays a key role in pioglitazone-induced apoptosis, the pan-caspase
inhibitor, z-VAD-fmk was used. As shown in Fig. 2A, the pioglitazone-induced apoptosis
was significantly blocked by pretreatment with z-VAD-fmk.
Furthermore, treatment with z-VAD-fmk inhibited the cleavage of
PARP and cleaved-caspase-3, and recovered c-FLIP(L), but
not Bcl-2 expression (Fig. 2B).
These findings indicate that pioglitazone-mediated apoptosis in
Caki cells is regulated by a caspase-dependent pathway via
downregulation of c-FLIP(L).
Pioglitazone-mediated apoptosis is not
associated with ROS
Studies have shown that ROS can modulate apoptosis
by regulating the expression levels of pro-apoptotic proteins, such
as caspases or anti-apoptotic proteins such as c-FLIP and Bcl-2
(26). We investigated whether ROS
plays a role in pioglitazone-induced apoptosis. Caki cells were
pretreated with NAC for 1 h and incubated with pioglitazone for 24
h. Pretreatment with NAC failed to inhibit pioglitazone-mediated
apoptosis (Fig. 3A). Additionally,
NAC did not affect PARP cleavage, caspase activation,
c-FLIP(L) and Bcl-2 expression levels in
pioglitazone-treated cells (Fig.
3B). Therefore, these results indicate that
pioglitazone-mediated apoptosis is not associated with ROS.
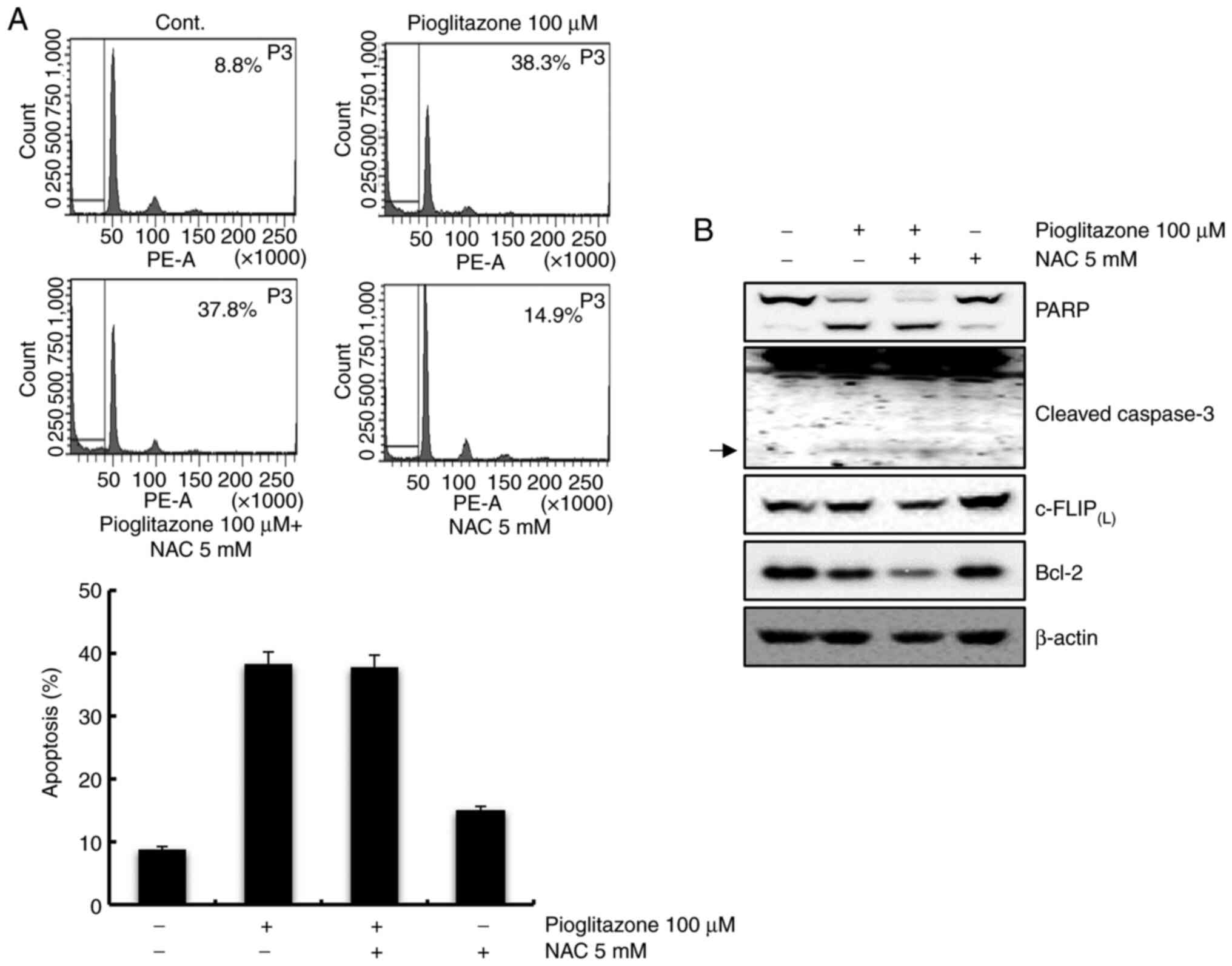 | Figure 3.Pioglitazone-induced apoptosis is not
mediated by ROS. (A) Caki cells were treated with 5 mM NAC or a
vehicle for 1 h before treatment with 100 µM pioglitazone. After 24
h, apoptosis was measured by flow cytometry. Representative FACS
histograms are presented in the upper panel and cumulative data in
the lower panel. (B) Cells were pretreated with 5 mM NAC or a
solvent for 1 h and treated with pioglitazone for 24 h. The
expression levels of PARP, cleaved-caspase-3, c-FLIP(L),
Bcl-2 and β-actin proteins were detected by western blot analysis.
