Introduction
Pancreatic cancer is one of the most lethal cancers,
with a 5-year survival rate of <9%. While its incidence is
increasing, its mortality rate has remained unchanged (1). Patient response to existing therapy is
limited (2). Therefore, to overcome
these issues, there is an increasing and urgent need to discover
new treatment options.
In clinical practice, the traditional drug discovery
process is expensive and takes over a decade (3). Thus, repositioning clinically evaluated
drugs can be a strategy for the rapid detection and application of
new alternative therapeutic approaches (4).
Pancreatic stellate cells (PSCs) are
myofibroblast-like cells and are located in the pancreatic stroma
(5). PSCs have the ability to
produce components of the extracellular matrix (ECM) and make the
stroma stiff. During pancreatic damage due to inflammation and
metabolic stress, PSCs are activated by growth factors/cytokines
released from neighboring cells (6).
Activated PSCs support the proliferation of cancer cells, as shown
by some studies assessing the relationship between cancer cells and
PSCs (7). Therefore, inactivating
PSCs could potentially be a novel approach for pancreatic cancer
treatment.
The aim of this study was to determine a promising
compound for inactivating PSCs. To achieve this, gene expression
profiles of cancer-associated fibroblasts (CAF) and normal
fibroblasts were analyzed, and neuroactive ligand-receptor
interaction pathway related to the cancer-associated PSCs was
identified. We focused on duloxetine, a serotonin-noradrenaline
reuptake inhibitor, as a potential compound for suppressing the
activity of PSCs.
Materials and methods
Cells and culture conditions
SUIT-2 [Japan Health Science Research Resources Bank
(JCRB), Osaka, Japan] was purchased and maintained in Dulbecco's
modified Eagle's medium (DMEM) (Sigma-Aldrich Co.), supplemented
with 10% fetal bovine serum (FBS). Human PSCs were generated in our
laboratory from two fresh surgical specimens of pancreatic ductal
adenocarcinoma (PDAC), using the outgrowth method (8), and were maintained in DMEM with 10%
FBS. Human Pancreatic Stellate Cells (HPaSteC cells, cat. no. 3830;
ScienCell Research Laboratories) were maintained in Stellate Cell
Medium (cat. no. 5301; ScienCell Research Laboratories), according
to the manufacturer's instructions. All cells were free of
mycoplasma contamination. The cells were authenticated using short
tandem repeat (STR) profiling. Cells at passages 3–8 were used for
assays.
Human PDAC organoid were established as described
previously (9,10). A PDAC sample from a 75-year-old male
who underwent pancreatoduodenectomy in 2015 at Kyushu University
was collected. Surgical specimens of PDAC were minced and digested
with a Tumor Dissociation Kit (human, cat. no. 130-095-929;
Miltenyi Biotec). Then, the tissue was embedded in growth
factor-reduced (GFR) Matrigel (cat. no. 356231; BD Bioscience) and
cultured in a human complete medium at 37°C for 14 days. The human
complete medium used was Advanced DMEM/F12 (cat. no. 12634-010;
Invitrogen), supplemented with 1M HEPES (Invitrogen), GlutaMax
(cat. no. 35050-061; Invitrogen), penicillin/streptomycin (cat. no.
15140122; Invitrogen), B27 (cat. no. 17504044; Invitrogen),
N-acetyl-L-cysteine (cat. no. A9165; Sigma-Aldrich Co.), Wnt-3a
(cat. no. 5036-WN-010; R&D Systems), R-Spondin 1 (cat. no.
120-38; Peprotech), Noggin (cat. no. 120-10C; Invitrogen),
epidermal growth factor (EGF, cat. no. AF-100-15; Peprotech),
fibroblast growth factor (FGF, cat. no. 100-26; Peprotech),
Nicotinamide (cat. no. N0636; Sigma-Aldrich Co.), Y-27263 (cat. no.
Y0503; Sigma-Aldrich Co.), and A83-01 (cat. no. 2939/10; R&D
Systems). Organoid area measurements were performed using KEYENCE
BZ-X analyzer.
Identification of differentially
expressed genes from public microarray data
Public gene expression profiles for GSE53524 were
obtained from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo). Data was analyzed,
and a set of genes differentially expressed in CAFs and normal
fibroblasts were identified. P-values <0.05 for microarray data
were selected. Pathway enrichment analysis of targets was performed
using the DAVID v6.8 (https://david.ncifcrf.gov/). The Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway enrichment analysis was performed
for identified up- and down-regulated genes. P<0.05 was
considered to indicate statistical significance.
Drugs
Duloxetine hydrochloride (cat. no. 040-34071, Wako
Pure Chemicals) was dissolved in dimethyl sulfoxide (DMSO) at a
final concentration of 10 mM and stored at −20°C. Paroxetine
hydrochloride (cat. no. 168-24431, Wako Pure Chemicals),
desloratadine (cat. no. D3787, TCI Chemicals), promethazine,
amitriptyline hydrochloride (cat. no. 013-12882, Wako Pure
Chemicals), and chlorpromazine hydrochloride (cat. no. 033-10581,
Wako Pure Chemicals) were dissolved in DMSO at a concentration of
10 mM and stored at −20°C.
Lipid accumulation assay
BODIPY® staining was used for fixed cells
as described previously (11). PSCs
were seeded on glass-bottomed multi-well plates (P06G-1.5-14-F,
MatTek Corporation) at 1×105 cells/well. After 24 h of
incubation, the medium was aspirated and fresh DMEM containing 10%
FBS was added. For the drug treatment group, each drug was
dissolved in the same DMEM at a concentration of 10 µM. We have
previously reported that chloroquine inhibits PSC activation
(11). We conducted a drug screening
for PSCs to select other candidate drugs that inhibit PSC
activation (12). Chloroquine had
been shown to inhibit PSC activity at a concentration of 10 µM, and
since this concentration was used as the standard for our previous
screening, we used this concentration as well. After 48 h of
incubation, cells were washed with PBS and fixed with 4%
paraformaldehyde phosphate buffer solution and stained with 1 mg/ml
4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene
(BODIPY® 493/503, cat. no. D-3922; Life Technologies)
and Hoechst 33342 solution (1 mg/ml, cat. no. H342; Dojindo) for 30
min in the dark and at room temperature. Cells were washed with
phosphate-buffered saline, and images were captured using a
fluorescent microscope.
For live cells, lipid accumulation was stained by
Lipidye® according to the manufacturer's instructions.
Cells were seeded in glass-bottomed multi-well plates at a density
of 1×105 cells/well. After 24 h of incubation, the
medium was aspirated, and a fresh medium with the indicated
concentrations of drugs or DMSO was added. After culturing for each
period, cells were stained with 1 mg/ml
1,3-Diphenyl-2-[4-(N,N-diphenylamino)phenyl] benzo[b]
phosphole-P-oxide (Lipidye®, 405/520) and 1 mg/ml
Hoechst 33342 for 2 h in the dark at 37°C. Images were captured
using a fluorescence microscope.
