Introduction
Prostate cancer (PCa) is one of the most prevalent
malignancies in men, the severity of which is heterogeneous,
ranging from indolent to lethal (1). The main therapeutic strategy for
metastatic PCa and castration-naïve recurrence is androgen
deprivation therapy (ADT) (2). By
reducing androgen levels, ADT blocks the activation of the androgen
signaling cascade and androgen receptor (AR)-mediated gene
expression (3). However, after a
period of ADT, evolution to castration-resistant prostate cancer
(CRPC) frequently occurs (4).
Treatment options for CRPC are limited, since the majority of the
second-generation anti-androgen therapeutic agents target the AR
(5). One option for treating this
type of cancer is enzalutamide, which was approved by the Food and
Drug Administration in 2018 (6).
It is an AR antagonist that also blocks its nuclear translocation
and AR-mediated DNA binding (7).
Despite the availability of this second-generation anti-androgen, a
proportion of tumors will develop resistance to enzalutamide
(8). By investigating causes and
characteristics underlying enzalutamide resistance in PCa, novel
therapeutic strategies can be discovered.
One reported cause of therapeutic resistance is
epithelial-mesenchymal transition (EMT), which is a fundamental
process of embryogenesis (9).
Resistance to oxaliplatin has been previously found in colon
carcinoma epithelial cell lines with mesenchymal morphology
(10). In addition, loss of
epithelial phenotype have also been reported to associate with
resistance to paclitaxel in ovarian carcinoma epithelial cell lines
(11). In a similar manner, EMT
has been found to promote the conversion to androgen-independent
PCa (12). EMT is also a key
process in promoting cancer cell invasiveness, since it disrupts
cell-to-cell or cell-to-extracellular matrix adherence (13). It serve a role in the metastasis of
certain malignancies by inducing the loss of E-cadherin expression
whilst increasing N-cadherin expression (13). A number of factors have been
documented to be involved in this mechanism (14). Snail, Slug and Twist are among the
number of E-cadherin transcriptional repressors that can induce the
epigenetic silencing of the E-cadherin promoter (14). Furthermore, α-smooth muscle actin
(α-SMA) is a myofibroblast marker that can be used as a marker of
cancer-associated fibroblasts (15). α-SMA-positive myofibroblasts can
promote the metastasis of oral tongue squamous cell carcinoma cells
by promoting EMT (15). Previous
studies have also shown that regulating particular markers, such as
Snail and Slug, may facilitate prostate cancer metastasis (16,17).
Cadherin-2 (CDH2), also known as N-cadherin, is
highly expressed in the nervous system and vascular endothelium
(18). It is a member of the
cadherin family and is involved in various intracellular signaling
pathways, such as the PI3K/Akt signaling pathway (19,20).
It also serves an important role in EMT. CDH2 expression has been
found to serve a role in several human cancers, including bladder,
colorectal, lung and gastric cancer (20–23).
Since it can weaken intercellular interactions and form homophilic
interactions with other CDH2-expressing tissues, CDH2 has been
shown to be a key component in mediating cancer cell invasion and
metastasis (24,25). In PCa, CDH2 expression is typically
higher in patients with high-grade primary tumors or lymph node
metastasis compared with that in patients with low-grade tumors
(26,27). In addition, CDH2 expression was
found to positively correlated with the Gleason score (26,27).
This increased CDH2 expression is sufficient for EMT as well as
prostate cancer invasion and metastasis (25). Furthermore, it was found to be
necessary for the proliferation of CRPC cells and causes CRPC
development (12,25). In a previous study performed by
Tanaka et al (25),
N-cadherin was present in a number of castration-resistant cell
lines but was absent in the hormone-sensitive LNCaP cell line
(25). The difference in molecular
expression between castration-resistant and hormone-sensitive cell
lines suggests CDH2 to be a possible target for CRPC treatment.
In the present study, two different prostate cancer
cell lines, LNCaP and enzalutamide-resistant LNCaP cells (LNCaP
EnzaR cells), were chosen. Compared with LNCaP cells, LNCaP EnzaR
cells display a similar morphology but heterogeneous proliferative
characteristics (28). LNCaP EnzaR
cells also display increased metastatic colonization potential in a
number of clinically relevant organs in vivo, including
bone, brain and the adrenal glands (28). By comparing the properties of these
two cell lines, the aim was to explore a novel strategy to
manipulate prostate cancer cell physiology. The expression levels
of CDH2 in these two PCa cell lines were first measured.
Subsequently, CDH2 expression was upregulated before assessing its
possible effects on cell viability, migratory capability and the
expression of EMT markers in LNCaP and LNCaP EnzaR cells. Finally,
to investigate the influence of CDH2 on cell viability and
migration, the same assays were performed on cells with LNCaP and
LNCaP EnzaR cells transfected with small interfering RNA
(siRNA)-CDH2 cells to downregulate CDH2.
Materials and methods
Cell culture
LNCaP cells were obtained from the American Tissue
Culture Collection and cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C with 5% CO2. LNCaP
cells were exposed to different concentrations of enzalutamide
(1–10 µM; cat. no. S1250; Selleck Chemicals). At each concentration
of enzalutamide, the cells were grown in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) under 5% CO2 at 37°C for
1 week to allow them to acclimatize and then proliferate for a ≥6
months. Since there is no consensus on the concentrations required
to generate EnzaR cells, the cells were treated under 5%
CO2 at 37°C with 10 µM enzalutamide in accordance with
previous studies (28,29). The LNCaP EnzaR cells generated from
10 µM enzalutamide treatment were maintained in the aforementioned
media containing 5 µM enzalutamide.
Transfection
To create the pCMV-CDH2 plasmid, the CDH2 (accession
no. NM_001792) open reading frame (ORF) sequence was cloned into
the Human-Tagged ORF Clone plasmid (cat. no. RC207170; Origene
Technologies, Inc.). Cells were cultured in six-well plates and
treated with the pCMV-CDH2 (2 µg/ml) (cat. no. RC207170; Origene
Technologies, Inc.) or pCMV-GFP plasmid (2 µg/ml) (cat. no.
PS100010; Origene Technologies, Inc.). Plasmid transfections were
performed using FuGENE HD Transfection Reagent (Roche Diagnostics,
Inc.) and incubated for 37°C and 5% CO2 for 24 h
according to the manufacturer's protocols. LNCaP and LNCaP EnzaR
cell lines were each divided into the following three groups:
Untreated cells (pCMV-GFP:-, pCMV-CDH2:-); empty vector-transfected
cells (pCMV-GFP:+, pCMV-CDH2:-); and CDH2 transfected cells
(pCMV-GFP:-, pCMV-CDH2:+).
For CDH2 knockdown, the CDH2 gene was
silenced using ON-TARGETplus CDH2 siRNA SMARTpool (siRNA-CDH2; cat.
no. L-011605-00-0005), which was purchased from Dharmacon, Inc.;
Cytiva. The sequences of CDH2 siRNA and the ON-TARGETplus
non-targeting pool (siRNA-control; Dharmacon, Inc.; Cytiva; cat.
no. D-001810-10-05) are listed in Table I. CDH2 siRNAs (5 pmol) were
transfected into the cells using Lipofectamine RNAiMAX reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) under 5%
CO2 at 37°C for 24 h. LNCaP and LNCaP EnzaR cell lines
were also divided into the following groups: Untreated cells
(siRNA-control:-, siRNA-CDH2:-), siRNA-control-transfected cells
(siRNA-control:+, siRNA-CDH2:-) and siRNA-CDH2-transfected cells
(siRNA-control:-, siRNA-CDH2:+).
 | Table I.CDH2 siRNA and siRNA-control target
sequence. |
Table I.
CDH2 siRNA and siRNA-control target
sequence.
|
Oligonucleotide | Target sequence 1
(5′-3′) | Target sequence 2
(5′-3′) | Target sequence 3
(5′-3′) | Target sequence 4
(5′-3′) | Cat. no. |
|---|
| ON-TARGETplus
CDH2 | GUGCAACAGUAUAC | GGACCCAGAUCGAU | CAUAGUAGCUAAUC | GACAGCCUCUUCUC |
L-011605-00-0005 |
| siRNA
SMARTpool | GUUAA | AUAUG | UAACU | AAUGU |
|
| ON-TARGETplus | UGGUUUACAUGUC | UGGUUUACAUGUUG | UGGUUUACAUGUUU | UGGUUUACAUGUUU | D-001810-10-05 |
| Non-targeting
pool | ACUAA | UGUGA | UCUGA | UCCUA |
|
After 24 h of transfection, the cells were subjected
to reverse transcription PCR (RT-PCR) and western blot (WB)
analysis.