β-actin was used as a loading control protein. Arrows indicate
cleaved forms of caspase-3. Data was representative from three
independent experiments. Data are expressed as the mean ± SD (n=3).
ROS, reactive oxygen species; NAC, N-acetyl-L-cysteine; FACS,
Fluorescence-activated cell sorting; PARP, poly(ADP-ribose)
polymerase; c-FLIP, cellular FLICE (FADD-like IL-1β-converting
enzyme)-inhibitory protein; Bcl-2, B cell lymphoma 2. |
Downregulation of c-FLIP(L)
contributes to pioglitazone-mediated apoptosis
We examined whether downregulation of
c-FLIP(L) by pioglitazone-induced apoptosis in Caki
cells overexpressing c-FLIP(L). Overexpression of
c-FLIP(L) significantly decreased pioglitazone-induced
apoptosis, whereas treatment with pioglitazone induced significant
apoptosis in Caki/vector cells (Fig.
4A). Expression of cleaved PARP and cleaved-caspase-3 induced
by pioglitazone treatment was also significantly inhibited by
overexpression of c-FLIP(L) (Fig. 4B). Therefore, these findings indicate
that the downregulation of c-FLIP(L) contributes to
pioglitazone-mediated apoptosis. To confirm the functional role of
downregulated Bcl-2 in pioglitazone-treated cells, Caki cells
engineered for Bcl-2 overexpression were used. As shown in Fig. 4C, overexpression of Bcl-2 was not
associated with pioglitazone-mediated apoptosis. Expression of
cleaved PARP and caspase-3 induced by pioglitazone-treated cells
was not affected by overexpression of Bcl-2. (Fig. 4D), suggesting that the downregulation
of Bcl-2 was not related to pioglitazone-induced apoptosis.
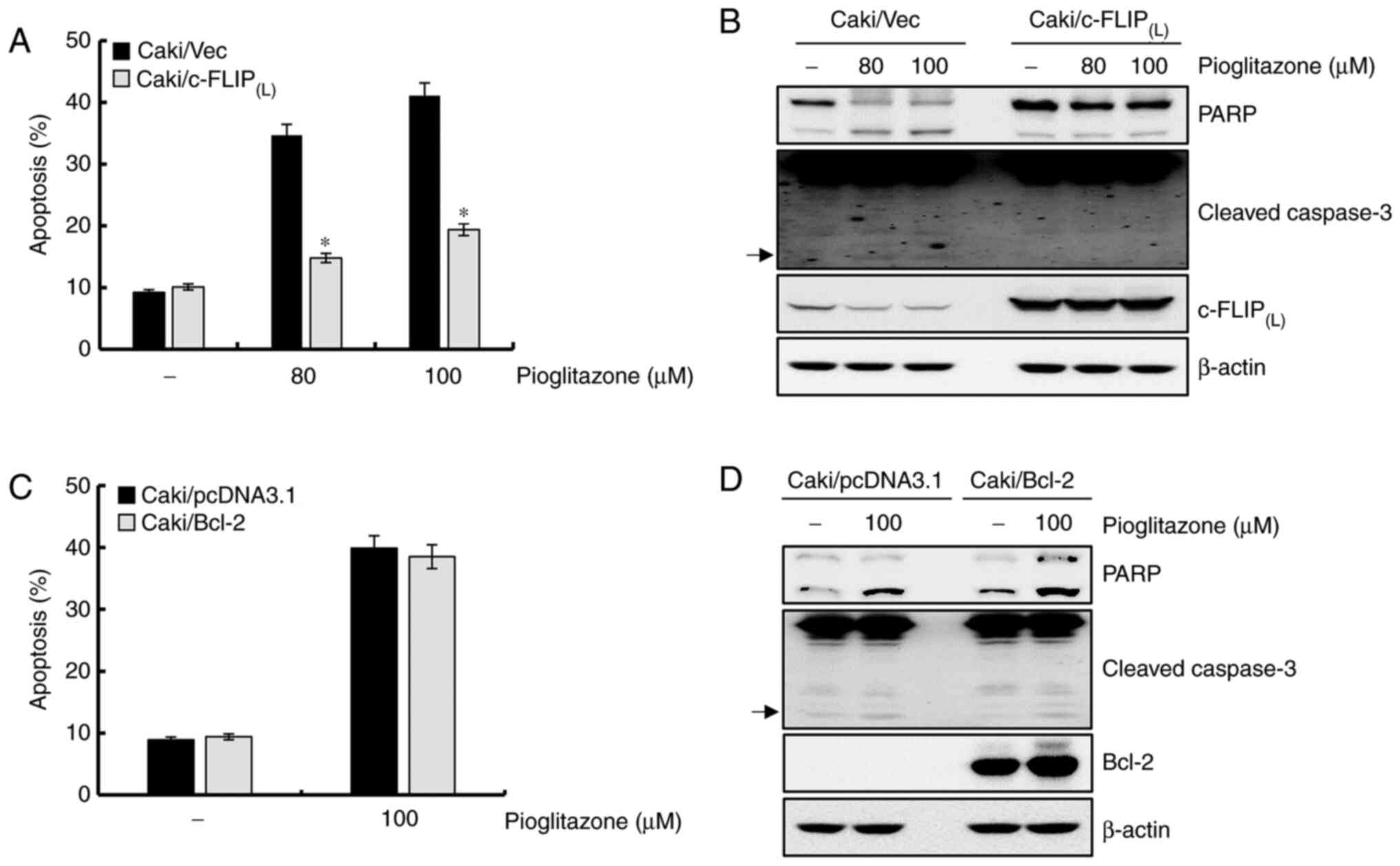 | Figure 4.Downregulation of
c-FLIP(L) plays a critical role in pioglitazone-mediated
apoptosis. (A) Caki/vector and Caki/c-FLIP(L) cells were
treated for 24 h with pioglitazone (80 and 100 µM). Apoptosis was
analyzed as a sub-G1 cell fraction by FACS. (B) Caki/vector and
Caki/c-FLIP(L) cells were treated with 80 and 100 µM
pioglitazone for 24 h. Expression levels of PARP,
cleaved-caspase-3, c-FLIP(L) and β-actin proteins were
detected by western blot analysis. (C) After transient transfection