Cell viability assay
Cell Titer-Glo Luminescent Cell Viability Assay
(Promega Corp.) was used for assaying. PSCs (1×103
cells/well) were seeded in 96-well polystyrene cell culture
microplates (cat. no. 655083, Greiner Bio-One International) and
incubated at 37°C in a humidified atmosphere with 10%
CO2. After 24 h, the medium was aspirated and fresh DMEM
containing 10% FBS and the candidate drugs at each concentration
were added. Fluorescence was measured after 48 h. The emission
value was measured as a control using a microplate reader (Infinite
F200; Tecan), according to the manufacturer's instructions. Cell
viability was evaluated as the ratio to the control.
Reverse transcription-quantitative
polymerase chain reaction
For duloxetine treatment, we used the concentration
of 10 µM. Total RNA was extracted from PSCs using a High Pure RNA
Isolation kit (cat. no. 11828665001, Roche), according to the
manufacturer's instructions. Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) was performed using STBR Green
RT-PCR kit (cat. no. 170-8892; Bio-Rad Laboratories) and the CFX96
Real-Time PCR System (Bio-Rad Laboratories). Primers were purchased
from Takara (Shiga, Japan). The primer sequences are listed in
Table SI. The reactions were
incubated at 50°C for 10 min, 95°C for 1 min, 95°C for 10 sec, 60°C
for 30 sec for 39 cycles, 60°C for 5 sec, and 95°C for 5 sec. The
quantity of messenger RNA (mRNA) was calculated using the
2−ΔΔCq method (13). mRNA
expression was normalized using glyceraldehyde-3-phosphate
dehydrogenase.
Western blotting
Cells were incubated for 48 h with each
concentration of duloxetine (0, 1, 5 and 10 µM). Proteins were
extracted using PRO-PREP Protein Extraction Solution (cat. no.
17081, iNtRON Biotechnology), according to the manufacturer's
instructions. The protein concentration was measured using a
NanoDrop 1000 Spectrophotometer (version 3.8.1; Thermo Fisher
Scientific). After 48 h of incubation, cells were washed, and the
medium was changed to DMEM with 0% FBS and was cultured overnight.
Culture supernatants were collected in a centrifuge tube (cat. no.
UFC900324, AmiconUltra; Merck Millipore) and centrifuged at 7,500
rpm for 30 min. Halt protease and phosphatase inhibitors (cat. no.
78442; Thermo Fisher Scientific) were added at a 1:1,000 dilution
into the concentrated supernatants. Collected proteins (20 µg per
lane from whole cell lysate) were fractionated on 4–15%
Mini-PROTEAN TGX Precast Gels (cat. no. 456-1084; Bio-Rad
Laboratories) and transferred to Trans-Blot Turbo Mini PVDF
Transfer Packs (cat. no. 170-4156, Bio-Rad Laboratories) using a
Trans-Blot Turbo Transfer Starter System (Bio-Rad Laboratories).
The membrane was incubated at 4°C overnight with anti-Akt (Rabbit
mAb, 1:1,000, cat. no. 4691; Cell Signaling Technology), anti-pAkt
(Rabbit mAb, 1:1,000, cat. no. 4060; Cell Signaling Technology),
anti-αSMA (Mouse mAb, 1:2,000, cat. no. M0851; Dako), anti-αtubulin
(Rabbit pAb, 1:2,000, cat. no. bs-50500R; Bioss), anti-Erk (Rabbit
mAb, 1:1,000, cat. no. 4695; Cell Signaling Technology), anti-pErk
(Rabbit mAb, 1:1,000, cat. no. 4370S; Cell Signaling Technology),
anti-fibronectin (Rabbit pAb, 1:1,000, cat. no. ab2413; Abcam),
anti-COL1A1 (Rabbit pAb, 1:1,000, cat. no. 84336S; Cell Signaling
Technology), anti-IL-6 (Mouse mAb, 1:1,000, ab9324; Abcam),
anti-periostin (POSTN; Rabbit pAb, 1:1,000, cat. no. ab14041;
Abcam), anti-PP2A (Rabbit mAb, 1:1,000, cat. no. ab32104; Abcam) or
mouse anti-β-actin (Mouse mAb, 1:10,000, cat. no. 81178; Santa Cruz
Biotechnology) antibodies. Membranes were then probed with
horseradish peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology). Immunoblots were detected by enhanced
chemiluminescence using the ChemiDoc XRS System (Bio-Rad
Laboratories) and were analyzed using Quantity One Software
(Bio-Rad Laboratories). Each experiment was repeated three
times.
Matrigel invasion and migration
assays
Cell invasion and migration were measured by
counting the number of cells through Transwell chambers with 8-µm
pores (cat. no. 353097; BD Biosciences). For invasion assays,
Transwell inserts were coated with 20 µg/well Matrigel (cat. no.
354234; Corning). After confirming the half-maximal inhibitory
concentration for PSCs, we used 10 µM for invasion and migration
assay, because this concentration did not exceed the
IC50 for PSCs. The procedure involved: a) control of
DMEM with 10% FBS, b) supernatant collected from PSC, c) DMEM with
10% FBS containing duloxetine at 10 µM, and d) supernatant
collected from PSC cultured with 10 µM duloxetine added in the
24-well culture plate (cat. no. 353504; Corning). SUIT-2 cells
(4×104) in 250 µl DMEM containing 2% FBS were seeded in
the upper chamber and placed in a 24-well culture plate. After 48 h
of incubation for invasion assays or 24 h for migration assays,
upper chambers were fixed with 70% ethanol. Cells were washed,
stained with hematoxylin and eosin, and counted in five random
fields at ×100 magnification using a BZ-X Analyzer (Keyence).
Samples were evaluated in a blinded manner. The experiments were
repeated three times.
Statistical analysis
All values are expressed as mean ± standard
deviation (SD). For comparison between two groups only, data
was analyzed with an unpaired two-tailed t-test. For comparison of
multiple groups, comparisons were conducted using ANOVA, followed
by Tukey-Kramer method. A P-value <0.05 was considered to
indicate a statistically significant difference.. Experiments were
repeated at least three times. All statistical analyses were
performed using GraphPad Prism 8.
