Introduction
Glioblastoma multiforme (GBM) is the most common type of primary brain tumor in adults. GBM has a high degree of malignancy and strong invasive ability and, therefore, usually has a poor prognosis. The overall survival time following diagnosis is ~12 months (1). The prognosis remains unchanged following maximal surgical resection, chemotherapy and radiotherapy (2). Therefore, in order to investigate and discover new therapeutic targets and develop more optimized and effective therapeutic strategies, it is necessary to further understand the molecular mechanisms underlying the development and progression of GBM (3). According to The Cancer Genome Atlas, GBM may be genetically classified into the following four subtypes: Mesenchymal, classical, neural and proneural (4). Previous studies have reported that the mesenchymal is the most aggressive subtype (5) and is associated with an inflammatory microenvironment, angiogenesis and drug resistance (6). Radiotherapy and chemotherapy can increase GBM resistance and invasiveness by promoting proneural-to-mesenchymal transition (5). Furthermore, numerous factors are involved in gliomagenesis, including numerous signaling pathways that are crucial for GBM development, such as the JAK/STAT (4,7,8), p53, retinoblastoma (RB) and receptor tyrosine kinase (RTK) signaling pathways (9). Mutations in isocitrate dehydrogenase 1 and 2 (IDH1/2) can reduce enzymatic activity and induce the development of GBM (10). Moreover, the deletion of chromosome 1p36 and 19q13 can cause oligodendroglioma (11). However, current research on the mechanisms underlying gliomagenesis remains insufficient and further investigation is needed for future diagnosis, classification and treatment of GBM (12).
Suppressor of cytokine signaling (SOCS) proteins are cytokine-inducible proteins that inhibit cytokine signaling in a number of different signaling pathways. The SOCS protein family comprises eight members: SOCS1-SOCS7 and cytokine-inducible SH2-containing protein (CIS). Structurally, these proteins are similar, as they each contain the Src homology 2 (SH2) domain, a variable amino-terminal domain and a carboxy-terminal SOCS box (13). Functionally, it was previously indicated that cytokine signaling may serve potentially negative roles in GBM, such as promoting GBM cell proliferation, migration and angiogenesis via the C-X-C motif chemokine ligand 2/IL-8/C-X-C motif chemokine receptor 2 pathway (14), while the SOCS family may serve a positive role in GBM mediated by certain signaling pathways or immune molecules; for example, SOCS can inhibit GBM cell proliferation and angiogenesis by negatively regulating the JAK/STAT3 signaling pathway (15). Therefore, the role of SOCS proteins in GBM has recently become a focus in GBM research.
The occurrence, progression, diagnosis and prognosis of numerous human tumors, including GBM, are associated with the SOCS protein family. SOCS3, which is one of the most well-studied members of the SOCS family, inhibits the JAK/STAT signaling pathway by binding to an activated receptor (16). Furthermore, SOCS3 inhibits the NF-κB signaling pathway, antagonizing cAMP-mediated signal transduction and enhancing signal transduction via the MAPK signaling pathway (17–19). In GBM, E3 ubiquitin ligases regulate important cellular functions, including the regulation of the RTK survival signal. Overexpression or mutation of numerous kinase family members, including RTK, may cause the development of malignancies; however, SOCS proteins serve a negative regulatory role in RTK signaling (20). The PI3K/Akt signaling pathway is abnormally activated in the majority of patients with GBM (21). Furthermore, casein kinase 1 (CK1) and glycogen synthase kinase-3β (GSK-3β) promote cytoplasmic degradation of IκB and NF-κB-mediated nuclear transcription to inhibit apoptosis (22). NF-κB can induce the expression of SOCS3 and activate the NF-κB signaling pathway by binding to the SOCS promoter and initiating gene transcription (23). SOCS3 ultimately acts as a negative regulator of the JAK/STAT3 signaling pathway (24). Overall, the SOCS protein family may be implicated in the occurrence, progression and prognosis of numerous tumors; SOCS3, in particular, may function as a tumor suppressor (25).
All cullin-RING E3 ligases (CRLs) share a conserved molecular structure. SOCS proteins are important substrate recognition modules for CRLs, which inhibit cytokine-induced signal transduction cascades by promoting ubiquitination of signal transduction intermediates that are then degraded by targeted proteasomes. For example, in the case of CRL5, SOCS interacts with the JAK/STAT signaling pathway to mediate the ubiquitination and degradation of cytokines and exert a tumor-inhibitory effect (26).
Furthermore, SOCS3 hypermethylation may be an important predictor of poor prognosis (27). The SOCS family has been proven to be involved in the invasion and metastasis of numerous types of tumors (28), but their role in the invasion and metastasis of GBM remains unclear. In addition, the significance of SOCS proteins in the treatment of patients with GBM, such as their roles in postoperative radiotherapy and chemotherapy, remains to be further investigated. The aim of the present review was primarily to summarize the regulatory role of SOCS proteins in GBM and their clinical significance, the interaction of E3 ubiquitin ligase and SOCS proteins and their role in GBM, the modified SOCS protein substrates and the therapeutic value of targeting SOCS proteins.
SOCS proteins
Biogenesis of SOCS proteins
In 1995, Yoshimura et al (29) discovered that a cytokine-responsive gene was upregulated during IL-3 treatment of hematopoietic stem cells. This gene was later named CIS. Starr et al in 1997 (30) and Nicholson and Hilton in 1998 (31) identified a new family of negative regulators in the monocytic leukemia M1 cell line via retrovirus expression in a hematopoietic cDNA library. SOCS1 was initially screened and cloned, and it was found that SOCS1 could bind to JAK to inhibit cytokine signaling. Similar to SOCS1, the other seven cDNAs (CIS and SOCS2-SOCS7) were shown to encode proteins containing a SOCS box. Members of the SOCS protein family have a conserved structure, including an N-terminal region, a central SH2 homologous region and a C-terminal region containing the SOCS box. Furthermore, both SOCS1 and SOCS3 genes encode a pseudo-substrate, which is a unique kinase-inhibitory region (KIR) and exists upstream of the central SH2 domain (25). SOCS protein activity is closely associated with the specificity of the SOCS-SH2 domain, which interacts with signaling molecules and, therefore, determines the signaling pathways that are regulated (28). Structurally, the C-terminal regions of SOCS proteins contain a sequence of 40 conserved amino acids, with the N-terminal regions varying in length, ranging from 50 to 380 amino acids. The N-terminal regions of the SOCS proteins contain no recognizable sequences, and only SOCS7 contains a nuclear localization signal and multiple proline-rich regions (Fig. 1) (32). It has also been determined that the expression levels of SOCS3 and SOCS1 are the highest in central nervous system cells, such as microglia and astrocytes, and the abnormal expression of SOCS3 and SOCS1 is associated with the poor prognosis of GBM (25). Furthermore, previous studies have reported that other SOCS family members can change the phenotype of GBM cells via signaling pathways such as the JUN/microRNA (miRNA/miR)-494/SOCS6 (33) and miR-599/androgen receptor (AR)/SOCS2-antisense RNA 1 (AS1) signaling pathways (34). However, further in vivo and in vitro experiments must be performed.
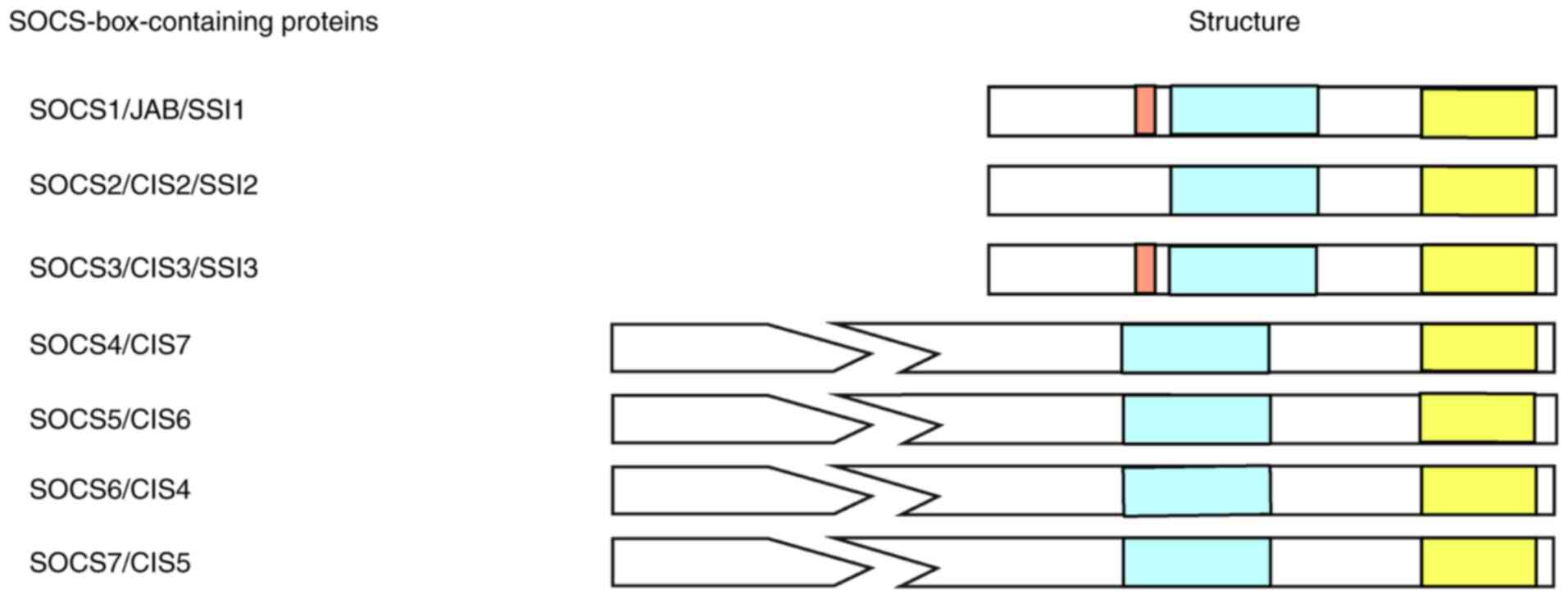 |
Figure 1.