with empty vector or the Bcl-2 expression vector, the cells were
treated with 100 µM pioglitazone for 24 h. The sub-G1 cell fraction
was measured by flow cytometry. (D) After transient transfection
with an empty vector or the Bcl-2 expression vector, the cells were
treated with pioglitazone. After 24 h, expression levels of PARP,
cleaved-caspase-3, Bcl-2 and β-actin proteins were detected by
western blotting. β-actin was used as a control for western blot
analysis. Arrows indicate cleaved forms of caspase-3. Data were
obtained from three independent experiments. The data are expressed
as the mean ± SD (n=3). *P<0.05 vs. pioglitazone-treated
Caki/vector cells. c-FLIP, cellular FLICE (FADD-like
IL-1β-converting enzyme)-inhibitory protein; FACS,
Fluorescence-activated cell sorting; PARP, poly(ADP-ribose)
polymerase; Bcl-2, B cell lymphoma 2. |
Pioglitazone attenuates the expression
of Bcl-2 caused by the reduction of protein stability
Whether the pioglitazone-induced decrease in Bcl-2
was regulated at the transcriptional level was evaluated next. As
shown in Fig. 5A, the Bcl-2 mRNA
level remained constant following treatment with pioglitazone,
suggesting that pioglitazone-mediated downregulation of Bcl-2
protein is regulated at the post-transcriptional level. To further
clarify the mechanisms underlying the decreased Bcl-2 expression
level in pioglitazone-treated cells, a protein stability assay for
Bcl-2 was performed. Cells were pretreated with cycloheximide (CHX)
for 1 h and then treated with pioglitazone for studying the
kinetics. As shown in Fig. 5B, the
protein expression level of Bcl-2 reduced more rapidly with the
co-treatment of CHX and pioglitazone compared with CHX treatment
alone. We also investigated whether treatment with pioglitazone
affects the induction of apoptosis in human normal kidney HK-2
cells. However, the sensitivity to apoptosis by pioglitazone was
markedly reduced in HK-2 cells, compared with pioglitazone-treated
Caki cells (Fig. 5C and D). These
results showed that the degradation of Bcl-2 protein was
facilitated by pioglitazone treatment and that pioglitazone
treatment reduced Bcl-2 protein stability.