Results
Gene expression profiles of
cancer-associated PSCs
To identify the potential therapeutic targets of
cancer-associated PSCs, we examined the gene expression profiles
from public microarray data obtained from murine pancreatic CAFs
and normal fibroblasts (GSE53524) (14). To identify the genes associated with
CAFs in comparison to normal fibroblasts, probe sets with a P-value
<0.05 for microarray data were selected. We found that many
up-regulated genes were associated with several pathways of the
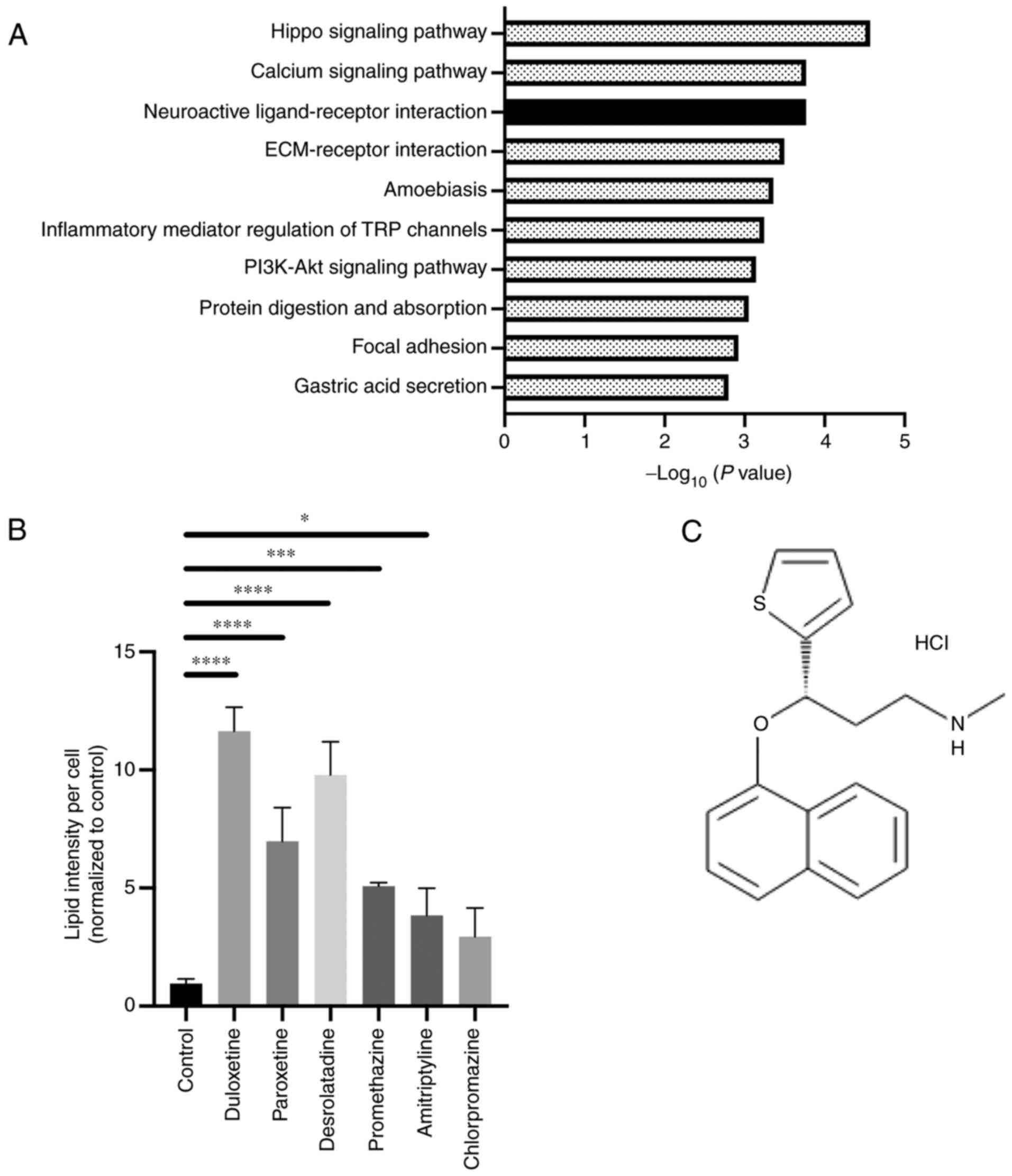
KEGG pathway enrichment analysis (Fig.
1A). The hippo-signaling pathway and calcium-signaling pathway
are reported potential target pathways in PSC activation (15,16). On
the other hand, neuroactive ligand-receptor signaling in PSCs has
not been studied. Therefore, we focused on this pathway.
To select candidate compounds, we referred to
previous studies and focused on the gene targets in this pathway
and selected compounds related to the pathway (17). In addition, we previously
demonstrated high-throughput drug screening for detecting compounds
that suppress the activity of PSCs (12). The result from this screening showed
that these six compounds were included as candidate drugs for
inactivating PSC function. Therefore, we selected these six
compounds and confirmed their role in PSC inactivation. The
following drugs were selected and evaluated: duloxetine, a
5-hydroxytryptamine and noradrenaline reuptake inhibitor;
paroxetine, a selective serotonin (5-HT) reuptake inhibitor;
desloratadine and promethazine, first-generation antihistamines;
amitriptyline, a tricyclic antidepressant; and chlorpromazine, a
dopamine receptor antagonist. These compounds are G-protein coupled
receptor (GPCR) inhibitors, and they showed an affinity to GPCR
(Table SII). Next, to assess the
effect of these candidates on the PSC state, lipid droplet
accumulation assaying for detecting quiescent PSCs was performed,
as previously reported (11). Lipid
accumulation in the cytoplasm was observed following treatment with
five out of six compounds, and duloxetine showed the highest
accumulation of lipid droplets (Fig.
1B). Therefore, duloxetine (Fig.
1C) was selected as a potential compound for suppressing the
activity of PSCs.
Duloxetine suppressed the activity of
PSCs
These results led us to further investigate the
effects of duloxetine on PSC activation. First, we investigated the
half-maximal inhibitory concentration of duloxetine for
patient-derived PSCs and HPaSteC cells (hPSC). These concentrations
ranged from 19.2 to 25.2 µM (Fig.
2A). Next, we investigated whether duloxetine suppresses the
PSC activity, according to the previous report (6,11). The
quiescent state is a reversible state (18). Therefore, we used live cells and
demonstrated the changes of lipid droplets in the cytoplasm between
duloxetine treatments. To determine the changes in lipid droplets
in live PSCs, Lipidye® staining was used for
immunofluorescent staining (19).
Lipid accumulation was observed with duloxetine treatment. Further,
after washout and replacement of the drug-free medium, the
intracellular lipid droplets disappeared (Fig. 2B). Next, we determined the expression
of the PSC activation markers, alpha-smooth muscle actin (αSMA),
and ECM proteins (collagen I, fibronectin and POSTN) by RT-qPCR and
western blotting of whole-cell lysates. Duloxetine reduced the
protein and mRNA expression of these markers in PSCs (Fig. 2C and D), suggesting that duloxetine
suppressed the activity of PSCs. This effect was reversible,
suggesting that duloxetine induced PSC quiescence.