RT-PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). In total, 1 µg total
RNA was subjected to reverse transcription into cDNA using
SuperScript™ III First Strand Synthesis System for RT-PCR
(Invitrogen; Thermo Fisher Scientific, Inc.). From 5 µg RNA, cDNA
was prepared using oligo dT (50 µM) and 10 mM dNTP. For the PCR
reactions, 1 µl oligo dT primer and 1 µl 10 mM dNTP mix were added
to 8 µl RNA, incubated for 5 min at 65°C and then placed on ice for
≥1 min. Subsequently, 10 µl cDNA synthesis mix was added [2 µl 10X
RT buffer, 4 µl 25 mM MgCl2, 2 µl 0.1 M DTT, 1 µl RNase
OUT™ (40 U/µl) and 1 µl SuperScript™ III RT (200 U/µl)] to each
RNA/primer mixture and incubated for 50 min at 50°C, followed by
reaction termination at 85°C for 5 min. For each reaction, 10X PCR
buffer (cat. no. 18067-017; Invitrogen; Thermo Fisher Scientific,
Inc.), MgCl2 (50 mM), dNTP mix (10 mM), cDNA, Taq DNA
polymerase (5 U/µl) and each pair of primers were added. The
resultant product was stored at −20°C. Reactions containing 5 µl
10X PCR buffer, 1.5 µl 50 mM MgCl2, 1 µl 10 mM dNTP mix,
2 µg cDNA, 10 µM of each pair of primers, 0.4 µl Taq DNA polymerase
and 38.1 µl DEPC water were first incubated for initial
denaturation at 94°C for 2 min. PCR was then performed for 35
cycles. For all PCR programs, an annealing temperature of 55°C for
30 sec and denaturation and extension temperatures of 94°C and
72°C, respectively, for 30 sec.
RNA was used as a template for reverse transcription
(Invitrogen; Thermo Fisher Scientific, Inc.) followed by PCR
analysis using specific primers for N-cadherin (forward,
5′-AGCCTGGAACATATGTGATGA-3′ and reverse,
5′-CCATAAAACGTCATGGCAGTAA-3′); GAPDH forward,
5′-ATGTGTCCGTCGTGGATCTGAC-3′ and reverse,
5′-AGACAACCTGGTCCTCAGTGTAG-3′. The expression levels of total RNA
were normalized to the expression of gene GAPDH (assay ID,
Hs03929097_g1; Thermo Fisher Scientific, Inc.). DNA (0.5 µg/lane)
was visualized using gel electrophoresis on a 2% agarose gel
stained with SafeView™ Classic staining (Applied Biological
Materials, Inc.).
WB
The cells were lysed using RIPA buffer (50 mM
Tris/pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate,
0.1% SDS, 1 mM sodium orthovanadate, 1 mM sodium fluoride and 1 mM
EDTA). A BSA standard curve was used to detect protein
concentration, which was used to analyze cell lystates. Protein
lysates (20 µg) were separated by 10% SDS-PAGE and then transferred
onto polyvinylidene fluoride membranes (EMD Millipore). After
blocking the membranes with 5% non-fat milk for 1 h at 25°C, they
were incubated with 1:2,000 dilutions of specific primary
antibodies against E-cadherin (CDH1; cat. no. ab53033; Abcam), CDH2
(cat. no. ab18203; Abcam), α-SMA (cat. no. ab5694; Abcam), Snail
(cat. no. ab85931; Abcam), Slug (cat. no. sc-166476; Santa Cruz
Biotechnology, Inc.) and β-actin (cat. no. A5441; Sigma-Aldrich;
Merck KGaA) at 4°C overnight. The membranes were then washed in
Tris-buffered saline with 0.1% Tween 20 and incubated with
HRP-conjugated secondary antibodies (cat. no. ab6721; Abcam;
1:4,000) for 1 h at room temperature. The Pierce™ ECL Western
Blotting Substrate enhanced chemiluminescence system (cat. no.
32209; Thermo Fisher Scientific, Inc.) was used for visualization
and detection using a Multi-function Gel Image System (cat. no.
MQIS-21-C2; Tangshan Top Bio Technology, Co., Ltd.).
Immunofluorescence staining
To perform immunocytochemistry analysis,
1×104 cells grown in two-well chamber slides were
transfected and incubated for 24 h at 37°C in a humidified
atmosphere with 5% CO2. Cells were washed with PBS two
times, fixed and permeabilized with 99.9% ice-cold methanol for 15
min at 4°C before blocking with 2% BSA in PBS for 1 h at 4°C. Cells
were then incubated with primary antibodies against CDH2 (cat. no.
ab18203; Abcam; 1:2,000) for 1 h at 4°C, washed with PBS three
times and incubated with Alexa Fluor® 488-labeled,
species-specific secondary antibodies (cat. no. ab150077; Abcam).
Before mounting, the slides were washed with PBS, counterstained
with 1.5 µg/ml DAPI for nuclear staining at room temperature for 1
h and then observed under a fluorescent microscope (Olympus
Corporation; magnification, ×200) using the ipwin32 (Image-Pro Plus
version no. 6; Media Cybernetics, Inc.) software.
MTT assay
Cells were grown in RPMI containing 10% FBS, which
were then plated at a density of 5×104 cells/well in
24-well plates overnight and incubated with pCMV-CDH2 for 24 h,
each at 37°C and 5% CO2. Cell viability was assessed
using MTT assay. After transfection for 24 h, MTT solution was
added into each well and incubated for 3 h. Then, 50 µl 5 mg/ml MTT
solution was added into each well containing 500 µl medium and
incubated at 37°C for 3 h, followed by the addition of 500 µl
isopropyl alcohol to dissolve the reduced formazan product. The
absorbance at 590 nm in each well was measured using a
spectrophotometer (Sunrise-Basic; Tecan Group, Ltd.) before cell
viability was examined. Values calculated represent the mean
OD590 ± SD from ≥ three independent reaction wells.
Cell Counting Kit-8 (CCK-8) assay
Cells were seeded into 96-well plates and allowed to
grow to 60–75% confluence before treatment. Cells were then
transfected with pCMV-GFP, siRNA-control, pCMV-CDH2 or siRNA-CDH2
in RPMI 1640 with 10% FBS at 37°C for 24 h. Cell viability was
evaluated using the CCK8 assay (cat. no. ab228554; Abcam).
Afterwards, the medium was aspirated, rinsed with PBS and treated
with CCK-8 at 10 µl/well for 2 h at 37°C. Absorbance was measured
at 450 nm using a spectrophotometer (Sunrise-Basic; Tecan Group,
Ltd.). The percentage of LNCaP and LNCaP EnzaR cell viability in
cell lines, as well as their transfected cell lines, was calculated
using the following formula: Viability (%)=(optical density of
sample/optical density of control) ×100.
Gap closure assay
Cell migration by LNCaP and LNCaP EnzaR cells was
examined using a gap closure assay with ibidi Culture-Insert 2 Well
system (Cat.No:80209, Ibidi, Gräfelfing, Germany) according to the
manufacturer's protocols. Cells were seeded overnight at a
concentration of 1.75×104/100 µl/well in each individual
compartment of the Ibidi culture insert. The culture plate was then
filled with RPMI complete medium as previously described (30,31),
before the Ibidi culture inserts were removed. A live cell imaging
light microscope (Leica AF 6000 LX; Leica Microsystems, GmbH;
magnification, ×200) was used to monitor and capture images of the
cells at 0 h and after 24 h of incubation at 37°C. For each image,
areas between one side of the gap and the other were measured using
Quantity One software (version 4.6.6; Bio-Rad Laboratories, Inc.).
Migration rate was quantified by dividing the change in wound area
by the time spent in migration and was expressed as a percentage.
To quantify the effects of CDH2 overexpression or knockdown on
migration, the percentage of gap closure after 24 h was
analyzed.
