Introduction
Gastric cancer (GC) has a highly aggressive clinical
course making it one of the most lethal malignant tumors with a
discouraging prognosis. Comparing published data by GLOBOCAN in
2012, the updated 2018 edition data showed an increase in incidence
and mortality with 1,033.7 thousand new patients and 782.7 thousand
deaths per year worldwide (1,2).
More advanced gastric endoscopic imaging provides an opportunity
for clinicians to detect the precancer lesions and treat the tumor
at an endoscopically curable stage. The strategy of early detection
and endoscopic resection has lowered morbidity in the countries
with a high prevalence of GC, including Japan and South Korea
(3–5). However, in other regions, a shortage
of properly trained endoscopy operators and inconsistencies in
diagnosis between pathologists contribute to the fact that a
significant proportion of patients are diagnosed at advanced stage.
Accurate diagnosis, especially for those who cannot be biopsied, is
an urgent problem that must be overcome for treatment of GC.
Although individual treatments, including surgery,
chemotherapy, radiotherapy and molecular targeted therapy, have
made great improvements, those with advanced-stage GC still have a
poor 5-year survival rate (6).
These poor outcomes have resulted in research focusing on
identifying prognostic related factors, including age, gender,
tumor grade and pathological molecular subtypes (7,8). In
addition, accumulating studies identify non-coding RNAs including
micro (mi)RNA, circular (circ)RNA and long-noncoding RNA as having
prognostic value. Although studies highlight the value of
biomarkers, some limitations cannot be ignored, such as conclusions
based on a single research cohort, inadequate multi-center
validation, single marker, and small sample sizes (9–12).
Therefore, new biomarkers with high accuracy and specificity are
needed to improve the diagnosis and prognosis of GC.
The present study used publicly available gene
expression profiles from The Cancer Genome Atlas (TCGA) and Gene
Expression Omnibus (GEO) datasets including mRNA, miRNA and circRNA
of GC to establish a competing endogenous (ce)RNA network. The
prediction models based on 5-mRNA signature and 5-miRNA signature
were generated by least absolute shrinkage and selection operation
(LASSO) penalized regression. The prediction models showed a good
capacity for diagnosis and prognosis in both internal validation
groups and external validation sets. Thus, the present study
identified and validated new candidate genes to diagnose and
prognose GC by assigning a patient to high-risk or low-risk
group.
Materials and methods
Patients and datasets
All the gene expression profiles were obtained from
The Cancer Genome Atlas (TCGA) Gene Expression Omnibus (GEO;
http://www.ncbi.nlm.nih.gov/geo)
database or Genotype-Tissue Expression (GTEx; http://gtexportal.org/home/) project. The information
of all selected datasets including sample size and sequencing form
are listed in Table I. GTEx data
was used to verify the diagnostic model as a supplement to normal
samples.
 | Table I.Datasets used in this study and their
sample distribution. |
Table I.
Datasets used in this study and their
sample distribution.
| Dataset | Experiment
type | RNA type | Tumor | Normal |
|---|
| TCGA | RNA-seq | mRNA | 375 | 32 |
| GSE54129 | Expression
profiling by array | mRNA | 111 | 21 |
| GTEx | RNA-seq | mRNA | - | 207 |
| TCGA | miRNA-Seq | miRNA | 436 | 41 |
| GSE106817 | Non-coding RNA
profiling by array | miRNA | 115 | 2,759 |
| GSE112264 | Non-coding RNA
profiling by array | miRNA | 50 | 41 |
| GSE83521 | Non-coding RNA
profiling by array | circRNA | 6 | 6 |
| GSE93541 | Non-coding RNA
profiling by array | circRNA | 3 | 3 |
Identification of differentially
expressed genes (DEGs)
DEGs, including mRNA, miRNA and circRNA were
identified from the aforementioned datasets. Significant DEGs
(|log2FC| > 2, adjusted P-value <0.05) were
identified by using the Limma package (https://www.bioconductor.org/packages/release/bioc/html/limma.html)
of R 4.0.3 (13,14).
Weighted correlation network analysis
(WGCNA)
Hub genes were identified using WGCNA. WGCNA
networks were constructed for mRNA, miRNA and circRNA using the
GSE54129, GSE106817 and GSE93541 datasets, respectively. First, the
similarity matrix was constructed based on the expression data by
calculating the Pearson correlation coefficient between two genes,
the top 20% differentially expressed mRNA, the top 50% of miRNA and
the top 75% of circRNA were chosen for further study. Next,
clustering detection was performed to exclude outlier samples.
Also, an appropriate power of β was adopted as a soft-thresholding
parameter through network topological analysis to construct
scale-free networks. An adjacency matrix was next transformed into
a topological overlap matrix (TOM), 1-TOM was used as the distance
to cluster the genes and a dynamic pruning tree was built to
identify the modules. Finally, the correlation between phenotypes
(tumor or normal tissue) and modules was calculated to recognize
the most clinically significant ones (15).
Construction of ceRNA network
RNAhybrid (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid)
was used to predict the interaction between circRNA and miRNA
(16). miRWalk 3.0 (http://mirwalk.umm.uni-heidelberg.de/)
was used to predict the interaction between mRNA and miRNA
(17), while the
circRNA-miRNA-mRNA network was built using Cytoscape 3.8.2
(18).
Functional annotation of hub mRNA
Node mRNAs in ceRNA network was conducted to Gene
Ontology (GO) molecular function enrichment and Kyoto Encyclopedia
of Genes and Genomes (KEGG) pathway enrichment using
clusterProfiler package of R4.0.3 (https://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html).
P<0.05 was defined as the threshold of statistically
significance (19).
Screening diagnostic and prognostic
signature
To screen diagnostic and prognostic signatures,
LASSO regression analysis was performed using the glmnet
(https://cran.r-project.org/web/packages/glmnet/index.html)
package of R 4.0.3 in TCGA dataset. After 10-fold cross validation,
the top 5 mRNA and miRNA ranked by absolute value of regression
coefficient were token as diagnostic and prognostic signature for
further study (20).
Construction of support vector machine
(SVM) diagnostic model
SVM diagnostic model was constructed to predict
carcinoma and non-carcinoma by using scikit-learn package supplied
by python v3.8 (https://www.python.org/downloads/release/python-3812/)
(21,22). Grid search with 3-fold cross
validation was conducted to test all parameters values shown in
Table II, then the diagnostic
model was constructed based on best parameter combination. Finally,
the model was verified by 10-fold cross validation and receiver
operating characteristic (ROC) curve was drawn to evaluate the
classification efficiency of the model (23).
| Table II.Support vector machine model
parameter options. |
Table II.
Support vector machine model
parameter options.
| Parameter name | Parameter value
range |
|---|
| Penalty C | 0.01-30 |
| Gamma |
1×10−10−1 |
| Kernel | rbf, linear |
Construction of prognostic model
The prognostic model was used to predict prognosis
based on RiskScore value. RiskScore values were calculated using a
linear combination of gene expression values weighted by univariate
Cox regression coefficients. The standard form was defined as
RiskScore=∑(βi × Xi), where i is the number of prognostic
signatures, β is the correlation coefficient of prognostic
signatures in univariate Cox regression analysis and X is the
expression value of prognostic signatures (24).