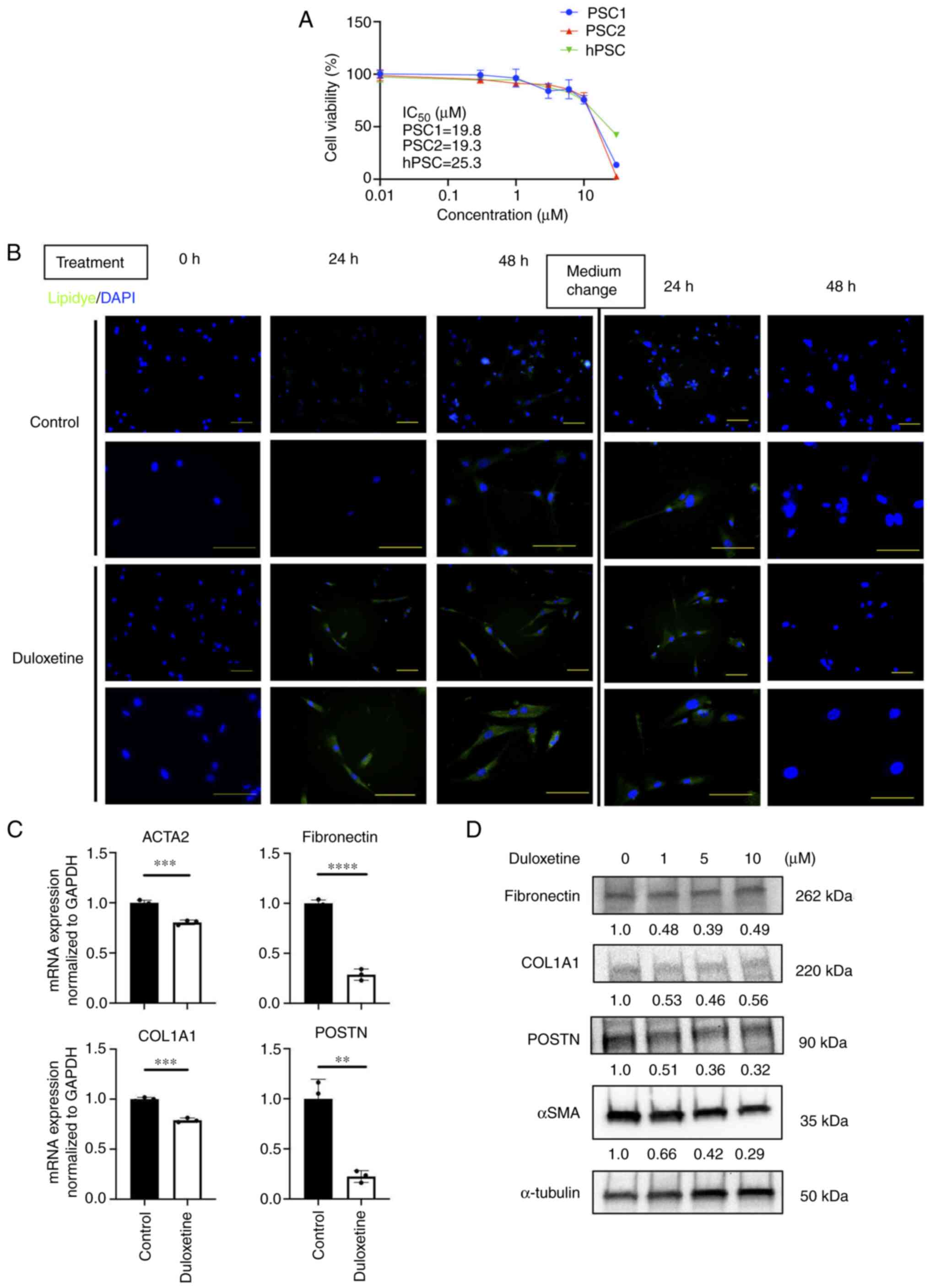 | Figure 2.Duloxetine induces PSCs into a
quiescent state. (A) Cell viability of PSCs in vitro. Each
IC50 value is presented in the graph. (B) Representative
photomicrographs of LipiDye staining of PSCs taken 0, 24 and 48 h
after duloxetine treatment. DAPI was used for nuclear staining.
After 48 h, the medium was washed off, fresh Dulbecco's modified
Eagle's medium with 10% FBS was added, and the culture was
continued. Magnifications, ×200 (upper panels) and ×400 (lower
panels). Scale bar, 100 µm. (C) mRNA expression levels of αSMA and
ECM proteins in PSCs. mRNA expression levels were normalized to
GAPDH expression and are presented as the fold-change in gene
expression relative to control PSCs. (D) Western blotting of αSMA
and ECM proteins from whole cell lysate of PSCs. Values indicate
densitometric ratios normalized to α-tubulin. **P<0.01,
***P<0.001, ****P<0.0001 vs. control group. PSCs, pancreatic
stellate cells; αSMA, α-smooth muscle actin; ACTA2, actin α2 smooth
muscle; ECM, extracellular matrix; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; hPSC, human pancreatic
stellate cells; COL1A1, α-1 type-1 collagen; POSTN, periostin. |
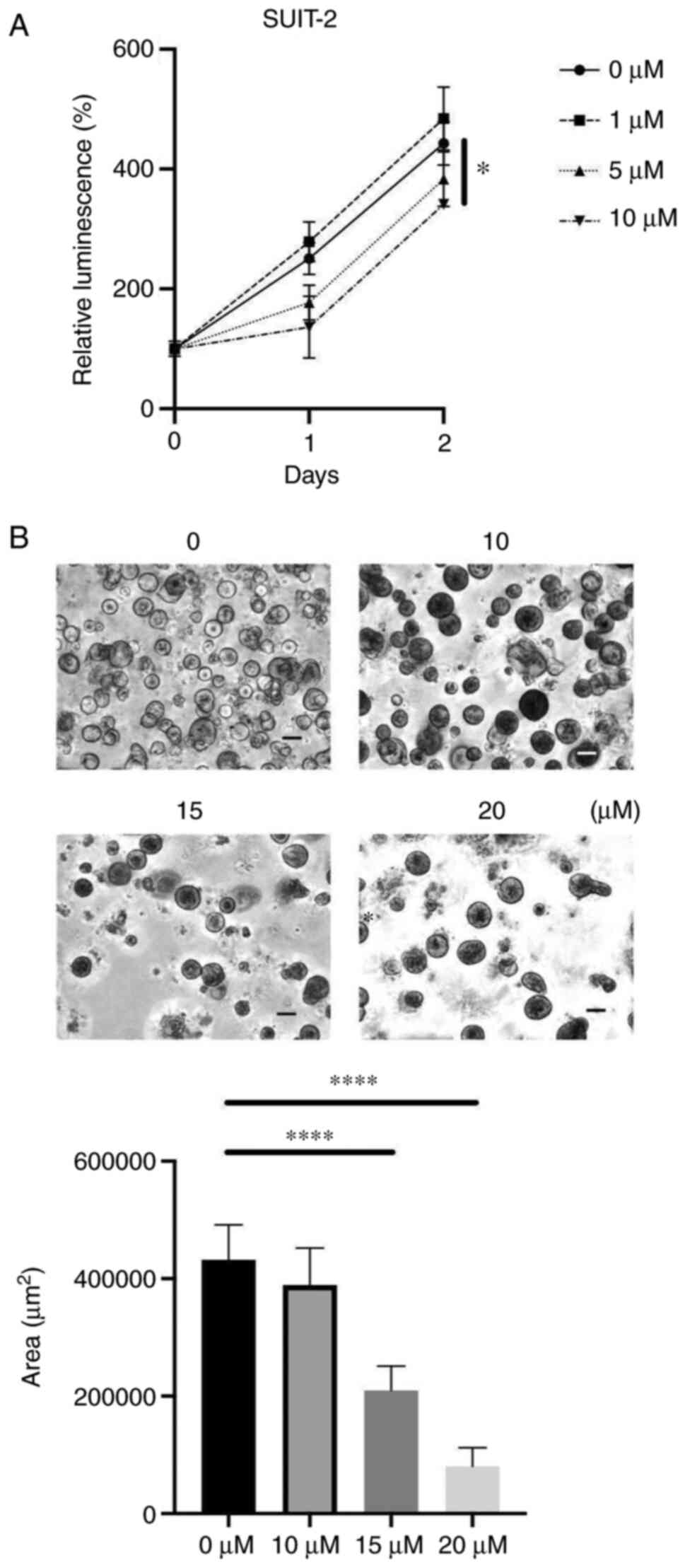
Duloxetine suppressed the
proliferation of patient-derived organoids
Next, to determine the effects of duloxetine on
cancer cell growth, we used a PCC 2D culture and proliferation of
human PCC cell line was directly attenuated by duloxetine treatment