The SOCS box motif of the SOCS protein family. SOCS family members (SOCS1-SOCS7 and CIS) contain a SOCS box, as well as a central SH2 homology region, which is shown in yellow. The kinase inhibitory region of SOCS1 and SOCS3 is marked in red, which is located in the upstream of the central SH2 domain. The N-terminal regions of SOCS proteins are relatively non-conserved in length, ranging between 50 and 380 amino acids. SOCS, suppressor of cytokine signaling; CIS, cytokine-inducible SH2-containing protein.
|
Mechanism of action of SOCS proteins
SOCS boxes consist of ~40 amino acids, have strong homology and are highly conserved. The SOCS-SH2 domain is located directly upstream of the SOCS box and is distinguished by an N-terminal α-helix known as the extended SH2 subdomain, which stabilizes the phosphorylated tyrosine-binding loop (35,36). By binding the SH2 domain to phosphorylated residues in the receptor complex, the SOCS box-associated E3 ubiquitin ligase becomes attached to its substrate (37). Functionally, SOCS proteins are hypothesized to negatively regulate the JAK/STAT signaling pathway by binding to the JAK/cytokine receptors or by competing with the STAT protein for phosphorylation docking sites on the receptor. Moreover, the KIR of SOCS1 and SOCS3 is crucial for inhibiting JAK. In addition, KIR serves an important regulatory role by interacting with elongin B, cullin-5 or cullin-2, RING box protein (RBX) 1 and E2 to degrade signaling proteins that bind to them, preventing E3 ubiquitin ligase targeting, while preventing the degradation of the proteins themselves, thereby regulating the stability of signaling molecules in the cell and inhibiting cell signal transduction (38–40). In order to mediate proteasomal degradation of related proteins, C-terminal SOCS boxes interact with components of the ubiquitin ligase mechanism (41). For certain endogenous SOCS proteins, due to the lack of a reliable detection method, the protein and mRNA expression levels of different subtypes of SOCS and the relationship between mRNA and protein expression levels remain unclear (42).
Functions of SOCS proteins
SOCS1 and SOCS3
SOCS1 inhibits IFN signaling by interacting with the IFN-α/β receptor subunit 1 and the IFN-γ receptor subunits, limiting the IFN activation of STAT3 (43). SOCS1 can also interfere with a wide range of signaling pathways by promoting the degradation of p65 of the NF-κB signaling pathway and kinase apoptosis signal-regulating kinase 1 upstream of the JNK and p38 signaling pathways (25). SOCS3 inhibits the signaling of the IL-6 cytokine family (44), and signaling mediated by lipopolysaccharide, type I and II IFN, growth hormone, IL-2, IL-3 and IL-4, participating in a wide range of immune responses (24,45–47). SOCS3 also inhibits the NF-κB signaling pathway, antagonizing cAMP-mediated signaling (26) and enhancing signaling via the MAPK pathway (43).
It was previously demonstrated that reduced SOCS1 and SOCS3 expression levels in human astrocytoma can lead to the phosphorylation of STAT3, which activates the JAK/STAT3 signaling pathway and indicates a poor prognosis (48). The mutual regulation of SOCS1 and SOCS3 can enhance GBM resistance to ionizing radiation (49); SOCS1 and SOCS3 may also serve as epigenetic regulators of GBM hypermethylation, which is associated with a poor GBM prognosis (50). However, the pathogenic role of SOCS1 and SOCS3 remains incompletely understood and further research is needed.
Other SOCS family members: SOCS2, 4, 5, 6, 7 and CIS
At present, there are relatively few studies on the role of the remaining SOCS family members (CIS and SOCS2, 4, 5, 6 and 7) in GBM, whereby most studies regulate the phenotypes of GBM cell lines using the targeted action of miRNAs. It was previously demonstrated that methylation of the SOCS2 promoter may be associated with a poor GBM prognosis (50). The circular (circ)RNA-aspartyl/asparaginyl β-hydroxylase (circ-ASPH) can promote the invasion of GBM cells by targeting the miR-599/AR/SOCS2-AS1 signaling pathway (34). Furthermore, miR-296-3p is regulated by neurofibromatosis 2 (NF2) and enhances the invasiveness of GBM cells via the SOCS2/STAT3 signaling pathway. Overexpression of miR-296-3p can inhibit the expression of STAT5A, inducing STAT3 phosphorylation by downregulating SOCS2 and, therefore, increasing the invasiveness of T98G cells (51). It was also previously demonstrated that SOCS6 can cause a change in the GBM cell phenotype via the JUN/miR-494/SOCS6 signaling pathway (33). Khalighfard et al (52) reported that miR-1290 directly targets the 3′-untranslated region (UTR) of the SOCS4 gene and inhibits its expression, which increases GBM cell proliferation, migration, invasion and resistance to chemotherapy and radiation. Moreover, Liu et al (53) determined that long intergenic non-protein coding RNA (LINC)00668 promoted glioma cell proliferation by regulating SOCS5 expression via competitively sponging miR-518c-3p. However, SOCS7 and CIS have rarely been studied in GBM. Therefore, more in vitro and in vivo experiments are needed to explore the role of these SOCS family members in GBM.
SOCS proteins and E3 ubiquitin ligase
Roles of E3 ubiquitin ligase
E3 ligases can be divided into three subtypes, including homologous to the E6-associated protein C-terminus domain ligases, RING E3 ligases, and RING-in-between-RING E3 ligases (54). In humans, most E3 ubiquitin ligases are of the RING subtype, with >600 falling into this category. In GBM, certain E3 ubiquitin ligases are involved in regulating the ubiquitination of SOCS proteins, such as the CRL E3 ligase family, which contains the cullin scaffold protein CRL5 complex, cullin 5-elongin B/C-SOCS (55,56). SOCS proteins link the BC complex and cullin 5 to promote JAK ubiquitination, thereby inhibiting the JAK/STAT signaling pathway (57). The ubiquitin-proteasome system (UPS) is an important protein degradation pathway in eukaryotes (58). Abnormalities in proteins regulated by UPS are closely associated with numerous types of human cancer (59), including GBM (60). Ubiquitin modification is a multi-stage process. The E1-activating enzyme is activated by ATP, and then transferred to the E2 ubiquitin-binding enzyme, which is connected to the active site of E2 by a thioester bond. E2 is activated by binding to a specific E3 ubiquitin ligase (Fig. 2) (61). E3 ubiquitin ligase serves an important role in determining the specificity of the UPS. E3 ubiquitin ligase is involved in numerous cellular processes, such as signal transduction, DNA repair and replication, by regulating the ubiquitination of proteins (62).
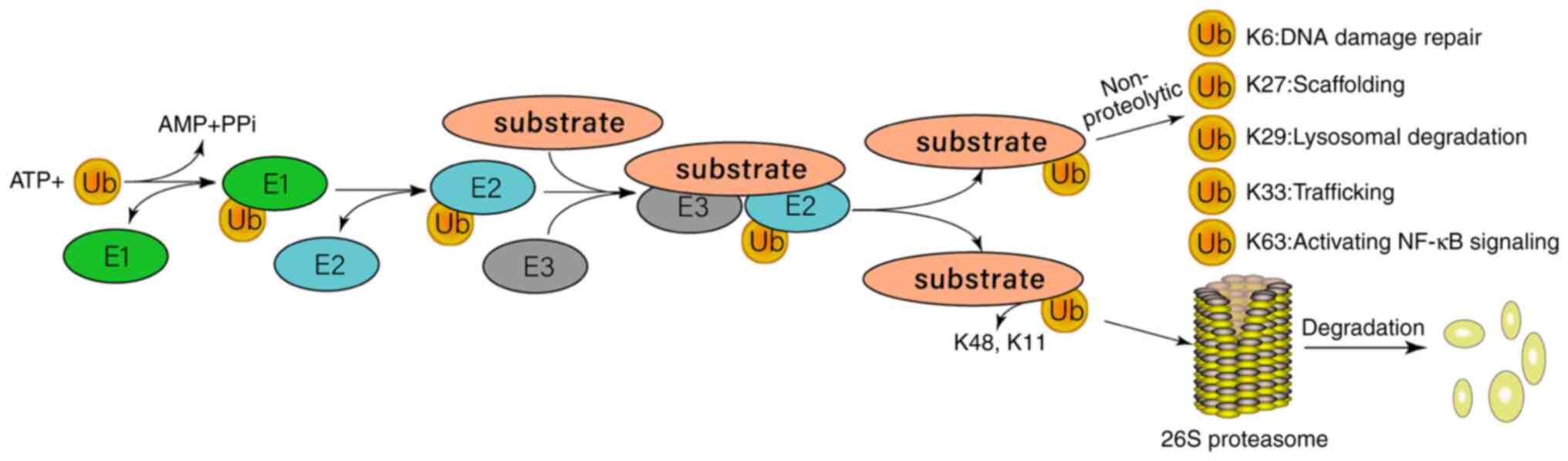 |
Figure 2.
Overview of the ubiquitin-proteasome system response process. Ubiquitin can help with protein degradation or regulate other biological functions via E1-E2-E3 catalytic cascade reaction. Ubiquitin is connected to E1 in an ATP-dependent manner and is activated by forming a ubiquitin thioester. E2 carries ubiquitin via thioester bonds. Finally, E3 transfers the activated ubiquitin to the lysine residue of the substrate. Ubiquitin binding to K48 or K11 causes substrate degradation. Ubiquitin binding to K63 represents non-protein ubiquitination and regulates a variety of biological processes, including DNA repair and replication, transcriptional regulation and signal transduction. K6, K11, K27, K29, K33, K48 and K63 indicate the lysine residues that serve as points of ubiquitination.
|
Relationship between E3 ubiquitin ligases and SOCS proteins
All CRLs possess a conserved molecular structure that recognizes a range of substrate compositions, has different modalities of regulation and unique ways of connecting the UB component (Fig. 3). The two most characteristic subfamilies of CRLs are the Skp1-cullin-F-box (SCF) and elongin-C-cullin-SOCS-box families, with the latter including the SOCS box-containing proteins and the von Hippel-Lindau (VHL) tumor suppressor. VHL and SOCS box proteins bind to cullin scaffolds using heterodimers of elongin B and C. SOCS box proteins are also recognized as important substrate recognition modules for CRLs (63). SOCS proteins partially inhibit the cytokine-induced signal transduction cascade by promoting ubiquitination of signal transduction intermediates, which are then targeted for proteasomal degradation. The process relies on interactions between the SOCS box domain, the adapter complex elongin B and elongin C and the E3 ubiquitin ligase scaffold protein cullin family (64).