Results
Differential expression analysis
The expression data between cancerous and
non-cancerous samples were compared and DEGs were defined according
to the standard of |log2FC| >2 and adjusted P-value
<0.05. The number of DEGs in each dataset were summarized in
Table III, the detail
information about up and downregulated RNAs can be viewed in
Tables SI,SII,SIII,SIV,SV,SVI,SVII. The log2FC and P-value
distribution of top 100 differentially expressed mRNAs were shown
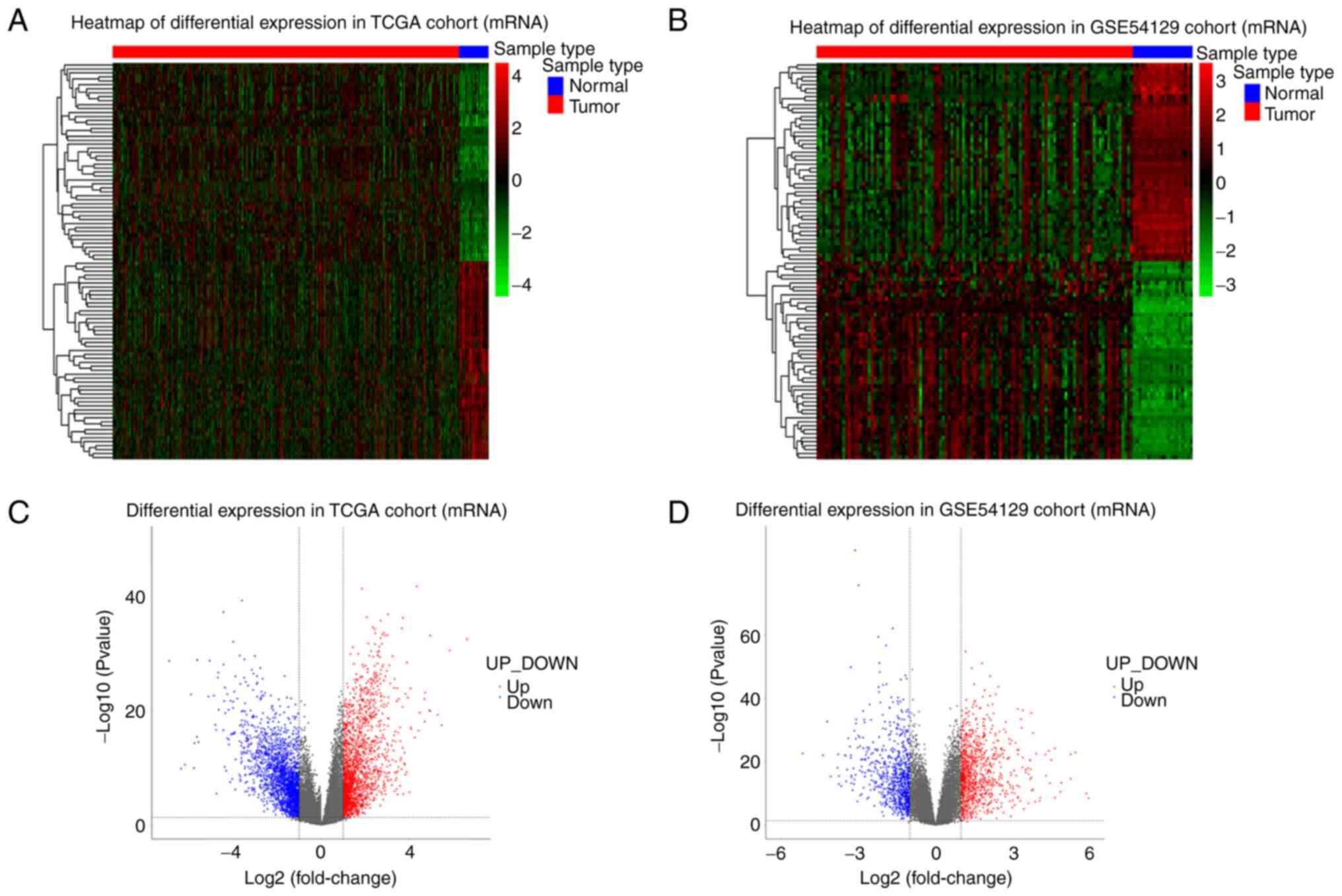
by heatmaps and volcano plot (Fig.
1). The heatmaps and volcano plots for miRNAs and circRNAs were
presented in Figs. S1 and
S2. The expression level of DEGs
in homogenous samples were consistent as seen in heatmaps and the
RNA expression levels in GC and normal tissue were significantly
different, indicating samples used in this cohort had good
uniformity and the DEGs screened were reliable. Volcano plots
illustrated the log2FC values of all differentially
expressed mRNAs were distributed between −6 and 6, with the
majority being distributed between −3 and 3. For miRNA,
log2FC values were distributed between −5 and 5 and most
distributed between −1 and 1. For circRNA, log2FC values
were distributed between −8 and 8, with most being distributed
between −2 and 2.
| Table III.Summary of the number of DEGs in this
study. |
Table III.
Summary of the number of DEGs in this
study.
| Dataset | Type | UP_DEGs | DOWN_DEGs |
|---|
| TCGA | mRNA | 2,087 | 2,322 |
| GSE54129 | mRNA | 998 | 830 |
| TCGA | miRNA | 72 | 81 |
| GSE106817 | miRNA | 569 | 428 |
| GSE112264 | miRNA | 602 | 428 |
| GSE83521 | circRNA | 70 | 80 |
| GSE93541 | circRNA | 202 | 216 |
WGCNA analysis
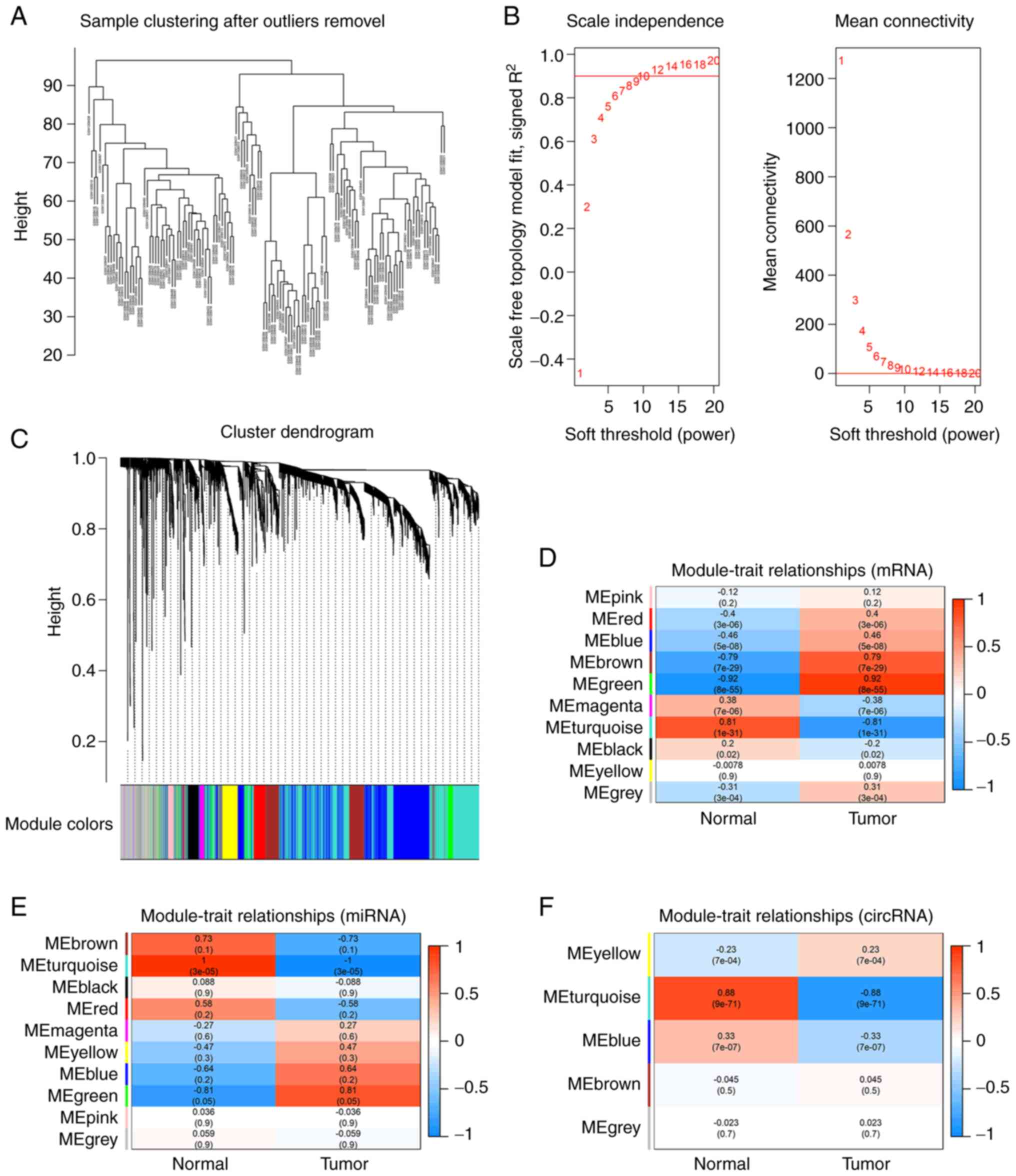
A total of 4,193 mRNAs (the top 20%) were identified
in GES54129 dataset ranked by the Pearson correlation coefficient.