Statistical analysis
Each experiment was performed ≥ three times and
representative images are shown. The results were expressed as the
mean ± standard error of the mean (SEM). Statistical analyzes with
GraphPad Prism 9 (GraphPad Software, Inc.) were performed using
one-way ANOVA followed by Tukey's post hoc test as appropriate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
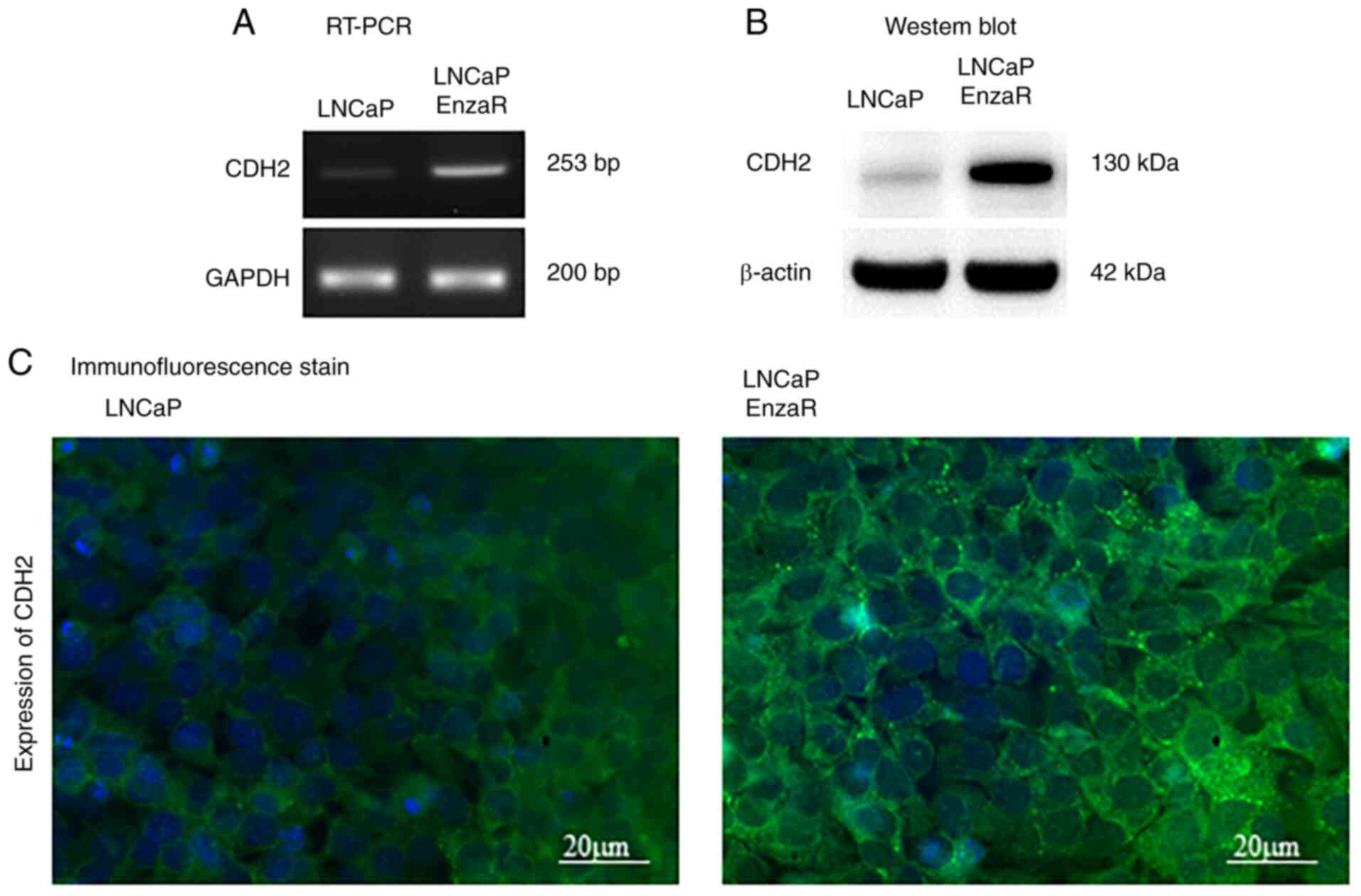
CDH2 expression is increased in LNCaP
EnzaR cells
To determine the role of CDH2 in LNCaP and LNCaP
EnzaR cells, RT-PCR was used to first measure the expression levels
of CDH2. CDH2 expression was markedly higher in the LNCaP EnzaR
cell line, which was almost absent in the sensitive LNCaP cell line
(Fig. 1A). Protein expression of
CDH2 was next evaluated by WB, where markedly higher CDH2
expression levels were also observed in LNCaP EnzaR cells compared
with those in LNCaP cells (Fig.
1B). In addition, immunofluorescence staining revealed high
CDH2 protein expression levels in LNCaP EnzaR cells, suggesting
that LNCaP EnzaR cells express CDH2 at higher levels compared with
that in LNCaP cells (Fig. 1C).
CDH2 was found to be localized at the surfaces of LNCaP EnzaR
cells, but exhibited low expression levels in the hormone-sensitive
LNCaP cells according to immunostaining (Fig. 1C). These observations suggest that
the expression of CDH2 is increased during the development of
enzalutamide resistance.
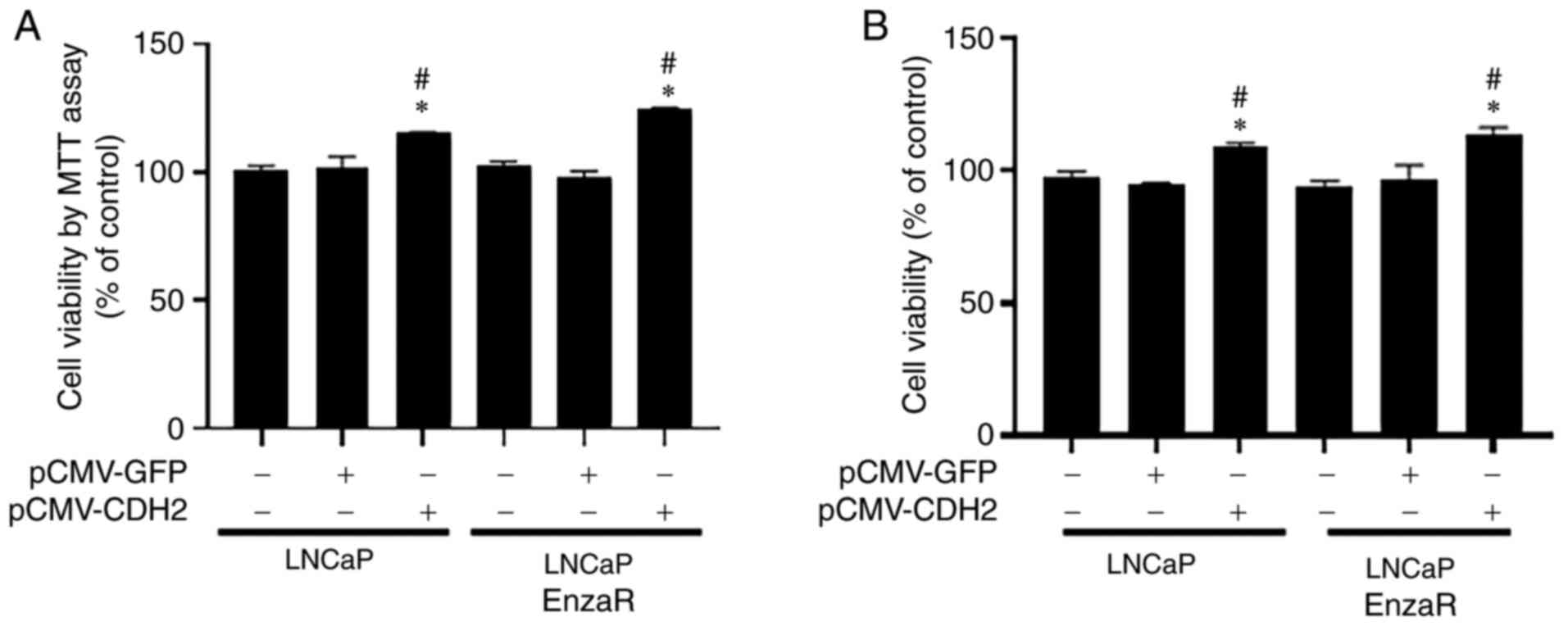
CDH2 overexpression increases LNCaP
and LNCaP EnzaR cell viability
To evaluate the effects of CDH2 on prostate cancer
cells, MTT and CCK-8 assays were used to measure cell viability.
pCMV-CDH2 plasmid transfection efficiency was confirmed by RT-PCR,
which markedly increased CDH2 expression in both LNCaP and LNCaP
EnzaR cells compared with that in cells transfected with the empty
vector (Fig. S1A).