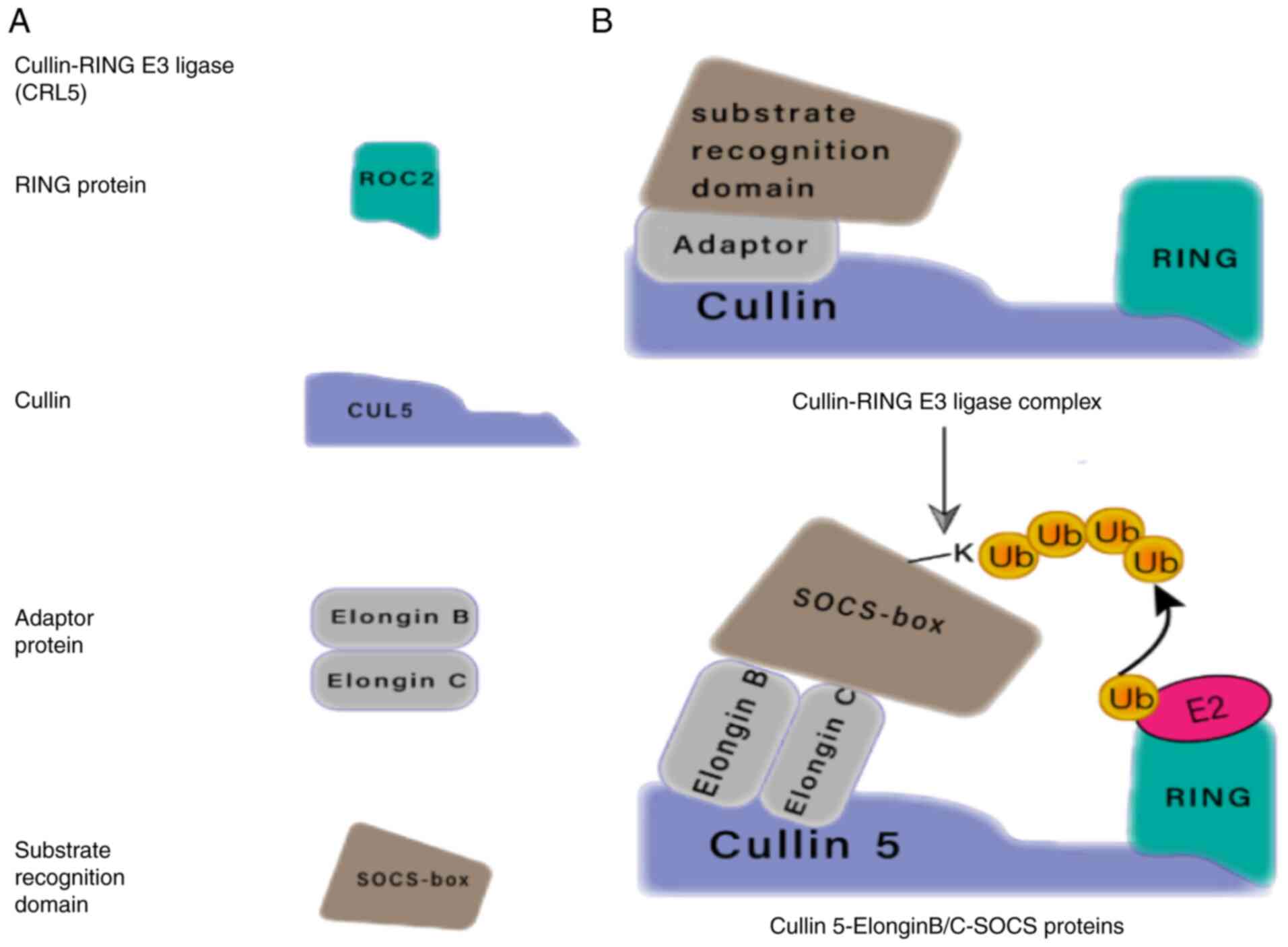 |
Figure 3.
Modularity of CRLs. (A) Components of the CRL5. (B) Ring E3 ligases can be organized into multi-subunit complexes consisting of cullin, E2-binding Ring-box proteins, and the connector (adaptor) proteins that mediate substrate recognition. For CRL5, the RING E3 ligase connector may be elongin B/C and the substrate-binding protein may be SOCS-box. SOCS, suppressor of cytokine signaling; CRLs, cullin-RING E3 ligases.
|
The CRL5 cullin 5-elongin B/C-SOCS ubiquitin ligase complex consists of cullin 5, RBX2, elongin B/C and SOCS proteins, among which the SOCS proteins have a substrate recognition function that enables the complex to interact with JAK proteins in the JAK/STAT signaling pathway (65). Furthermore, SOCS proteins inhibit the kinase activity of the JAK protein, which prevents the STAT protein from binding to the corresponding cytokine receptors and mediates the ubiquitination and degradation of cytokines (26). In summary, the CRL5 cullin 5-Ring E3 ubiquitin ligase complex is negatively associated with innate immunity. The occurrence and development of tumors are often associated with the inflammatory response, and SOCS proteins are believed to serve a tumor suppressive role by inhibiting the chronic inflammatory response (66). The expression of SOCS proteins has been found to be significantly downregulated in tumor tissues and the expression level of SOCS proteins has been shown to be positively correlated with the survival of patients with glioma (50). However, the molecular mechanism of the role of SOCS proteins in the CRL5 complex (cullin 5-elongin B/C-SOCS-E3 ubiquitin ligase), as well as in the initiation and progression of GBM, is not well understood and therefore needs to be explored further.
RTK signaling regulation in GBM by SOCS proteins and E3 ubiquitin ligases
In GBM, E3 ubiquitin ligases regulate important cellular functions, including RTK pro-survival signaling (67). Members of the RTK family are associated with numerous malignancies caused by the overexpression or mutation of RTK proteins, and have therefore become potential targets for cancer therapeutics (67). In RTK signal transduction, the activation of RTK results in the transcriptional activation of SOCS coding genes; therefore, SOCS proteins serve an important regulatory role (68). The SH2 domain of SOCS proteins interacts with RTK, which mobilizes the E3 ubiquitination mechanism to induce ubiquitination and limit receptor stability. SOCS proteins serve a negative regulatory role in RTK signaling (20). Furthermore, EGF signaling is a typical example of RTK signal changes in GBM (67). Phosphorylation of EGF results in the activation of the JAK/STAT and PI3K/Akt signaling pathways, thus generating downstream signals (69). Numerous studies have demonstrated that SOCS can regulate EGFR (26). For example, SOCS1 and SOCS3 can block EGF-induced activation of STAT1 and STAT3 signaling pathways (70), whereas SOCS2 can inhibit EGF-induced phosphorylation of the STAT5 signaling pathway (71), and SOCS4 can reduce EGFR expression levels (72). CIS negatively regulates EGF-induced phosphorylation of STAT3 (73). On the cell membrane, E3 ubiquitin ligase reduces carcinogenic signals by promoting EGFR ubiquitination, whereas in the nucleus, the E3 ubiquitin ligase TRIM24 regulates the JAK/STAT signaling pathway (Fig. 4) (67).
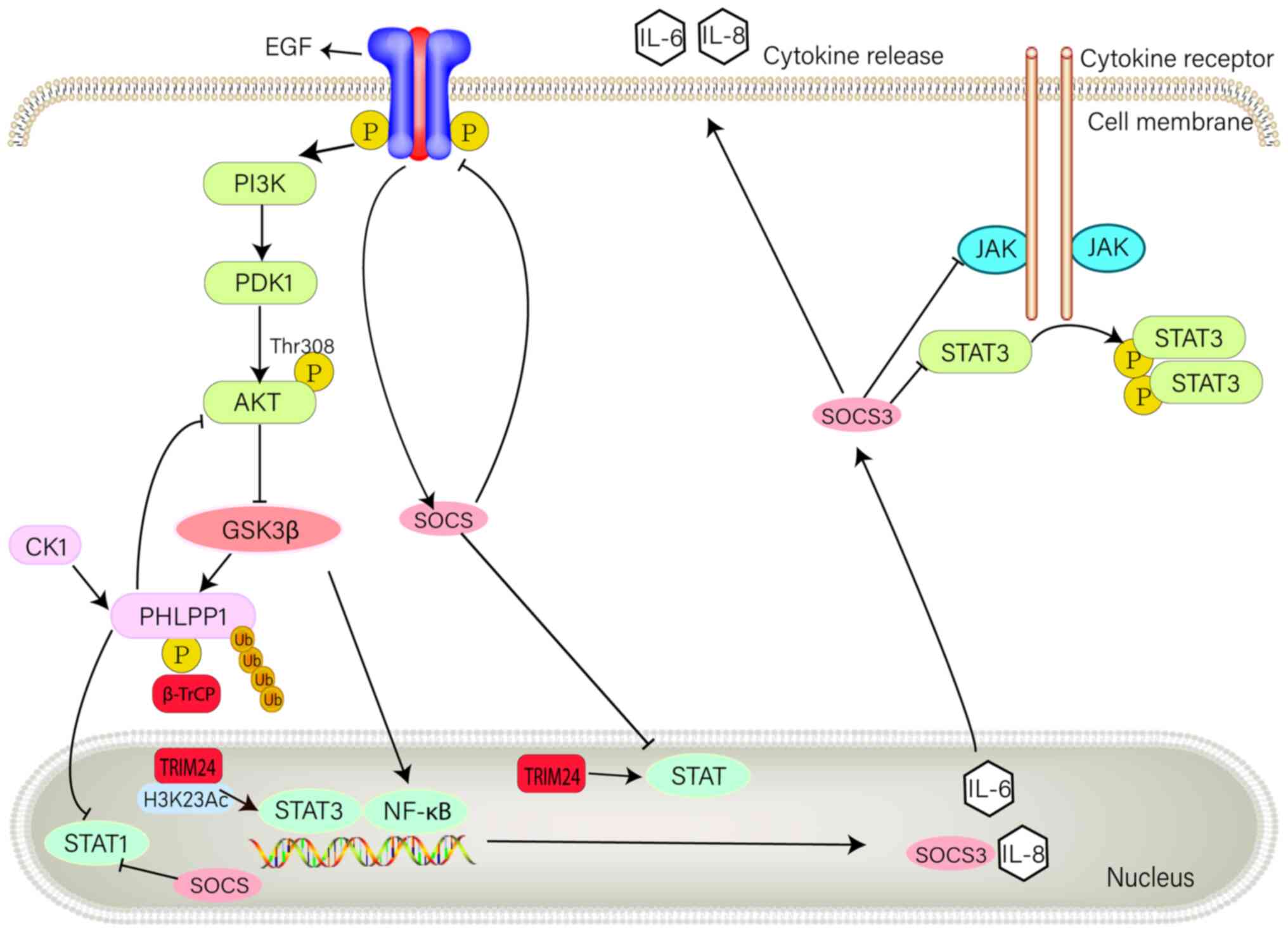 |
Figure 4.