Sample clustering analysis showed three obvious outlier samples
with heights >100, the remaining samples after pruning
(cutHeight=100) were reserved for following analysis. Network
topological analysis indicated the appropriate power of β was 9
(Fig. 2B) and mRNAs with similar
expression levels were categorized into the same module. Finally,
the dataset was divided into 10 modules (Fig. 2C). The correlation analysis between
phenotypes and modules indicated that green and turquoise modules
were the most clinically significant (|R|>0.8, P<0.05;
Fig. 2D).
The top 50% of miRNAs (1,282) were identified in the GSE106817
dataset. Sample clustering analysis showed there were no outlier
samples. Topological analysis indicated the appropriate power of β
was 3. All candidate miRNAs were categorized into 5 modules
(Fig. S3A-C) and turquoise was
the most clinically significant (|R|>0.8, P<0.05; Fig. 2E).
The top 75% of circRNAs (1,313) were observed in GSE93541 for
further analysis. Sample clustering analysis showed that there were
no outlier samples. Topological network analysis showed the
appropriate power of β was 7. circRNAs were classified into 10
modules (Fig. S3D-F) and
turquoise was the most clinically significant module (|R|>0.8,
P<0.05; Fig. 2F). The number of
mRNA, miRNA and circRNA in each module were listed in Table IV.
| Table IV.Number of RNAs contained in each
module in weighted correlation network analysis. |
Table IV.
Number of RNAs contained in each
module in weighted correlation network analysis.
| Module | GSE54129
(mRNA) | GSE106817
(miRNA) | GSE93541
(circRNA) |
|---|
| Black | 130 | - | 49 |
| Blue | 1,110 | 87 | 312 |
| Brown | 395 | 55 | 141 |
| Green | 183 | - | 76 |
| Grey | 423 | 544 | 5 |
| Magenta | 66 | - | 35 |
| Pink | 70 | - | 45 |
| Red | 164 | - | 52 |
| Turquoise | 1,410 | 550 | 507 |
| Yellow | 242 | 46 | 91 |
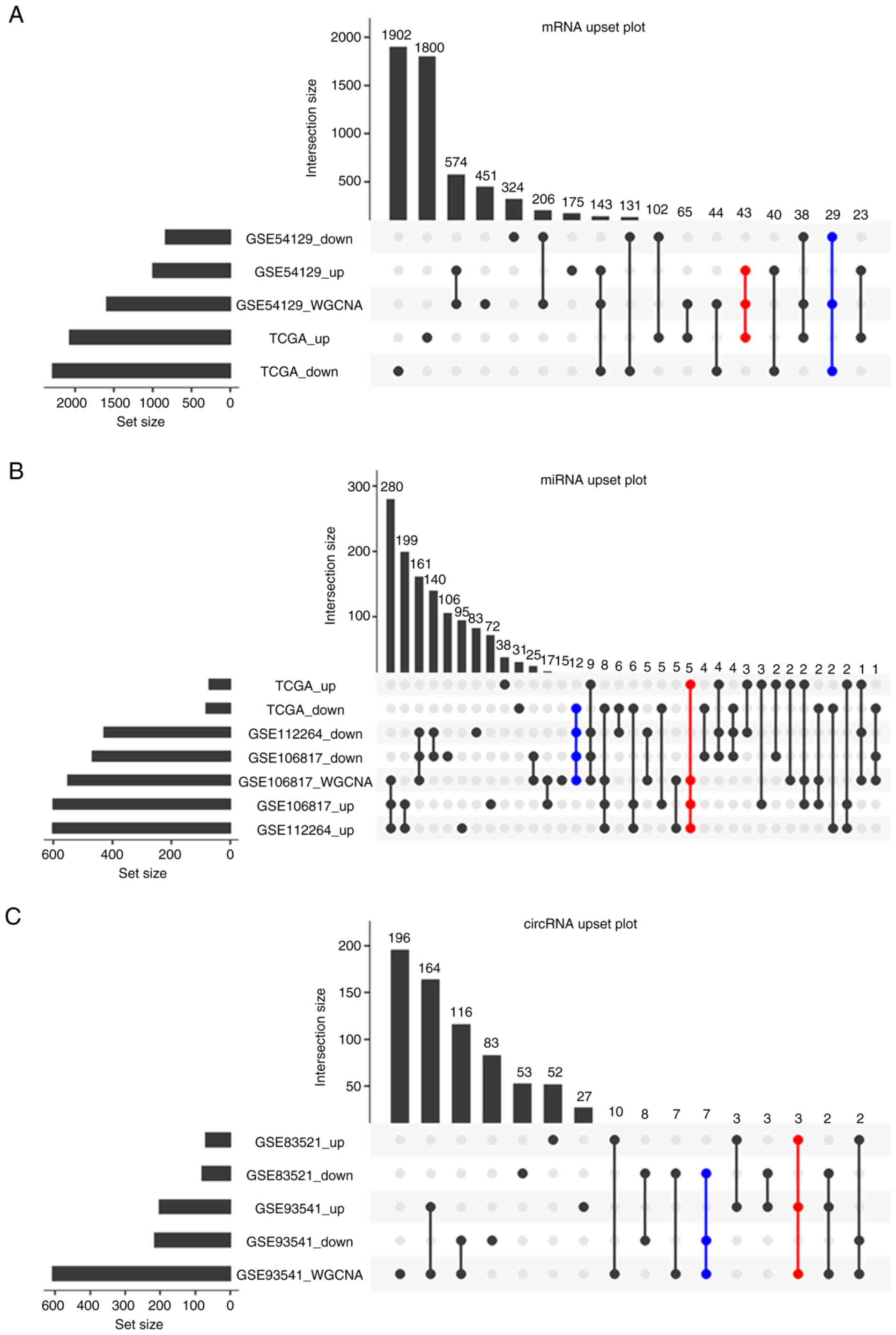
Candidate RNAs screening
To promote the reliability and applicability of the
ceRNA network, the present study took the RNAs obtained from WGCNA
analysis intersected with the DEGs in TCGA and GEO datasets. The DE
mRNAs in turquoise and green modules were intersected with DEGs
from GSE54129 and TCGA, the DE miRNAs in the turquoise modules were
intersected with GSE106817, GSE112264 and TCGA datasets. Similarly,
the DE circRNAs in turquoise were intersected with GSE93541 and
GSE83521 datasets. 72 mRNAs (43 upregulated and 29 downregulated),
17 miRNAs (5 upregulated and 12 downregulated) and 10 circRNAs (3
upregulated and 7 downregulated) were obtained as candidate RNAs to
construct ceRNA in next step (Fig.
3). The DEGs after intersection are shown in Tables SVIII-SX.