(Fig. 3A). It has been widely
suggested that organoid models can epitomize in vivo-like
growth and differentiation of tissues as compared to 2D cultures
(20). We found that duloxetine
suppressed organoid formation and growth in organoids developed
from the human surgical specimens (Fig.
3B), suggesting that duloxetine inhibits the growth of tumor
organoids.
Duloxetine suppressed tumor-stromal
interaction
Previous data showed that activated PSCs affect
pancreatic cancer cell invasion and migration (21). To confirm whether duloxetine
suppresses tumor-stromal interaction between pancreatic cancer
cells (PCCs) and PSCs, an invasion and migration assay was
performed. Compared to the control group, duloxetine itself did not
attenuate PCC invasion and migration (Fig. 4A). Consistent with previous reports,
co-culture with PSC supernatant stimulated PCC invasion and
migration. However, this enhancement effect was abolished by
co-culturing with a duloxetine-treated PSC supernatant (Fig. 4A and B), supporting the hypothesis
that duloxetine suppresses the activity of PSCs and production of
growth factors and cytokines that promote tumor-stromal
interaction.
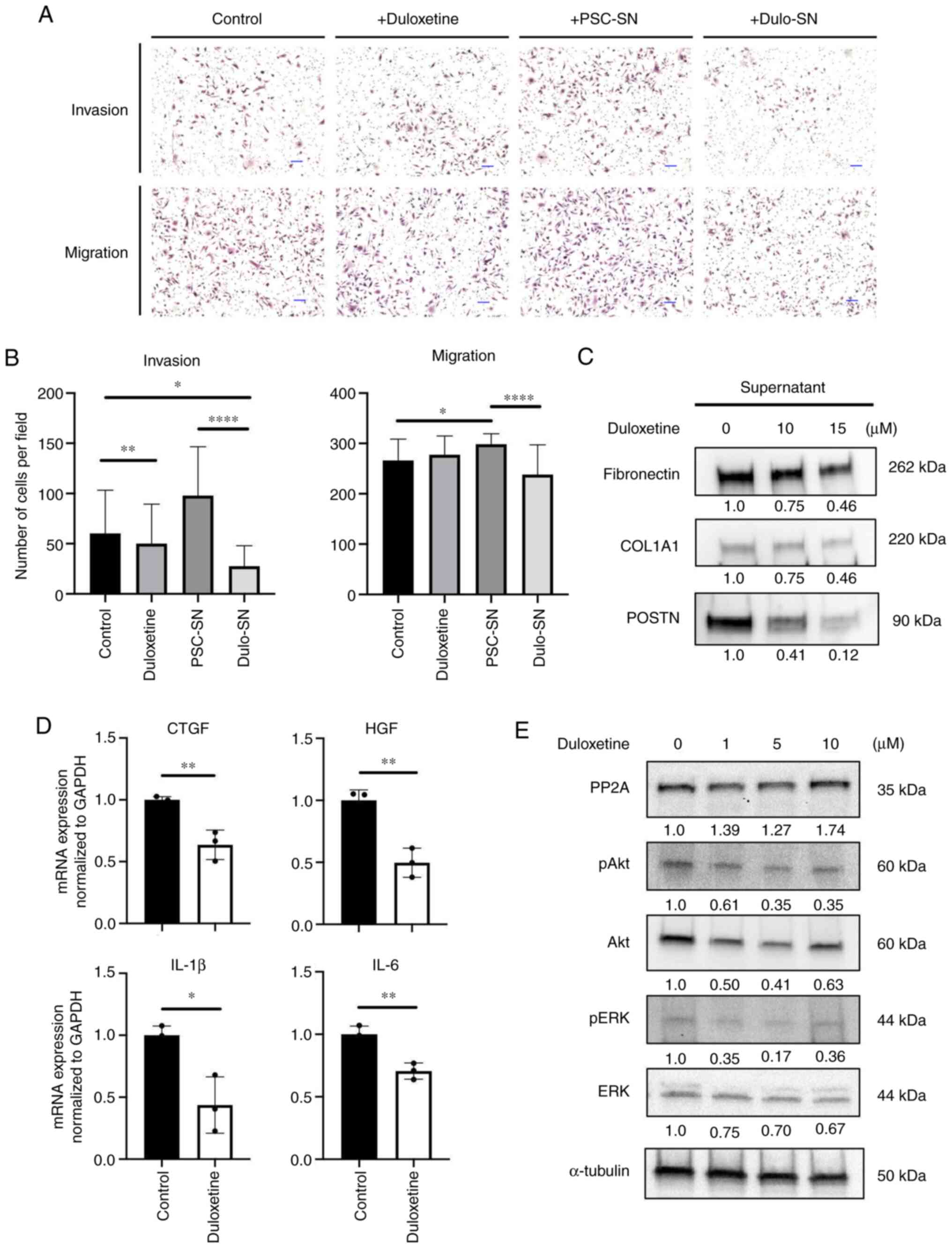 | Figure 4.Duloxetine suppresses tumor-stromal
interactions by attenuating secretomes from PSCs. (A)
Representative photomicrographs of invading and migrating SUIT-2
cells in monoculture and indirect coculture with drugs or
supernatants following hematoxylin and eosin staining.