Role of E3 ubiquitin ligases and SOCS3 in RTK signaling pathway in glioblastoma multiforme. RTK is a transmembrane receptor containing extracellular, transmembrane and intracellular parts. Extracellular domains interact with the extracellular environment, allowing expressed cells to respond and adapt to extracellular signals. Upon binding to their homologous ligands, RTKs undergo autophosphorylation, leading to downstream signaling. The role of RTK signaling in the PI3K/Akt pathway is shown in the figure, as well as its association with SOCS3. SOCS, suppressor of cytokine signaling; RTK, receptor tyrosine kinase; GSK, glycogen synthase kinase; PDK, pyruvate dehydrogenase kinase; PHLPP1, pleckstrin homology domain leucine-rich repeat protein phosphatase; CK1, casein kinase 1.
|
The majority of patients with GBM exhibit changes in the PI3K/Akt signaling pathway (74). Akt signaling is negatively regulated by the phosphorylation of Akt hydrophobic units by Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP1) (75). PHLPP1 is recognized by the substrate binding F-box protein and β-transducin repeat-containing protein (β-TrCP), following the phosphorylation of CK1 and GSK-3β. β-TrCP is the substrate recognition protein for SCFβ−TrCP (22). Recognition of PHLPP1 by SCFβ−TrCP results in its ubiquitination and subsequent degradation by the proteasome. It was previously demonstrated that deficient ubiquitin degradation of PHLPP1 in colon cancer cells can effectively dephosphorylate Akt and inhibit tumor cell proliferation. Therefore, E3 ubiquitin ligase β-TrCP serves an important role in regulating PHLPP1, which provides a novel and alternative focus for the study of GBM (76). Furthermore, PHLPP1 inhibits STAT1 phosphorylation and, therefore, its transcriptional activity in the nucleus, uncovering a potential mechanism for SOCS protein synergistic inhibition of STAT phosphorylation as a way to regulate GBM development (77).
Evidence suggests that inhibition of GSK-3β induces apoptosis in various types of cancer. GSK-3β-deficient mice display hepatocyte apoptosis and embryonic lethality, whereas cells from GSK-3β-deficient mice display reduced NF-κB signaling activity, which suggests that the inhibition of GSK-3β-induced apoptosis occurs via the NF-κB signaling pathway (78). GSK-3β phosphorylation of the NF-κB essential modulator has been reported to promote the nuclear localization and transcription of NF-κB (79). Therefore, GSK-3β may inhibit apoptosis by promoting cytoplasmic IκB degradation and NF-κB-mediated nuclear transcription (80). As NF-κB is inactivated in the cytoplasm, IκBα is degraded when p65 and the NF-κB p50 subunit are phosphorylated, which activates the NF-κB signaling pathway (81). When entering the nucleus, the phosphorylated dimer of NF-κB binds to the promoters of IL-6, IL-8 and SOCS3 to initiate gene transcription. IL-6 and IL-8 are translated into proteins and secreted. SOCS3 is also translated and acts as a negative regulator of the JAK/STAT3 signaling pathway, inhibiting STAT3 activation (25).
SOCS proteins and signal transduction
As NF-κB is inactivated in the cytoplasm, IκBα is degraded when p65 and p50 are phosphorylated, thereby activating the NF-κB signaling pathway (82). In GBM, RTKs, including EGFRs and platelet-derived growth factor (PDGF) receptors, emit abnormal signals, which promote cell proliferation and inhibit apoptosis while maintaining the tumor environment. Furthermore, uncontrolled activation of the MAPK, NF-κB, PI3K/Akt and JAK/STAT signaling pathways is associated with GBM development and progression (83–85).
SOCS proteins and the JAK/STAT signaling pathway
The JAK/STAT signaling pathway is important in the biogenesis of GBM (86). JAK/STAT signaling is involved in a variety of regulatory mechanisms, interacting with SOCS proteins and the protein inhibitor of activated STAT (PIAS) family (25). Inducible expression of SOCS proteins in the cytoplasm attenuates STAT signaling activity by inhibiting upstream JAK activation in a negative feedback loop (87). Activated STAT3 induces SOCS3 expression, which inhibits STAT3 signal transduction by binding and weakening the cytokine receptor associated with glycoprotein 130 and the associated JAK protein (30,88,89). In an immunohistochemical study, GBM cells have been determined to contain tyrosine-phosphorylated STAT3 (86). Activated STAT3 is localized to VEGFR2 in tumor endothelial cells (86,90–92) and is a major transcriptional regulator of GBM (26). Furthermore, experimental mouse gliomas expressed constitutively activated STAT3 and IL-6, which are therefore considered to be associated with brain tumors (90,91,93). In GBM cells, upregulation of IL-6 and oncostatin M modulates VEGF promoter activity, potentially promoting angiogenesis (93,94). Moreover, the PIAS protein interferes with DNA binding via its NH2 terminal domain, which inhibits the transcriptional activity of STAT (94). Data collected from cell lines, mouse models and patient samples suggest that high expression of IL-6 in GBM may be associated with the composition, induction and activation of STAT3 (Fig. 5) (93).
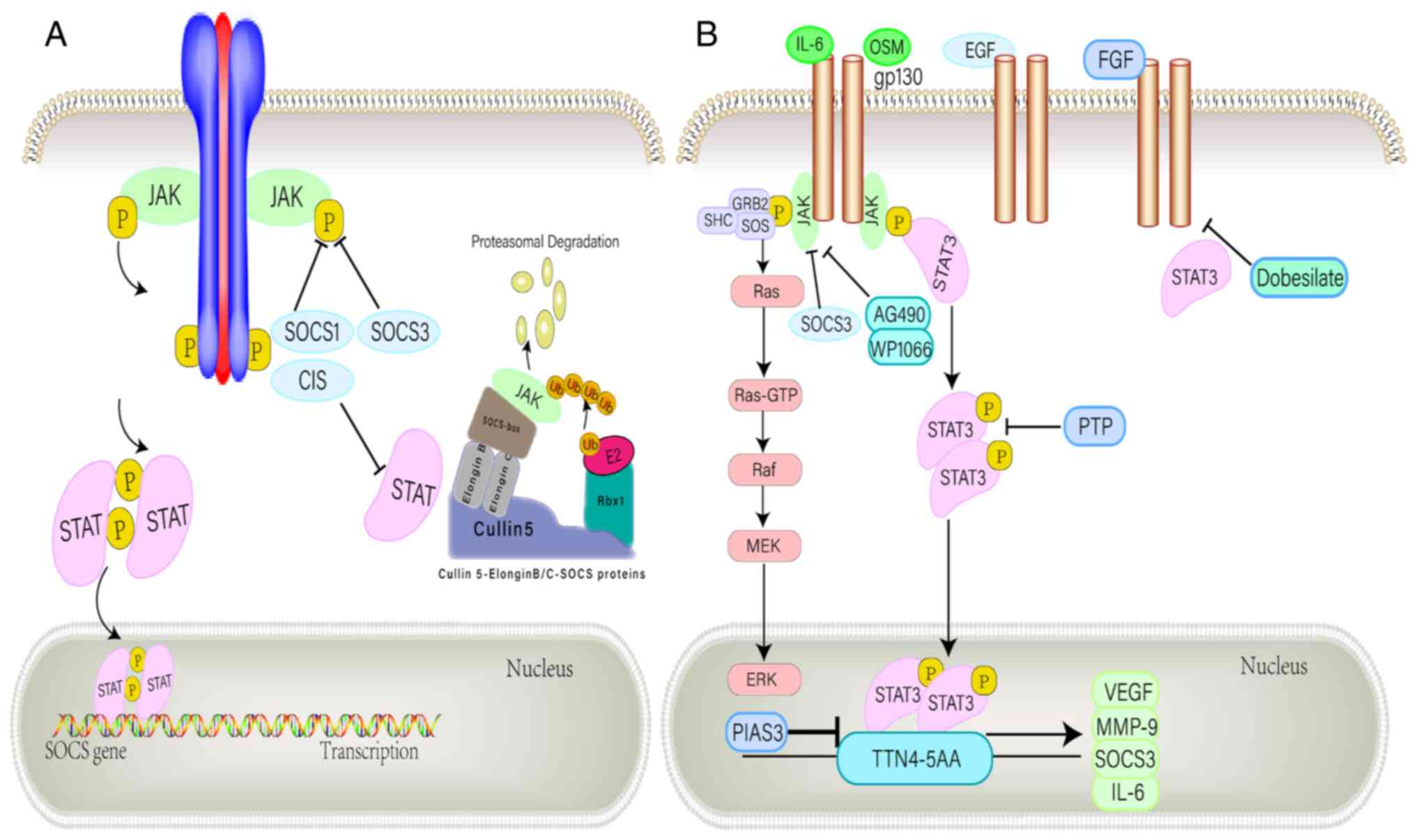 |
Figure 5.