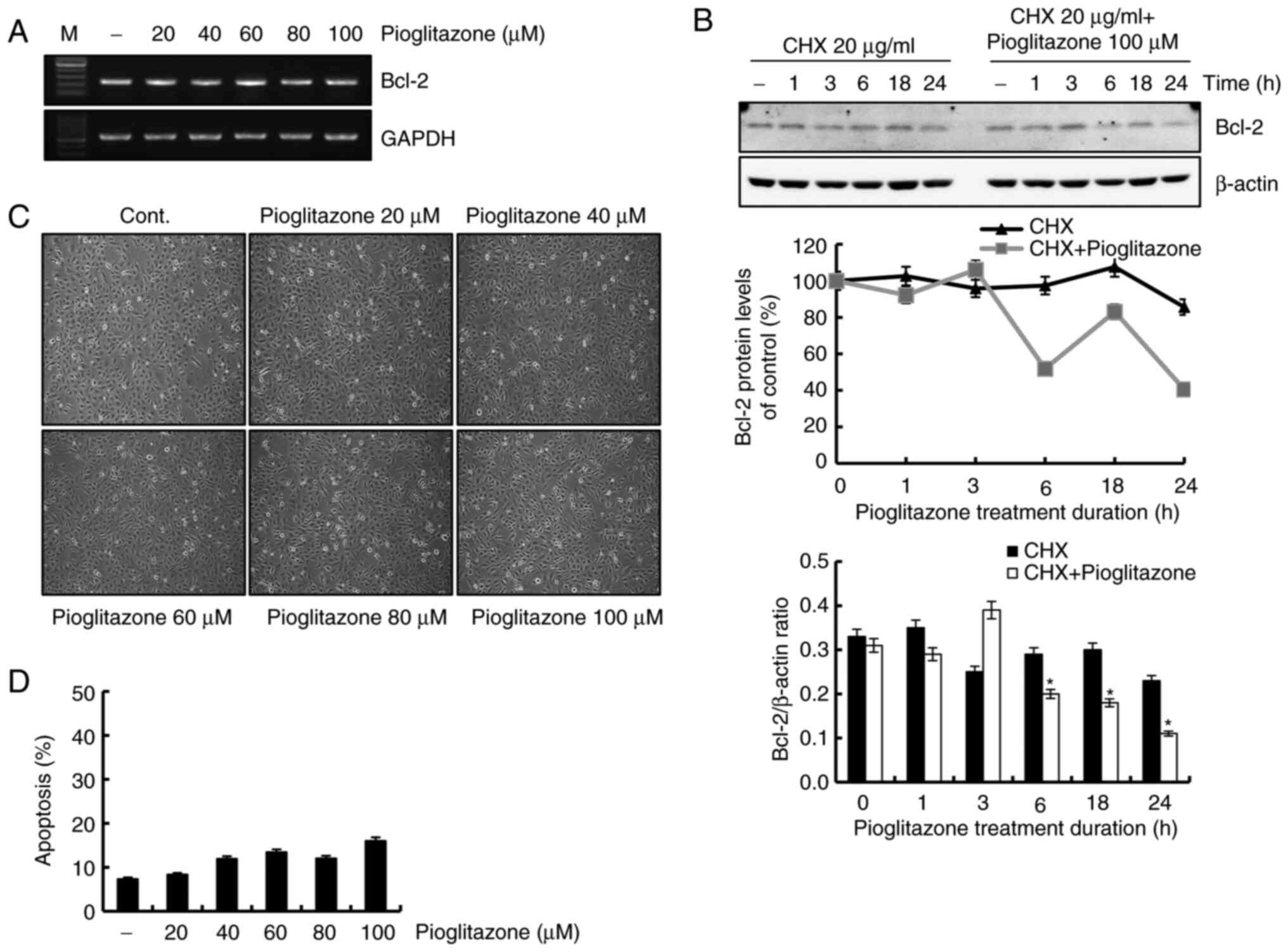 | Figure 5.Downregulation of Bcl-2 expression by
pioglitazone treatment results from decreased protein stability.