Magnification, ×100. Scale bar, 100 µm. (B) Graphs show the number
of invading and migrating SUIT-2 cells. (C) Western blotting of
extracellular matrix proteins in PSC supernatant and drug-treated
PSC supernatant. (D) Relative mRNA expression of growth factors and
cytokines associated with tumor-stromal interactions in PSCs
treated with duloxetine. The expression levels of each gene were
normalized to GAPDH. (E) Western blotting of PP2A and related
proteins in whole cell lysates of PSCs treated with duloxetine at
various concentrations (1, 5 and 10 µM). Values indicate
densitometric ratios normalized to α-tubulin. *P<0.05,
**P<0.01, ****P<0.0001 vs. control group. PSCs, pancreatic
stellate cells; PSN-SN, PSC supernatant; Dulo-SN, supernatant from
PSCs treated with duloxetine; PP2A, protein phosphatase 2A; COL1A1,
α-1 type-1 collagen; POSTN, periostin; CTGF, connective tissue
growth factor; HGF, hepatocyte growth factor; IL-1β,
interleukin-1β; IL-6, interleukin-6; p, phosphorylated. |
To further confirm the mechanism of duloxetine
action, we used western blotting to determine the major secreted
proteins and cytokines that attenuate tumor-stromal interaction
from the PSC supernatant and whole cell lysates. Growth factors and
pro-inflammatory cytokines released from adjacent cells cause PSC
activation (22). Activated PSCs
maintain their activated state by autocrine stimulation of
cytokines, leading to proliferation, migration, and overproduction
of the ECM (23). The
duloxetine-treated PSC supernatant had fewer ECM protein amounts
than those in the control (Fig. 4C).
mRNA expression of connective tissue growth factor (CTGF),
hepatocyte growth factor (HGF), interleukin-1β (IL-1β), and
interleukin-6 (IL-6) in PSCs decreased significantly with
duloxetine treatment (Fig. 4D).
These findings suggested that duloxetine suppressed PSC activation
and reduced the quantity of the proteins and mRNA expression that
contribute to stromal stiffness and tumor-stromal interactions.
Duloxetine suppressed the activation
of Akt-ERK pathway via PP2A activation
Protein phosphatase 2A (PP2A) is known as a tumor
suppressor and is involved in the regulation of many cellular
functions, including extracellular signal-regulated kinase (ERK)
signaling (24). A previous study
revealed that PP2A contributes to cellular quiescence (24,25). To
confirm that duloxetine affects PP2A and suppresses PSC activation,
the protein expression was confirmed. In the western blot analysis,
the PP2A expression level was increased by duloxetine treatment
(Fig. 4E). Also, duloxetine
treatment decreased in Akt and ERK phosphorylation (Fig. 4E). These data suggested that
duloxetine reversed PP2A activation, which led PSCs into a
quiescent state.
Discussion
In this study, the pathway analysis using a public
microarray data of CAFs showed aberration hubs in the ‘neuroactive
ligand-receptor interaction’ pathway. This led to the
identification of a promising compound, duloxetine. Duloxetine
suppressed PSC activation and disrupted tumor-stromal interaction.
Together, these experiments elucidated the potential role of
duloxetine as an alternative drug for pancreatic cancer
treatment.
From the microarray analysis, other pathways
identified in the top 10 list were previously reported as targets
of cell proliferation and adhesion. We focused on the neuroactive
ligand-receptor interaction pathway because it is the only pathway
whose relationship with PSC activation has not been reported. Lipid
accumulation was detected in cells treated with all compounds
related to this pathway; therefore, we selected duloxetine, which
showed the highest lipid accumulation. Most of the drugs' targets
related to this pathway are G protein-coupled receptors (GPCRs).
GPCRs are located on the cell surface and are related to tumor cell
proliferation and invasion in various types of tumors (26,27).
More than 30% of approved drugs exert their therapeutic effects by
acting on GPCRs (28). Therefore,
they are recognized as potential targets of anti-cancer drugs
(29). There are some reports that
show the relationship between PSC activation and GPCRs. Cortes
et al observed that G protein-coupled estrogen receptor, a
GPER, is a mechanosensor of PSCs and can remodel stromal tissue,
thereby preventing tissue stiffness (30). Other research indicated that GPR68, a
proton-sensing GPCR, was expressed in CAFs and acted as a mediator
of the tumor microenvironment (31).
These prior studies support targeting one of the GPCRs as a
promising approach for suppressing PSC activation. However, this
study did not clarify whether the suppression of activation is
mediated by GPCRs and what structural differences exist in the
GPCRs that exert therapeutic effects. Given that GPCRs are widely
expressed across tissues and their role is mediated through various
signaling pathways (32), further
experiments are required to elucidate the mechanism of GPCR in PSC
function.
The PSC supernatant enhanced the invasion and
migration of PCCs as previously reported. These findings are due to
enhanced tumor-stromal interactions (22). PCC invasion and migration were
interrupted to a further extent by co-culturing with the
supernatant from duloxetine-treated PSCs and not with duloxetine
itself. This suggested that cytokines or proteins secreted by PSCs
were suppressed by duloxetine. During activation, PSCs receive
various stimuli from adjacent cells, and activated PSCs are potent
to secrete inflammatory cytokines and ECM proteins (33). We calculated ECM proteins and
cytokines secreted by activated PSCs and observed that these
secretions were decreased by duloxetine. Together, we concluded
that duloxetine not only has a toxic effect on PSCs, but can induce
inactive PSC.
Treatment strategies for reshaping the tumor stroma
have been widely discussed and investigated in the past decades. In
clinical settings, expression of pro-tumorigenic markers and ECM
components is correlated with a worse prognosis (34). In this study, PSCs treated with
duloxetine produced less ECM components, including collagen and
POSTN that constitute the tumor-supportive microenvironment
(35). Therefore, converting
pro-tumor PSCs into tumor-suppressing cells may be an ideal
approach for treating pancreatic tumors. It has also been revealed
that different fibroblast subtypes have different roles in
tumorigenesis and treatment response (36). In our study, PSCs derived from
different patient specimens showed different responses to
duloxetine. However, we performed experiments with a limited number
of PSCs and organoids from one PDAC patient sample. Further study
is needed to investigate the response based on the clinical feature
and changes in specific fibroblast subtype with duloxetine
treatment.