In the LNCaP cell line, the pCMV-CDH2-transfected
cells exhibited the highest levels of cell viability compared with
those in the other two control groups 24 h after transfection,
according to results from MTT assay (Fig. 2). A similar result was observed in
the LNCaP EnzaRA cell line (Fig.
2A). Cell viability was next assessed in the both LNCaP and
LNCaP EnzaR cell lines using CCK-8 assay. Cells overexpressing CDH2
also showed the highest levels of cell viability in both LNCaP and
LNCaP EnzaR cell lines compared with those in the other two control
groups 24 h after transfection (Fig.
2B). These results suggest that CDH2 overexpression can
increase PCa cell viability.
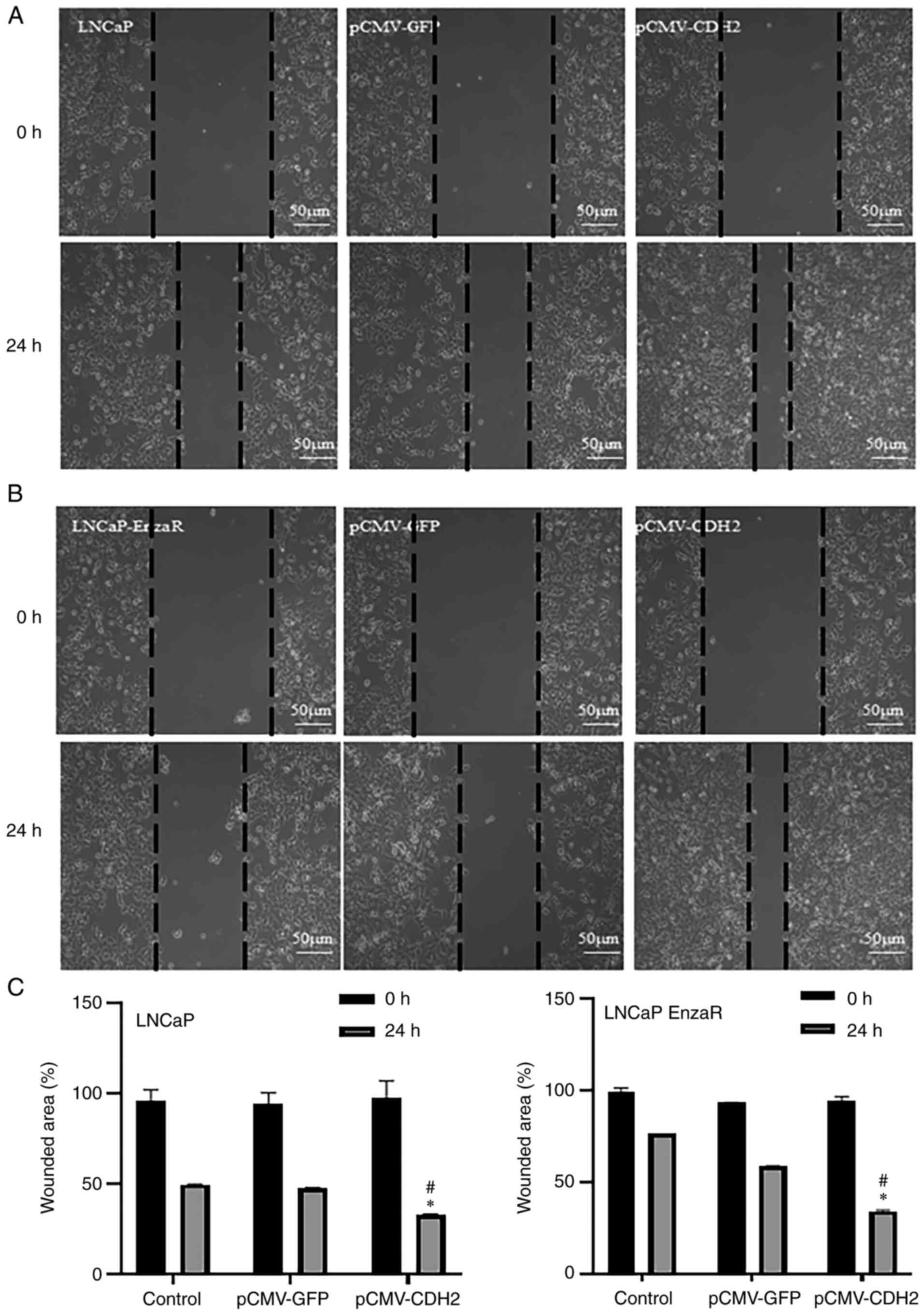
CDH2 overexpression increases LNCaP
and LNCaP EnzaR cell migration
Ibidi gap closure assays were performed to examine
the cell migratory capacity after transfection. After 24 h of
transfection, the migration capacity of untreated LNCaP and LNCaP
EnzaR cells was similar to that of empty vector-transfected cells.
However, pCMV-CDH2-transfected cells showed a significantly
increase in the capacity to migrate towards the center of the well
compared with that in cells transfected with the empty vector
(Fig. 3). These results were
observed in both the LNCaP and LNCaP EnzaR cell lines (Fig. 3).
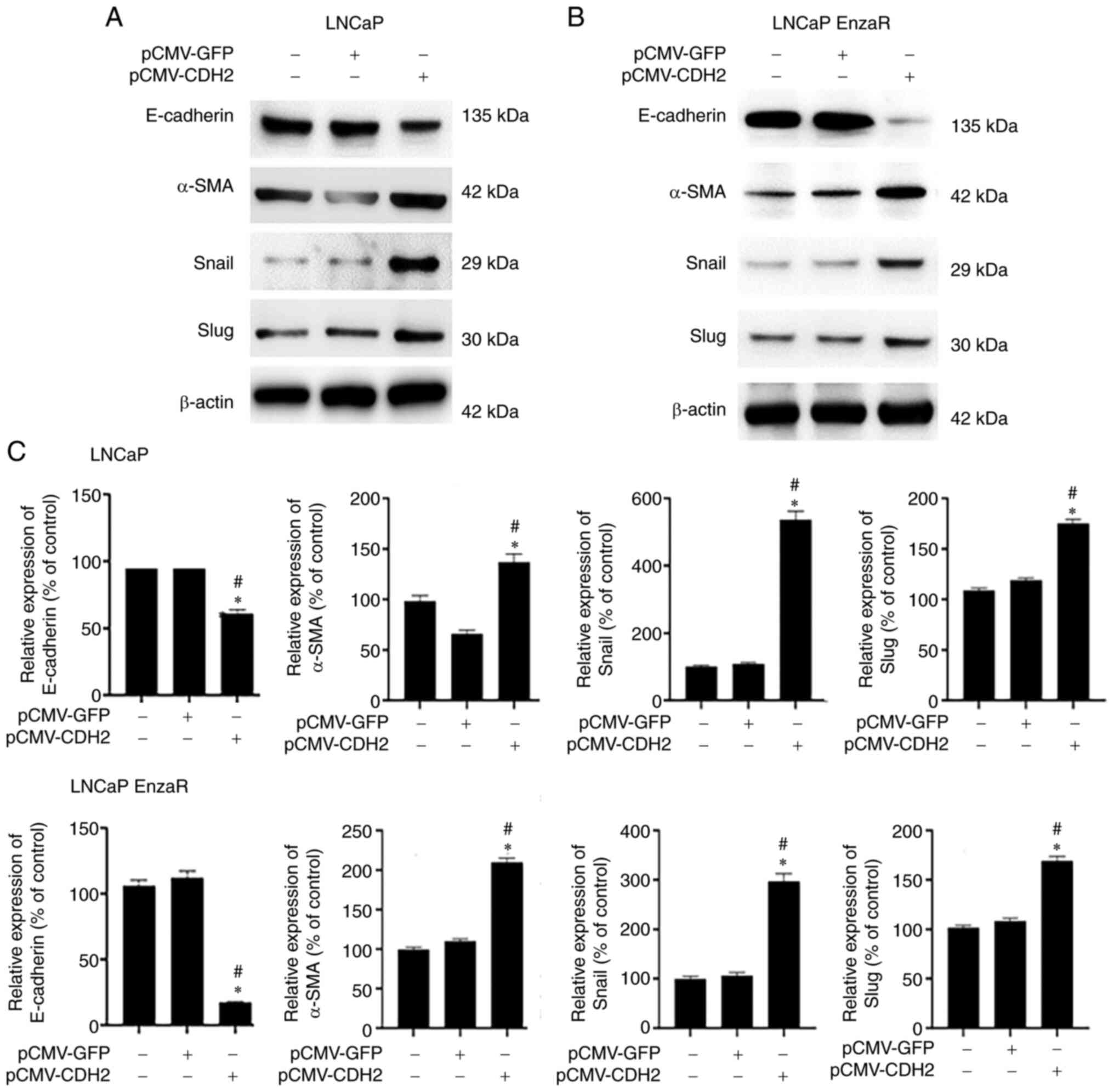
EMT is known to be associated with cancer cell
invasion and migration (13). To
clarify the mechanism underlying the increase in cell migration
after transfection with pCMV-CDH2, WB was used to measure the
expression of EMT markers E-cadherin, α-SMA, Snail and Slug. α-SMA,
Snail and Slug expression are positively correlated, whilst
E-cadherin expression is negatively correlated with EMT (13). The expression pattern of these four
markers in untreated cells was similar to that in empty
vector-transfected cells, which possibly explains the similar cell
migratory capacities between these two groups of cells. In
pCMV-CDH2-transfected LNCaP and LNCaP EnzaR cells, E-cadherin was
significantly downregulated, whilst the other three markers were
significantly upregulated (Fig.