(A) Caki cells were treated with pioglitazone. After 24 h, mRNA
expression of Bcl-2 was measured via reverse
transcription-semi-quantitative PCR. GAPDH was used as a loading
control. (B) Cells were treated with or without 100 µM pioglitazone
in the presence of 20 µg/ml CHX for the indicated duration. Western
blotting was performed to determine Bcl-2 protein expression levels
(upper panel). The Bcl-2 density was measured using ImageJ software
(middle panel). The data obtained from the western blot analyses of
Bcl-2 and β-actin were used to evaluate the effect of pioglitazone
on the Bcl-2/β-actin ratio (lower panel). β-actin was used as a
loading control protein. (C) HK-2 cells were treated with the
indicated concentrations of pioglitazone (0, 20, 40, 60, 80 and 100
µM) for 24 h. The morphological changes were examined under an
inverted microscope (magnification, ×200). (D) HK-2 cells were
treated with pioglitazone (0, 20, 40, 60, 80 and 100 µM) for 24 h.
Apoptosis was determined by flow cytometry. Data were obtained from
three independent experiments. The data are expressed as the mean ±
SD (n=3). *P<0.05 vs. CHX-treated Caki cells. GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; Bcl-2, B cell lymphoma 2;
CHX, cycloheximide. |
Discussion
In this study, it was shown that pioglitazone exerts
potent anticancer effects on human ccRCC Caki cells.
Pioglitazone-induced apoptosis was mediated by caspase-dependent
signaling pathways in treated cells. Moreover, the molecular
mechanism of pioglitazone-mediated apoptosis is ascribed to
caspase-mediated degradation of c-FLIP(L) protein and
reduction of Bcl-2 protein stability.
Pioglitazone, a PPARγ agonist, is used to lower
blood glucose levels in patients with type 2 diabetes (27). Studies have shown that pioglitazone
may exert antitumor effects in several human cancer cell types,
including bladder cancer, acute lymphocytic leukemia and glioma via
inducing apoptosis and cell growth inhibition (28–30).
Nevertheless, it has been shown that diabetic patients with
long-term and high-dose exposure to pioglitazone may increase the
risk of bladder cancer (31).
However, there is a conflicting study that pioglitazone treatment
does not increase the risk of bladder cancer in diabetic patients
and that bladder cancer risk does not correlate with cumulative
dose and duration of treatment, indicating that pioglitazone makes
it much more effective and safer for glycemic control in diabetic
patients (32). Additionally, in our
system, pioglitazone treatment did not induce cell proliferation in
bladder cancer T24 cells (data not shown). In this study, the
antitumor effect of pioglitazone on Caki cells was confirmed.
Consistent with previous studies, it was found that increasing
concentrations of pioglitazone led to an increased in the sub-G1
cell population.
Pioglitazone and other PPARγ agonists show
anticancer activity against several cancer types, such as non-small
cell lung carcinoma, acute promyelocytic leukemia, bladder cancer,
breast cancer, lung cancer and CRC via enhancing growth arrest,
upregulating the expression of DNA damage-inducible 153 gene and
PTEN, inactivating the PI3K-Akt pathway, sustaining activated MAPK,
modulating DR5 and c-FLIP(L) expression and
downregulating Bcl-2 expression (33–38).
Caspase activation regulates apoptotic-regulatory
proteins (39). However, there are
conflicting reports of caspase involvement in apoptosis induced by
PPARγ agonists in cancer cells. Pioglitazone-mediated apoptosis
occurs via a caspase-independent pathway in bladder cancer cells
(5). In contrast, pretreatment of
PC-3 cells with z-VAD-fmk inhibited PPARγ agonist-mediated
apoptosis, indicating the involvement of the caspase-dependent
pathway in prostate cancer (40). It
was also shown that pioglitazone-induced apoptotic cells were
remarkably inhibited by pretreatment with z-VAD-fmk. Thus,
pioglitazone-mediated apoptosis is regulated by the
caspase-dependent apoptotic pathway in Caki cells.
c-FLIP is an important modulator of anti-apoptotic
pathway and is expressed in a variety of cancer cell types
(41). Previous reports have
demonstrated that PPARγ ligands modulate apoptosis via
downregulating c-FLIP(L) expression in cervical cancer
cell lines (42). To determine
whether the downregulation of c-FLIP(L) was involved in
pioglitazone-induced apoptosis, c-FLIP(L)-overexpressing
cells were established in this study. Our results showed that
pioglitazone-induced apoptosis was blocked in
c-FLIP(L)-overexpressing cells, suggesting that
pioglitazone-mediated apoptosis occurred via downregulating
c-FLIP(L) expression.
Bcl-2 expression is regulated at the transcriptional
or post-transcriptional levels (43,44).