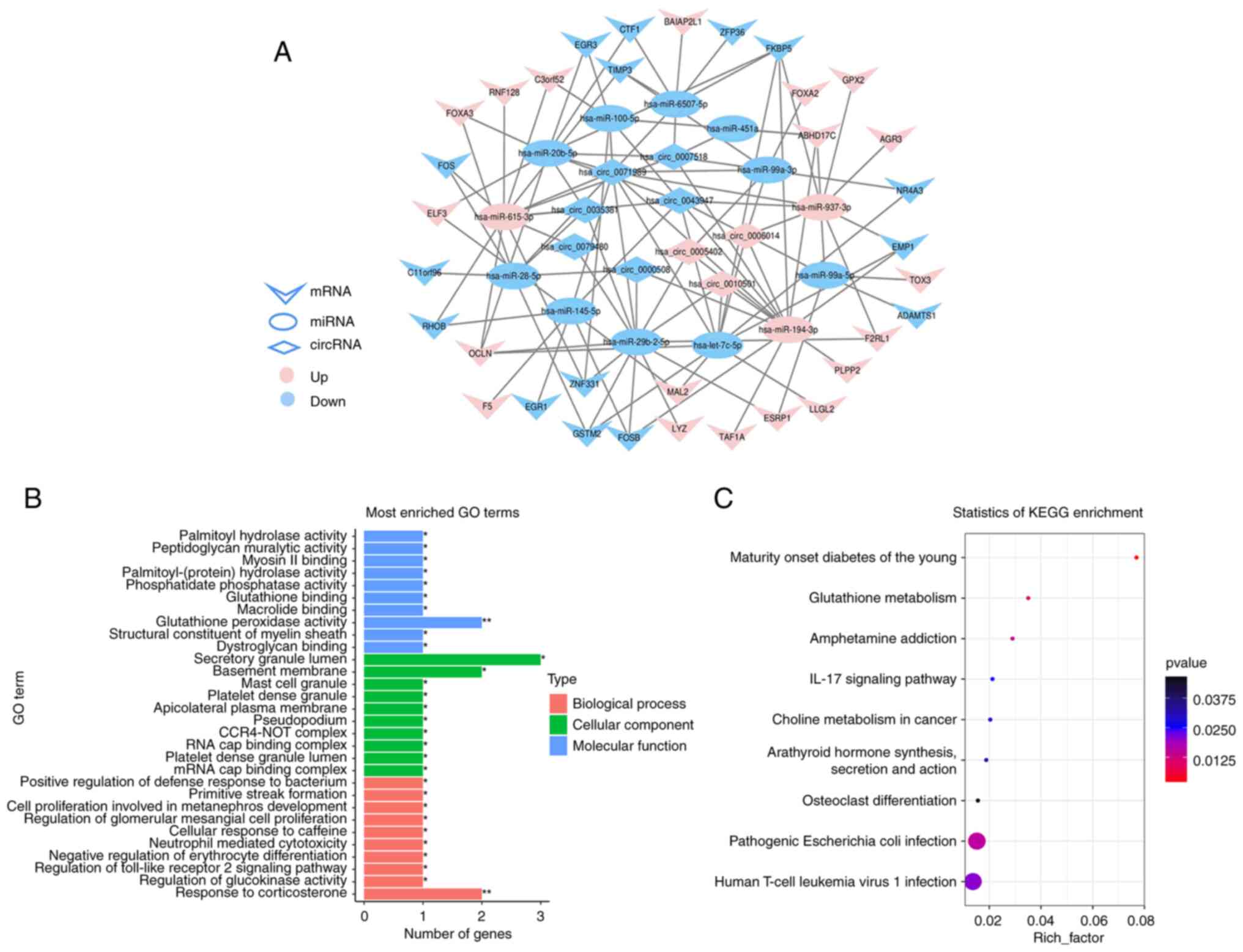
Construction of ceRNA network
The interaction relationship between hub circRNAs,
miRNAs and mRNAs were predicted. Next, the interacting connections
among hub RNAs were imaged by Cytoscape software. The GC ceRNA
network contained 109 edges and 56 nodes, including 13 miRNAs, 9
circRNAs and 34 mRNAs (Fig. 4A),
the details of 56 node RNAs are shown in Table SXI.
Functional annotation of hub
mRNAs
GO and KEGG analyses were performed on node mRNAs
identified in ceRNA network. GO enrichment analysis indicated that
biological processes mainly involved in metabolism, immunity, cell
proliferation and development. For cellular component, it mainly
participated in the formation of transcription complex, platelet
and lateral plasma membrane, etc. Molecular function mainly
involved in the activity of enzymes and the binding of substances
(Fig. 4B). For KEGG, there were 8
significantly enriched pathways (P<0.05), which mainly involved
in metabolism and immune-related pathways, such as glutathione
metabolism, pathogenic Escherichia coli infection and IL-17
signaling pathway, etc. (Fig.
4C).
Diagnostic and prognostic signature
identification
Referencing the mRNAs and miRNAs in ceRNA network,
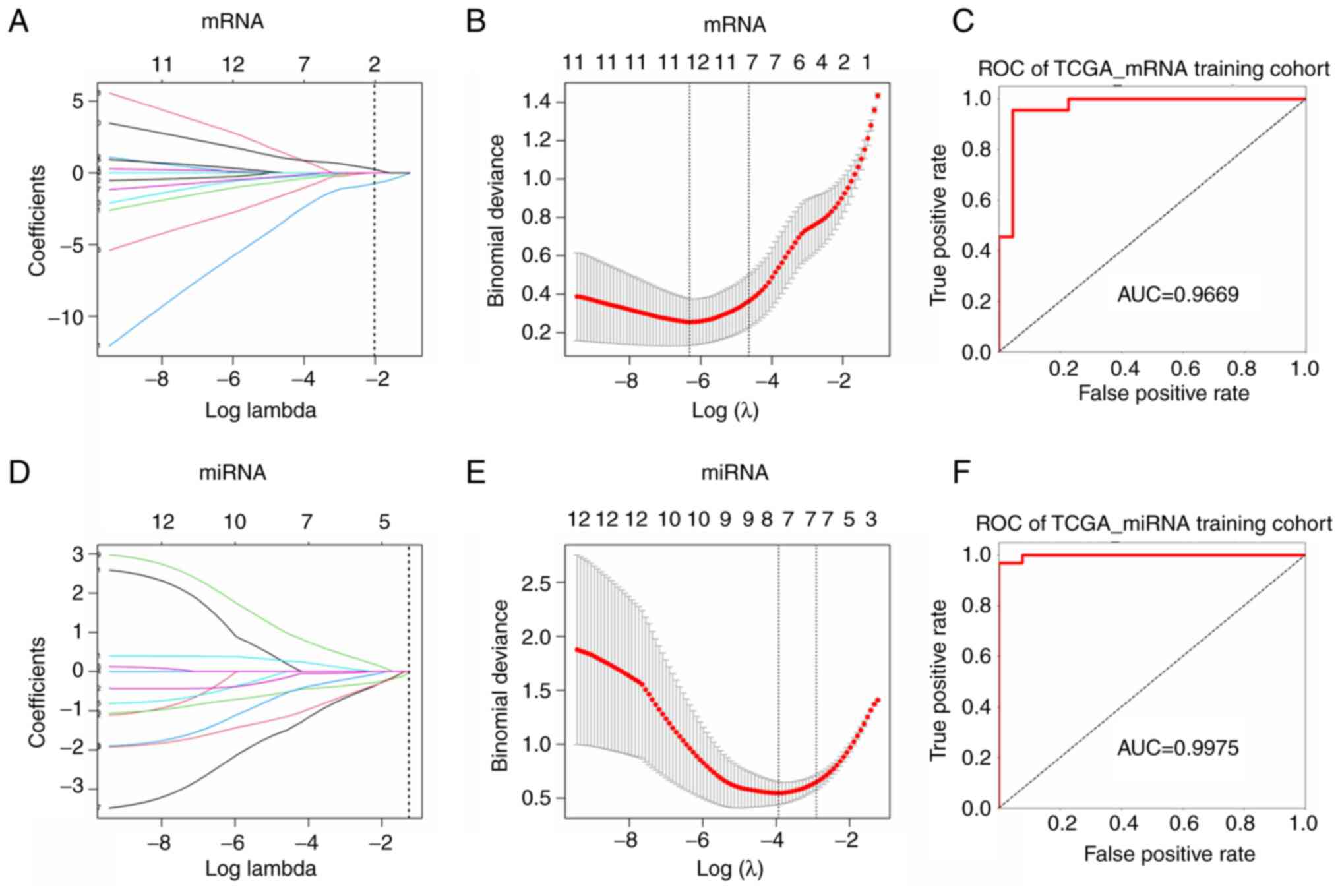
LASSO regression analysis was performed on TCGA to further screen
diagnostic and prognostic signature of mRNA (Fig. 5A and B) and miRNA (Fig. 5D and E). The top 5 mRNAs were CTF1,
FKBP5, RNF128, GSTM2 and ADAMTS1, the top 5 miRNA were miR-145-5p,
miR-615-3p, miR-6507-5p, miR-937-3p and miR-99a-3p. The value of
LASSO regression coefficient is listed in Table V.
| Table V.Least absolute shrinkage and
selection operation regression coefficient absolute value of the
top 5 RNAs. |
Table V.