4). This suggests that CDH2 overexpression may induce EMT in
LNCaP and LNCaP EnzaR cells, which may be the reason why
pCMV-CDH2-transfected cells exhibited the highest cell migration
levels. These results suggest that CDH2 overexpression can promote
EMT to increases the migratory capacity of PCa cells.
CDH2 knockdown reduces LNCaP and LNCaP
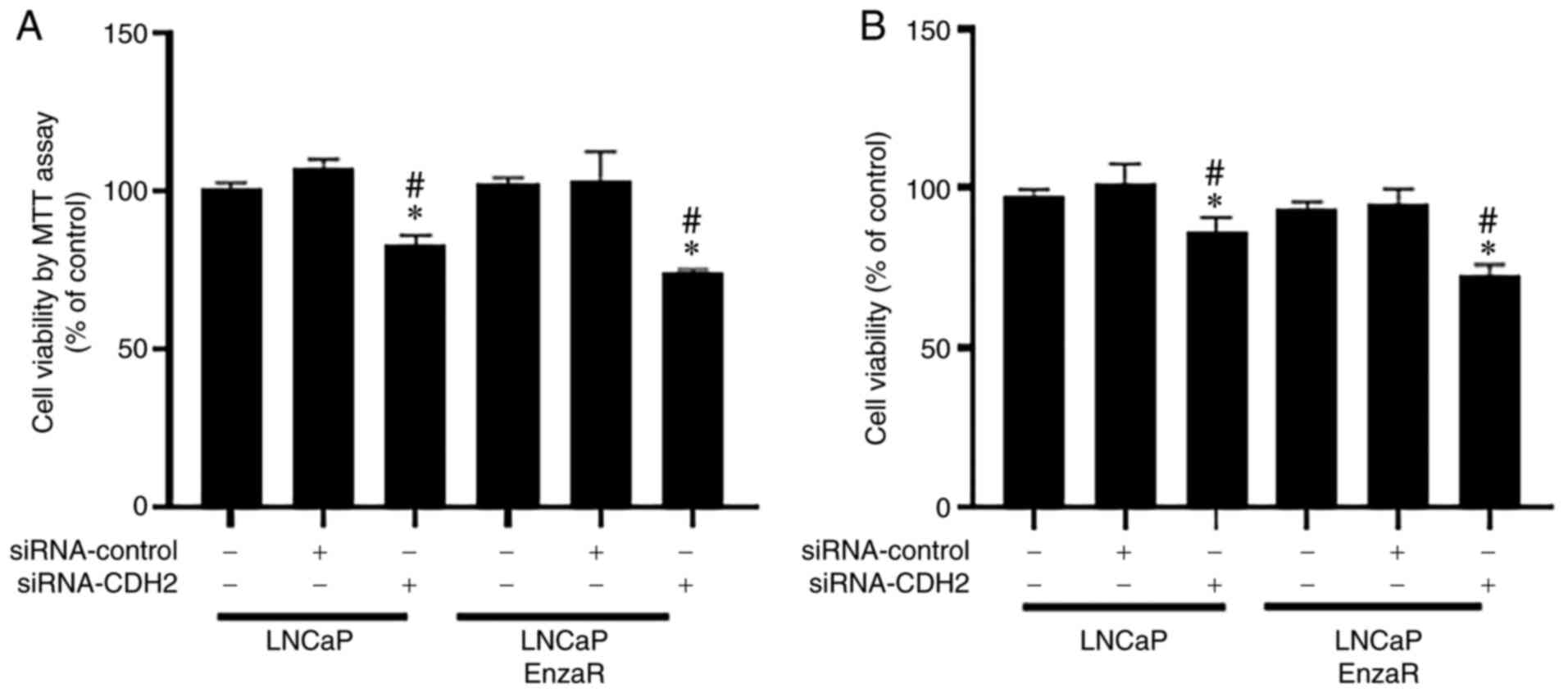
EnzaR cell viability
After overexpression, the possible effects of CDH2
knockdown on PCa cells were evaluated. MTT and CCK-8 assays were
performed to measure cell viability. siRNA-CDH2 transfection was
used to knock down CDH2 expression. siRNA-CDH2 transfection
efficiency in LNCaP and LNCaP EnzaR cells was verified using
RT-PCR, which markedly reduced CDH2 expression compared with that
in cells transfected with siRNA-control (Fig. S1). In terms of the LNCaP cell
line, siRNA-CDH2-transfected cells exhibited the lowest levels of
cell viability compared with that of the untreated cells and cells
transfected with the siRNA-control according to MTT assay (Fig. 5A). In addition, cell viability was
significantly reduced by siRNA-CDH2 transfection in the LNCaP EnzaR
cell line (Fig. 5A). Similar
results were observed according to CCK-8 assay. Specifically,
siRNA-CDH2-transfected cells showed the lowest cell viability in
both LNCaP and LNCaP EnzaR cells compared with that of the
untreated cells and cells transfected with the siRNA-control
(Fig. 5B). These results suggest
that CDH2 knockdown using siRNA reduced PCa cell viability.
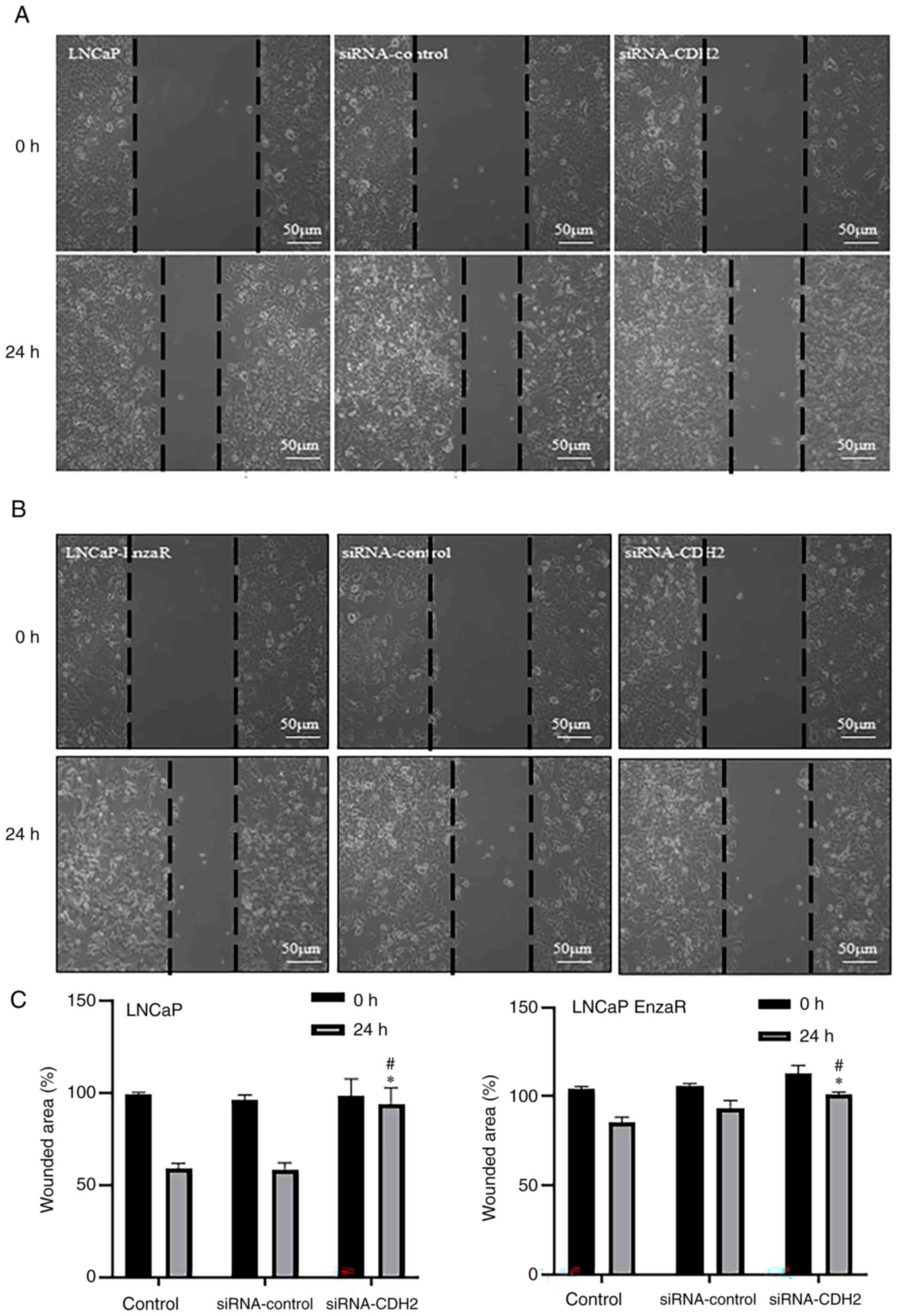
CDH2 knockdown inhibits LNCaP and
LNCaP EnzaR cell migration
According to the Ibidi gap closure assay, it was
observed that the level of migration in LNCaP cells transfected
with the siRNA-CDH2 was significantly slower compared with that in
untreated cells and cells transfected with the siRNA-control after
24 h (Fig. 6A and C). Similar
findings were made regarding the levels of LNCaP EnzaR cell
migration after 24 h (Fig. 6B and
C).