Studies have indicated that pioglitazone-mediated apoptosis is
regulated by suppressed Bcl-2 transcription in hepatocellular
carcinoma (45). In contrast, we
found that the degradation of Bcl-2 protein was facilitated by
pioglitazone treatment without affecting Bcl-2 mRNA expression
levels. Thus, our data indicate that pioglitazone-mediated decrease
in Bcl-2 protein is regulated at the post-transcriptional
level.
ROS are critical regulators of apoptosis in a wide
range of human cancer cells (46,47). It
has been reported that pioglitazone induces apoptosis by inducing
ROS production in lung cancer (48).
Thus, we confirmed whether pioglitazone-mediated apoptosis was
associated with ROS production. In our study, pretreatment with NAC
did not affect pioglitazone-treated cells, thereby providing
evidence that pioglitazone-induced apoptosis is independent of ROS
production in Caki cells.
Collectively, our results demonstrate that
pioglitazone-mediated apoptosis is facilitated by caspase-dependent
signaling pathways via downregulating c-FLIP(L)
expression and reducing Bcl-2 protein stability in human ccRCC Caki
cells. Therefore, based on our study outcomes, we propose that
pioglitazone may be a potential therapeutic agent for human RC.
Acknowledgements
Not applicable.
Funding
The present study was supported by Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no.
2020R1I1A1A01068857).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JYK conceived and designed the study. JHJ and TJL
conducted most of the experiments and data analysis, and wrote the
manuscript. EGS and IHS conducted data analysis of flow cytometry
and Annexin V/PI staining experiments, and wrote and revised the
manuscript. JHJ and JYK confirm the authenticity of all the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Elrod HA and Sun SY: PPARgamma and
apoptosis in cancer. PPAR Res. 2008:7041652008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang Y, Zhao LH, Huang B, Wang RY, Yuan
SX, Tao QF, Xu Y, Sun HY, Lin C and Zhou WP: Pioglitazone, a PPARγ
agonist, inhibits growth and invasion of human hepatocellular
carcinoma via blockade of the rage signaling. Mol Carcinog.
54:1584–1595. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heliövaara MK, Herz M, Teppo AM, Leinonen
E and Ebeling P: Pioglitazone has anti-inflammatory effects in
patients with type 2 diabetes. J Endocrinol Invest. 30:292–297.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dromparis P, Sutendra G, Paulin R, Proctor
S, Michelakis ED and McMurtry MS: Pioglitazone inhibits
HIF-1α-dependent angiogenesis in rats by paracrine and direct
effects on endothelial cells. J Mol Med (Berl). 92:497–507. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsubaki M, Takeda T, Tomonari Y, Kawashima
K, Itoh T, Imano M, Satou T and Nishida S: Pioglitazone inhibits
cancer cell growth through STAT3 inhibition and enhanced AIF
expression via a PPARγ-independent pathway. J Cell Physiol.
233:3638–3647. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lützen U, Zhao Y, Lucht K, Zuhayra M, Marx
M, Cascorbi I and Culman J: Pioglitazone induces cell growth arrest
and activates mitochondrial apoptosis in human uterine
leiomyosarcoma cells by a peroxisome proliferator-activated
receptor γ-independent mechanism. Naunyn Schmiedebergs Arch
Pharmacol. 390:37–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ozdemir Kutbay N, Biray Avci C, Sarer
Yurekli B, Caliskan Kurt C, Shademan B, Gunduz C and Erdogan M:
Effects of metformin and pioglitazone combination on apoptosis and
AMPK/mTOR signaling pathway in human anaplastic thyroid cancer
cells. J Biochem Mol Toxicol. 34:e225472020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koga H, Selvendiran K, Sivakumar R,
Yoshida T, Torimura T, Ueno T and Sata M: PPARgamma potentiates
anticancer effects of gemcitabine on human pancreatic cancer cells.