Negative regulation of the JAK/STAT pathway. (A) SOCS1 and SOCS3 bind directly to tyrosine-phosphorylated JAK via the SH2 domain, thereby inhibiting kinase activity. CIS inhibits STST by competing with STAT for docking position on receptors. An alternative regulatory approach is provided by the E3 ubiquitin ligase complex, which binds the SOCS box motif and ubiquitinates related proteins that target proteasomal degradation. (B) IL-6 family members bind to a common receptor subunit, gp130, which produce either homodimer or heterodimer when the ligand binds. JAK protein activation mediates downstream signal transduction through the MAPK and STAT3 pathways. The STAT3 dimer translocates to the nucleus and binds to common sequences in gene promoters including VEGF, MMP-9, IL-6 and SOCS3. SOCS3 inhibits cytoplasmic JAK activation and subsequent signal transduction, while PIAS3 inhibits DNA binding of STAT3 in the nucleus. The pharmacological inhibition of JAK activity by AG490 and WP1066 suppressed the signal leading to STAT3 activation, and various PTPs endogenously inhibit STAT3 activity. Hydroxobenzene sulfonate inhibits STAT3-mediated gene expression by attenuating STAT3-activated upstream kinase signaling. SOCS, suppressor of cytokine signaling; CIS, cytokine-inducible SH2-containing protein; PTP, protein tyrosine phosphatase; FGF, fibroblast growth factor; PIAS, protein inhibitor of activated STAT; OSM, oncostatin M.
|
As the JAK/STAT signaling pathway is abnormally regulated in GBM, it is possible to identify potential therapeutic targets for GBM by identifying negative regulators of this pathway, such as SOCS proteins (95). For example, it was previously demonstrated that hypoxia activates the JAK/STAT signaling pathway by reducing SOCS3 expression levels. Moreover, activated STAT3 combines with hypoxia-related elements located upstream of the transcription start site of the VEGF promoter to regulate VEGF gene expression and promote angiogenesis in GBM (96). Lee et al (51) reported that overexpression of miR-296-3p could inhibit the expression of STAT5A, activate the JAK/STAT3 signaling pathway by downregulating SOCS2, and ultimately increase the invasiveness of GBM cells. Furthermore, SOCS may serve as novel therapeutic targets to improve the radiotherapy response of GBM through the JAK/STAK signaling pathway. Ventero et al (77) reduced SOCS1 and SOCS3 expression via small interfering RNAs and reported that inhibition of SOCS3 increased the radiation resistance of GBM cell lines, whereas silencing of SOCS1 had no effect. It was predicted that this result was related to the JAK/STAK signaling pathway, but further work is needed to verify this hypothesis. Overall, the JAK/STAT3 signaling pathway can markedly promote tumor cell survival and invasion while inhibiting antitumor immunity (97).
SOCS proteins and the NF-κB signaling pathway
NF-κB proteins are a family of transcription factors that mediate immune and inflammatory responses (98–100). Within this group, NF-κB dimers can contain p65, RelB and c-Rel subunits (101). In the central nervous system, the p65 and p50 heterodimer NF-κB proteins are dominant. Inactive NF-κB can be activated via several signaling pathways associated with GBM, particularly canonical signaling pathways (102). NF-κB is constitutively activated in GBM and is positively associated with increasing grades of astrocytic tumors. The mesenchymal subclass of GBM is characterized by elevated levels of NF-κB signaling components, enhanced tolerance to chemotherapy and radiotherapy and poor prognosis (4). The major transcription factor of the mesenchymal subtype of GBM is STAT3. The STAT3 inhibitor JSI-124 activates the NF-κB signaling pathway in GBM via NF-κB p65 phosphorylation and nuclear translocation, which reduces JAK2 and STAT3 phosphorylation, decreases cell proliferation and induces apoptosis of GBM cells. Furthermore, JSI-124 induces the increased expression of IL-6, IL-8 and SOCS3 (26).
When JSI-124 activates intracellular signaling cascades including NF-κB, the expression of the downstream SOCS3 gene is increased within 2 h following JSI-124 treatment. SOCS3 is an endogenous negative regulator of the JAK/STAT3 signaling pathway and is often induced by JAK/STAT3 activation (97). STAT3 is not activated following JSI-124 treatment and, therefore, does not lead to increased expression levels of SOCS3. This result confirms that activated NF-κB p65 is recruited to the SOCS3 promoter during JSI-124 treatment and is responsible for the increases observed in gene expression levels (46,103,104). Driven by NF-κB, SOCS3 expression may serve as a negative regulator of STAT3, which neutralizes STAT3 activation and provides a mechanism to inhibit STAT3 following JSI-124 treatment. This ultimately exerts a potential inhibitory effect on GBM. JSI-124 inhibits STAT3 by activating the NF-κB signaling pathway and, although this process has potential antitumor effects, an off-target pro-inflammatory signaling pathway is also activated. Therefore, thorough clinical studies are needed to investigate and prevent potentially harmful side effects (26).
Characteristics of GBM regulation via SOCS proteins
SOCS proteins in the GBM microenvironment
GBM is highly malignant and is characterized by high heterogeneity at the tissue and molecular levels, as well as high intratumoral heterogeneity. At the molecular level, different GBM subtypes display significant differences in their microenvironments (105). Analysis of gene expression profiles and database mining of the Gene Expression Profiling Interactive Analysis (http://gepia.cancer-pku.cn/) database and the Chinese Glioma Genome Atlas (http://www.cgga.org.cn/) database demonstrated that the expression levels of SOCS in tumor tissues were significantly higher compared with those in healthy tissues in patients with GBM. Furthermore, low SOCS expression levels in GBM may be considered as indicative of a poor prognosis. For example, SOCS3 is highly expressed in GBM, and SOCS3 expression levels increase with an increase in tumor grade. Furthermore, higher wild-type IDH1 levels, 1p/19q co-deletion and wild-type patients with GBM have higher SOCS3 expression levels (Fig. 6). SOCS proteins can also regulate numerous signaling pathways via extracellular signals, thereby promoting gene transcription and tumor cell proliferation. For example, the JUN/miR-494/SOCS6 signaling pathway is involved in GBM cell proliferation, apoptosis and maintenance of stemness (33). Moreover, the activation of the JAK/STAT3/SOCS3 signaling pathway can promote the formation of the GBM inflammatory microenvironment and is involved in tumor angiogenesis, epithelial-to-mesenchymal transition and extracellular matrix degradation, which serves an important role in the invasion and metastasis of tumors (15). SOCS proteins can also participate in the epigenetic regulation of GBM cells via methylation (106) and participate in GBM resistance to chemotherapy (50). In summary, the aforementioned results and studies suggest that SOCS proteins serve important roles in the regulation of the tumor microenvironment.
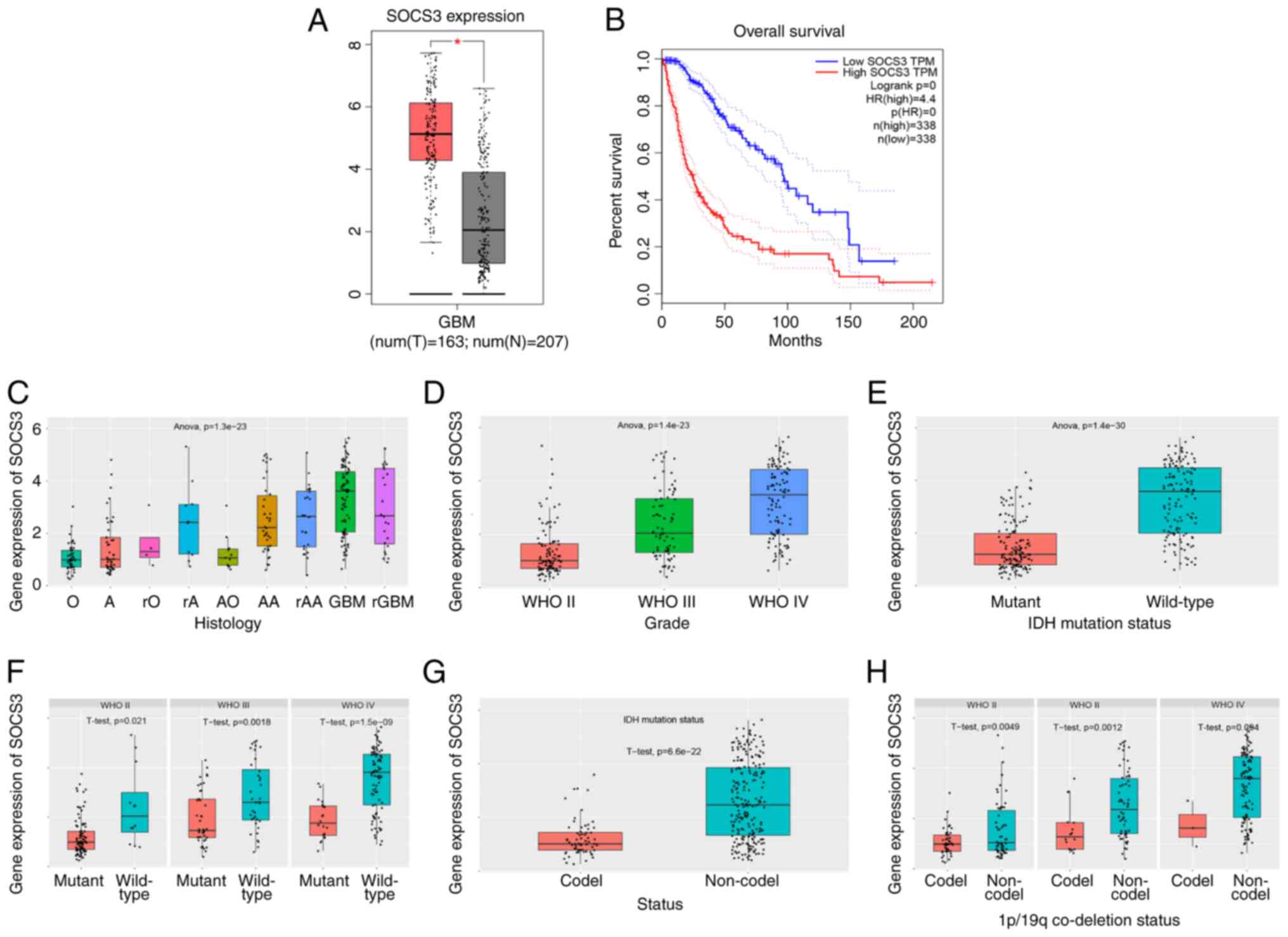 |
Figure 6.