To assess the association between decreased
migration capacity and EMT, the protein expression of E-cadherin,
α-SMA, Snail and Slug was measured in both LNCaP and LNCaP EnzaR
cells by WB. Consistent with this hypothesis, cells with CDH2
expression knocked down appeared to exhibit reduced EMT induction.
Specifically, in siRNA-CDH2-transfected LNCaP and LNCaP EnzaR
cells, E-cadherin expression was significantly increased, whilst
the expression of α-SMA, Snail and Slug was significantly decreased
compared with that in untreated cells and cells transfected with
the siRNA-control (Fig. 7). These
results suggest that CDH2 knockdown can inhibit EMT and EMT-related
protein expression to suppress PCa cell migration.
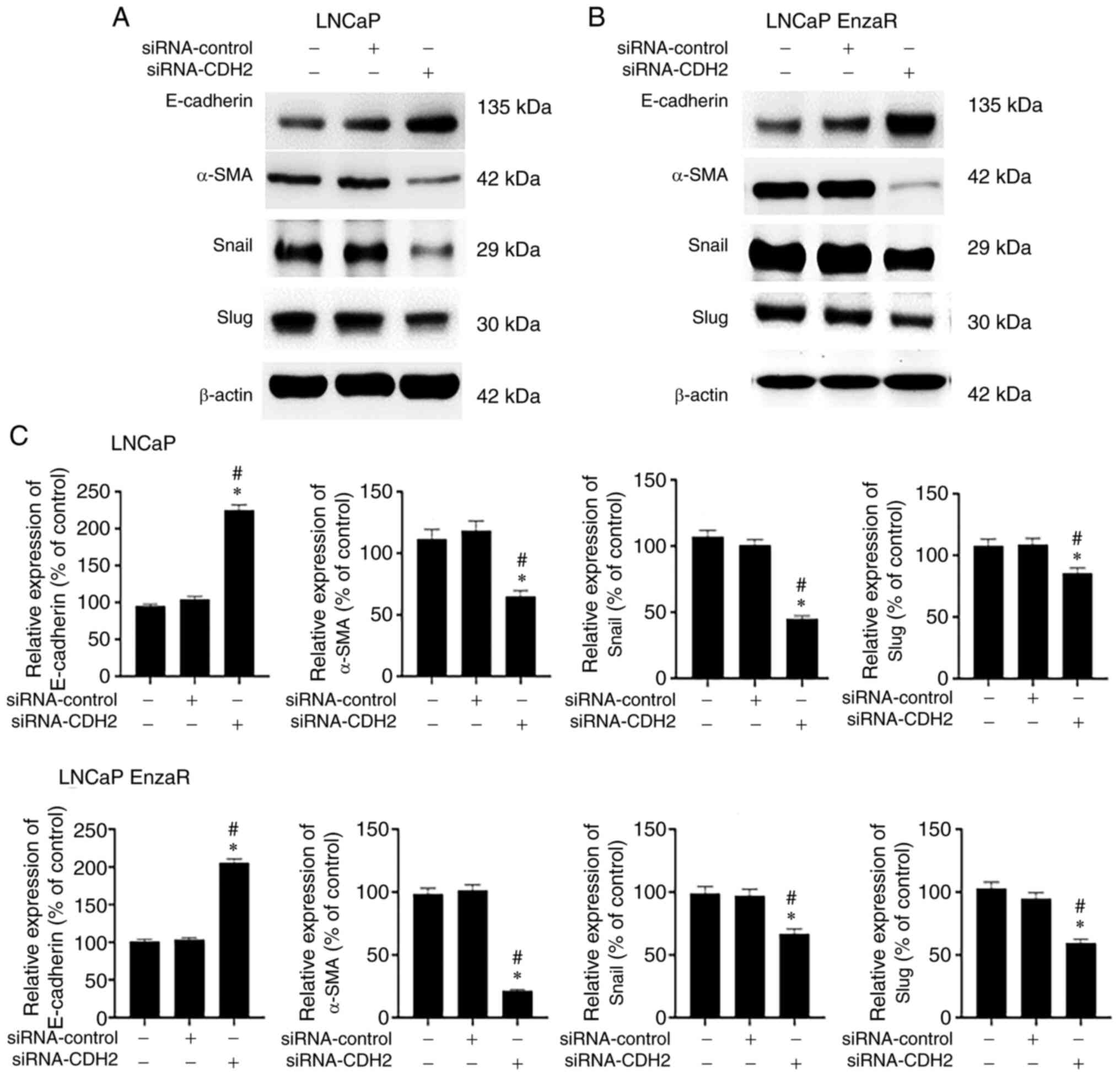 | Figure 7.Measurement of epithelial-mesenchymal
transition markers in PCa cells with CDH2 knockdown. Western blot
analysis measuring E-cadherin, α-SMA, Snail and slug expression in
(A) LNCaP cells and (B) LNCaP EnzaR cells, (C) which were then
quantified. α-SMA, Snail and slug protein expression in cells
transfected with siRNA-CDH2 was reduced, whereas E-cadherin
expression was increased compared with that in cells transfected
with siRNA-control or the control group. *P<0.05 vs. Control;
and #P<0.05 vs. siRNA-control. PCa, prostate cancer;
CDH2, N-cadherin; α-SMA, α-smooth muscle actin; EnzaR,
enzalutamide-resistant; siRNA, small interfering RNA |
Discussion
In the present study, it was found that CDH2 was
expressed at higher levels in LNCaP EnzaR cells compared with that
in LNCaP cells in vitro. This finding is not unexpected,
because CDH2 expression has been previously reported to be
increased in poorly differentiated PCa and positively correlate
with the Gleason score (26,27,32).
Similar observations in terms of the difference in CDH2 expression
were obtained by Tanaka et al (25) Jennbacken et al (33) and Nalla et al (34), where androgen-dependent cell lines
(LNCaP and LAPC4-AD) and androgen-independent cell lines (LNCaP-19,
PC-3 and LAPC4-CR) were assessed.
In terms of cell viability, increased viability was
noted after CDH2 overexpression, whereas the opposite was observed
after siRNA-CDH2 transfection. This suggests that CDH2 exerts a key
influence on PCa cell survival and proliferation. A similar finding
was reported in a previous study by Gao et al (35), where microRNA-194 overexpression,
which targeted CDH2, was used to regulate PCa cells to reduce cell
viability whilst increasing the rate of apoptosis (35). In addition, Wang et al
(36) performed colony formation
assays to explore the effect of CDH2 on the proliferation of PCa
cells and demonstrated a positive association between CDH2
expression and PCa cell proliferation (36). Tanaka et al (25) also previously showed that
N-cadherin-positive LAPC9 cells tended to proliferate more rapidly
compared with that in N-cadherin-negative cells (25). The RAS/Raf signaling cascade
following the cross-talk of CDH2 with other membrane proteins, such
as integrins, may be the cause of tumor cell proliferation
(37). However, the underlying
mechanism of this was not evaluated in the present study.