Least absolute shrinkage and
selection operation regression coefficient absolute value of the
top 5 RNAs.
| A, mRNA |
|---|
|
|---|
| Symbol | β |
|---|
| GSTM2 | −3.513723131 |
| ADAMTS1 | −1.606401793 |
| FKBP5 | 1.462560621 |
| RNF128 | 1.065936307 |
| CTF1 | −0.587182395 |
|
| B,
miRNA |
|
| Symbol | β |
|
| hsa-miR-615-3p | −0.63417029 |
| hsa-miR-937-3p | −0.58439658 |
| hsa-miR-99a-3p | 0.39320561 |
|
hsa-miR-6507-5p | −0.35158869 |
| hsa-miR-145-5p | −0.18756029 |
Diagnostic model construction and
validation
Considering the cancer and para-cancer sample
imbalance in TCGA (375 vs. 32), para-cancer samples were all
retained, and 32 cancer samples were randomly selected from 375,
then 64 samples were randomly assigned to training group (44
samples) and internal validation group (20 samples) according to
ratio of 7:3. The expression data of the top 5 mRNAs from training
group were entered into the training model. Following 5-fold cross
validation, the optimal penalty C was set as 3.0702, gamma was set
as 1.2648 and kernel was set as rbf. Model accuracy (ACC) was 0.91
and area under curve (AUC) value was 0.9669 (Fig. 5C). Then the internal validation
showed ACC was 0.95 and the AUC value was 1.0000 (Fig. S4A). To further validate the
robustness of the model and diagnostic ability of candidate mRNAs,
21 pairs of cancerous and non-cancerous samples from the GSE54129
dataset and all samples from GTEx and TCGA datasets were used as
external data to further verify the model. The results showed ACC
and AUC values of these two datasets are both greater than 0.8
(Fig. S4B and C). The above
results indicated that the 5 characteristic mRNA had strong ability
to distinct gastric cancer from non-cancer.
For miRNA, 41 cancer samples from TCGA GC dataset
(41/436) were selected randomly to balance 41 para-cancer samples,
82 samples were randomly assigned to the training group (57
samples) and the internal validation group (25 samples) according
to ratio of 7:3. The expression data of the 5 signature miRNAs from
the training group were entered into the model for training and the
optimal penalty C was set as 3.0702, gamma was set as 1.2648 and
kernel was set as rbf. Results indicated ACC was 0.93 and AUC value
was 0.9975 (Fig. 5F). Internal
validation group indicated the ACC was 0.92 and AUC value was
0.9733 (Fig. S4D). GSE106817 and
GSE112264 datasets were used as external validation data and
results showed that the ACC and AUC values of these two datasets
are both more than 0.8 (Fig. S4E and
F) (25,26).
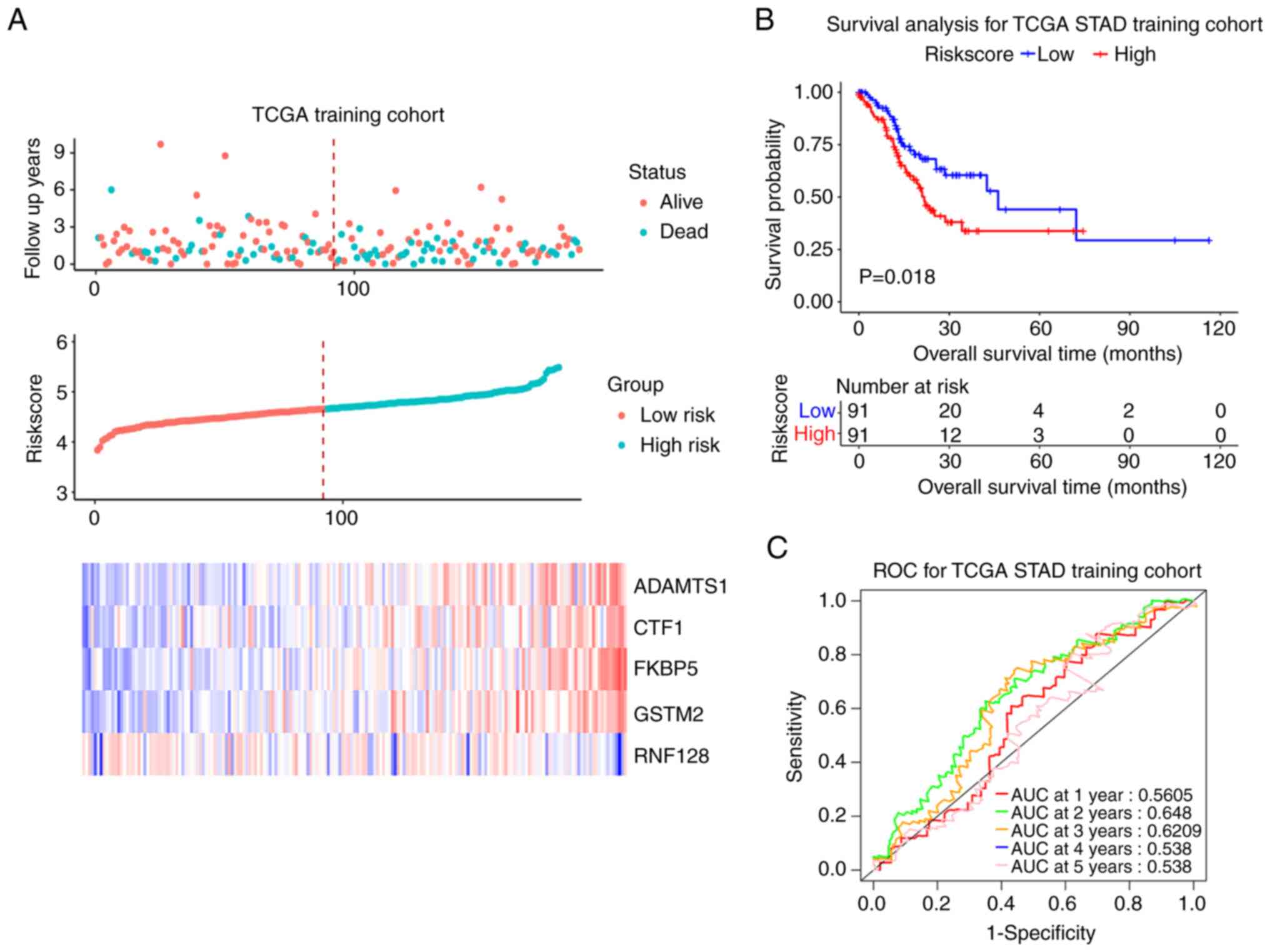
Prognostic model construction and
validation
Patients with both overall survival (OS) information
and expressed data of the 5 candidate mRNAs were selected and
finally 375 cases were recruited. The results of Univariate Cox
regression analysis are summarized in Table VI. The 375 samples were randomly
divided into training and test cohort (188 vs. 187). In training
cohort, the median RiskScore value was taken as the cutoff point
(4.664). Patients with RiskScore >4.664 were defined as a
high-risk group (94 cases) and those with RiskScore ≤4.664 were
defined as a low-risk group. The survival status, RiskScore,
candidate genes expression levels and the survival curves of
training cohort were shown in Fig.
6A. The prognosis of the high-risk group was worse compared
with that of low-risk group (P<0.05; Fig. 6B). The ROC curve showed that the
signature had good prognosis accuracy for 1–5 years in the training
test, the AUC values were >0.5 (Fig. 6C). The same trend was seen from
both the test set and the entire set (P<0.05; AUC >0.5;
Figs. S5 and S6).
| Table VI.Univariate Cox regression analysis of
RNAs. |
Table VI.