SOCS3 in GBM microenvironment. (A) SOCS3 is highly expressed in GBM. (B) GBM with high SOCS3 expression is associated with a lower survival rate. (C) SOCS3 expression by histological type. (D-F) SOCS3 expression increases with the increase of GBM grade and mutation status, with higher expression of SOCS3 in wild-type GBM. (G and H) GBM with 1p/19q co-deletion has higher expression levels of SOCS3. SOCS, suppressor of cytokine signaling; GBM, glioblastoma multiforme; IDH, isocitrate dehydrogenase; O, oligodendroglioma; A, astrocytoma; rO, recurrence of oligodendroglioma; rA, recurrence of astrocytoma; AO, anaplastic oligodendro; AA, anaplastic astrocytoma; rAA, recurrence of anaplastic astrocytoma; GBM, glioblastoma; rGBM, recurrence of GBM.
|
SOCS proteins regulate GBM cell proliferation
The RTK signaling pathway facilitates the proliferation and migration of tumor cells and regulates tumor progression (107). Activation of RTK signaling increases STAT3 gene expression and induces SOCS3 expression, which inhibits the proliferation of GBM cells (30,88,89). Multiple growth factors, such as EGF, fibroblast growth factor (FGF) and PDGF, can also affect SOCS3 transcriptional activation levels (25). SOCS3 binds constitutively to the cytoplasmic domain of EGFR and this binding leads to the negative regulation of EGF-induced STAT1 and STAT3 phosphorylation in GBM cells. In the RTK signaling pathway, FGFR3 is constitutively related to SOCS3 and is independent of FGF stimulation or receptor kinase activity (26). Therefore, SOCS3 and RTKs are strongly associated with SOCS3 regulating downstream signaling via ubiquitination-dependent receptor degradation. PDGF induces SOCS3 tyrosine phosphorylation of the Y202 and Y221 residues in the mouse embryonic fibroblast NIH3T3 cell line and recruits p120 Ras GTPase-activating protein to activate ERK kinase (43). Overexpression of SOCS3 mutants lacking two tyrosine residues was shown to inhibit PDGF-induced cell proliferation, compared with wild-type SOCS3 that did not exert this effect. These previous findings demonstrate that tyrosine phosphorylation of RTKs promotes the adaptor function of SOCS3, which may be associated with the biological function of tumor cells (26).
SOCS proteins in cell migration and invasion
Tumor cells are characterized by their migration and invasion abilities (43). In recent years, numerous studies have shown that members of the SOCS family are involved in the metastasis of various types of tumors, which provides a potential novel approach to the control and improvement of GBM patients. For example, SOCS1 promoter methylation and expression were shown to be associated with colorectal cancer (108), whereas SOCS2 was found to inhibit the metastasis of liver cancer (109). It was also demonstrated that IL-23 selectively promoted the metastasis of colorectal carcinoma cells with SOCS3 dysfunction via the STAT5 signaling pathway (110). However, SOCS proteins regulate GBM invasion and metastasis via numerous pathways. For example, circ-ASPH regulates the miR-599/AR/SOCS2-AS1 signaling pathway, which promotes glioma cell proliferation and invasiveness (34). It has also been reported that miR-296-3p is regulated by NF2 and enhances the invasiveness of GBM cells via the SOCS2/STAT3 signaling pathway (111). Xu et al (15) reported that the miR-221/222 cluster downregulates the expression of the JAK/STAT signaling pathway by upregulation of SOCS3, and that this downregulation reduces the invasion, migration, proliferation and angiogenesis of GBM cells, as well as the protein expression levels of MMP-2, MMP-9 and VEGF. The hypermethylation of the SOCS3 promoter was shown to lead to SOCS3 inactivation and EGFR activation, thereby activating STAT3 and focal adhesion kinase (FAK), and promoting glioma cell invasion (27). Furthermore, Khalighfard et al (52) reported that miR-1290 can target the 3′-UTR of SOCS4 and inhibit its expression, which increases GBM cell proliferation, migration, invasion and resistance to chemotherapy and radiotherapy. LINC00668 can also regulate the expression of SOCS5 in a miR-518C-3p-dependent manner and promote the proliferation and invasion of glioma cells (53). Moreover, Ou-Yang et al (33) demonstrated that neurotensin receptor 1 inhibits GBM invasion via the Jun/miR-494/SOCS6 axis.
In summary, numerous studies have demonstrated that SOCS proteins are associated with the invasion and metastasis of GBM, which provides a new therapeutic target for the control of metastasis and improvement of GBM prognosis.
SOCS proteins function as diagnostic and prognostic biomarkers
Aberrant methylation of DNA is the most extensively investigated epigenetic abnormality in tumorigenesis, and promoter hypermethylation of tumor suppressor genes, including adenomatous polyposis coli, p16, BRCA1, RB and murine double minute 2, is often associated with cancer development (2). Similarly, in multiple myeloma, the human RAD23 homolog B (HHR23B) gene is key to the nucleotide excision repair promoter in the myeloma KAS-6/1 cell line. IL-6 enhances and maintains hypermethylation of the p53 tumor suppressor and HHR23B genes (23). Furthermore, IL-6-induced hypomethylation of EGFR leads to enhanced EGFR expression and proliferation of cholangiocarcinoma cells (23). These results of the aforementioned previous studies suggest that DNA hypo- and hypermethylation are important mechanisms that contribute to inflammation-associated tumorigenesis. As SOCS proteins have recently been identified as tumor suppressors, research has focused on the methylation status of SOCS in human tumors. Earlier studies demonstrated that patients with GBM and methylated SOCS proteins have a significantly worse prognosis compared with patients with unmethylated proteins. The association of methylation with prognosis is highly significant in the case of SOCS3 hypermethylation. These previous studies demonstrated that methylation of SOCS3 results in SOCS3 inactivation, which may serve as a significant predictor of an unfavorable prognosis (27,50,112).
It has also been reported that SOCS3 promoter hypermethylation in gliomas, activated by EGFR, is almost absent, and gliomas with EGFR inactivation display hypermethylation of the SOCS3 promoter. SOCS3 inactivation can therefore act as an alternative mechanism for EGFR activation in certain gliomas (27,113). Methylation of SOCS3 in EGFR-inactivated gliomas preferentially promotes glioma cell invasion via STAT3 and FAK activation, which further elucidates the pathogenic role of SOCS3 promoter methylation in GBM tumors (27). However, it was previously demonstrated that hypermethylation of different parts of the cancer genome does not necessarily result in an improved or worse patient outcome (114,115). SOCS3 promoter hypermethylation is only one example and its negative effects on tumor initiation or progression may be offset by the hypermethylation of other areas of the genome (114). In different types of colorectal cancer, CpG island methylator phenotype (CIMP) affects specific genes and their functions differently, either as favorable or unfavorable predictors for patient prognosis (116). Glioma-CIMPs (G-CIMPs) are enriched in the proneural subgroup (117). In G-CIMP-positive samples, patients with hypermethylation of the SOCS3 promoter generally had more favorable outcomes (114,115). However, the G-CIMP biomarker can only be used as a prognostic predictor for G-CIMP-positive but not-negative patients (115).
In summary, hypermethylation of tumor suppressor gene promoters and hypomethylation of oncogene promoters are important changes implicated in tumor development (118,119). Numerous studies have demonstrated that methylation of SOCS proteins predicts a poor prognosis (112,120). However, a few studies have found that hypermethylation of SOCS3 promoter is associated with a good prognosis in patients with GBM, such as in the case of G-CIMP (115). More research is needed to determine the role of SOCS3 in the diagnosis and prognosis of GBM.
SOCS proteins in GBM treatment
As a result of in-depth research on the role of SOCS in the occurrence and development of GBM, SOCS proteins may become a potential therapeutic target in the future. SOCS proteins mainly serve a tumor suppressor role in GBM and, therefore, a SOCS protein imbalance may affect GBM development. SOCS1 and SOCS3 are both involved in the JAK/STAT signaling pathway and constitutive activation of this pathway is considered as a hallmark of numerous types of cancer (57,121). SOCS1 and SOCS3 proteins are also associated with cell proliferation and apoptosis (122). For example, when miR-221/222 clusters are downregulated or silenced, the activity of the JAK/STAT3 signaling pathway is inhibited. Therefore, upregulation of SOCS3 expression with ensuing decreased migration, proliferation and angiogenesis of GBM cells ultimately significantly prolongs patient survival (15). It was previously demonstrated that, when human IFN-β is added to GBM cells, cell viability and miR-431 expression levels are significantly decreased, which therefore upregulates the expression of SOCS6, inhibiting the JAK1/STAT2 signaling pathway and reducing cell proliferation (123). Future SOCS-based therapies may be divided into the following two categories: i) SOCS activators and ii) SOCS mimics, both aimed at controlling the tumor suppressor function of SOCS proteins.
Although radiotherapy, chemotherapy and targeted drugs serve an important role in the treatment of GBM, GBM has been clinically proven to be resistant to a number of these treatments in recent years (124,125). Recently, SOCS proteins have been demonstrated to regulate GBM drug and radiation resistance. The epigenetic silencing of the SOCS1 locus induced by CIMP inhibits SOCS1 expression in GBM (49). Moreover, the constitutive expression of SOCS3 is dependent on the STAT3 transcription factor (115). Defects in SOCS proteins expression can increase radiation resistance and, thus, affect the biological response of GBM cells to insulin resistance (49). Evidence suggests that SOCS1 and SOCS3 may be associated with tumor aggressiveness and radiation tolerance and that SOCS protein expression profiles may have predictive value in evaluating tumor cell response to insulin resistance (49). In GBM, ERK MAPK is activated by the oncogenic EGFR and the radioprotective effect of EGFR is considered to involve activation of the ERK/MAPK cascade (55,126). SOCS3 can enhance ERK signal transduction, while SOCS1 blocks ERK activation, which is consistent with the concept that SOCS1 and SOCS3 enhance ERK signal transduction and radiation resistance in GBM. Overexpression or inhibition of SOCS proteins may provide new effective targets for the future treatment of patients with GBM. Drugs that prevent SOCS3 expression and function or reactivate SOCS1 transcription can sensitize GBM cells to insulin resistance and ultimately improve the effectiveness of radiation therapy (49). Furthermore, previous studies have demonstrated that SOCS proteins may also affect the sensitivity of GBM to chemotherapy. Khalighfard et al (52) demonstrated that downregulation or silencing of miR-1290 can target the 3′-UTR of the SOCS4 gene and promote its expression, which results in the increased sensitivity of GBM cells to chemotherapy drugs.
In summary, the overexpression of SOCS proteins can potentially regulate the proliferation of GBM cells and tumor growth, as well as GBM drug and radiation resistance, and is therefore expected to become a new therapeutic target for GBM treatment in the future.