Cancer metastasis is a process that requires
multiple steps, with migration being a key step (38). In the present study, CDH2
expression in LNCap cells was found to be associated with cell
migration. Using overexpression and gene silencing methods to
manipulate CDH2 expression, LNCap and LNCap EnzaR cells with higher
CDH2 expression were found to have higher migratory capabilities.
In previous studies, CDH2 has been frequently reported as a factor
that can promote liver, lung, bladder, renal, colorectal, breast,
prostate and brain cancer cell migration and metastasis (25,33,36,37).
The Rac signaling pathway was found to be one of the underlying
mechanistic causes (37).
Furthermore, EMT has been found to be highly associated with cell
migration (13). Several signaling
pathways, including Wnt/β-catenin, PI3K/AKT, T-cell factor/lymphoid
enhancer-binding factor and RhoA, can become activated following
cadherin switching (39–43). Crosstalk between these signaling
pathways can increase the expression of a number of EMT
transcription factors, including Snail, Slug and Twist (39). The increase of EMT transcription
factor expression was previously demonstrated in PC3, LNCaP and
DU145 cell lines (17,44–45).
After determining the interaction between transcription factors,
CDH2, EMT and cancer cell migration, a possible connection was
found between EMT and PCa cell migration by measuring the
expression levels of these transcription factors following CDH2
regulation in the present study.
A number of factors and compounds that can target
EMT have been previously demonstrated to modulate PCa cells. Li
et al (46) found that
resveratrol can reverse EMT through the Hedgehog pathway in PCa
(46). In addition, curcumin was
found to inhibit PCa cell EMT and invasion through the monoamine
oxidase A/mTOR/hypoxia-inducible factor-1α signaling pathway
(47). MicroRNAs are short,
non-coding and single-stranded RNA molecules that have been
previous assessed as a potential biomarker in many types of cancer,
where they serve a key regulatory functions in PCa progression
(48,49). MicroRNA-205, microRNA-143,
microRNA-145 have been found to inhibit the EMT process by
negatively regulating the expression of several transcription
factors (50). Another possible
target for PCa inhibition is CDH2. Using overexpression and
knockdown approaches, Tanaka et al (25) were able to regulate CDH2 expression
in the presence or absence of monoclonal CDH2 antibodies and
determine its involvement in PCa. After monoclonal antibody
inhibition, decreased cell proliferation and invasion in
vitro and decreased growth and metastasis in vivo were
observed (25). Similarly, the
present study demonstrated that siRNA-CDH2 transfection reduced
cell viability and migration in both LNCaP and LNCaP EnzaR cells
in vitro. To the best of our knowledge, the present study is
also the first to use siRNA for the downregulation of CDH2
expression in EnzaR PCa cells.
The present study is a preliminary study. Therefore,
there are a number of limitations. Unlike other studies that used a
panel of PCa cell lines, only LNCaP cells were used whereas its
subline, LNCaP EnzaR, were used for examination. Furthermore, only
in vitro experiments were performed to observe the effect of
CDH2 knockdown. Since no in vivo experiments were performed,
the precise mechanism underlying the effects of CDH2 knockdown on
prostate tissues could not be verified. The only conclusion that
can be drawn from the present study was that PCa cell migration,
one of the key steps of metastasis, was impaired as a result of
CDH2 knockdown. To further understand the effect of CDH2 knockdown
on metastasis, invasion and the extent of mesenchymal-epithelial
transition should be examined. In addition, results in the present
study showed that CDH2 expression is positively associated with PCa
cell viability, proliferation, migration and EMT. Further studies
are warranted to determine the underlying mechanisms. Subsequent
experiments should be focused on treating different PCa cell lines
and in in vivo animal models.
According to the present study, it was demonstrated
that EMT served an important role in modulating PCa cell
proliferation and migration. In addition, the expression of CDH2,
which significantly influences EMT, could be manipulated to reduce
PCa cell viability and migration. These findings raise the
possibility that CDH2 may be key to controlling CRPC and can be
exploited in clinical practice.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr See-Tong Pang
(Department of Uro-Oncology, Chang Gung Memorial Hospital, Taiwan),
for providing the Enzalutamide-resistant LNCaP cells.
Funding
The present study was supported by E-Da Hospital Research (grant
no. NCKUEDA10906) and National Science Council Grants, Taiwan
(grant no. MOST 109-2314-B-650-013-MY2).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
VCHL and CHO conceived and designed the study. CHL
and CHW were the major contributors in writing the manuscript. CHL
and PFH analyzed and interpreted the data. CHW, PFH, CYW and WWTK
performed the literature review and conducted the experiments. All
authors read and approved the final manuscript. VCHL, CHO, CHL and
CHW confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CDH2
|
N-cadherin
|
|
EnzaR
|
enzalutamide-resistant
|
|
siRNA
|
small interfering RNA
|
|
EMT
|
epithelial-mesenchymal transition
|
|
PCa
|
prostate cancer
|
|
ADT
|
androgen deprivation therapy
|
|
CRPC
|
castration-resistant prostate
cancer
|
|
α-SMA
|
α-smooth muscle actin
|
|
WB
|
western blotting
|
References
|
1
|
Cooperberg MR, Cowan J, Broering JM and
Carroll PR: High-risk prostate cancer in the United States,
1990-2007. World J Urol. 26:211–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pagliarulo V, Bracarda S, Eisenberger MA,
Mottet N, Schröder FH, Sternberg CN and Studer UE: Contemporary
role of androgen deprivation therapy for prostate cancer. Eur Urol.
61:11–25. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu T, Wu LY, Fulton MD, Johnson JM and
Berkman CE: Prolonged androgen deprivation leads to downregulation
of androgen receptor and prostate-specific membrane antigen in
prostate cancer cells. Int J Oncol. 41:2087–2092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chandrasekar T, Yang JC, Gao AC and Evans
CP: Mechanisms of resistance in castration-resistant prostate
cancer (CRPC). Transl Androl Urol. 4:365–380. 2015.PubMed/NCBI
|
|
5
|
Teo MY, Rathkopf DE and Kantoff P:
Treatment of advanced prostate cancer. Annu Rev Med. 70:479–499.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
FDA approves enzalutamide for
castration-resistant prostate cancer, . https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-enzalutamide-castration-resistant-prostate-cancerDecember
1–2021
|
|
7
|
Wengner AM, Scholz A and Haendler B:
Targeting DNA damage response in prostate and breast cancer. Int J
Mol Sci. 21:82732020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beer TM, Armstrong AJ, Rathkopf DE, Loriot
Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J,
Chowdhury S, et al: Enzalutamide in metastatic prostate cancer
before chemotherapy. N Engl J Med. 371:424–433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim YS, Yi BR, Kim NH and Choi KC: Role of
the epithelial-mesenchymal transition and its effects on embryonic
stem cells. Exp Mol Med. 46:e1082014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.PubMed/NCBI
|
|
12
|
Jennbacken K, Gustavsson H, Welén K,
Vallbo C and Damber JE: Prostate cancer progression into androgen
independency is associated with alterations in cell adhesion and
invasivity. Prostate. 66:1631–1640. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Heerboth S, Housman G, Leary M, Longacre
M, Byler S, Lapinska K, Willbanks A and Sarkar S: EMT and tumor
metastasis. Clin Transl Med. 4:62015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vesuna F, van Diest P, Chen JH and Raman
V: Twist is a transcriptional repressor of E-cadherin gene
expression in breast cancer. Biochem Biophys Res Commun.
367:235–241. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vered M, Dayan D, Yahalom R, Dobriyan A,
Barshack I, Bello IO, Kantola S and Salo T: Cancer-associated
fibroblasts and epithelial-mesenchymal transition in metastatic
oral tongue squamous cell carcinoma. Int J Cancer. 127:1356–1362.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Randle DD, Clarke S, Henderson V and
Odero-Marah VA: Snail mediates invasion through uPA/uPAR and the
MAPK signaling pathway in prostate cancer cells. Oncol Lett.