Univariate Cox regression analysis of
RNAs.
| Symbol | β | Hazard ratio (95%
CI) | Wald test | P-value |
|---|
| ADAMTS1 | 0.065 | 1.1 (0.94-1.2) | 0.94 | 0.33 |
| CTF1 | 0.054 | 1.1 (0.95-1.2) | 1 | 0.31 |
| FKBP5 | 0.15 | 1.2 (0.99-1.4) | 3.4 | 0.067 |
| GSTM2 | 0.13 | 1.1 (0.97-1.3) | 2.5 | 0.11 |
| RNF128 | 0.056 | 1.1 (0.94-1.2) | 0.93 | 0.34 |
| miR-145-5p | 0.13 | 1.1 (1-1.3) | 6.8 | 0.0091 |
| miR-615-3p | −0.045 | 0.96 (0.88-1) | 1.1 | 0.29 |
| miR-6507-5p | 0.2 | 1.2 (0.93-1.6) | 2.1 | 0.14 |
| miR-937-3p | −0.054 | 0.95 (0.86-1) | 1.2 | 0.28 |
| miR-99a-3p | 0.11 | 1.1 (1-1.2) | 5.9 | 0.015 |
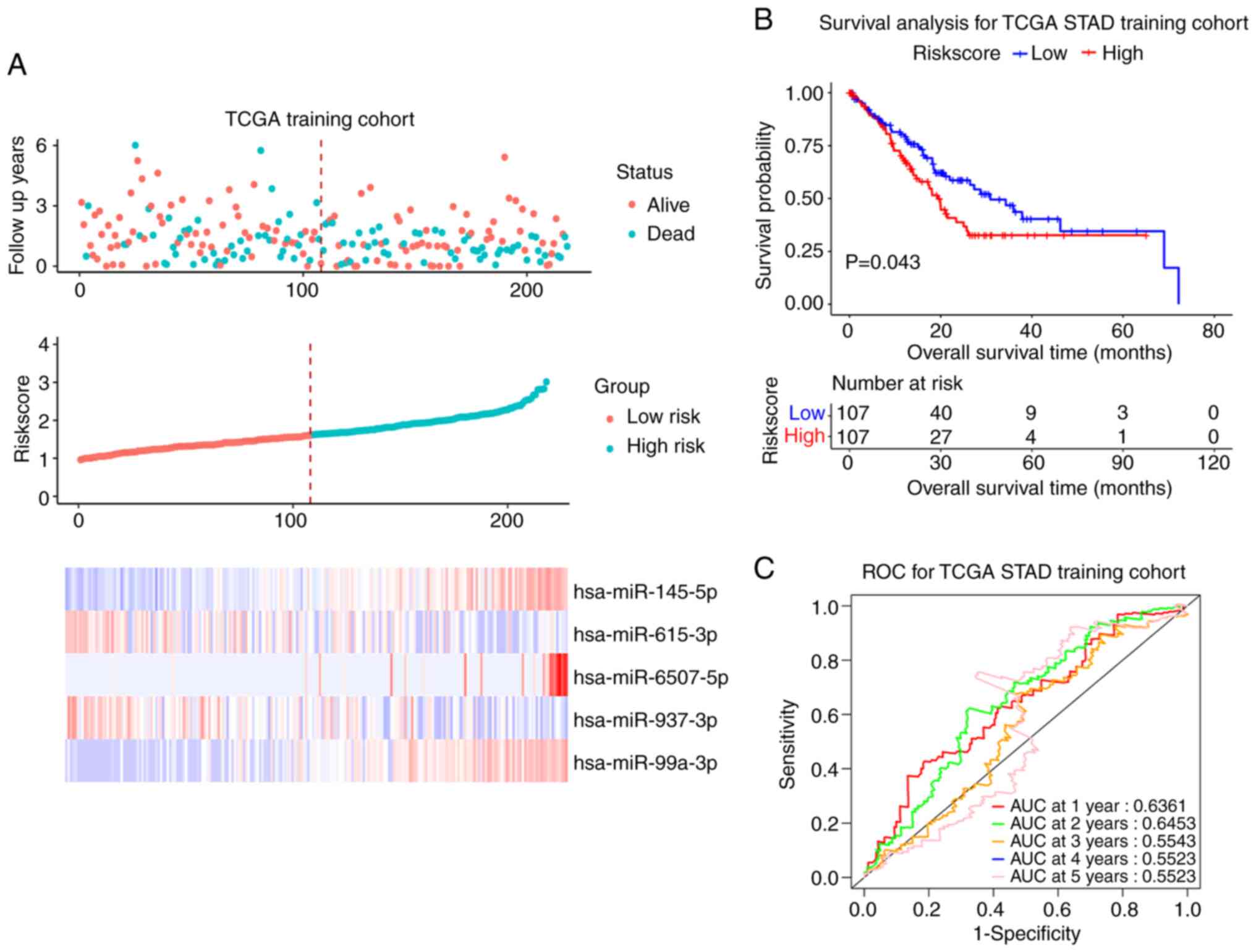
For miRNA, the present study used the same
validation method as for mRNA; 436 patients with OS information
were finally recruited. The results of Univariate Cox regression
analysis are summarized in Table
VI. The 436 samples were randomly divided into the training set
and the test set (218 vs. 218) according to ratio of 1:1. The
median value of RiskScore was 1.621. The training cohort was
divided into a high-risk group (RiskScore >1.621) and a low-risk
group (RiskScore ≤1.621). Survival status, RiskScore value,
candidate miRNAs expression levels and the survival curve in the
training cohort are shown in Fig.
7A. The prognosis of high-risk patients was worse compared with
that of low-risk patients (P<0.05; Fig. 7B). The ROC curve showed that the
miRNA signature had good prognosis accuracy for 1–5 years in the
training test, the AUC values were >0.5 (Fig. 7C). The same conclusion was also
obtained from the test set and the entire sample set (Figs. S7 and S8).
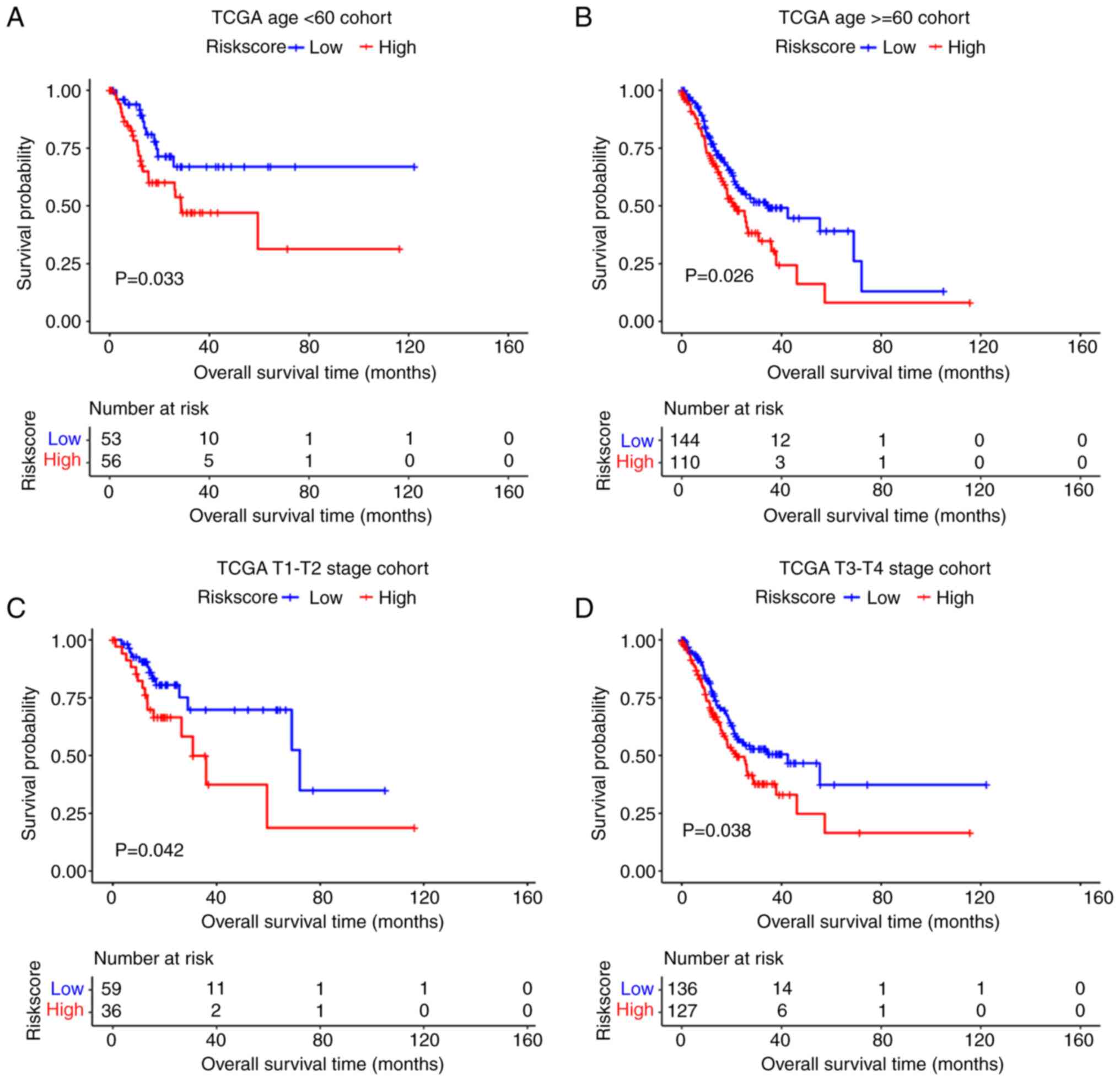
Stratified analysis of the prognostic
signature
The present study investigated the predictive power
of the mRNA prognostic model in different clinical subgroups in
TCGA dataset. The results showed that the prognostic model had good
predictive power t in subgroups <65 and ≥65, T1-T2 and T3-T4
(P<0.05; Fig. 8).