Int J Oncol. 40:679–685. 2012.PubMed/NCBI
|
|
9
|
Jiao XX, Lin SY, Lian SX, Qiu YR, Li ZH,
Chen ZH, Lu WQ, Zhang Y, Deng L, Jiang Y and Hu GH: The inhibition
of the breast cancer by PPARγ agonist pioglitazone through
JAK2/STAT3 pathway. Neoplasma. 67:834–842. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Safa AR: c-FLIP, a master anti-apoptotic
regulator. Exp Oncol. 34:176–184. 2012.PubMed/NCBI
|
|
11
|
Safa AR, Day TW and Wu CH: Cellular
FLICE-like inhibitory protein (C-FLIP): A novel target for cancer
therapy. Curr Cancer Drug Targets. 8:37–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bagnoli M, Canevari S and Mezzanzanica D:
Cellular FLICE-inhibitory protein (c-FLIP) signalling: A key
regulator of receptor-mediated apoptosis in physiologic context and
in cancer. Int J Biochem Cell Biol. 42:210–213. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Poukkula M, Kaunisto A, Hietakangas V,
Denessiouk K, Katajamäki T, Johnson MS, Sistonen L and Eriksson JE:
Rapid turnover of c-FLIPshort is determined by its unique
C-terminal tail. J Biol Chem. 280:27345–27355. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou XD, Yu JP, Liu J, Luo HS, Chen HX and
Yu HG: Overexpression of cellular FLICE-inhibitory protein (FLIP)
in gastric adenocarcinoma. Clin Sci (Lond). 106:397–405. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ullenhag GJ, Mukherjee A, Watson NF,
Al-Attar AH, Scholefield JH and Durrant LG: Overexpression of FLIPL
is an independent marker of poor prognosis in colorectal cancer
patients. Clin Cancer Res. 13:5070–5075. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korkolopoulou P, Goudopoulou A, Voutsinas
G, Thomas-Tsagli E, Kapralos P, Patsouris E and Saetta AA: c-FLIP
expression in bladder urothelial carcinomas: Its role in resistance
to Fas-mediated apoptosis and clinicopathologic correlations.
Urology. 63:1198–1204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang W, Wang S, Song X, Sima N, Xu X, Luo
A, Chen G, Deng D, Xu Q, Meng L, et al: The relationship between
c-FLIP expression and human papillomavirus E2 gene disruption in
cervical carcinogenesis. Gynecol Oncol. 105:571–577. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Llambi F and Green DR: Apoptosis and
oncogenesis: Give and take in the BCL-2 family. Curr Opin Genet
Dev. 21:12–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kale J, Osterlund EJ and Andrews DW: BCL-2
family proteins: Changing partners in the dance towards death. Cell
Death Differ. 25:65–80. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kalkavan H and Green DR: MOMP, cell
suicide as a BCL-2 family business. Cell Death Differ. 25:46–55.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ludwig LM, Maxcy KL and LaBelle JL: Flow
cytometry-based detection and analysis of BCL-2 family proteins and
mitochondrial outer membrane permeabilization (MOMP). Methods Mol
Biol. 1877:77–91. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Knight T, Luedtke D, Edwards H, Taub JW
and Ge Y: A delicate balance-The BCL-2 family and its role in
apoptosis, oncogenesis, and cancer therapeutics. Biochem Pharmacol.
162:250–261. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Inada T, Kikuyama S, Ichikawa A, Igarashi
S and Ogata Y: Bcl-2 expression as a prognostic factor of survival
of gastric carcinoma. Anticancer Res. 18:2003–2010. 1998.PubMed/NCBI
|
|
24
|
Binder C, Marx D, Overhoff R, Binder L,
Schauer A and Hiddemann W: Bcl-2 protein expression in breast
cancer in relation to established prognostic factors and other
clinicopathological variables. Ann Oncol. 6:1005–1010. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee KH, Im SA, Oh DY, Lee SH, Chie EK, Han
W, Kim DW, Kim TY, Park IA, Noh DY, et al: Prognostic significance
of bcl-2 expression in stage III breast cancer patients who had
received doxorubicin and cyclophosphamide followed by paclitaxel as
adjuvant chemotherapy. BMC Cancer. 7:632007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Azad N and Iyer AKV: Reactive oxygen
species and apoptosis. Systems Biol Free Radicals Antioxid.
113–135. 2014. View Article : Google Scholar
|
|
27
|
Han S and Roman J: Peroxisome
proliferator-activated receptor gamma: A novel target for cancer
therapeutics? Anticancer Drugs. 18:237–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kostapanos MS, Elisaf MS and Mikhailidis
DP: Pioglitazone and cancer: Angel or demon? Curr Pharm Des.