6:1767–1773. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Uygur B and Wu WS: SLUG promotes prostate
cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol
Cancer. 10:1392011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Navarro P, Ruco L and Dejana E:
Differential localization of VE- and N-cadherins in human
endothelial cells: VE-cadherin competes with N-cadherin for
junctional localization. J Cell Biol. 140:1475–1484. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hatta K, Takagi S, Fujisawa H and Takeichi
M: Spatial and temporal expression pattern of N-cadherin cell
adhesion molecules correlated with morphogenetic processes of
chicken embryos. Dev Biol. 120:215–227. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Derycke LD and Bracke ME: N-cadherin in
the spotlight of cell-cell adhesion, differentiation,
embryogenesis, invasion and signalling. Int J Dev Biol. 48:463–476.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma T, Zhao Y, Wei K, Yao G, Pan C, Liu B,
Xia Y, He Z, Qi X, Li Z, et al: MicroRNA-124 functions as a tumor
suppressor by regulating CDH2 and epithelial-mesenchymal transition
in non-small cell lung cancer. Cell Physiol Biochem. 38:1563–1574.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van der Horst G, Bos L, van der Mark M,
Cheung H, Heckmann B, Clément-Lacroix P, Lorenzon G, Pelger RC,
Bevers RF and van der Pluijm G: Targeting of alpha-v integrins
reduces malignancy of bladder carcinoma. PLoS One. 9:e1084642014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Markou A, Lazaridou M, Paraskevopoulos P,
Chen S, Świerczewska M, Budna J, Kuske A, Gorges TM, Joosse SA,
Kroneis T, et al: Multiplex gene expression profiling of in vivo
isolated circulating tumor cells in high-risk prostate cancer
patients. Clin Chem. 64:297–306. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sandig M, Voura EB, Kalnins VI and Siu CH:
Role of cadherins in the transendothelial migration of melanoma
cells in culture. Cell Motil Cytoskeleton. 38:351–364. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tanaka H, Kono E, Tran CP, Miyazaki H,
Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, et
al: Monoclonal antibody targeting of N-cadherin inhibits prostate
cancer growth, metastasis and castration resistance. Nat Med.
16:1414–1420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tomita K, van Bokhoven A, van Leenders GJ,
Ruijter ET, Jansen CF, Bussemakers MJ and Schalken JA: Cadherin
switching in human prostate cancer progression. Cancer Res.
60:3650–3654. 2000.PubMed/NCBI
|
|
27
|
Jaggi M, Nazemi T, Abrahams NA, Baker JJ,
Galich A, Smith LM and Balaji KC: N-cadherin switching occurs in
high gleason grade prostate cancer. Prostate. 66:193–199. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kregel S, Chen JL, Tom W, Krishnan V, Kach
J, Brechka H, Fessenden TB, Isikbay M, Paner GP, Szmulewitz RZ and
Griend DJV: Acquired resistance to the second-generation androgen
receptor antagonist enzalutamide in castration-resistant prostate
cancer. Oncotarget. 7:26259–26274. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee GT, Rosenfeld JA, Kim WT, Kwon YS,
Palapattu G, Mehra R, Kim WJ and Kim IY: TCF4 induces enzalutamide
resistance via neuroendocrine differentiation in prostate cancer.
PLoS One. 14:e02134882019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pangestu NS, Chueakwon P, Talabnin K,
Khiaowichit J and Talabnin C: RNF43 overexpression attenuates the
wnt/β-catenin signalling pathway to suppress tumour progression in
cholangiocarcinoma. Oncol Lett. 22:8462021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang YH, Kuo HC, Yang YL and Wang FS:
MicroRNA-29a is a key regulon that regulates BRD4 and mitigates
liver fibrosis in mice by inhibiting hepatic stellate cell
activation. Int J Med Sci. 16:212–220. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bussemakers MJ, Van Bokhoven A, Tomita K,
Jansen CF and Schalken JA: Complex cadherin expression in human
prostate cancer cells. Int J Cancer. 85:446–450. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jennbacken K, Tesan T, Wang W, Gustavsson
H, Damber JE and Welén K: N-cadherin increases after androgen
deprivation and is associated with metastasis in prostate cancer.
Endocr Relat Cancer. 17:469–479. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nalla AK, Estes N, Patel J and Rao JS:
N-cadherin mediates angiogenesis by regulating monocyte
chemoattractant protein-1 expression via PI3K/Akt signaling in
prostate cancer cells. Exp Cell Res. 317:2512–2521. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gao S, Zhao Z, Wu R, Wu L, Tian X and
Zhang Z: MicroRNA-194 regulates cell viability and apoptosis by
targeting CDH2 in prostatic cancer. Onco Targets Ther.
11:4837–4844. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang M, Ren D, Guo W, Huang S, Wang Z, Li
Q, Du H, Song L and Peng X: N-cadherin promotes
epithelial-mesenchymal transition and cancer stem cell-like traits
via ErbB signaling in prostate cancer cells. Int J Oncol.
48:595–606. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mariotti A, Perotti A, Sessa C and Rüegg
C: N-cadherin as a therapeutic target in cancer. Expert Opin
Investig Drugs. 16:451–465. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Loh CY, Chai JY, Tang TF, Wong WF, Sethi
G, Shanmugam MK, Chong PP and Looi CY: The E-cadherin and
N-cadherin switch in epithelial-to-mesenchymal transition:
Signaling, therapeutic implications, and challenges. Cells.
8:11182019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jiang YG, Luo Y, He DL, Li X, Zhang LL,
Peng T, Li MC and Lin YH: Role of Wnt/beta-catenin signaling
pathway in epithelial-mesenchymal transition of human prostate
cancer induced by hypoxia-inducible factor-1alpha. Int J Urol.
14:1034–1039. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chang L, Graham PH, Hao J, Ni J, Bucci J,
Cozzi PJ, Kearsley JH and Li Y: Acquisition of
epithelial-mesenchymal transition and cancer stem cell phenotypes
is associated with activation of the PI3K/Akt/mTOR pathway in
prostate cancer radioresistance. Cell Death Dis. 4:e8752013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liang J, Li Y, Daniels G, Sfanos K, De
Marzo A, Wei J, Li X, Chen W, Wang J, Zhong X, et al: LEF1
targeting EMT in prostate cancer invasion is regulated by miR-34a.
Mol Cancer Res. 13:681–688. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen X, Cheng H, Pan T, Liu Y, Su Y, Ren
C, Huang D, Zha X and Liang C: mTOR regulate EMT through RhoA and
Rac1 pathway in prostate cancer. Mol Carcinog. 54:1086–1095. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Baygi ME, Soheili ZS, Essmann F, Deezagi
A, Engers R, Goering W and Schulz WA: Slug/SNAI2 regulates cell
proliferation and invasiveness of metastatic prostate cancer cell
lines. Tumour Biol. 31:297–307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stylianou N, Lehman ML, Wang C, Fard AT,
Rockstroh A, Fazli L, Jovanovic L, Ward M, Sadowski MC, Kashyap AS,
et al: A molecular portrait of epithelial-mesenchymal plasticity in
prostate cancer associated with clinical outcome. Oncogene.
38:913–934. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li J, Chong T, Wang Z, Chen H and Li H,
Cao J, Zhang P and Li H: A novel anti-cancer effect of resveratrol:
Reversal of epithelial-mesenchymal transition in prostate cancer
cells. Mol Med Rep. 10:1717–1724. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Du Y, Long Q, Zhang L, Shi Y, Liu X, Li X,
Guan B, Tian Y, Wang X, Li L and He D: Curcumin inhibits
cancer-associated fibroblast-driven prostate cancer invasion
through MAOA/mTOR/HIF-1α signaling. Int J Oncol. 47:2064–2072.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vanacore D, Boccellino M, Rossetti S,
Cavaliere C, D'Aniello C, Di Franco R, Romano FJ, Montanari M, La
Mantia E, Piscitelli R, et al: Micrornas in prostate cancer: An
overview. Oncotarget. 8:50240–50251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lo UG, Yang D and Hsieh JT: The role of
microRNAs in prostate cancer progression. Transl Androl Urol.
2:228–241. 2013.PubMed/NCBI
|
|
50
|
Cochetti G, Rossi de Vermandois JA, Maulà
V, Giulietti M, Cecati M, Del Zingaro M, Cagnani R, Suvieri C,
Paladini A and Mearini E: Role of miRNAs in prostate cancer: Do we
really know everything? Urol Oncol. 38:623–635. 2020. View Article : Google Scholar : PubMed/NCBI
|