The relationship between antidepressants and cancer
has been discussed for over a decade. There is no strong evidence
of a relationship between them; however, the relationship between
antidepressants and carcinogenesis of colorectal cancer has been
reported (37). Other cancers showed
a tumor-suppressive effect by exhibiting antidepressant-like
properties in a mouse experiment (38,39).
Duloxetine has already been used in patients with chronic pains or
painful chemotherapy-induced peripheral neuropathy (40,41);
therefore, it is well-tolerated for clinical use. Kajiwara et
al recently demonstrated that duloxetine treatment improved
cancer-associated pain in a PDAC mouse model (42). They stated that duloxetine inhibited
the proliferation of PCCs and CAFs, which was consistent with our
report. Based on our results, duloxetine is a potential drug for
reducing the adverse effects of chemotherapy and enhancing the
effect of chemotherapy by targeting stromal remodeling.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms. Emiko Manabe and
Ms. Shoko Sadatomi (Department of Surgery and Oncology, Kyushu
University Hospital, Fukuoka, Japan) for their technical assistance
during experiments.
Funding
This study was supported in part by the Japan
Society for the Promotion of Science Grants-in-Aid (B) and (C), and
a Young Scientists Grant (grant nos. JP18H02880, JP19H03732,
JP19K18153 and JP20H03754), the Takeda Science Foundation,
Kobayashi Foundation for Cancer Research and The Shinnihon
Foundation of Advanced Medical Treatment Research.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AS, KN and MN conceived and designed the study. AS
performed the experiments, data collection and wrote the
manuscript. SM designed the experimental approach and contributed
to data interpretation and discussion. WG, TS and CI helped with
the investigation and formal analysis of data. NI and KO
contributed to the methodology and data analysis. KN and MN
supervised the project, obtained funding and confirm the
authenticity of all the raw data. AS and KN revised the manuscript.
MN approved the final version of the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
participants prior to using their pancreatic cancer surgical
specimens for the establishment of pancreatic stellate cells and
human organoids. This study was approved by the Ethics Committee of
Kyushu University (approval no. IRB: 28-189; Fukuoka, Japan), and
all experiments were conducted according to the Ethical Guidelines
for Human Genome/Gene Research enacted by the Japanese Government
and the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
αSMA
|
α-smooth muscle actin
|
|
CAF
|
cancer-associated fibroblast
|
|
COL1
|
type-1 collagen
|
|
CTGF
|
connective tissue growth factor
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
ECM
|
extracellular matrix
|
|
FBS
|
fetal bovine serum
|
|
HGF
|
hepatocyte growth factor
|
|
IL-1β
|
interleukin-1β
|
|
IL-6
|
interleukin-6
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GPCR
|
G-protein coupled receptor
|
|
mRNA
|
messenger RNA
|
|
PBS
|
phosphate-buffered saline
|
|
PCCs
|
pancreatic cancer cells
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
POSTN
|
periostin
|
|
PSCs
|
pancreatic stellate cells
|
|
PP2A
|
protein phosphatase 2A
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
References
|
1
|
Rawla P, Sunkara T and Gaduputi V:
Epidemiology of pancreatic cancer: Global trends, etiology and risk
factors. World J Oncol. 10:10–27. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Binenbaum Y, Na'ara S and Gil Z:
Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug
Resist Updat. 23:55–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parvathaneni V, Kulkarni NS, Muth A and
Gupta V: Drug repurposing: A promising tool to accelerate the drug
discovery process. Drug Discov Today. 24:2076–2085. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pushpakom S, Iorio F, Eyers PA, Escott KJ,
Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, et
al: Drug repurposing: Progress, challenges and recommendations. Nat
Rev Drug Discov. 18:41–58. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Apte MV, Haber PS, Darby SJ, Rodgers SC,
McCaughan GW, Korsten MA, Pirola RC and Wilson JS: Pancreatic
stellate cells are activated by proinflammatory cytokines:
Implications for pancreatic fibrogenesis. Gut. 44:534–541. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Apte MV, Wilson JS, Lugea A and Pandol SJ:
A starring role for stellate cells in the pancreatic cancer
microenvironment. Gastroenterology. 144:1210–1219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bachem MG, Schünemann M, Ramadani M, Siech
M, Beger H, Buck A, Zhou S, Schmid-Kotsas A and Adler G: Pancreatic
carcinoma cells induce fibrosis by stimulating proliferation and
matrix synthesis of stellate cells. Gastroenterology. 128:907–921.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bachem MG, Schneider E, Gross H,
Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grünert A and
Adler G: Identification, culture, and characterization of
pancreatic stellate cells in rats and humans. Gastroenterology.
115:421–432. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boj SF, Hwang CI, Baker LA, Chio II, Engle
DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al:
Organoid models of human and mouse ductal pancreatic cancer. Cell.
160:324–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koikawa K, Ohuchida K, Ando Y, Kibe S,
Nakayama H, Takesue S, Endo S, Abe T, Okumura T, Iwamoto C, et al:
Basement membrane destruction by pancreatic stellate cells leads to
local invasion in pancreatic ductal adenocarcinoma. Cancer Lett.
425:65–77. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Endo S, Nakata K, Ohuchida K, Takesue S,
Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, et al:
Autophagy is required for activation of pancreatic stellate cells,
associated with pancreatic cancer progression and promotes growth
of pancreatic tumors in mice. Gastroenterology. 152:1492–1506.e24.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sagara A, Nakata K, Yamashita T, Guan W,
Zhong P, Matsumoto S, Endo S, Iwamoto C, Shindo K, Ikenaga N, et
al: New high-throughput screening detects compounds that suppress
pancreatic stellate cell activation and attenuate pancreatic cancer
growth. Pancreatology. Apr 20–2021.(Epub ahead of print). doi:
10.1016/j.pan.2021.04.002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Özdemir BC, Pentcheva-Hoang T, Carstens
JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C,
Novitskiy SV, et al: Depletion of carcinoma-associated fibroblasts
and fibrosis induces immunosuppression and accelerates pancreas
cancer with reduced survival. Cancer Cell. 25:719–734. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiao Y, Zhang H, Ma Q, Huang R, Lu J,
Liang X, Liu X, Zhang Z, Yu L, Pang J, et al: YAP1-mediated
pancreatic stellate cell activation inhibits pancreatic cancer cell
proliferation. Cancer Lett. 462:51–60. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jakubowska MA, Ferdek PE, Gerasimenko OV,
Gerasimenko JV and Petersen OH: Nitric oxide signals are
interlinked with calcium signals in normal pancreatic stellate
cells upon oxidative stress and inflammation. Open Biol.