Conclusion
In conclusion, the SOCS protein family is involved in numerous cellular processes, such as cell proliferation, migration and apoptosis, via the JAK/STAT signaling pathway. Promoted by the NF-κB protein, SOCS3 expression neutralizes STAT3 activation following JSI-124 treatment and provides a mechanism for STAT3 inhibition, with a potential inhibitory effect on GBM progression. RTKs have been extensively studied, as they comprise a family of cell surface receptors involved in the development of a variety of solid tumors, including GBM. SOCS proteins serve a negative regulatory role in RTK signaling. Furthermore, SOCS proteins may function as E3 ubiquitin ligases, which accelerate the ubiquitination and degradation of SOCS binding partners. SOCS proteins regulate the biological activity of GBM by targeting the RTK signaling pathway. Furthermore, SOCS3 hypermethylation may be an important predictor of a poor prognosis and serves as a predictor of genome-wide methylation. Cell migration and invasion are also important biological processes in tumors. Evidence suggests that members of the SOCS family are involved in the invasion and metastasis of numerous types of tumors, including GBM. As a predictive biomarker, SOCS may help predict the response of patients with GBM to postoperative chemoradiotherapy. Therefore, SOCS proteins are expected to become a novel target for GBM therapeutics in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 81972361).
Availability of data and materials
Not applicable.
Authors' contributions
LD wrote the manuscript. ZL, WL, WH, SZ, ZY, YT, XH, ZX, JM and ZS revised it critically for important intellectual content and gave important advice. XW provided the overall idea of the article and revised the original manuscript. Data authentication is not applicable. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Yang P, Wang Y, Peng X, You G, Zhang W, Yan W, Bao Z, Wang Y, Qiu X and Jiang T: Management and survival rates in patients with glioma in China (2004–2010): A retrospective study from a single-institution. J Neurooncol. 113:259–266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zheng S and Li Z: Identification of a cullin5-RING E3 ligase transcriptome signature in glioblastoma multiforme. Aging (Albany NY). 12:17380–17392. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu B, Mei J, Ji W, Huo Z, Bian Z, Jiao J, Li X, Sun J and Shao J: MicroRNAs involved in the EGFR pathway in glioblastoma. Biomed Pharmacother. 134:1111152021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al: Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fedele M, Cerchia L, Pegoraro S, Sgarra R and Manfioletti G: Proneural-mesenchymal transition: Phenotypic plasticity to acquire multitherapy resistance in glioblastoma. Int J Mol Sci. 20:27462019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamini B: NF-κB, mesenchymal differentiation and glioblastoma. Cells. 7:1252018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al: The transcriptional network for mesenchymal transformation of brain tumours. Nature. 463:318–325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, James JD, Gumin J, Diefes KL, Kim SH, et al: The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 25:2594–2609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al: An integrated genomic analysis of human glioblastoma multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dimitrov L, Hong CS, Yang C, Zhuang Z and Heiss JD: New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int J Med Sci. 12:201–213. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wesseling P, van den Bent M and Perry A: Oligodendroglioma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 129:809–827. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kondo T: Molecular mechanisms involved in gliomagenesis. Brain Tumor Pathol. 34:1–7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HM and Hilton DJ: The SOCS box: A tale of destruction and degradation. Trends Biochem Sci. 27:235–241. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Urbantat RM, Blank A, Kremenetskaia I, Vajkoczy P, Acker G and Brandenburg S: The CXCL2/IL8/CXCR2 pathway is relevant for brain tumor Malignancy and Endothelial cell function. Int J Mol Sci. 22:26342021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu CH, Liu Y, Xiao LM, Chen LK, Zheng SY, Zeng EM, Li DH and Li YP: Silencing microRNA-221/222 cluster suppresses glioblastoma angiogenesis by suppressor of cytokine signaling-3-dependent JAK/STAT pathway. J Cell Physiol. 234:22272–22284. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I and Yoshimura A: CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. 275:29338–29347. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baetz A, Frey M, Heeg K and Dalpke AH: Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 279:54708–54715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bellezza I, Neuwirt H, Nemes C, Cavarretta IT, Puhr M, Steiner H, Minelli A, Bartsch G, Offner F, Hobisch A, et al: Suppressor of cytokine signaling-3 antagonizes cAMP effects on proliferation and apoptosis and is expressed in human prostate cancer. Am J Pathol. 169:2199–2208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cacalano NA, Sanden D and Johnston JA: Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 3:460–465. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kazi JU, Kabir NN and Soh JW: Bioinformatic prediction and analysis of eukaryotic protein kinases in the rat genome. Gene. 410:147–153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Wu C, Chen N, Gu H, Yen A, Cao L, Wang E and Wang L: PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 7:33440–33450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ and Harper JW: The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 13:270–283. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McFarland BC, Gray GK, Nozell SE, Hong SW and Benveniste EN: Activation of the NF-κB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol Cancer Res. 11:494–505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qin H, Wilson CA, Roberts KL, Baker BJ, Zhao X and Benveniste EN: IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 177:7761–7771. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baker BJ, Akhtar LN and Benveniste EN: SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 30:392–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kazi JU, Kabir NN, Flores-Morales A and Rönnstrand L: SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell Mol Life Sci. 71:3297–3310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lindemann C, Hackmann O, Delic S, Schmidt N, Reifenberger G and Riemenschneider MJ: SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol. 122:241–251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dai L, Li Z, Tao Y, Liang W, Hu W, Zhou S, Fu X and Wang X: Emerging roles of suppressor of cytokine signaling 3 in human cancers. Biomed Pharmacother. 144:1122622021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, Hara T and Miyajima A: A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 14:2816–2826. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA and Hilton DJ: A family of cytokine-inducible inhibitors of signalling. Nature. 387:917–921. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nicholson SE and Hilton DJ: The SOCS proteins: A new family of negative regulators of signal transduction. J Leukoc Biol. 63:665–668. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D and Nicola NA: Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci USA. 95:114–119. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ou-Yang Q, He X, Yang A, Li B and Xu M: Interference with NTSR1 expression exerts an anti-invasion effect via the Jun/miR-494/SOCS6 axis of glioblastoma cells. Cell Physiol Biochem. 49:2382–2395. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qu Y, Qi L, Hao L and Zhu J: Upregulation of circ-ASPH contributes to glioma cell proliferation and aggressiveness by targeting the miR-599/AR/SOCS2-AS1 signaling pathway. Oncol Lett. 21:3882021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bullock AN, Debreczeni JE, Edwards AM, Sundström M and Knapp S: Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc Natl Acad Sci USA. 103:7637–7642. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, Willson TA, Hilton DJ, Nicola NA, Baca M, et al: The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell. 22:205–216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Linossi EM and Nicholson SE: The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life. 64:316–323. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Piessevaux J, Lavens D, Peelman F and Tavernier J: The many faces of the SOCS box. Cytokine Growth Factor Rev. 19:371–381. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bulatov E, Martin EM, Chatterjee S, Knebel A, Shimamura S, Konijnenberg A, Johnson C, Zinn N, Grandi P, Sobott F and Ciulli A: Biophysical studies on interactions and assembly of full-size E3 ubiquitin ligase: Suppressor of cytokine signaling 2 (SOCS2)-elongin BC-cullin 5-ring box protein 2 (RBX2). J Biol Chem. 290:4178–4191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cooper JA, Kaneko T and Li SS: Cell regulation by phosphotyrosine-targeted ubiquitin ligases. Mol Cell Biol. 35:1886–1897. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Babon JJ, Sabo JK, Soetopo A, Yao S, Bailey MF, Zhang JG, Nicola NA and Norton RS: The SOCS box domain of SOCS3: Structure and interaction with the elonginBC-cullin5 ubiquitin ligase. J Mol Biol. 381:928–940. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Linossi EM, Calleja DJ and Nicholson SE: Understanding SOCS protein specificity. Growth Factors. 36:104–117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rahaman SO, Vogelbaum MA and Haque SJ: Aberrant Stat3 signaling by interleukin-4 in malignant glioma cells: Involvement of IL-13 Ralpha2. Cancer Res. 65:2956–2963. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shuai K and Liu B: Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 3:900–911. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qin H, Niyongere SA, Lee SJ, Baker BJ and Benveniste EN: Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 181:3167–3176. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG and Ludwig S: Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 4:e10001962008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cohney SJ, Sanden D, Cacalano NA, Yoshimura A, Mui A, Migone TS and Johnston JA: SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 19:4980–4988. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu LH, Li H, Cheng XX, Kong QY, Chen XY, Wu ML, Li Y, Liu J and Li C: Correlative analyses of the expression levels of PIAS3, p-SHP2, SOCS1 and SOCS3 with STAT3 activation in human astrocytomas. Mol Med Rep. 15:847–852. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou H, Miki R, Eeva M, Fike F, Seligson D, Yang L, Yoshimura A, Teitell MA, Jamieson CA and Cacalano NA: Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res. 13:2344–2353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Martini M, Pallini R, Luongo G, Cenci T, Lucantoni C and Larocca LM: Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int J Cancer. 123:2955–2960. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lee H, Hwang SJ, Kim HR, Shin CH, Choi KH, Joung JG and Kim HH: Neurofibromatosis 2 (NF2) controls the invasiveness of glioblastoma through YAP-dependent expression of CYR61/CCN1 and miR-296-3p. Biochim Biophys Acta. 1859:599–611. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Khalighfard S, Kalhori MR, Haddad P, Khori V and Alizadeh AM: Enhancement of resistance to chemo-radiation by hsa-miR-1290 expression in glioblastoma cells. Eur J Pharmacol. 880:1731442020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu Z, Wang J, Tong H, Wang X, Zhang D and Fan Q: LINC00668 Modulates SOCS5 expression through competitively sponging miR-518c-3p to facilitate glioma cell proliferation. Neurochem Res. 45:1614–1625. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang D, Ma L, Wang B, Liu J and Wei W: E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 36:683–702. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lammering G, Valerie K, Lin PS, Hewit TH and Schmidt-Ullrich RK: Radiation-induced activation of a common variant of EGFR confers enhanced radioresistance. Radiother Oncol. 72:267–273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhao Y and Sun Y: Cullin-RING Ligases as attractive anti-cancer targets. Curr Pharm Des. 19:3215–3225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rawlings JS, Rosler KM and Harrison DA: The JAK/STAT signaling pathway. J Cell Sci. 117((Pt 8)): 1281–1283. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wilkinson KD: Protein ubiquitination: A regulatory post-translational modification. Anticancer Drug Des. 2:211–229. 1987.PubMed/NCBI