19:4913–4929. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zang C, Liu H, Posch MG, Waechter M,
Facklam M, Fenner MH, Ruthardt M, Possinger K, Phillip Koeffler H
and Elstner E: Peroxisome proliferator-activated receptor gamma
ligands induce growth inhibition and apoptosis of human B
lymphocytic leukemia. Leuk Res. 28:387–397. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wan Z, Shi W, Shao B, Shi J, Shen A, Ma Y,
Chen J and Lan Q: Peroxisome proliferator-activated receptor γ
agonist pioglitazone inhibits β-catenin-mediated glioma cell growth
and invasion. Mol Cell Biochem. 349:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang H, Shi W, Fu S, Wang T, Zhai S, Song
Y and Han J: Pioglitazone and bladder cancer risk: A systematic
review and meta-analysis. Cancer Med. 7:1070–1080. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Agrawal P, Jain A, Gautam A, Nigam AK,
Pursnani N and Farooqui M: A retrospective study to assess the risk
of bladder cancer in type-2 diabetic patients treated with
pioglitazone. Perspect Clin Res. 12:9–13. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Esmaeili S, Safaroghli-Azar A,
Pourbagheri-Sigaroodi A, Salari S, Gharehbaghian A, Hamidpour M and
Bashash D: Stimulation of peroxisome proliferator-activated
receptor-gamma (PPARγ) using pioglitazone decreases the survival of
acute promyelocytic leukemia cells through up-regulation of PTEN
expression. Anticancer Agents Med Chem. 21:108–119. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lv S, Wang W, Wang H, Zhu Y and Lei C:
PPARγ activation serves as therapeutic strategy against bladder
cancer via inhibiting PI3K-Akt signaling pathway. BMC Cancer.
19:2042019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kole L, Sarkar M, Deb A and Giri B:
Pioglitazone, an anti-diabetic drug requires sustained MAPK
activation for its anti-tumor activity in MCF7 breast cancer cells,
independent of PPAR-γ pathway. Pharmacol Rep. 68:144–154. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zou W, Liu X, Yue P, Khuri FR and Sun SY:
PPARgamma ligands enhance TRAIL-induced apoptosis through DR5
upregulation and c-FLIP downregulation in human lung cancer cells.
Cancer Biol Ther. 6:99–106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee CJ, Han JS, Seo CY, Park TH, Kwon HC,
Jeong JS, Kim IH, Yun J, Bae YS, Kwak JY and Park JI: Pioglitazone,
a synthetic ligand for PPARgamma, induces apoptosis in RB-deficient
human colorectal cancer cells. Apoptosis. 11:401–411. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Satoh T, Toyoda M, Hoshino H, Monden T,
Yamada M, Shimizu H, Miyamoto K and Mori M: Activation of
peroxisome proliferator-activated receptor-gamma stimulates the
growth arrest and DNA-damage inducible 153 gene in non-small cell
lung carcinoma cells. Oncogene. 21:2171–2180. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shiau CW, Yang CC, Kulp SK, Chen KF and
Chen CS, Huang JW and Chen CS: Thiazolidenediones mediate apoptosis
in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2
functions independently of PPARgamma. Cancer Res. 65:1561–1569.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fulda S: Targeting c-FLICE-like inhibitory
protein (CFLAR) in cancer. Expert Opin Ther Targets. 17:195–201.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Plissonnier ML, Fauconnet S, Bittard H,
Mougin C, Rommelaere J and Lascombe I: Cell death and restoration
of TRAIL-sensitivity by ciglitazone in resistant cervical cancer
cells. Oncotarget. 8:107744–107762. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cui J and Placzek WJ: Post-transcriptional
regulation of anti-apoptotic BCL2 family members. Int J Mol Sci.
19:3082018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen N, Hu T, Gui Y, Gao J, Li Z and Huang
S: Transcriptional regulation of Bcl-2 gene by the PR/SET domain
family member PRDM10. PeerJ. 7:e69412019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shim J, Kim BH, Kim YI, Kim KY, Hwangbo Y,
Jang JY, Dong SH, Kim HJ, Chang YW and Chang R: The peroxisome
proliferator-activated receptor gamma ligands, pioglitazone and
15-deoxy-Delta(12,14)-prostaglandin J(2), have antineoplastic
effects against hepatitis B virus-associated hepatocellular
carcinoma cells. Int J Oncol. 36:223–231. 2010.PubMed/NCBI
|
|
46
|
Sheikh BY, Sarker MMR, Kamarudin MNA and
Mohan G: Antiproliferative and apoptosis inducing effects of citral
via p53 and ROS-induced mitochondrial-mediated apoptosis in human
colorectal HCT116 and HT29 cell lines. Biomed Pharmacother.
96:834–846. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yodkeeree S, Sung B, Limtrakul P and
Aggarwal BB: Zerumbone enhances TRAIL-induced apoptosis through the
induction of death receptors in human colon cancer cells: Evidence
for an essential role of reactive oxygen species. Cancer Res.
69:6581–6589. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Srivastava N, Kollipara RK, Singh DK,
Sudderth J, Hu Z, Nguyen H, Wang S, Humphries CG, Carstens R,
Huffman KE, et al: Inhibition of cancer cell proliferation by PPARγ
is mediated by a metabolic switch that increases reactive oxygen
species levels. Cell Metab. 20:650–661. 2014. View Article : Google Scholar : PubMed/NCBI
|