6:1601492016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jahchan NS, Dudley JT, Mazur PK, Flores N,
Yang D, Palmerton A, Zmoos AF, Vaka D, Tran KQT, Zhou M, et al: A
drug repositioning approach identifies tricyclic antidepressants as
inhibitors of small cell lung cancer and other neuroendocrine
tumors. Cancer Discov. 3:1364–1377. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marescal O and Cheeseman IM: Cellular
mechanisms and regulation of quiescence. Dev Cell. 55:259–271.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamaguchi E, Wang C, Fukazawa A, Taki M,
Sato Y, Sasaki T, Ueda M, Sasaki N, Higashiyama T and Yamaguchi S:
Environment-sensitive fluorescent probe: A benzophosphole oxide
with an electron-donating substituent. Angew Chemie Int Ed.
54:4539–4543. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pampaloni F, Reynaud EG and Stelzer EHK:
The third dimension bridges the gap between cell culture and live
tissue. Nat Rev Mol Cell Biol. 8:839–845. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kozono S, Ohuchida K, Eguchi D, Ikenaga N,
Fujiwara K, Cui L, Mizumoto K and Tanaka M: Pirfenidone inhibits
pancreatic cancer desmoplasia by regulating stellate cells. Cancer
Res. 73:2345–2356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bynigeri RR, Jakkampudi A, Jangala R,
Subramanyam C, Sasikala M, Rao GV, Reddy DN and Talukdar R:
Pancreatic stellate cell: Pandora's box for pancreatic disease
biology. World J Gastroenterol. 23:382–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu LG, Packman LC, Weldon M, Hamlett J and
Rhodes JM: Protein phosphatase 2A, a negative regulator of the ERK
signaling pathway, is activated by tyrosine phosphorylation of
putative HLA class II-associated protein I (PHAPI)/pp32 in response
to the antiproliferative lectin, jacalin. J Biol Chem.
279:41377–41383. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Janssens V and Goris J: Protein
phosphatase 2A: A highly regulated family of serine/threonine
phosphatases implicated in cell growth and signalling. Biochem J.
353:417–439. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Naetar N, Soundarapandian V, Litovchick L,
Goguen KL, Sablina AA, Bowman-Colin C, Sicinski P, Hahn WC,
DeCaprio JA and Livingston DM: PP2A-mediated regulation of ras
signaling in G2 is essential for stable quiescence and normal G1
length. Mol Cell. 54:932–945. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li L and Hanahan D: Hijacking the neuronal
NMDAR signaling circuit to promote tumor growth and invasion. Cell.
153:86–100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arakaki AKS, Pan WA and Trejo JA: GPCRs in
cancer: Protease-activated receptors, endocytic adaptors and
signaling. Int J Mol Sci. 19:18862018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shiraishi Y, Natsume M, Kofuku Y, Imai S,
Nakata K, Mizukoshi T, Ueda T, Iwaï H and Shimada I:
Phosphorylation-induced conformation of β2-adrenoceptor
related to arrestin recruitment revealed by NMR. Nat Commun.
9:1942018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nieto Gutierrez A and McDonald PH: GPCRs:
Emerging anti-cancer drug targets. Cell Signal. 41:65–74. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cortes E, Sarper M, Robinson B, Lachowski
D, Chronopoulos A, Thorpe SD, Lee DA and Hernández AE: GPER is a
mechanoregulator of pancreatic stellate cells and the tumor
microenvironment. EMBO Rep. 20:e465562019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiley SZ, Sriram K, Liang W, Chang SE,
French R, McCann T, Sicklick J, Nishihara H, Lowy AM and Insel PA:
GPR68, a proton-sensing GPCR, mediates interaction of
cancer-associated fibroblasts and cancer cells. FASEB J.
32:1170–1183. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lappano R and Maggiolini M: G
protein-coupled receptors: Novel targets for drug discovery in
cancer. Nat Rev Drug Discov. 10:47–60. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Omary MB, Lugea A, Lowe AW and Pandol SJ:
The pancreatic stellate cell: A star on the rise in pancreatic
diseases. J Clin Invest. 117:50–59. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miyamoto H, Murakami T, Tsuchida K, Sugino
H, Miyake H and Tashiro S: Tumor-stroma interaction of human
pancreatic cancer: Acquired resistance to anticancer drugs and
proliferation regulation is dependent on extracellular matrix
proteins. Pancreas. 28:38–44. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Erkan M, Kleeff J, Gorbachevski A, Reiser
C, Mitkus T, Esposito I, Giese T, Büchler MW, Giese NA and Friess
H: Periostin creates a tumor-supportive microenvironment in the
pancreas by sustaining fibrogenic stellate cell activity.
Gastroenterology. 132:1447–1464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Öhlund D, Handly-Santana A, Biffi G,
Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA,
Lee EJ, et al: Distinct populations of inflammatory fibroblasts and
myofibroblasts in pancreatic cancer. J Exp Med. 214:579–596. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu W, Tamim H, Shapiro S, Stang MR and
Collet JP: Use of antidepressants and risk of colorectal cancer: A
nested case-control study. Lancet Oncol. 7:301–308. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tegowski M, Fan C and Baldwin AS:
Thioridazine inhibits self-renewal in breast cancer cells via
DRD2-dependent STAT3 inhibition, but induces a G1 arrest
independent of DRD2. J Biol Chem. 293:15977–15990. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gwynne WD, Hallett RM, Girgis-Gabardo A,
Bojovic B, Dvorkin-Gheva A, Aarts C, Dias K, Bane A and Hassell JA:
Serotonergic system antagonists target breast tumor initiating
cells and synergize with chemotherapy to shrink human breast tumor
xenografts. Oncotarget. 8:32101–32116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Smith EML, Pang H, Cirrincione C,
Fleishman S, Paskett ED, Ahles T, Bressler LR, Fadul CE, Knox C,
Le-Lindqwister N, et al: Effect of duloxetine on pain, function,
and quality of life among patients with chemotherapy-induced
painful peripheral neuropathy: A randomized clinical trial. JAMA.
309:1359–1367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hirayama Y, Ishitani K, Sato Y, Iyama S,
Takada K, Murase K, Kuroda H, Nagamachi Y, Konuma Y, Fujimi A, et
al: Effect of duloxetine in Japanese patients with
chemotherapy-induced peripheral neuropathy: A pilot randomized
trial. Int J Clin Oncol. 20:866–871. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kajiwara I, Sano M, Ichimaru Y, Oshima Y,
Kitajima O, Hao H, Masamune A, Kim J, Ishii Y, Ijichi H, et al:
Duloxetine improves cancer-associated pain in a mouse model of
pancreatic cancer through stimulation of noradrenaline pathway and
its antitumor effects. Pain. 161:2909–2919. 2020. View Article : Google Scholar : PubMed/NCBI
|