Discussion
Accumulating high-throughput sequencing evidence has
revealed that global transcriptome deregulation is associated with
tumorigenesis and the development of GC. However, the molecular
mechanisms underlying gastric carcinogenesis remain to be
elucidated. The present study explored the circRNA-miRNA-mRNA
interacting axis by constructing ceRNA network. Finally, the
visible network contained 109 edges and 56 nodes, of which 34
mRNAs, 13 miRNAs and 9 circRNAs were included. Several promising
interacting axes, such as circ_0007518/
circ_0071989-miR-6507-5p-CTF1 were further identified and the RNAs,
including circRNA, miRNA and mRNA, identified in these axes
provided a basis and direction for further mechanism research.
Based on the hub RNAs involved in ceRNA, LASSO
regression analysis was performed to screen the prediction model of
mRNA signature and miRNA signature. This indicated that both the 5
mRNA-based signature (CTF1, FKBP5, RNF128, GSTM2 and ADAMTS1) and 5
miRNA-based signature (miR-145-5p, miR-615-3p, miR-6507-5p,
miR-937-3p and miR-99a-3p) had good prediction capacity of
diagnosis and prognosis for GC patients. The use of LASSO
regression analyses allowed for more automated setting of weights
to zero, which was needed for this high-dimensional data.
Additionally, LASSO allowed for easy interpretation of the data
that enabled the present study to screen quickly for the most
crucial information in the model (27,28).
Yan et al (29)
successfully built a more extensive ceRNA network for
hepatocellular carcinoma and identified 4 gene-based signatures
(PBK, CBX2, CLSPN and CPEB 3) using a LASSO regression model, which
predicted the overall survival of hepatocellular carcinoma
effectively. Li et al (30)
also used LASSO regression analysis to screen an immune-related
prognostic signature involving 24 genes to predict the OS and
immune status in colorectal cancer, which was conducive to better
stratification and more precise immunotherapy for patients.
Only the CTF1 gene identified in this cohort were
reported in GC research, Pan et al (31) reported that CTF1 combining with
BTN3A3 and ADA2 genes as a prognostic model to predict the survival
state of GC patients with fluorouracil-bases chemotherapy and help
clinicians develop personalized treatment. By contrast, studies on
the pathogenesis of GC concerning the CTF1 gene have not been
reported. The other mRNAs identified in the present study that have
been reported in studies not related to GC include FK506 binding
protein 5 (FKBP5); a regulatory protein of the
hypothalamic-pituitary-adrenal (HPA) axis, which mainly has
functions in various stress-related psychiatric disorders and which
is seldom reported in GC (32). In
in vitro experiments, Zou et al (33) demonstrated that GSTM2 might
correlated with the cisplatin resistance of GC cells. A disintegrin
and metalloprotease with thrombospondin motifs (ADAMTS) is a family
of 19 secreted membrane-anchored proteases, Kilic et al
(34) reported that ADAMTS1
protease was highly expressed in GC and nodal metastases,
indicating important role in carcinogenesis and lymphatic
metastasis, however the specific regulatory mechanism of ADAMTS1
has not been studied.
In the cohort of the present study, the combination
of 5 miRNAs identified could distinguish GC patients from the
healthy controls and predict survival when patients were placed
into high-risk or low-risk groups according to their risk value.
miRNAs are small endogenous non-coding regulatory RNAs, which take
a vital part in the progression of tumor by depredating the target
mRNA, while circRNA functions as an miRNA sponge to regulate
selective splicing, expression and translation of host genes
through endogenous competing miRNA (35–38).
In GC, multiple miRNAs are differentially expressed and showed
evidence of a function in tumorigenesis. Zhong et al
(39) revealed that the expression
levels of miR-145-5p are significantly decreased in GC cells and
correlate with the expression of KCNQ1OT1 in tumors, which promotes
disease progression through the miR-145-5p/ARF6 axis. Wang et
al (40) report that
miR-615-3p promotes GC proliferation and migration by deregulating
CELF2 expression in vitro and in vivo. The visible
competing network in the present study displayed the interacting
control between hub RNAs and the specific regulatory link between
the mentioned RNAs have not been reported as so far, which will be
the direction for further research.
miRNAs are found in serum, plasma and other body
fluids because of their ability to avoid degradation, therefore,
they become an ideal noninvasive biomarker to diagnose and predict
survival rates. The 5-miRNA signature reported in the present study
has promising clinical application. The miRNA biomarker panel assay
was also found by other studies, So et al (11) recently developed a valid risk
assessment tool composed of 12 serum miRNAs able to detect GC.
Similarly, Japanese studies by Abe et al (41) developed a novel combination of four
serum miRNAs (miR-4257, miR-6785-5p, miR-187-5p and miR-5739) to
discriminate early GC from normal tissue lesions with high
accuracy. The present study performed a similar analysis on circRNA
with the expectation of constructing a prediction model of it, but
the limited sample size of the original circRNA datasets hampered
the panel which did not display strong diagnostic and prognostic
capacity. Although the present study designed internal and external
validations, it also had some limitations, including the fact that
all conclusions were obtained from already published bioinformatic
data and the lack of in vitro validation experiments; the
functional mechanism study was the main direction of the present
study.
In conclusion, the present study constructed ceRNA
network of gastric cancer using circRNA, miRNA and mRNA public
datasets, and the interaction between hub RNAs provided the basis
for the further molecular pathogenesis research. In addition, the
present study developed and validated 5 mRNA-based signature and 5
miRNA-based signature that have the potential to be useful tools to
diagnose GC in patients and to predict their survival rates.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the Science Foundation of Guangzhou
First People's Hospital (grant no. M2019002).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WD, HD designed the study and confirmed the
authenticity of all the raw data. WD drafted the manuscript,
tables, and figures. LW, XL, LC, GL performed the bioinformatic
analysis. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Ethics Committee of Guangzhou First People's Hospital (approval no.