|
|
59
|
Mosesson Y, Mills GB and Yarden Y: Derailed endocytosis: An emerging feature of cancer. Nat Rev Cancer. 8:835–850. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jin WL, Mao XY and Qiu GZ: Targeting deubiquitinating enzymes in glioblastoma multiforme: Expectations and challenges. Med Res Rev. 37:627–661. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Scholz N, Kurian KM, Siebzehnrubl FA and Licchesi JDF: Targeting the ubiquitin system in glioblastoma. Front Oncol. 10:5740112020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
d'Azzo A, Bongiovanni A and Nastasi T: E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic. 6:429–441. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, Conaway JW and Nakayama KI: VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 18:3055–3065. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kile BT and Alexander WS: The suppressors of cytokine signalling (SOCS). Cell Mol Life Sci. 58:1627–1635. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhao Y, Xiong X and Sun Y: Cullin-RING Ligase 5: Functional characterization and its role in human cancers. Semin Cancer Biol. 67:61–79. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Inagaki-Ohara K, Kondo T, Ito M and Yoshimura A: SOCS, inflammation, and cancer. JAKSTAT. 2:e240532013.PubMed/NCBI
|
|
67
|
Humphreys LM, Smith P, Chen Z, Fouad S and D'Angiolella V: The role of E3 ubiquitin ligases in the development and progression of glioblastoma. Cell Death Differ. 28:522–537. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, et al: The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. 96:2071–2076. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Scaltriti M and Baselga J: The epidermal growth factor receptor pathway: A model for targeted therapy. Clin Cancer Res. 12:5268–5272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Xia L, Wang L, Chung AS, Ivanov SS, Ling MY, Dragoi AM, Platt A, Gilmer TM, Fu XY and Chin YE: Identification of both positive and negative domains within the epidermal growth factor receptor COOH-terminal region for signal transducer and activator of transcription (STAT) activation. J Biol Chem. 277:30716–30723. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Goldshmit Y, Walters CE, Scott HJ, Greenhalgh CJ and Turnley AM: SOCS2 induces neurite outgrowth by regulation of epidermal growth factor receptor activation. J Biol Chem. 279:16349–16355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bullock AN, Rodriguez MC, Debreczeni JE, Songyang Z and Knapp S: Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure. 15:1493–1504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kario E, Marmor MD, Adamsky K, Citri A, Amit I, Amariglio N, Rechavi G and Yarden Y: Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem. 280:7038–7048. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al: The somatic genomic landscape of glioblastoma. Cell. 155:462–77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gao T, Furnari F and Newton AC: PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 18:13–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li X, Liu J and Gao T: Beta-TrCP-mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol Cell Biol. 29:6192–6205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ventero MP, Fuentes-Baile M, Quereda C, Perez-Valeciano E, Alenda C, Garcia-Morales P, Esposito D, Dorado P, Manuel Barbera V and Saceda M: Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS One. 14:e02125812019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O and Woodgett JR: Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 406:86–90. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
King TD, Bijur GN and Jope RS: Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res. 919:106–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lin J, Song T, Li C and Mao W: GSK-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim Biophys Acta Mol Cell Res. 1867:1186592010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lawrence T: The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 1:a0016512009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ohgaki H and Kleihues P: Epidemiology and etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Rao RD and James CD: Altered molecular pathways in gliomas: An overview of clinically relevant issues. Semin Oncol. 31:595–604. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Brantley EC and Benveniste EN: Signal transducer and activator of transcription-3: A molecular hub for signaling pathways in gliomas. Mol Cancer Res. 6:675–684. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, Roarty K and Benveniste EN: Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: Implications for STAT-3 activation and gene expression. Clin Cancer Res. 14:4694–4704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Schaefer LK, Ren Z, Fuller GN and Schaefer TS: Constitutive activation of Stat3alpha in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene. 21:2058–2065. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yoshimura A, Naka T and Kubo M: SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 7:454–465. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO and Benveniste EN: Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 179:5966–5976. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ramana CV, Kumar A and Enelow R: Stat1-independent induction of SOCS-3 by interferon-gamma is mediated by sustained activation of Stat3 in mouse embryonic fibroblasts. Biochem Biophys Res Commun. 327:727–733. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA and Haque SJ: Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 21:8404–8413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN and Nutt CL: Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 65:1181–1188. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Weissenberger J, Loeffler S, Kappeler A, Kopf M, Lukes A, Afanasieva TA, Aguzzi A and Weis J: IL-6 is required for glioma development in a mouse model. Oncogene. 23:3308–3316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Repovic P, Fears CY, Gladson CL and Benveniste EN: Oncostatin-M induction of vascular endothelial growth factor expression in astroglioma cells. Oncogene. 22:8117–8124. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Loeffler S, Fayard B, Weis J and Weissenberger J: Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int J Cancer. 115:202–213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Keewan E and Matlawska-Wasowska K: The emerging role of suppressors of cytokine signaling (SOCS) in the development and progression of leukemia. Cancers (Basel). 13:40002021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Yokogami K, Yamashita S and Takeshima HJBtp: Hypoxia-induced decreases in SOCS3 increase STAT3 activation and upregulate VEGF gene expression. Brain Tumor Pathol. 30:135–143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yu H, Pardoll D and Jove R: STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer. 9:798–809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bassères DS and Baldwin AS: Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 25:6817–6830. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hoffmann A and Baltimore D: Circuitry of nuclear factor kappaB signaling. Immunol Rev. 210:171–186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Karin M: NF-kappaB and cancer: Mechanisms and targets. Mol Carcinog. 45:355–361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Perkins ND: The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 12:121–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Gray GK, McFarland BC, Nozell SE and Benveniste EN: NF-κB and STAT3 in glioblastoma: Therapeutic targets coming of age. Expert Rev Neurother. 14:1293–1306. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Akhtar LN, Qin H, Muldowney MT, Yanagisawa LL, Kutsch O, Clements JE and Benveniste EN: Suppressor of cytokine signaling 3 inhibits antiviral IFN-beta signaling to enhance HIV-1 replication in macrophages. J Immunol. 185:2393–2404. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J and Marcu KB: IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 277:45129–45140. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chen Z and Hambardzumyan D: Immune microenvironment in glioblastoma subtypes. Front Immunol. 9:10042018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Tamiya T, Kashiwagi I, Takahashi R, Yasukawa H and Yoshimura A: Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol. 31:980–985. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang Z: ErbB receptors and cancer. Methods Mol Biol. 1652:3–35. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kang XC, Chen ML, Yang F, Gao BQ, Yang QH, Zheng WW and Hao S: Promoter methylation and expression of SOCS-1 affect clinical outcome and epithelial-mesenchymal transition in colorectal cancer. Biomed Pharmacother. 80:23–29. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cui M, Sun J, Hou J, Fang T, Wang X, Ge C, Zhao F, Chen T, Xie H, Cui Y, et al: The suppressor of cytokine signaling 2 (SOCS2) inhibits tumor metastasis in hepatocellular carcinoma. Tumour Biol. 37:13521–13531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Zhang L, Li J, Li L, Zhang J, Wang X, Yang C, Li Y, Lan F and Lin P: IL-23 selectively promotes the metastasis of colorectal carcinoma cells with impaired Socs3 expression via the STAT5 pathway. Carcinogenesis. 35:1330–1340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lee H, Shin CH, Kim HR, Choi KH and Kim HH: MicroRNA-296-5p promotes invasiveness through downregulation of nerve growth factor receptor and caspase-8. Mol Cells. 40:254–261. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Sutherland KD, Lindeman GJ, Choong DY, Wittlin S, Brentzell L, Phillips W, Campbell IG and Visvader JE: Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. 23:7726–7733. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Quesnelle KM, Boehm AL and Grandis JR: STAT-mediated EGFR signaling in cancer. J Cell Biochem. 102:311–319. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
You JS and Jones PA: Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell. 22:9–20. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Feng Y, Wang Z, Bao Z, Yan W, You G, Wang Y, Hu H, Zhang W, Zhang Q and Jiang T: SOCS3 promoter hypermethylation is a favorable prognosticator and a novel indicator for G-CIMP-positive GBM patients. PLoS One. 9:e918292014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Barreau O, Assié G, Wilmot-Roussel H, Ragazzon B, Baudry C, Perlemoine K, René-Corail F, Bertagna X, Dousset B, Hamzaoui N, et al: Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J Clin Endocrinol Metab. 98:E174–E184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al: Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 17:510–522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Kloten V, Becker B, Winner K, Schrauder MG, Fasching PA, Anzeneder T, Veeck J, Hartmann A, Knüchel R and Dahl E: Promoter hypermethylation of the tumor-suppressor genes ITIH5, DKK3, and RASSF1A as novel biomarkers for blood-based breast cancer screening. Breast Cancer Res. 15:R42013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA and Liang G: Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 6:e10009172010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Fourouclas N, Li J, Gilby DC, Campbell PJ, Beer PA, Boyd EM, Goodeve AC, Bareford D, Harrison CN, Reilly JT, et al: Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. Haematologica. 93:1635–1644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Thomas SJ, Snowden JA, Zeidler MP and Danson SJ: The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 113:365–371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Trengove MC and Ward AC: SOCS proteins in development and disease. Am J Clin Exp Immunol. 2:1–29. 2013.PubMed/NCBI
|
|
123
|
Tanaka T, Arai M, Jiang X, Sugaya S, Kanda T, Fujii K, Kita K, Sugita K, Imazeki F, Miyashita T, et al: Downregulation of microRNA-431 by human interferon-β inhibits viability of medulloblastoma and glioblastoma cells via upregulation of SOCS6. Int J Oncol. 44:1685–1690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Cao H, Li X, Wang F, Zhang Y, Xiong Y and Yang Q: Phytochemical-mediated glioma targeted treatment: Drug resistance and novel delivery systems. Curr Med Chem. 27:599–629. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Chistiakov DA and Chekhonin VP: Contribution of microRNAs to radio- and chemoresistance of brain tumors and their therapeutic potential. Eur J Pharmacol. 684:8–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Lammering G, Hewit TH, Valerie K, Contessa JN, Amorino GP, Dent P and Schmidt-Ullrich RK: EGFRvIII-mediated radioresistance through a strong cytoprotective response. Oncogene. 22:5545–5553. 2003. View Article : Google Scholar : PubMed/NCBI
|