K-2020-010-01)
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Colombet M, Soerjomataram I,
Mathers C, Parkin DM, Piñeros M, Znaor A and Bray F: Estimating the
global cancer incidence and mortality in 2018: GLOBOCAN sources and
methods. Int J Cancer. 144:1941–1953. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young E, Philpott H and Singh R:
Endoscopic diagnosis and treatment of gastric dysplasia and early
cancer: Current evidence and what the future may hold. World J
Gastroenterol. 27:5126–5151. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang H, Leung C, Saito E, Katanoda K, Hur
C, Kong CY, Nomura S and Shibuya K: Effect and cost-effectiveness
of national gastric cancer screening in Japan: A microsimulation
modeling study. BMC Med. 18:2572020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suh Y, Lee J, Woo H, Shin D, Kong SH, Lee
HJ, Shin A and Yang HK: National cancer screening program for
gastric cancer in Korea: Nationwide treatment benefit and cost.
Cancer. 126:1929–1939. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smyth EC, Nilsson M, Grabsch HI, van
Grieken NC and Lordick F: Gastric cancer. Lancet. 396:635–648.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Sethi NS, Hinoue T, Schneider BG,
Cherniack AD, Sanchez-Vega F, Seoane JA, Farshidfar F, Bowlby R,
Islam M, et al: Comparative molecular analysis of gastrointestinal
adenocarcinomas. Cancer Cell. 33:721–735.e728. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shimozaki K, Nakayama I, Takahari D,
Kamiimabeppu D, Osumi H, Wakatsuki T, Ooki A, Ogura M, Shinozaki E,
Chin K and Yamaguchi K: A novel clinical prognostic index for
patients with advanced gastric cancer: Possible contribution to the
continuum of care. ESMO Open. 6:1002342021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu C, Ren C, Han J, Ding Y, Du J, Dai N,
Dai J, Ma H, Hu Z, Shen H, et al: A five-microRNA panel in plasma
was identified as potential biomarker for early detection of
gastric cancer. Br J Cancer. 110:2291–2299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen D, Ping S, Xu Y, Wang M, Jiang X,
Xiong L, Zhang L, Yu H and Xiong Z: Non-coding RNAs in gastric
cancer: From malignant hallmarks to clinical applications. Front
Cell Dev Biol. 9:7320362021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
So J, Kapoor R, Zhu F, Koh C, Zhou L, Zou
R, Tang YC, Goo PC, Rha SY, Chung HC, et al: Development and
validation of a serum microRNA biomarker panel for detecting
gastric cancer in a high-risk population. Gut. 70:829–837. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Imaoka H, Toiyama Y, Okigami M, Yasuda H,
Saigusa S, Ohi M, Tanaka K, Inoue Y, Mohri Y and Kusunoki M:
Circulating microRNA-203 predicts metastases, early recurrence, and
poor prognosis in human gastric cancer. Gastric Cancer. 19:744–753.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Madar V and Batista S: FastLSU: A more
practical approach for the Benjamini-Hochberg FDR controlling
procedure for huge-scale testing problems. Bioinformatics.
32:1716–1723. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rehmsmeier M, Steffen P, Hochsmann M and
Giegerich R: Fast and effective prediction of microRNA/target
duplexes. RNA. 10:1507–1517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sticht C, De La Torre C, Parveen A and
Gretz N: miRWalk: An online resource for prediction of microRNA
binding sites. PLoS One. 13:e02062392018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. Omics. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bac J, Mirkes EM, Gorban AN, Tyukin I and
Zinovyev A: Scikit-Dimension: A python package for intrinsic
dimension estimation. Entropy (Basel). 23:13682021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Daberdaku S and Ferrari C: Antibody
interface prediction with 3D Zernike descriptors and SVM.
Bioinformatics. 35:1870–1876. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baldi P, Brunak S, Chauvin Y, Andersen CA
and Nielsen H: Assessing the accuracy of prediction algorithms for
classification: An overview. Bioinformatics. 16:412–424. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiong Y, Wang R, Peng L, You W, Wei J,
Zhang S, Wu X, Guo J, Xu J, Lv Z and Fu Z: An integrated lncRNA,
microRNA and mRNA signature to improve prognosis prediction of
colorectal cancer. Oncotarget. 8:85463–85478. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yokoi A, Matsuzaki J, Yamamoto Y, Yoneoka
Y, Takahashi K, Shimizu H, Uehara T, Ishikawa M, Ikeda SI, Sonoda
T, et al: Int extracellular microRNA profiling for ovarian cancer
screening. Nat Commun. 9:43192018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Urabe F, Matsuzaki J, Yamamoto Y, Kimura
T, Hara T, Ichikawa M, Takizawa S, Aoki Y, Niida S, Sakamoto H, et
al: Large-scale circulating microRNA profiling for the liquid
biopsy of prostate cancer. Clin Cancer Res. 25:3016–3025. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ren S, Huang S, Ye J and Qian X: Safe
feature screening for generalized LASSO. IEEE Trans Pattern Anal
Mach Intell. 40:2992–3006. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Waldmann P, Ferenčaković M, Mészáros G,
Khayatzadeh N, Curik I and Sölkner J: AUTALASSO: An automatic
adaptive LASSO for genome-wide prediction. BMC Bioinformatics.
20:1672019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan Y, Lu Y, Mao K, Zhang M, Liu H, Zhou
Q, Lin J, Zhang J, Wang J and Xiao Z: Identification and validation
of a prognostic four-genes signature for hepatocellular carcinoma:
Integrated ceRNA network analysis. Hepatol Int. 13:618–630. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li M, Wang H, Li W, Peng Y, Xu F, Shang J,
Dong S, Bu L, Wang H and Wei W: Identification and validation of an
immune prognostic signature in colorectal cancer. Int
Immunopharmacol. 88:1068682020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pan J, Dai Q, Xiang Z, Liu B and Li C:
Three biomarkers predict gastric cancer patients' susceptibility to
fluorouracil-based chemotherapy. J Cancer. 10:2953–2960. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang JI, Chung HC, Jeung HC, Kim SJ, An SK
and Namkoong K: FKBP5 polymorphisms as vulnerability to anxiety and
depression in patients with advanced gastric cancer: A controlled
and prospective study. Psychoneuroendocrinology. 37:1569–1576.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zou M, Hu X, Xu B, Tong T, Jing Y, Xi L,
Zhou W, Lu J, Wang X, Yang X and Liao F: Glutathione S-transferase
isozyme alpha 1 is predominantly involved in the cisplatin
resistance of common types of solid cancer. Oncol Rep. 41:989–998.
2019.PubMed/NCBI
|
|
34
|
Kilic M, Aynekin B, Kara A, Icen D and
Demircan K: Differentially regulated ADAMTS1, 8, and 18 in gastric
adenocarcinoma. Bratisl Lek Listy. 118:71–76. 2017.PubMed/NCBI
|
|
35
|
Liu L, Tian YC, Mao G, Zhang YG and Han L:
MiR-675 is frequently overexpressed in gastric cancer and enhances
cell proliferation and invasion via targeting a potent anti-tumor
gene PITX1. Cell Signal. 62:1093522019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bartel D: Metazoan microRNAs. Cell.
173:20–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Slack FJ and Chinnaiyan AM: The role of
non-coding RNAs in oncology. Cell. 179:1033–1055. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dragomir M and Calin GA: Circular RNAs in
cancer-lessons learned from microRNAs. Front Oncol. 8:1792018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhong X, Wen X, Chen L, Gu N, Yu X and Sui
K: Long non-coding RNA KCNQ1OT1 promotes the progression of gastric
cancer via the miR-145-5p/ARF6 axis. J Gene Med. 23:e33302021.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang J, Liu L, Sun Y, Xue Y, Qu J, Pan S,
Li H, Qu H, Wang J and Zhang J: miR-615-3p promotes proliferation
and migration and inhibits apoptosis through its potential target
CELF2 in gastric cancer. Biomed Pharmacother. 101:406–413. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Abe S, Matsuzaki J, Sudo K, Oda I, Katai
H, Kato K, Takizawa S, Sakamoto H, Takeshita F, Niida S, et al: A
novel combination of serum microRNAs for the detection of early
gastric cancer. Gastric Cancer. 24:835–843. 2021. View Article : Google Scholar : PubMed/NCBI
|